

Abstract
Background
Doyne honeycomb retinal dystrophy (DHRD)/malattia leventinese (ML) is an autosomal dominant, progressive retinal disorder characterized by massive central retinal drusen often partly coalescent forming a characteristic honeycomb‐like pattern. Debut of vision loss often occurs in early to mid‐adulthood, and the degree varies. A single variant in EFEMP1: c.1033C>T (R345W) has been identified as the cause in all cases.
Methods
Following DNA isolation, exome sequencing was performed in seven genes associated with flecked retina. Direct sequencing was used for variant verification.
Results
We report the first Scandinavian case of molecular genetically verified DHRD/ML: a 57‐year‐old woman debuting with vision loss and metamorphopsia. On both eyes, ophthalmological findings included massive hard drusen in the macular region and nasal to the optic disc as well as macular hyperpigmentation. Secondary choroidal neovascularizations were identified on both eyes, and anti‐vascular endothelial growth factor was administered, without effect.
Conclusion
Molecular genetic investigation revealed heterozygosity for the known pathogenic missense variant in EFEMP1: c.1033C>T (R345W) previously reported in relation to DHRD/ML. Family history revealed no other cases of similar visual impairment suggesting a de novo mutation. Furthermore, there was no correlation between the unique DHRD/ML haplotypes reported in the literature and our patient.
Keywords: Doyne honeycomb retinal dystrophy, EFEMP1, Malattia leventinese
To the best of our knowledge, this case represents the first molecular genetically verified case of DHRD/ML in Scandinavia. A haplotype comparison to the originally described Swiss R345W haplotype suggest a de novo mutation in our index.
1. INTRODUCTION
Doyne honeycomb retinal dystrophy (DHRD) and malattia leventinese (ML) refer to autosomal dominant progressive retinal diseases characterized by the accumulation of macular and peripapillary yellow‐white deposits (drusen) beneath the retinal pigment epithelium in Bruch's membrane (Doyne, 1899; Evans et al., 1997; Piguet et al., 1995). With age, drusen increase in size and number, often to form a characteristic honeycomb‐like pattern (Doyne, 1899; Evans et al., 1997; Piguet et al., 1995; Scarpatetti et al., 1978). Massive drusen, geographic retinal atrophy, and macular hyperpigmentation eventually cause visual symptoms such as decreased visual acuity, metamorphopsia, photophobia, and paracentral scotoma often debuting around age of 40–50 years (Evans et al., 1997; Heon et al., 1996; Scarpatetti et al., 1978). Complicating secondary choroidal neovascularizations (CNVs) and hemorrhage can lead to rapid progression (Pager et al., 2001; Piguet et al., 1995; Sohn et al., 2011; Zech et al., 1999).
Complete penetrance in DHRD/ML and a great variability in phenotype/severity have been described (Evans et al., 1997; Marmorstein, 2004; Takeuchi et al., 2010).
DHRD was first reported by Doyne in 1899 (Doyne, 1899), and ML was initially described in patients with familial drusen living in the Levantine Valley in Switzerland in 1925 (Vogt, 1925). An autosomal dominant inheritance and the overlapping phenotype suggested that the two retinal diseases represented the same disorder (Forni & Babel, 1962; Waardenburg, 1948). However, subsequent funduscopic descriptions revealed differences in composition and distribution of retinal drusen, for example, with drusen appearing in a more radial pattern in ML, which was not a prominent feature in DHRD (Evans et al., 1997; Piguet et al., 1995). Despite these small discrepancies possibly explained by different genetic backgrounds and/or environmental influences, a single pathogenic variant in EFEMP1 (MIM 601548): c.1033C>T (R345W) was found by Stone et al. in 1999, to be the cause of both conditions (Stone et al., 1999). Consequently, in the following, they will be referred to as the same disease.
The EFEMP1 gene is localized on chromosome 2p16.1 and encodes the epidermal growth factor‐containing fibrillin‐like extracellular matrix protein 1 (Efemp1, fibulin 3), the function of which remains to be fully elucidated. Protein misfolding and basal deposit formation in retinal pigment epithelium containing Efemp1 have been observed in EFEMP1 R345W affected and is believed to be responsible for drusen formation (Fu et al., 2007; Marmorstein et al., ,2002, 2007; Roybal et al., 2005). In 2005, Roybal et al. discovered that the R345W variant led to increased expression of vascular endothelial growth factor (VEGF), known to play a pivotal role in the development of complicating CNVs also observed in DHRD/ML. Furthermore, dense confluent drusen formation can possibly induce hypoxia, which is known to stimulate VEGF expression in retinal pigment epithelium promoting CNV development (Mousa et al., 1999).
The disease‐causing mechanism of the EFEMP1 R345W mutation is not thought to be caused by a loss‐of‐function effect based on the complete absence of histopathological features compatible with DHRD/ML in EFEMP1 knockout mouse (McLaughlin et al., 2007). In EFEMP1 knockout mouse, however, early aging and multiple hernias have been observed, suggesting connective tissue disorder (McLaughlin et al., 2007). This is in contrast to EFEMP1 knock‐in mouse carrying the R345W mutation, where no other apparent extraretinal pathology has been described (Fu et al., 2007; Marmorstein et al., 2007). This is supported by current knowledge on DHRD/ML where disease is limited to the retina. In a few case reports regarding pronounced connective tissue disorder, biallelic variants in EFEMP1 with suspected loss of function have been described as the cause (Bizzari et al., 2020; Driver et al., 2020). In both these cases, several phenotypic similarities with Marfan syndrome exist; however, EFEMP1 is currently not associated with any connective tissue disorder.
In the 39 families (Switzerland, the United States, and Australia) studied by Stone et al. (1999), the absence of de novo mutations and areas with loss of heterozygosity (complete sharing of alleles of four intragenic EFEMP1 polymorphisms) suggested a common ancestor in all affected individuals. Since 1999, a three‐digit number of genetically verified DHRD/ML has been reported in populations around the world, most cases being in the United Kingdom, the United States, Australia, Switzerland, and neighboring countries (Cusumano et al., 2019; Gregory et al., 1996; Heon et al., 1996; Matsumoto & Traboulsi, 2001; Narendran et al., 2005; Serra et al., 2017; Sohn et al., 2011).
2. MATERIALS AND METHODS
2.1. Clinical report
We present the molecular genetic findings of a 57‐year‐old Scandinavian woman of non‐consanguineous Danish Caucasian parents debuting with vision loss and distorted vision primarily on the left eye at age of 47 years. Corrected visual acuity was reduced by 0.8/0.5 cc, respectively. On the left eye, metamorphopsia was reported using an Amsler grid. Ophthalmological examination revealed radially oriented massive hard drusen partly coalescent on both eyes in the macular region and nasal to the disc as well as macular hyperpigmentation (Figure 1). Optical coherence tomography (OCT) on both eyes show foveal drusen and retinal atrophy on the left eye (Figure 2). Fluorescein angiography (FLU‐A) and indocyanine green angiography (ICG‐A) showed no signs of secondary CNVs. The patient was clinically diagnosed with DHRD/ML and followed annually with stable conditions.
FIGURE 1.
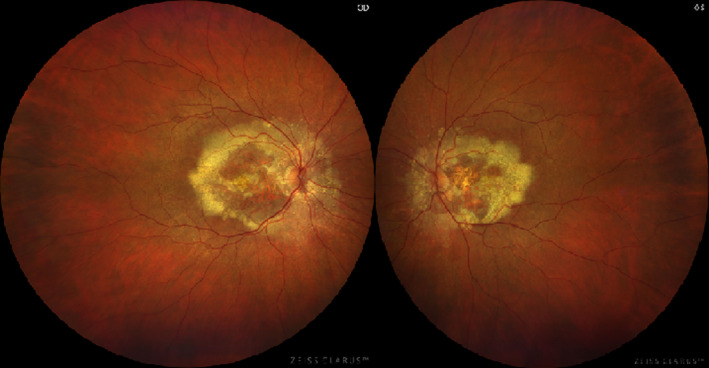
Fundus photography show massive, partly coalescent drusen in the macular area and nasal to the optic disc on both eyes. OD = right eye. OS = left eye
FIGURE 2.
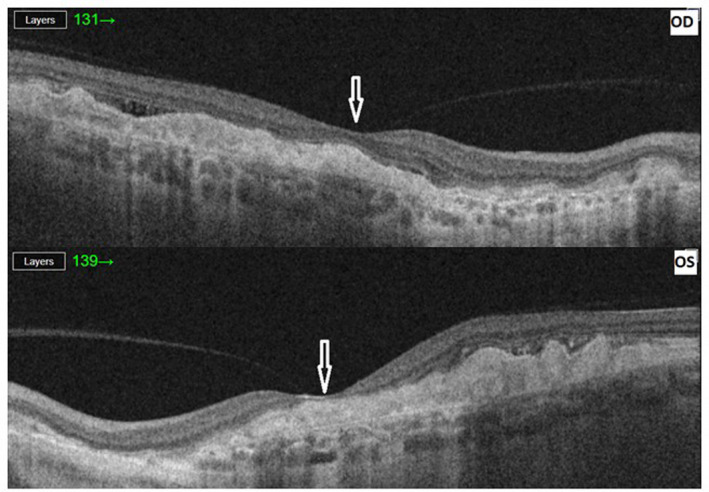
Optical coherence tomography (OCT). OCT on both eyes show foveal drusen and retinal atrophy on the left eye. Arrowheads indicate the location of fovea. OD = right eye. OS = left eye
Ten years later, reduced visual acuity (0.2/0.63 cc) and increased atrophy with subretinal fluid were observed. FLU‐A/ICG‐A indicated secondary CNVs on both eyes, and anti‐vascular endothelial growth factor (anti‐VEGF) was administrated in three consecutive treatments on both eyes, without effect. A follow‐up FLU‐A indicated, however, that the condition was stable.
Family history revealed no other cases of reduced vision, and both the patient's parents are currently in their nineties. The patient's 45‐year‐old younger sister underwent ophthalmological examination with normal findings and was thus not genotyped.
2.2. Editorial policies and ethical considerations
Oral informed consent was obtained by the patient.
2.3. Molecular evaluation and verification
Genomic DNA was obtained from blood leukocytes using standard procedures.
The sequencing analysis of seven genes associated with flecked retina was done by Amplexa genetics (Amplexa FleckRet7 Panel). A heterozygous mutation in EFEMP1 (NM_001039348.3) c.1033C>T (R345W) was detected (Figure 3). No pathogenic variants were found in the rest of the sequenced genes. The detected mutation was verified by direct sequencing analysis of exon 10 of the EFEMP1 gene using the following primers: forward primer 5′‐TGCTCTCACACCTCCTTCCT‐3′ and reverse primer, 5′‐CACATGGCATTTGAGACTGG‐3′. The PCR amplification was performed according to the following protocol: 95°C for 2 min, followed by 30 cycles of 95°C for 15 s, 55°C for 30 s, 72°C for 30 s, and a final step of extension for 5 min at 72°C. Employed microsatellite markers included D2S378, D2S2352, and intragenic SNPs rs1346786, rs3791676, and rs1430197 (primers are available upon request) (Fu et al., 2007; Stone et al., 1999). For sequencing and genotyping, samples from patient and controls were amplified by PCR and loaded on an ABI 3100 Genetic Analyzer System following manufacturer's instructions. Fragment analysis was done using GeneMarker software 2.6.4 (SoftGenetics, LLC).
FIGURE 3.
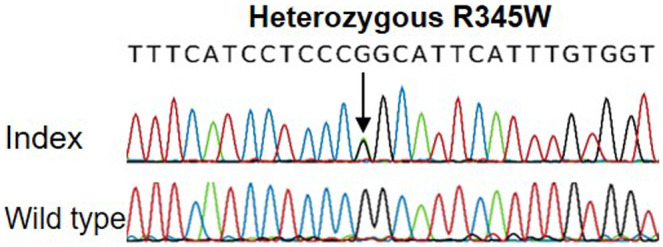
Partial sequence electropherograms of exon 10 of EFEMP1 from our index patient and a control individual (wild type)
3. RESULTS
A known pathogenic variant NM_001039348 (EFEMP1): c.1033C>T (R345W) previously seen in relation to DHRD/ML was identified in a heterozygote state in the index. No other causative variants were identified.
Haplotype analysis indicated a different haplotype compared with the original study by Stone et al. (1999) (Table 1), suggesting that the mutation arose independently, although predictive testing in the family was not performed and parental data therefore remain unknown.
TABLE 1.
Haplotype data for two microsatellite markers flanking the EFEMP1 gene and three SNPs within the EFEMP1 gene
| Index | Swiss R345W haplotype | Wild type | |
|---|---|---|---|
| D2S2352 a | 321 | 319/321 | 315/321 |
| R345W | M/+ | M/+ | +/+ |
| RS1346786 | T/C | T/C | C/C |
| RS3791676 | C/C | C/T | T/T |
| RS1430197 | A/G | A/G | G/G |
| D25378 a | 368/381 | 370 | 370/380 |
The index patient exhibits a different disease haplotype from the previously described Swiss R345W haplotype. + = wild type; M = p.R345W mutation.
Microsatellite markers analysis are presented here by length of generated fragments (bp).
4. DISCUSSION
To the best of our knowledge, this is the first case of molecular genetically verified DHRD/ML in Scandinavia. Heterozygosity for the known pathogenic EFEMP1 variant R345W was shown to be the cause of visual impairment and DHRD/ML in this 57‐year‐old Caucasian Scandinavian woman. In our case, no family history of DHRD/ML was reported. We then speculated that DHRD/ML in our index therefore had to be explained either by a great variability in disease phenotype with a mildly affected and undiagnosed parent, different paternal origin, or a R345W de novo mutation in our index. Since parental gene testing was not possible, a haplotype analysis was conducted in our index patient.
As previously mentioned, Stone et al. (1999) found evidence of a common ancestor in all affected individuals. However, in 2007, Fu et al. reported a consanguineous Indian family with DHRD/ML where the R345W mutation segregated with a different disease haplotype indicating that the mutation arose independently. Another unique haplotype was later in 2010 reported in a Japanese DHRD family (Takeuchi et al., 2010). In order to investigate a possible common ancestor of the mutation in our index patient, haplotype analysis was performed, revealing a haplotype different from the Swiss R345W haplotype (Stone et al., 1999), thus indicating a de novo mutation with the proviso of absence of confirmatory data from the patient's parents.
DHRD/ML has been of special interest to researchers due to several similarities with age‐related macular degeneration (AMD), a leading cause of blindness in developed countries (Edwards et al., 1998; Wong et al., 2014). Retinal drusen is a hallmark in both DHRD/ML and AMD, although differences in extracellular matrix proteins have been reported (Sohn et al., 2015). To date, no mutation in EFEMP1 has been reported in AMD patients (Stone et al., 1999).
No general recommendations regarding neither surgical nor medical treatment in DHRD/ML exist. Current research is primarily based on interventional case reports where minor beneficial effects of treatment with argon laser (Lenassi et al., 2013) and subthreshold nanolaser (Cusumano et al., 2019) have been proposed. Based on the experience in treating neovascular AMD (Lin & Rosenfeld, 2007), administration of anti‐VEGF (bevacizumab) has been tried in DHRD/ML when complicated by CNV development, suggesting some beneficial effect (Sohn et al., 2011). In this case, three consecutive treatments with anti‐VEGF (Eylea) were administrated in both eyes; however, no increase in visual acuity was found, and no change in the retinal condition was registered. The need for additional clinical trials with long‐term follow‐up is therefore greatly needed in order to establish a potential effect of the previously mentioned and other treatments in DHRD/ML. This, however, is made difficult by the low number of affected individuals.
5. CONCLUSION
To the best of our knowledge, this case represents the first molecular genetically verified case of DHRD/ML in Scandinavia. Stone et al. (1999) found evidence of a common ancestor in all cases in the original study; however, haplotype comparison suggested a de novo mutation in our index, although parental data remain unknown.
On both eyes, our index developed complicating CNVs, and treatment with anti‐VEGF was administered, without a beneficial effect.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
INS and MBP drafted the manuscript. Laboratory work including haplotype analysis and result interpretation were conducted by IBL and HO. MBP, LPK, and SKA provided clinical input. All authors reviewed the results and contributed to critical revision of the manuscript.
DATA AVAILIBILITY STATEMENT
The data that support the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
ACKNOWLEDGEMENTS
We would like to thank the patient involved. We acknowledge Amplexa Genetics for the sharing of exome data of our index for the purpose of determining disease haplotype. In addition to this, Francis L. Munier and Daniel Schorderet are acknowledged for supplying DNA from one of the originally affected DHRD/ML patients from the Levantine Valley in Switzerland (Stone et al., 1999), which enabled a comparison of the haplotype of our case to the Swiss DHRD/ML haplotype.
Funding information
The Obel Family Foundation grant number 27915.
REFERENCES
- Bizzari, S. , El‐Bazzal, L. , Nair, P. , Younan, A. , Stora, S. , Mehawej, C. , El‐Hayek, S. , Delague, V. , & Mégarbané, A. (2020). Recessive marfanoid syndrome with herniation associated with a homozygous mutation in Fibulin‐3. European Journal of Medical Genetics, 63, 103869. 10.1016/j.ejmg.2020.103869 [DOI] [PubMed] [Google Scholar]
- Cusumano, A. , Falsini, B. , Giardina, E. , Cascella, R. , Sebastiani, J. , & Marshall, J. (2019). Doyne honeycomb retinal dystrophy – Functional improvement following subthreshold nanopulse laser treatment: A case report. J Med Case Rep, 13(1), 5. 10.1186/s13256-018-1935-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyne, R. (1899). Peculiar condition of choroiditis occuring in several members of the same family. Transactions of the Ophthalmological Societies of the United Kingdom, 19, 71. [Google Scholar]
- Driver, S. G. W. , Jackson, M. R. , Richter, K. , Tomlinson, P. , Brockway, B. , Halliday, B. J. , Markie, D. M. , Robertson, S. P. , & Wade, E. M. (2020). Biallelic variants in EFEMP1 in a man with a pronounced connective tissue phenotype. European Journal of Human Genetics, 28(4), 445–452. 10.1038/s41431-019-0546-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, A. O. , Klein, M. L. , Berselli, C. B. , Hejtmancik, J. F. , Rust, K. , Wirtz, M. K. , Weleber, R. G. , & Acott, T. S. (1998). Malattia leventinese: Refinement of the genetic locus and phenotypic variability in autosomal dominant macular drusen. American Journal of Ophthalmology, 126(3), 417–424. 10.1016/s0002-9394(98)00097-x [DOI] [PubMed] [Google Scholar]
- Evans, K. , Gregory, C. Y. , Wijesuriya, S. D. , Kermani, S. , Jay, M. R. , Plant, C. , & Bird, A. C. (1997). Assessment of the phenotypic range seen in Doyne honeycomb retinal dystrophy. Archives of Ophthalmology, 115(7), 904–910. 10.1001/archopht.1997.01100160074012 [DOI] [PubMed] [Google Scholar]
- Forni, S. , & Babel, J. (1962). Clinical and histological study of the disease of Leventina. Disease belonging to the group of hyaline degenerescences of the posterior pole. Ophthalmologica, 143, 313–322. 10.1159/000304248 [DOI] [PubMed] [Google Scholar]
- Fu, L. I. , Garland, D. , Yang, Z. , Shukla, D. , Rajendran, A. , Pearson, E. , Stone, E. M. , Zhang, K. , & Pierce, E. A. (2007). The R345W mutation in EFEMP1 is pathogenic and causes AMD‐like deposits in mice. Human Molecular Genetics, 16(20), 2411–2422. 10.1093/hmg/ddm198 [DOI] [PubMed] [Google Scholar]
- Gregory, C. Y. , Evans, K. , Wijesuriya, S. D. , Kermani, S. , Jay, M. R. , Plant, C. , Cox, N. , Bird, A. C. , & Bhattacharya, S. S. (1996). The gene responsible for autosomal dominant Doyne's honeycomb retinal dystrophy (DHRD) maps to chromosome 2p16. Human Molecular Genetics, 5(7), 1055–1059. 10.1093/hmg/5.7.1055 [DOI] [PubMed] [Google Scholar]
- Heon, E. , Piguet, B. , Munier, F. , Sneed, S. R. , Morgan, C. M. , Forni, S. , & Stone, E. M. (1996). Linkage of autosomal dominant radial drusen (malattia leventinese) to chromosome 2p16‐21. Archives of Ophthalmology, 114(2), 193–198. 10.1001/archopht.1996.01100130187014 [DOI] [PubMed] [Google Scholar]
- Lenassi, E. , Troeger, E. , Wilke, R. , Tufail, A. , Hawlina, M. , Jeffery, G. , & Webster, A. R. (2013). Laser clearance of drusen deposit in patients with autosomal dominant drusen (p.Arg345Trp in EFEMP1). American Journal of Ophthalmology, 155(1), 190–198. 10.1016/j.ajo.2012.07.003 [DOI] [PubMed] [Google Scholar]
- Lin, R. C. , & Rosenfeld, P. J. (2007). Antiangiogenic therapy in neovascular age‐related macular degeneration. International Ophthalmology Clinics, 47(1), 117–137. 10.1097/IIO.0b013e31802bd873 [DOI] [PubMed] [Google Scholar]
- Marmorstein, L. (2004). Association of EFEMP1 with malattia leventinese and age‐related macular degeneration: A mini‐review. Ophthalmic Genetics, 25(3), 219–226. 10.1080/13816810490498305 [DOI] [PubMed] [Google Scholar]
- Marmorstein, L. Y. , McLaughlin, P. J. , Peachey, N. S. , Sasaki, T. , & Marmorstein, A. D. (2007). Formation and progression of sub‐retinal pigment epithelium deposits in Efemp1 mutation knock‐in mice: A model for the early pathogenic course of macular degeneration. Human Molecular Genetics, 16(20), 2423–2432. 10.1093/hmg/ddm199 [DOI] [PubMed] [Google Scholar]
- Marmorstein, L. Y. , Munier, F. L. , Arsenijevic, Y. , Schorderet, D. F. , McLaughlin, P. J. , Chung, D. , Traboulsi, E. , & Marmorstein, A. D. (2002). Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age‐related macular degeneration. Proceedings of the National Academy of Sciences of the United States of America, 99(20), 13067–13072. 10.1073/pnas.202491599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, M. , & Traboulsi, E. I. (2001). Dominant radial drusen and Arg345Trp EFEMP1 mutation. American Journal of Ophthalmology, 131(6), 810–812. 10.1016/s0002-9394(00)00926-0 [DOI] [PubMed] [Google Scholar]
- McLaughlin, P. J. , Bakall, B. , Choi, J. , Liu, Z. , Sasaki, T. , Davis, E. C. , Marmorstein, A. D. , & Marmorstein, L. Y. (2007). Lack of fibulin‐3 causes early aging and herniation, but not macular degeneration in mice. Human Molecular Genetics, 16(24), 3059–3070. 10.1093/hmg/ddm264 [DOI] [PubMed] [Google Scholar]
- Mousa, S. A. , Lorelli, W. , & Campochiaro, P. A. (1999). Role of hypoxia and extracellular matrix‐integrin binding in the modulation of angiogenic growth factors secretion by retinal pigmented epithelial cells. Journal of Cellular Biochemistry, 74(1), 135–143. [PubMed] [Google Scholar]
- Narendran, N. , Guymer, R. H. , Cain, M. , & Baird, P. N. (2005). Analysis of the EFEMP1 gene in individuals and families with early onset drusen. Eye (London, England), 19(1), 11–15. 10.1038/sj.eye.6701435 [DOI] [PubMed] [Google Scholar]
- Pager, C. K. , Sarin, L. K. , Federman, J. L. , Eagle, R. , Hageman, G. , Rosenow, J. , & Donoso, L. A. (2001). Malattia leventinese presenting with subretinal neovascular membrane and hemorrhage. American Journal of Ophthalmology, 131(4), 517–518. 10.1016/s0002-9394(00)00821-7 [DOI] [PubMed] [Google Scholar]
- Piguet, B. , Haimovici, R. , & Bird, A. C. (1995). Dominantly inherited drusen represent more than one disorder: A historical review. Eye (London, England), 9(Pt 1), 34–41. 10.1038/eye.1995.5 [DOI] [PubMed] [Google Scholar]
- Roybal, C. N. , Marmorstein, L. Y. , Vander Jagt, D. L. , & Abcouwer, S. F. (2005). Aberrant accumulation of fibulin‐3 in the endoplasmic reticulum leads to activation of the unfolded protein response and VEGF expression. Investigative Ophthalmology and Visual Science, 46(11), 3973–3979. 10.1167/iovs.05-0070 [DOI] [PubMed] [Google Scholar]
- Scarpatetti, A. , Forni, S. , & Niemeyer, G. (1978). The function of the retina in malattia leventinese (dominant drusen) (author's transl). Klinische Monatsblatter Für Augenheilkunde, 172(4), 590–597. [PubMed] [Google Scholar]
- Serra, R. , Coscas, F. , Messaoudi, N. , Srour, M. , & Souied, E. (2017). Choroidal neovascularization in Malattia Leventinese diagnosed using optical coherence tomography angiography. American Journal of Ophthalmology, 176, 108–117. 10.1016/j.ajo.2016.12.027 [DOI] [PubMed] [Google Scholar]
- Sohn, E. H. , Patel, P. J. , MacLaren, R. E. , Adatia, F. A. , Pal, B. , Webster, A. R. , & Tufail, A. (2011). Responsiveness of choroidal neovascular membranes in patients with R345W mutation in fibulin 3 (Doyne honeycomb retinal dystrophy) to anti‐vascular endothelial growth factor therapy. Archives of Ophthalmology, 129(12), 1626–1628. 10.1001/archophthalmol.2011.338 [DOI] [PubMed] [Google Scholar]
- Sohn, E. H. , Wang, K. , Thompson, S. , Riker, M. J. , Hoffmann, J. M. , Stone, E. M. , & Mullins, R. F. (2015). Comparison of drusen and modifying genes in autosomal dominant radial drusen and age‐related macular degeneration. Retina, 35(1), 48–57. 10.1097/iae.0000000000000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, E. M. , Lotery, A. J. , Munier, F. L. , Héon, E. , Piguet, B. , Guymer, R. H. , Vandenburgh, K. , Cousin, P. , Nishimura, D. , Swiderski, R. E. , Silvestri, G. , Mackey, D. A. , Hageman, G. S. , Bird, A. C. , Sheffield, V. C. , & Schorderet, D. F. (1999). A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nature Genetics, 22(2), 199–202. 10.1038/9722 [DOI] [PubMed] [Google Scholar]
- Takeuchi, T. , Hayashi, T. , Bedell, M. , Zhang, K. , Yamada, H. , & Tsuneoka, H. (2010). A novel haplotype with the R345W mutation in the EFEMP1 gene associated with autosomal dominant drusen in a Japanese family. Investigative Ophthalmology and Visual Science, 51(3), 1643–1650. 10.1167/iovs.09-4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt, A. (1925). Die Ophthalmoskopie im rotfreien Licht (3rd ed.). Springer Verlag. [Google Scholar]
- Waardenburg, P. J. (1948). On Macula‐degeneration. Ophthalmologica, 115(2), 115. [PubMed] [Google Scholar]
- Wong, W. L. , Su, X. , Li, X. , Cheung, C. M. , Klein, R. , Cheng, C. Y. , & Wong, T. Y. (2014). Global prevalence of age‐related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta‐analysis. The Lancet Global Health, 2(2), e106–e116. 10.1016/s2214-109x(13)70145-1 [DOI] [PubMed] [Google Scholar]
- Zech, J. C. , Zaouche, S. , Mourier, F. , Placuchu, H. , Grange, J. D. , & Trepsat, C. (1999). Macular dystrophy of malattia leventinese. A 25 year follow up. British Journal of Ophthalmology, 83(10), 1195–1196. 10.1136/bjo.83.10.1194b [DOI] [PMC free article] [PubMed] [Google Scholar]