

Abstract
Background
Orofacial clefts (OFCs) are congenital malformations of the face and palate, with an incidence of 1 per 700 live births. Clubfoot or congenital talipes equinovarus (CTEV) is a three‐dimensional abnormality of the leg, ankle, and feet that leads to the anomalous positioning of foot and ankle joints and has an incidence of 1 per 1000 live births. OFCs and CTEV may occur together or separately in certain genetic syndromes in addition to other congenital abnormalities. Here, we sought to decipher the genetic etiology of OFC and CTEV that occurred together in six probands.
Methods
At the time of recruitment, the most clinically obvious congenital anomalies in these individuals were the OFC and CTEV. We carried out whole‐exome sequencing (WES) on DNA samples from probands and available parents employing the Agilent SureSelect XT kit and Illumina HiSeq2500 platform, followed by bioinformatics analyses. WES variants were validated by clinical Sanger Sequencing.
Results
Of the six probands, we observed probable pathogenic genetic variants in four. In three probands with probable pathogenic genetic variants, each individual had variants in three different genes, whereas one proband had probable pathogenic variant in just one gene. In one proband, we observed variants in DIS3L2, a gene associated with Perlman syndrome. A second proband had variants in EPG5 (associated with Vici Syndrome), BARX1 and MKI67, while another proband had potentially etiologic variants in FRAS1 (associated with Fraser Syndrome 1), TCOF1 (associated with Treacher Collins Syndrome 1) and MKI67. The last proband had variants in FRAS1, PRDM16 (associated with Cardiomyopathy, dilated, 1LL/Left ventricular noncompaction 8) and CHD7 (associated with CHARGE syndrome/Hypogonadotropic hypogonadism 5 with or without anosmia).
Conclusion
Our results suggest that clubfoot and OFCs are two congenital abnormalities that can co‐occur in certain individuals with varying genetic causes and expressivity, warranting the need for deep phenotyping.
Keywords: clubfoot, genetic syndromes, orofacial clefts, whole‐exome sequencing
Whole‐exome sequencing was applied to hunt for possible etiologic variants in six individuals that presented with orofacial clefts and clubfoot. Our genomics and bioinformatics analyses revealed many genetic syndromes in four out of the six probands, with multiple variants in genes associated with certain genetic syndromes being observed in some probands.

1. INTRODUCTION
Orofacial clefts (OFCs) are congenital malformations of human face and palate, presenting as either nonsyndromic or syndromic, with the syndromic forms presenting with additional congenital malformations (Dixon et al., 2011). These structural birth defects may present as cleft lip only (CL), cleft palate only (CP), and cleft lip and palate (CLP). About 70% of CL and CLP are nonsyndromic (Mossey & Modell, 2012). About 50% of CP cases are thought to be syndromic (Leslie et al., 2015). OFCs have multifactorial etiology, with genetic, stochastic, environmental, and gene‐environment factors playing crucial roles (Dixon et al., 2011). Various studies have demonstrated that both syndromic and nonsyndromic forms of OFCs may exhibit variable penetrance and expressivity, with molecular genetic mechanisms such as compound heterozygosity and gene‐gene interactions being suggested as possible underlying mechanisms (Dixon et al., 2011; Gowans et al., 2017). Moreover, genes for syndromic cases, such as IRF6 (OMIM:607199), have been implicated in the etiology of nonsyndromic cases (Kondo et al., 2002; Zucchero et al., 2004).
Varied approaches have been employed to tease out genetic factors and/or gene‐environment interactions associated with OFCs. To date, over 60 genetic loci have been associated with nonsyndromic OFCs (NSOFCs) at genome‐wide significance level employing techniques such as genome‐wide association studies (GWAS), GWAS meta‐analyses and imputation, replication studies of GWAS‐hit loci, re‐sequencing of risk loci and large scale linkage analysis (Buniello et al., 2019; Butali et al., 2019; Carlson et al., 2019; Gowans et al., 2018; Huang et al., 2019; van Rooij et al., 2019). An interesting observation from most of these studies is that a significant number of candidate genes at loci for NSOFCs have also been implicated in the etiology of syndromic forms of these structural birth defects (Beaty et al., 2016; Dixon et al., 2011). A recent study that utilized whole genome sequencing of case‐parent trios followed by genome‐wide association studies employing the transmission disequilibrium test (TDT) revealed population‐specific risk locus in Columbians that had never been reported in studies involving Latin American cohorts (Mukhopadhyay et al., 2020). Whole‐exome sequencing studies have also facilitated the identification of genetic risk loci for OFCs cases that largely follow Mendelian inheritance patterns (Bureau et al., 2014; Cox et al., 2019). Risk variants identified by whole‐exome sequencing (WES) are functional and have large effect sizes than those loci identified through GWAS (Cox et al., 2018). Interestingly, many studies have demonstrated that OFCs largely do not follow Mendelian patterns of inheritance and most cases are sporadic and non‐familial (Dixon et al., 2011). This makes the detection of etiologic genetic variants very challenging.
Congenital talipes equinovarus (CTEV) or clubfoot is a three‐dimensional abnormality of the leg, ankle, and feet which leads to the anomalous positioning of foot and ankle joint with an incidence of 1 per 1000 live births (Pavone et al., 2018). About 80% of cases are nonsyndromic and has multifactorial etiology (O’Shea & Sabatini, 2016). The remaining 20% of CTEV cases are syndromic as they present with other congenital malformations that emanate from chromosomal abnormalities and genetic syndromes (Basit & Khoshhal, 2018). Only one GWAS has been published on nonsyndromic CTEV and this yielded suggestive loci at 12q24.31 between NCOR2 (OMIM:600848) and ZNF664 (OMIM:617890; Zhang et al., 2014). A study that employed WES implicated FLNB (OMIM:603381) in the etiology of nonsyndromic CTEV (Yang et al., 2016). Most WES studies on CTEV have involved syndromic forms of the condition, which implicated syndromes such oculofaciocardiodental (OFCD) Syndrome—caused by variants in BCOR, OMIM:300485 (Zhou et al., 2018), spondyloepiphyseal dysplasia and hearing loss—caused by variants in CHST 3, OMIM:603799 (Waryah et al., 2016), distal arthrogryposis type 5—caused by variants in ECEL1, OMIM:605896 (Umair et al., 2019), TARP syndrome—caused by variants in RBM10, OMIM:300080 (Niceta et al., 2019), distal arthrogryposis with impaired proprioception and touch—caused by variants in PIEZO2, OMIM:613629 (Behunova et al., 2019), and severe disorder of brain development and arthrogryposis—caused by variants in KIAA1109, OMIM:611565 (Gueneau et al., 2018). Other genes implicated in the etiology of nonsyndromic CTEV through targeted gene sequencing and other approaches include those in pathways or processes such as PITX1(OMIM:602149)‐TBX4 (OMIM:601719), HOX genes, muscle contractile genes (e.g., TNNC2 [OMIM:191039], TPM1 [OMIM:191010] and TPM2 [OMIM:190990]), N‐acetylation genes (e.g., NAT 1 [OMIM:108345], NAT 2 [OMIM: 612182] and CYP1A1 [OMIM:108330]) and apoptotic genes (e.g., CASP8 [OMIM:601763], CASP10 [OMIM: 601762] and CFLAR [OMIM:603599] (Sadler et al., 2019). One of the most common syndromic causes of CTEV is distal arthrogryposis (DA) which results from variants in sarcomeric muscle proteins, such as PIEZO2, MYH3 (OMIM:160720), and FBN 2 (OMIM:612570), that partake in muscle contraction (Hall, 2014). Apart from DA, over 30 genetic syndromes often have phenotype sequelae that include clubfoot (Basit & Khoshhal, 2018). These include syndromes that emanate from variants in genes that play cardinal roles in phenomena or structures such as TGF‐β signaling pathway (e.g., TGFBR1 [OMIM:190181], TGFBR2 [OMIM:190182], SMAD3 [OMIM:603109], TGFB2 [OMIM:190220], TGFB3 [OMIM:190230], GDF5 [OMIM:601146] and SKI [OMIM:164780]), extracellular matrix (e.g., COL 9A1 [OMIM:120210], COL 9A2 [OMIM:120260], COL 9A3 [OMIM:120270], COL 3A1 [OMIM:120180], COMP [OMIM:600310], MATN3 [OMIM:602109], SLC26A2 [OMIM:606718] and FBN 1 [OMIM:134797]), peroxisomal defects (e.g., GDAP1 [OMIM:606598] and PEX 26 [OMIM:608666]), and proteoglycans (e.g., CHST 14 [OMIM:608429], COG 4 [OMIM:606976], TGDS [OMIM:616146] and DSE [OMIM:605942]; Sadler et al., 2019). Importantly, certain genetic syndromes, such as Loeys‐Dietz, Marfan, and Moebius syndromes, may present with either OFCor clubfoot, or both of these two structural birth defects may co‐occur as associated phenotypes (Basit & Khoshhal, 2018; Sadler et al., 2019; Slavotinek & Tifft, 2002). Notably, CTEV may co‐occur with a number of multiple congenital anomalies (MCAs). For example, CTEV is seen as a rare or occasional finding in about 13% of syndromic cleft lip with or without palate (CL ± P), 20% of syndromic CP, and 13% of non‐OFC birth defects (Rittler et al., 2008). To ascertain the preferential association of OFCs with clubfoot, Rittler et al., (2008) also demonstrated that CTEV was positively associated with CP but was negatively associated with CL ± P; however, when both CP and CL ± P were combined, all OFCs were negatively associated with CTEV.
As part of our OFC studies in Ghana, we recruited six probands that presented with CL‐clubfoot, and CLP‐clubfoot phenotypes, with no additional clinically identifiable malformations at the time of recruitment. Subsequent to this observation, we sought to identify causal coding and splice site variants that will help us ascertain whether these OFC‐clubfoot phenotypes were syndromic or that the clubfoot phenotype was just an associated anomaly. We achieved our aim by conducting whole‐exome sequencing of affected probands and available relatives. This was followed by the validation of implicated variants using Sanger Sequencing.
2. MATERIALS AND METHODS
2.1. Ethical considerations and DNA processing
The study was approved by the Institutional Review Board (IRB) at Kwame Nkrumah University of Science and Technology (KNUST), Ghana, the approval number being CHRPE/RC/018/13. Patient recruitment was carried out at Komfo Anokye Teaching Hospital (KATH), Kumasi, Ghana. Since this is collaborative research, the IRB at The University of Iowa, Iowa, USA, also gave approval for the study. Written informed consent was obtained from all participating families before sample and data collection. All probands and their family members recruited for this study were born to Ghanaian parents and no Caucasian or Asian families were recruited for the study. Probands were individuals that presented with cleft lip (CL) or cleft lip and palate (CLP) together with club foot. Subsequent to the collection of clinical and phenotypic data, cheek swab or saliva samples were collected from study participants using Oragene saliva kits and sponges (http://www.dnagenotek.com). Cheek swab and saliva samples were shipped to the Butali Laboratory at the University of Iowa, where DNA processing and genetic analyses took place.
The detailed protocol for DNA processing has been published elsewhere (Gowans et al., 2016). In summary, DNA was extracted from saliva and cheek swab samples using Oragene DNA saliva processing kits and a modified protocol. The amount of DNA obtained for each sample was quantified using Qubit Assay with Qubit 2.0 Fluorometer (http://www.invitrogen.com/site/us/en/home/brands/Product‐Brand/Qubit.html). As a quality control step, we verified the sexes of participants using XY Genotyping on Real‐Time PCR machine employing TaqMan genotyping assay kits.
2.2. Whole‐exome sequencing and bioinformatics analyses
We carried out WES on DNA samples from six probands and available parents as described elsewhere (Al Dhaheri et al., 2020). In summary, DNA samples used for WES had over 1 µg of DNA. The Agilent SureSelect XT kit (Agilent Technologies, Santa Clara, CA) was used to capture the consensus coding sequence (CCDS) of exonic regions and flanking intronic regions which totaled about 51 Mb. Subsequently, the Illumina HiSeq2500 platform (Illumina) was utilized to generate paired‐end reads of 100 base pairs (bp). WES was carried out at the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG) at John Hopkins University, USA, and the raw data are available under controlled‐access to other investigators who may apply to the Center for the data.
We carried out both individual and cohort bioinformatics analyses of the WES data generated. Our individual analysis of WES data was carried out in order to determine rare missense, nonsense, splicing, and indel variants that may be inherited as either autosomal dominant or autosomal recessive, including homozygous or compound heterozygous models, as well as de novo analysis. The Burrows‐Wheeler Alignment (BWA) v.0.5.10‐tpx (Li & Durbin, 2009) was employed to align FASTQ files to the reference human genome (GRCh37; Ensembl core database release 50_361; 1000 genomes 2012); this resulted in SAM/BAM output. Polymerase chain reaction (PCR) duplicates were flagged with Picard (http://broadinstitute.github.io/picard/). The Genome Analysis Toolkit (GATK) v.2.3‐9‐ge5ebf34 (McKenna et al., 2010) was utilized to perform local realignment around indels, base call quality score recalibration, as well as reduced‐read BAMs. Multi‐sample single‐nucleotide variant (SNV) and indel calling were carried out on the reduced‐read BAM files using UnifiedGenotyper tool embedded in GATK. We employed the Variant Quality Score Recalibration (VQSR) method embedded in GATK (DePristo et al., 2011) to filter the variant sites. Based on VQSR analyses, all heterozygous genotypes that did not have at least five alternate allele reads were excluded from further analysis. Sequel to this, we employed the Variant Analysis Tool of PhenoDB (Sobreira et al., 2015) to analyze the annotated files (ANNOVAR) by prioritizing for heterozygous, homozygous, compound heterozygous, and hemizygous rare variants, including missense, nonsense, splice site variants, as well as indels, that potentially had functional effects on phenotypes.
All variants with minor allele frequency (MAF) >0.01 based on 1000 Genomes Project (www.internationalgenome.org), dbSNP 126, 129 and 131 (www.ncbi.nlm.nih.gov/sns/), Exome Variant Server (release ESP6500SI‐V2; Lek et al., 2016), ExAC (http://exac.broadinstitute.org/), gnomAD (https://gnomad.broadinstitute.org/), as well as our in‐house Baylor‐Hopkins Center for Mendelian Genomics (BHCMG) samples, were excluded from further analysis. This was performed in line with the entire data set in these databases as well as data specific to populations of African ancestry. Our WES variant filtering process also excluded variants in genes that were highly mutated or presented with many false positives (Supplemental material S1). We ascertained probable candidate genes that may be responsible for the etiology of the cleft‐clubfoot phenotypes by highlighting variants in genes known to be associated with phenotypes related to the one being investigated using Online Mendelian Inheritance in Man [OMIM] (www.omim.org), mouse models (www.informatics.jax.org), the NHGRI‐EBI Catalog of published Genome‐Wide Association Studies [GWAS] (www.ebi.ac.uk), Human Gene Mutation Database [HGMD] (www.hgmd.cf.ac.uk) and ClinVar (www.ncbi.nlm.gov/clinvar/). The probable deleteriousness or otherwise of candidate variants, as well as their functional impacts on the mutant proteins, were determined using in silico algorithmic tools such as CADD v1.3 (http://cadd.gs.washington.edu; Rentzsch et al., 2019), SIFT (http://sift.jcvi.org/; Kumar et al., 2009), Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/; Adzhubei et al., 2010) and HOPE (http://www.cmbi.ru.nl/hope; Venselaar et al., 2010). HOPE was used to model only the novel variants observed in this study; however, HOPE could not model the effect of the variants in FRAS1 (OMIM:607830) due to the large size of the FRAS1 protein. Moreover, HOPE was unable to simulate the effects of the indels observed in TCOF1 (OMIM:606847) and CHD7 (OMIM:608892) genes as the tool was developed to simulate the effects of missense variants.
In our cohort analysis pipeline, the initial steps were similar to individual analysis (Figure S1). In addition, we highlighted families that had two or more loss of function (LOF) variants, including stop‐gain or stop‐loss, frameshift, splicing, exonic, splicing variants, and de novo variants. We filtered out variants with tolerance score (CCDS) >10%. We also looked for variants that interact with other genes that are known to play a role in the etiology of OFCs. However, we were unable to ascertain any candidate causal variants that exhibited autosomal dominant or autosomal recessive modes of Mendelian inheritance in our cohort analyses, possibly due to the fact that our study involved simplex families and the determination of the carrier status of other unaffected family members was not possible.
2.3. Confirmation of etiologic variants using Sanger sequencing
Due to the 99.99% high accuracy of the Sanger sequencing platform which makes it the gold standard for clinical research, we employed this technology to validate the variants that were implicated by our WES analyses. The gene reference sequence transcripts whose variants were validated by Sanger sequencing included NM_001257281.2 (DIS3L2), NM_020964.3 (EPG5), NM_021570.4 (BARX1), NM_001145966.2 (MKI67), NM_025074.7 (FRAS1), NM_001008657.3 (TCOF1), NM_022114.4 (PRDM16) and NM_017780.4 (CHD7). The procedure for Sanger sequencing and bioinformatics analyses have been published elsewhere (Gowans et al., 2016), but a summary is presented here. We employed Primer3 (www.primer3.ut.ee) to design primers to cover the exonic regions which harbored the implicated variants. PCR was subsequently used to amplify implicated exonic regions and the success of the PCR reaction based on the size of the amplicon was verified using agarose gel electrophoresis. Correctly amplified products were sequenced using an ABI 3730XL DNA Sequencer (http://www.appliedbiosystems.com/absite/us/en/home.html) at Functional Biosciences in Madison, Wisconsin, US. Chromatograms generated were transferred to a Unix workstation, PHRED (http://www.phrap.org/phredphrapconsed.html, v.0.961028) was used for base calling, PHRAP (http://www.phrap.org/, v.0.960731) was utilized to assemble bases which were subsequently scanned by POLYPHRED (http://droog.gs.washington.edu/polyphred/, v. 0.970312) and were viewed with CONSED (http://www.phrap.org/consed/consed.html, v. 4.0). Reads were aligned to the reference human genome build GRCh37 using the “Blat” function of UCSC Genome Browser (https://genome.ucsc.edu/).
3. RESULTS
3.1. Description of probands and case families
In total, we recruited six probands that presented with clubfoot with CL or CLP. The probands comprised five males and one female. All the affected families were simplex families, where there was no other affected family member except the proband. We also followed up on these families after our WES studies, which were over 2 years after the patient recruitment. This was performed through phone calls, as well as the review of medical records at KATH Cleft‐Craniofacial Clinic. The reason for our follow‐up was twofold: (a) to ascertain if any additional phenotypic features had developed after our patient recruitment, and (b) to determine the survival rate of children that are born with cleft‐clubfoot phenotypes. We were able to re‐contact three (50%) of our case families after the WES analyses.
Family 1: proband is a male with right unilateral incomplete CL and left unilateral clubfoot and was 1 week old at the time of subject recruitment. We obtained DNA samples and DNA sequences from the proband and both parents. The father is a peasant farmer whereas the mother is a petty trader and a peasant farmer. As a peasant farmer, the mother was exposed to pesticides and nitrate compounds, particularly fertilizer, during gestation, and accessed antenatal care after three months of conception. A follow‐up on this family after the WES analyses revealed that there was no record of the patient undergoing reconstructive surgery to correct the CL at KATH. Moreover, the family could not be traced through phone call or any other means.
Family 2: the probands is a female with bilateral complete CLP and left unilateral clubfoot and was aged two weeks at the time of subject recruitment. DNA samples and DNA sequences were obtained from both parents as well as the proband. Mother is a teacher and had once lived in a mining community and accessed antenatal care within the first trimester. After the WES analyses, our follow‐up on this family demonstrated that the clinical assessment that was carried out later showed that proband had facial asymmetry and hypotonia. Family was successfully contacted through a phone call and the feedback was that the patient is still alive with no additional phenotypic features or health problems.
Family 3: this family had a male proband with left unilateral complete CL and bilateral clubfoot and was aged 4 years old at the time of subject recruitment. We obtained DNA samples, and subsequently DNA sequences, from the mother and proband. Mother is a trader whereas the father is an architect. Mother was exposed to alcohol, skin lightening creams, and enemas during gestation, and as well had antenatal care after 3 months of gestation. We also followed up on this family after our WES study. It was observed that clinical examinations that were later carried out about two years after sample collection revealed that the proband also had hypotonia (weak muscle tone) and sickling trait. The proband is currently doing very well, with no additional phenotypes being observed.
Family 4: this family had a male proband with left unilateral complete CL, bilateral CP, and right clubfoot who was aged one week at the time of subject recruitment. We obtained DNA samples and DNA sequences from proband and mother. The father is a steel bender and the mother is a hairdresser. Mother delivered the proband at 45 weeks of gestation by a cesarean section and was diagnosed with hepatitis B during gestation. Mother was also exposed to air pollution through firewood usage, as well as herbal remedies during gestation, and had antenatal care after three months of gestation. Five years after patient recruitment and upon the completion of our WES analyses, the family was re‐contacted through a phone call, with the feedback that the proband is currently doing well. The family also re‐visited KATH Cleft‐Craniofacial Clinic in January 2019 and clinical assessment of the proband during that period revealed that the proband had normal intelligence both at school and home, and had no hearing or eye problems. Other phenotypes that were observed in this later clinical assessment included flat, sunken or sad look to the face, mild micrognathia, sparse eyelashes, and down‐slanting eyes.
Family 5: the male proband was 2 weeks old at the time of subject recruitment and had bilateral CLP and bilateral clubfoot, as well as syndactyly of the third, fourth, and fifth toes of the left foot (Figure 1). DNA samples and DNA sequences were obtained from proband and both parents. The proband is a same‐sex twin who was born prematurely at 28 weeks of gestation with a very low birth weight of 1.2 kg. According to the parents, the other twin was clinically normal, being discordant for the CLP phenotype and any other congenital malformations observed in the proband. However, we could not obtain the DNA sample from the unaffected twin because the parents were unwilling to bring him to the Clinic. DNA sample from the unaffected twin would have enhanced our variant filtering process as well as our ability to genetically determine whether these same‐sex twins were monozygotic or dizygotic. The mother is a petty trader whereas the father is a cloth weaver. Mother utilized enemas during gestation, had one miscarriage prior to the conception of the proband, and antenatal care after three months of gestation. Our follow‐up after our WES studies showed that there was no record of repair of the cleft on the proband and the family could not be traced through phone call or other means. The last time the family ever visited the Cleft‐Craniofacial Clinic at KATH was when the proband was one month three weeks and had a weight of 3.30 kg.
FIGURE 1.
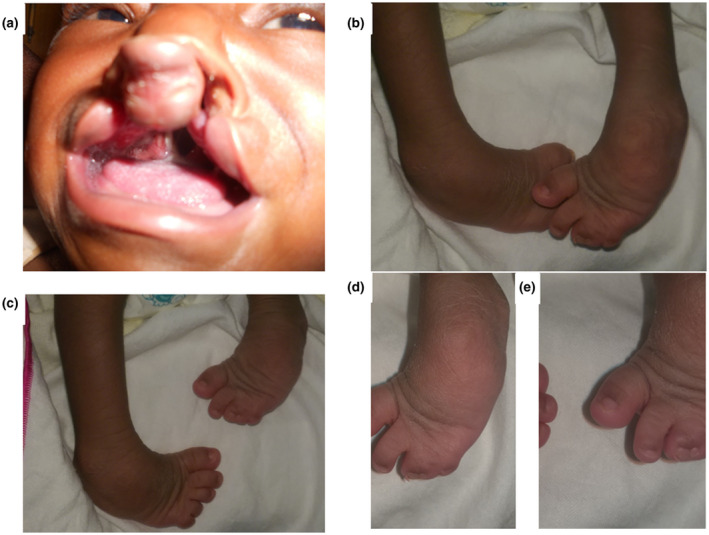
Phenotypes observed in the proband of Family 5. (a) Bilateral cleft lip and palate. (b) Bilateral clubfoot phenotype with one foot slightly superimposed on the other to depict the severity of the defect. (c) Two feet standing in isolation to depict the syndactyly on left foot but lack of same in the right foot. (d and e) syndactyly of third, fourth, and fifth toes of the left foot
Family 6: Proband is a male with right unilateral incomplete CL and left clubfoot who was aged two weeks at the time of subject recruitment. DNA samples and DNA sequences were obtained from proband and mother, with both parents being peasant farmers. Mother was exposed to hypertensive drugs and enemas during gestation, and had antenatal care after the first trimester. Our follow‐up after the WES analysis showed that there was no record of cleft repair on the proband. However, later clinical assessment after sample collection showed that the proband had some genital abnormalities, including unusually small penis (micropenis) and undescended testes (cryptorchidism). This notwithstanding, the family could not be traced for further updates.
3.2. Syndromes and gene variants implicated by WES analyses
Many variants in genes, some of whom have been associated with known genetic syndromes, were observed in four out of six probands in the individual analyses (Table 1; Figure 2). For both Families 2 and 5, we did not observe any candidate variants in genes whose variants have been associated with any of the phenotypes under consideration when our data were compared to that in OMIM, GWAS catalog, mouse models, HGMD, and ClinVar. About 67% of the observed variants in this study are among the top 1% of most deleterious variants in the human genome, with PHRED‐scaled CADD scores within the range of 20 to 29. Furthermore, approximately 27% of the observed variants are among the top 10% of most deleterious variants in the human genome with PHRED‐scaled CADD scores within 10 to 19.
TABLE 1.
Variants that were observed in four out of the six probands through whole‐exome sequencing
| Family ID | Gene name | RefSeq | Exon harboring variant | HGVSc | HGVSp | MAF | Reason | Polyphen−2 | SIFT | Scaled CADD score |
|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | DIS3L2 | NM_001257281.2 | 8 | c.878C>A | p.Pro293His (rs187563594) | 3.4 × 10−3 | OMIM Phenotype | Probably Damaging | Deleterious | 24.6 |
| 13 | c.1570G>A | p.Glu524Lys (rs201308521) | 4.1 × 10−3 | Possibly Damaging | Deleterious | 24.4 | ||||
| Family 3 | EPG5 | NM_020964.3 | 7 | c.1594C>T | p.His532Tyr | Novel | OMIM Phenotype | Probably damaging | Deleterious | 25.9 |
| 3 | c.1048C>T | p.Arg350Cys (rs761594653) | 0.00 | Benign | Tolerated | 25.2 | ||||
| BARX1 | NM_021570.4 | 2 | c.423G>C | p.Lys141Asn | Novel | Mouse model | Probably damaging | Deleterious | 25.3 | |
| MKI67 | NM_001145966.2 | 12 | c.6911G>A | p.Arg2304Lys (rs138997814) | 2.8 × 10−3 | GWAS | Possibly Damaging | Tolerated | 18.2 | |
| Family 4 | FRAS1 | NM_025074.7 | 58 | c.8644G>C | p.Val2882Leu | Novel | OMIM Phenotype | Benign | Tolerated | 22.7 |
| 65 | c.10160T>C | p.Leu3387Pro (rs137982616) | 1.2 × 10−2 | Possibly Damaging | Deleterious | 23.4 | ||||
| TCOF1 | NM_001008657.3 | 18 | c.2864_2865insGG | p.Glu956GlyfsTer31 | Novel | OMIM Phenotype | N/A | N/A | N/A | |
| MKI67 | NM_001145966.2 | 12 | c.6740G>A | p.Arg2247His (rs34688192) | 9.4 × 10−3 | GWAS | Probably damaging | Deleterious | 16.1 | |
| Family 6 | FRAS1 | NM_025074.7 | 68 | c.10594A>G | p.Ile3532Val (rs144715071) | 7.5 × 10−3 | OMIM Phenotype | Benign | Deleterious | 23.6 |
| 74 | c.11605A>G | p.Ile3869Val (rs145035489) | 7.7 × 10−3 | Benign | Tolerated | 14.4 | ||||
| 74 | c.11907C>G | p.His3969Gln (rs140492803) | 8.7 × 10−3 | Benign | Tolerated | 5.8 | ||||
| PRDM16 | NM_022114.4 | 9 | c.1188T>C | p.Cys396Cys (rs201309284) | 5.3 × 10−3 | Mouse model | N/A | N/A | 12.7 | |
| CHD7 | NM_017780.4 | 18 | c.4248delA | p.Arg1417GlufsTer14 | Novel | OMIM Phenotype | N/A | N/A | N/A |
Abbreviations: GWAS, genome‐wide association studies; MAF, Minor Allele Frequency as reported for populations of African ancestry in gnomAD; N/A, not applicable; OMIM, Online Mendelian Inheritance in Man.
FIGURE 2.
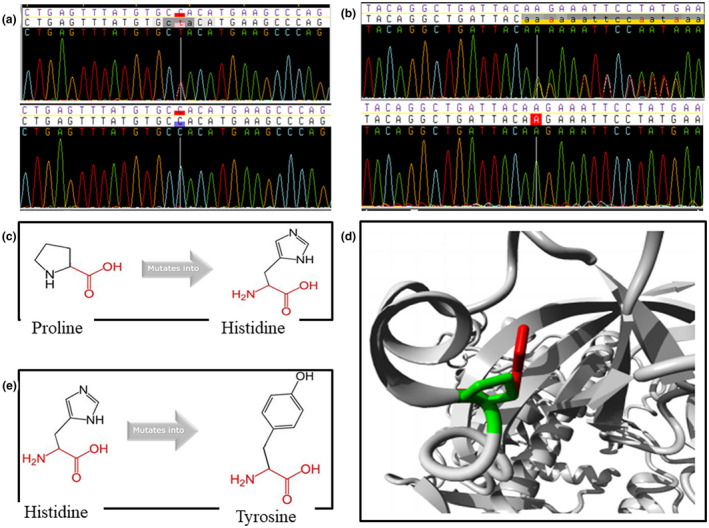
Electropherogram and simulation of the effect of mutations on protein structure by HOPE for the novel variants. (a) electropherogram for c.1594C>T variant in EPG5 (NM_020964.3), with a white vertical line indicating the position of the variant—top electropherogram shows the heterozygous variant in proband and bottom one is for the mother who lacked the variant. (b) electropherogram for c.4248delA in CHD7 (NM_017780.4), with a white vertical line indicating the position of the variant—top electropherogram indicates the heterozygous deletion in the proband whereas the bottom is for the mother who did not have the variant. (c) Schematic simulation of mutant protein for p.Pro293His in DIS3L2 (NM_001257281.2) by HOPE: proline is more hydrophobic and smaller than histidine. (d) Ribbon simulation of mutant protein for p.Pro293His in DIS3L2 by HOPE: green and red indicate wild‐type and mutant residues, respectively. (e) Schematic representation of p.His532Tyr variant in EPG5: the mutant residue, (tyrosine) is bigger and more hydrophobic than the wild‐type residue (histidine)
In Family 1, two compound heterozygote missense variants in DIS3 like 3'‐5' exoribonuclease 2 (DIS3L2) gene (OMIM:614184) were observed in the proband. These included p.Pro293His (rs187563594) which was inherited from the mother and p.Glu524Lys (rs201308521), inherited from the father. According to HOPE simulations of the p.Pro293His variant (Figure 2c,d), proline is more hydrophobic and smaller than histidine. Based on these, together with the high conservation score of the wild‐type residue, HOPE predicts that the p.Pro293His variant is probably damaging to the protein because it affects protein conformation and certain amino acid interactions that are crucial for the activity of the protein. Compound heterozygote missense variants in DIS3L2, such as those observed in this study, have been reported in individuals with autosomal recessive Perlman Syndrome (MIM:267000).
For Family 3, the proband had multiple rare and probably pathogenic variants in three different genes, one of which has been associated with a known genetic syndrome. Two compound heterozygous missense variants were observed in the ectopic P‐granules autophagy protein 5 homolog (EPG5) gene (OMIM:615068), a gene whose variants have been reported in individuals with autosomal recessive Vici Syndrome (MIM:242840). The EPG5 gene variants included the novel variant p.His532Tyr and p.Arg350Cys (rs761594653). Neither of these variants was observed in the mother but there was no paternal sample to ascertain whether any of the variants originated from the father or is a de novo variant. For the novel p.His532 Tyr variant, HOPE simulation (Figure 2e) demonstrated that the mutant residue, tyrosine, is bigger and more hydrophobic than the wild‐type residue, histidine. The bigger mutant residue may lead to bumps (protuberances) and its relatively higher hydrophobicity can result in loss of hydrogen bonds and/or disturb correct folding. These observations, together with the fact that the wild‐type residue is near a highly conserved position, is the basis for the HOPE prediction that this variant is probably damaging to protein structure and function. Two other missense variants in two other genes, including the novel p.Lys141Asn variant in BARX homeobox 1 (BARX1; OMIM:603260) and p.Arg2304Lys (rs138997814) in the marker of proliferation Ki‐67 (MKI67; OMIM:176741) genes, were also observed in the proband of Family 3. These two other variants were not observed in the mother. STRING analyses (https://string‐db.org/) demonstrated that BARX1 is a transcription factor that plays crucial roles in craniofacial development and odontogenesis, and interact with genes in the WNT signaling pathway, as well as genes which have been implicated in OFCs such as BMP4 (OMIM:112262), TBX22 (OMIM:300307), FGF8 (OMIM:600483), PAX9 (OMIM:167416) and MSX1, OMIM:142983 (Figure S2).
In Family 4, genetic variants in three different genes, two of which are associated with genetic syndromes, were observed in the proband. We observed that the proband harbored two compound heterozygote missense variants in Fraser extracellular matrix complex subunit 1 (FRAS1) gene (OMIM:607830), including the novel p.Val2882Leu variant and p.Leu3387Pro (rs137982616). Similar variants have been reported in individuals with autosomal recessive Fraser Syndrome 1 (MIM:219000). The p.Leu3387Pro variant in FRAS1 was inherited from the clinically unaffected mother but there was no paternal DNA sample to ascertain the origin of the p.Val2882Leu variant. However, the p.Val2882Leu variant was observed in the available DNA sample of the clinically unaffected paternal grandmother of the proband, suggesting that the proband may have inherited this variant from the father through the paternal grandmother. Moreover, a novel 2 base pair (bp) insertion that leads to a frameshift in the treacle ribosome biogenesis factor 1 (TCOF1) gene (OMIM:606847), c.2864_2865insGG (p.Glu956GlyfsTer31), was observed in the proband. Such frameshift variants have been reported to be responsible for the etiology of the majority of the cases of autosomal dominant Treacher Collins Syndrome 1 (MIM: 154500). The TCOF1 variant abolishes the canonical stop codon after the terminal amino acid residue at position 958, converting the stop codon (UGA) to a leucine codon (CUU). This insertion also increases the length of the polypeptide by 29 more amino acids before it is terminated, and as well alter the amino acid sequence and composition from position 956 to 958. Last, we also observed in this proband a missense variant, p.Arg2247His (rs34688192), in the MKI67 gene (OMIM:176741). Neither of the variants in TCOF1 and MK167 genes was observed in the available samples of the mother and paternal grandmother.
Interestingly, the proband in Family 6 had probable etiologic variants in three different genes whose variants may lead to certain genetic syndromes. For this proband, we observed three different missense variants in the FRAS1 gene, including p.Ile3532Val (rs144715071), p.Ile3869Val (rs145035489), and p.His3969Gln (rs140492803). Of the three variants in FRAS1, in silico analysis (Table 1) suggests that only the p.Ile3532Val and p.Ile3869Val compound heterozygote variants may be pathogenic. Segregation analysis showed that p.Ile3532Val was inherited from the mother but there was no paternal sample to confirm or otherwise the paternal origin of the p.Ile3869Val variant. This proband also harbored a synonymous single‐nucleotide variant (SNV), p.Cys396Cys (rs201309284), in the PR/SET domain 16 (PRDM16) gene (OMIM:605557). This PRDM16 variant alters a wild‐type splice donor site and most probably affects the splicing of this gene (Human Splice Finder; www. http://www.umd.be/HSF/). Similar variants in PRDM16 have been reported in individuals with autosomal dominant cardiomyopathy, dilated, 1LL (CMD1LL; MIM:615373) and/or left ventricular noncompaction 8 (LVNC8; MIM:615373) syndromes. Last, we observed in this proband a novel frameshift deletion, c.4248delA (p.Arg1417GlufsTer14), in chromodomain helicase DNA binding protein 7 (CHD7) gene (OMIM:605806). Pathogenic variants in CHD7 cause autosomal dominant coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities (CHARGE) Syndrome (MIM:214800) and/or hypogonadotropic hypogonadism 5 with or without anosmia (HH5; MIM: 612370). For the c.4248delA frameshift variant in CHD7, threonine (T) at amino acid position 1416 remained unchanged due to the degeneracy of the threonine codons; however, the effect of the variant was seen from amino acid position 1417 (arginine is replaced by glutamic acid) and other codons downstream. Neither of the variants in PRDM16 or CHD7 was observed in the mother but there was no paternal sample to determine whether these variants were de novo or segregated from the father to the proband.
4. DISCUSSION
OFCs and congenital clubfoot are structural birth defects that place a significant psycho‐social burden on affected individuals and families, with both conditions having multifactorial etiology (Dixon et al., 2011; Pavone et al., 2018). We sought to ascertain how genetic factors influence the pathogenesis of these two congenital anomalies that co‐occurred in six case families. We observed possibly etiologic genetic variants in four out of the six probands (~67%) studied in our WES analyses, including novel variants. A number of postulates may account for our inability to detect etiologic variants in all six probands: (a) since our WES analyses focused on coding part of the human genome, probable etiologic variants in regulatory regions, such as enhancers and promoters, may have been missed, (b) the multifactorial etiology of these two conditions suggests that apart from etiologic genetic variants, maternal peri‐conception environmental exposures may also play a role in the etiology of some cases of these two structural birth defects, and (c) our analysis pipeline for WES data may have missed genetic variants that may be etiologic, but eluded us due to limitations of and some level of inconsistencies in current algorithmic tools.
We could not re‐contact about 50% of our study cohort and there was also no evidence of the OFC being repaired in these patients at the treatment center. It is a common observation at Cleft‐Craniofacial Clinic at KATH that patients with multiple congenital anomalies that include OFCs have relatively low survival rate. This is probably due to the high risk of peri‐natal death that is characteristic of certain genetic syndromes, such as Perlman syndrome (Soma et al., 2017), as well as probable infanticide that is fueled by stigma together with psychosocial and socioeconomic challenges (Camille et al., 2014).
Ascertaining the genetic variant(s) that may be responsible for the etiology of both or either of the co‐occurring CL ± P and CTEV in our study cohort is quite challenging. An inference based on the Rittler et al., (2008) study, which suggested that CTEV was negatively associated with CL ± P suggests that these two phenotypes occur together in our study cohort purely by chance and it is possible that different genetic causes may be responsible for either of these two phenotypes. Alternatively, both CL ± P and CTEV may be part of sequelae of phenotypes common to some syndromes with a single or common genetic cause (Nayak et al., 2017; Slavotinek & Tifft, 2002). In harmony with these two postulates, we observed that in three out of the four probands with possible etiologic genetic variants, about two or three different genes were mutated in such individuals, suggesting that the co‐occurring CL ± P and CTEV in these three probands may either have a common or different genetic cause(s). However, further functional genomics studies on these variants are warranted before any definitive conclusion can be made.
The only proband in which we observed a probable etiologic variant in a single gene was from Family 1. We observed two compound heterozygous variants (p.Pro293His and p.Glu524Lys) in the DIS3L2 gene. DIS3L2 is an RNA‐binding protein with 3′‐5′ exoribonuclease activity, playing cardinal role in cytoplasmic RNA surveillance and decay (Labno et al., 2016). Thus, for uridylated RNAs that occur in the cytoplasm, such as mRNA and ncRNAs like pre‐miRNA and mature miRNA, their recognition, binding, and degradation are mediated by DIS3L2 (Luan et al., 2019). DIS3L2 also partake in several physiological and biological processes, including cell proliferation, differentiation, division, and apoptosis (Labno et al., 2016). Compound heterozygote variants in DIS3L2 cause Perlman Syndrome, an overgrowth syndrome characterized by the high neonatal mortality rate (Soma et al., 2017). Consistent with this high neonatal mortality rate, medical records at the Cleft‐Craniofacial Clinic at KATH suggest that this proband did not survive up to 3 months, after which cleft lip reconstructive surgery is normally carried out. Perlman syndrome presents with a number of phenotypes: polyhydramnios, characteristic facial dysmorphisms, renal dysplasia, macrosomia, a predisposition to Wilms’ tumor and, sometimes, orofacial clefts (www.omim.org). Here, we present a patient with two compound heterozygote missense variants in DIS3L2; variants of this nature are characteristic of the etiology of classical Perlman Syndrome. The patient presented with cleft lip and clubfoot, with the clubfoot being a rare presentation of Perlman syndrome. However, we did not observe most of the classical Perlman Syndrome phenotypes in the proband in that our study was deficient in phenotypic data because apart from the proband in Family 3, all other probands were recruited at 1 or 2 weeks after birth. At 1 or 2 weeks of age, not all phenotypes were clinically glaring or evident, suggesting the need for continuous clinical and phenotypic assessment at different time points in order to make a definite syndrome diagnosis.
In Family 3, we observed in the proband possible etiologic variants in three different genes, including EPG5, BARX1, and MKI67. Homozygous or compound heterozygous missense variants in EPG5 cause autosomal recessive Vici Syndrome (Byrne et al., 2016). EPG5 is involved in the regulation of autophagy, a fundamental cellular degradative pathway that is crucial for the removal of aberrant proteins and organelles, adaptation to varying metabolic requirements, and defense against infections (Vojcek et al., 2020). Vici Syndrome may presents with a number of multisystem abnormalities, including pigmentary anomalies, variable immunodeficiency (leading to recurrent infections), agenesis of the corpus callosum (ACC), progressive cardiomyopathy, cataracts, severe psychomotor retardation, myopathy that may lead to hypotonia, dysmorphic facial features, cleft lip and palate, toe syndactyly and failure to thrive (Byrne et al., 2016; Cullup et al., 2013). Here, we report a patient with EGP‐related presentation who had cleft lip, bilateral clubfoot, and hypotonia, as well as compound heterozygote missense variants in EPG5. Since most of the classical features of Vici Syndrome were absent in this proband, it is elusive to pinpoint if the observed phenotypes are an expansion of the sequelae of phenotypes of Vici Syndrome. Alternatively, the phenotypes observed in the proband may be due to a new syndrome if the variants observed in the EPG5 gene are later shown to be specific in similar cases. This notwithstanding, variants that were observed in the two other genes may also contribute to the phenotypes in the proband, though how these pathways and genes interact is unclear. For example, BARX1 is expressed in structures affected by CLP largely during the ages of mixed dentition in humans (Krivicka‐Uzkureleet al., 2008). In the chicken embryo, Wnt signaling pathway regulates maxillary growth and morphogenesis through many major morphogenetic signaling pathways that include genes such as Barx1 (Shimomura et al., 2019). In mouse and zebrafish, regulation of Barx1 by Gata4 is crucial for the development of neural crest and craniofacial skeleton (Guo et al., 2018). Our STRING analyses (https://string‐db.org/) demonstrated that BARX1 is relevant for craniofacial development and odontogenesis, and directly or indirectly interacts with several genes that have been implicated in the etiology of OFCs, including MSX1, BMP4, FGFR2, and WNT signaling pathway genes. As a homeobox gene, we also speculate that BARX1 may also play a role in the etiology of the clubfoot phenotype of this proband because: (a) in mouse, Barx1 is expressed in the proximal fore‐ and hind limbs (https://omim.org/entry/603260?search=BARX1&highlight=barx1), and (b) BARX1 is a member of homeobox gene family whose members are involved in body patterning and include some HOX genes that have been implicated in the etiology of clubfoot in humans (Sadler et al., 2019). Moreover, MKI67 gene product is necessary for cellular proliferation and organization, development of multiple organ systems and is expressed in an array of developing morphogenetic structures, including branchial arches and limbs (http://www.informatics.jax.org/marker/key/26677). In cleft affected lip tissues, it was observed that the expression of MKI67 correlated positively with that of Interleukin 8 (IL‐8); this observation was stronger in the epithelial tissue than the connective tissue (Pilmane et al., 2019).
Another interesting phenomenon in this study was the observation of compound heterozygote missense variants in the FRAS1 gene in the two probands of Families 4 and 6. Each of these probands had different compound heterozygous variants in the FRAS1 gene which is characteristic of most cases of Fraser Syndrome, though variants in other genes such as FREM2 (OMIM:608945) and GRIP1 (OMIM:604597) may also cause this syndrome (Bouaoud et al., 2020). FRAS1 is an extracellular matrix protein that is crucial for certain processes in embryonic development, including apoptosis and epithelial‐mesenchymal interactions (Pavlakis et al., 2011). Fraser syndrome may frequently present with cryptophthalmos, renal agenesis, genital anomalies, syndactyly, as well as many other less frequent anomalies such as hormonal, anorectal, ear‐nose‐throat (ENT), craniofacial and oro‐dental defects (van Haelst et al., 2008). Fraser Syndrome may also present with either or both OFC and clubfoot and has high perinatal mortality (Nayak et al., 2017; Slavotinek & Tifft, 2002). In consonance with this phenotypic spectrum of Fraser syndrome, we observed that OFC and clubfoot co‐occurred in both probands of families 4 and 6 together with other anomalies that were observed later after subject recruitment. The proband in Family 4 had other maxillofacial anomalies whereas the proband of Family 6 had genital anomalies and probably, perinatal death. This observation may depict the variable expressivity of Fraser syndrome as observed elsewhere (Midro et al., 2020; Nayak et al., 2017).
Though variants in FRAS1 may contribute to the etiology of the cleft‐clubfoot phenotype in the proband in Families 4 and 6, the other two mutated genes that were observed in each of these probands are also worthy of consideration with regard to the etiology of these congenital anomalies. This is supported by the fact that apart from a cardinal and most probable causative gene variant implicated, WES may also reveal other gene variants that may contribute to the phenotype at stake or such variants may be of unknown significance (Delplancq et al., 2020). For example, proband in Family 4 also had variants in TCOF1 and MKI67. Pathogenic variants in TCOF1, most of which are de novo and frameshift insertions and deletions, cause about 70–93% cases of Treacher Collins Syndrome (TCS) in an autosomal dominant pattern whereas about 11–23% cases of TCS are due to pathogenic variants in POLR1D (OMIM:613715) and POLR1C (OMIM:610060), though the molecular pathology of about 8%–11% of cases is unknown (Fan et al., 2019; Katsanis & Jabs, 2018). Mutant TCOF1 protein hamper ribosome biogenesis in neural crest cells (NCCs) and neuroepithelial cells, which ultimately reduce the number of NCCs that migrate to the developing craniofacial region resulting in hypoplasia of the first and second branchial arches (van Gijn et al., 2013). Significantly, the craniofacial dysmorphologies seen in the proband of Family 4 are common in patient with TCS, whereas limb abnormalities are extremely rare, occurring in less than 2% of cases (Vincent et al., 2016; Yin et al., 2019). Furthermore, the proband in Family 6 had other probable pathogenic variants in two other interesting genes, PRDM16 and CHD7. Heterozygous variants in PRDM16, which encodes a zinc finger transcription factor, cause cardiomyopathy, dilated, 1LL (CMD1LL), and left ventricular noncompaction 8 (LVNC8) which may present with facial dysmorphism (Delplancq et al., 2020). Common variants in PRDM16 have been associated with NSOFCs (Yin et al., 2018) and some other facial phenotypes (Li et al., 2019). Pathogenic heterozygous variants in CHD7 cause CHARGE syndrome and Hypogonadotropic hypogonadism 5 with or without anosmia (HH5), with some cases of CHARGE syndrome presenting with OFCs (Li et al., 2020; Siavrienė et al., 2019). Interestingly, certain genital anomalies that have been associated with both CHARGE syndrome and HH5 were observed in the proband, apart from the cleft‐clubfoot phenotype. Unfortunately, there is no evidence of this proband undergoing cleft repair at our recruitment center, presupposing he may have died from this avalanche of congenital conditions before the due date for cleft lip or palate repair.
5. CONCLUSION
In conclusion, we sought to decipher the genetic etiology of OFC and clubfoot phenotypes that co‐occurred in six probands from six affected families. Our results suggest that clubfoot and OFCs are two congenital abnormalities that can co‐occur in certain individuals with possible varying genetic causes. Observations made in this study also support the variable expressivity and phenotypic overlap in certain genetic syndromes, warranting the need for case reviews at different time points in the life of the proband. Thus, there is the need for deep phenotyping of affected individuals at different time points in order to delineate the additional phenotypes that distinguish cohorts with co‐occurring OFC‐clubfoot phenotypes from those with isolated OFCs or clubfoot. Our observations are crucial for syndrome diagnosis in OFC care, genetic counseling, and elucidating OFC and clubfoot pathogenesis.
CONFLICT OF INTERESTS
The authors declare no potential conflicts of or competing interest with respect to the research, authorship, and/or publication of this article.
AUTHOR CONTRIBUTIONS
LJJG was involved in the conception and design of the study, acquisition, analysis, and interpretation of whole‐exome and Sanger sequencing, as well as clinical data, and drafted and critically revised the manuscript. ML, TB, WAA, AAA, and TN were involved in acquisition, analysis, and interpretation of whole‐exome sequencing and Sanger sequencing data, and critically revised the manuscript. SOY, AAO, DKS, FKNA, PT, PA, and GPR were involved in patient phenotyping and recruitment, conception of study, acquisition, and interpretation of whole‐exome and Sanger sequencing, as well as clinical data, and critically revised the manuscript. PD, JCM, NLMS, and AB were involved in the conception and design of the study, acquisition, analysis, and interpretation of whole‐exome and Sanger sequencing, as well as the clinical data, and critically revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig S1
Fig S2
Supplementary Material
ACKNOWLEDGMENTS
We greatly appreciate the families that participated in this study as well as doctors and nurses at the Cleft‐Craniofacial Clinic at Komfo Anokye Teaching Hospital (KATH), Kumasi, Ghana. We want to appreciate the enormous support of Barbara Frimpong, the Nurse In‐Charge of the Cleft‐Craniofacial Clinic, KATH.
Funding information
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Dental and Craniofacial Research, NIDCR (R00 DE022378 and R01DE028300, A.B), Robert Wood Johnson Foundation (72429, A.B.); NIDCR/Fogarty International Center, FIC (K43DE029427, L.J.J.G) and the National Institutes of Health (R37 DE‐08559, J.C.M.). The funding agencies were not directly involved in the design of the study, collection, analysis and interpretation of data, and manuscript writing. All information on the manuscript, therefore, reflects the views of the authors but not necessarily that of the funding agencies.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaevet, S. R. (2010). A method and server for predicting damaging missense variants. Nature Methods, 7, 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Dhaheri, N. , Wu, N. , Zhao, S. , Wu, Z. , Blank, R. D. , Zhang, J. , Raggio, C. , Halanski, M. , Shen, J. , Noonan, K. , & Qiu, G. (2020). KIAA1217: A novel candidate gene associated with isolated and syndromic vertebral malformations. American Journal of Medical Genetics. Part A, 182(7), 1664–1672. 10.1002/ajmg.a.61607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit, S. , & Khoshhal, K. I. (2018). Genetics of clubfoot; recent progress and future perspectives. European Journal of Medical Genetics, 61, 107–113. 10.1016/j.ejmg.2017.09.006 [DOI] [PubMed] [Google Scholar]
- Beaty, T. H. , Marazita, M. L. , & Leslie, E. J. (2016). Genetic factors influencing risk to orofacial clefts: today's challenges and tomorrow's opportunities. F1000Research, 5, 2800. 10.12688/f1000research.9503.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behunova, J. , Gerykova Bujalkova, M. , Gras, G. , Taylor, T. , Ihm, U. , Kircher, S. , Rehder, H. , & Laccone, F. (2019). Distal Arthrogryposis with impaired proprioception and touch: description of an early phenotype in a boy with compound heterozygosity of PIEZO2 mutations and review of the literature. Molecular Syndromology, 9(6), 287–294. 10.1159/000494451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoud, J. , Olivetto, M. , Testelin, S. , Dakpe, S. , Bettoni, J. , & Devauchelle, B. (2020). Fraser syndrome: review of the literature illustrated by a historical adult case. International Journal of Oral and Maxillofacial Surgery, 49(10), 1245–1253. 10.1016/j.ijom.2020.01.007 [DOI] [PubMed] [Google Scholar]
- Buniello, A. , MacArthur, J. A. L. , Cerezo, M. , Harris, L. W. , Hayhurst, J. , Malangone, C. , McMahon, A. , Morales, J. , Mountjoy, E. , Sollis, E. , Suveges, D. , Vrousgou, O. , Whetzel, P. L. , Amode, R. , Guillen, J. A. , Riat, H. S. , Trevanion, S. J. , Hall, P. , Junkins, H. , … Parkinson, H. (2019). The NHGRI‐EBI GWAS Catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Research, 47, D1005–D1012. 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau, A. , Parker, M. M. , Ruczinski, I. , Taub, M. A. , Marazita, M. L. , Murray, J. C. , Mangold, E. , Noethen, M. M. , Ludwig, K. U. , Hetmanski, J. B. , Bailey‐Wilson, J. E. , Cropp, C. D. , Li, Q. , Szymczak, S. , Albacha‐Hejazi, H. , Alqosayer, K. , Field, L. L. , Wu‐Chou, Y.‐H. , Doheny, K. F. , … Beaty, T. H. (2014). Whole Exome sequencing of distant relatives in multiplex families implicates rare variants in candidate genes for oral clefts. Genetics, 197, 1039–1044. 10.1534/genetics.114.165225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butali, A. , Mossey, P. A. , Adeyemo, W. L. , Eshete, M. A. , Gowans, L. J. J. , Busch, T. D. , Jain, D. , Yu, W. , Huan, L. , Laurie, C. A. , Laurie, C. C. , Nelson, S. , Li, M. , Sanchez‐Lara, P. A. , Magee, W. P. , Magee, K. S. , Auslander, A. , Brindopke, F. , Kay, D. M. , … Adeyemo, A. A. (2019). Genomic analyses in African populations identify novel risk loci for cleft palate. Human Molecular Genetics, 28(6), 1038–1051. 10.1093/hmg/ddy402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne, S. , Dionisi‐Vici, C. , Smith, L. , Gautel, M. , & Jungbluth, H. (2016). Vici syndrome: a review. Orphanet Journal of Rare Diseases, 11, 21. 10.1186/s13023-016-0399-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camille, A. , Evelyne, A. K. , Martial, A. E. , Denise, K. , Marie‐Josée, T. A. , & Emmanuel, K. (2014). Advantages of early management of facial clefts in Africa. International Journal of Pediatric Otorhinolaryngology, 78(3), 504–506. 10.1016/j.ijporl.2013.12.031 [DOI] [PubMed] [Google Scholar]
- Carlson, J. C. , Anand, D. , Butali, A. , Buxo, C. J. , Christensen, K. , Deleyiannis, F. , Hecht, J. T. , Moreno, L. M. , Orioli, I. M. , Padilla, C. , Shaffer, J. R. , Vieira, A. R. , Wehby, G. L. , Weinberg, S. M. , Murray, J. C. , Beaty, T. H. , Saadi, I. , Lachke, S. A. , Marazita, M. L. , … Leslie, E. J. (2019). A systematic genetic analysis and visualization of phenotypic heterogeneity among orofacial cleft GWAS signals. Genetic Epidemiology, 43, 704–716. 10.1002/gepi.22214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, L. L. , Cox, T. C. , Moreno Uribe, L. M. , Zhu, Y. , Richter, C. T. , Nidey, N. , Standley, J. M. , Deng, M. , Blue, E. , Chong, J. X. , Yang, Y. , Carstens, R. P. , Anand, D. , Lachke, S. A. , Smith, J. D. , Dorschner, M. O. , Bedell, B. , Kirk, E. , Hing, A. V. , … Roscioli, T. (2018). Mutations in the epithelial cadherin‐p120‐catenin complex cause mendelian non‐syndromic cleft lip with or without cleft palate. American Journal of Human Genetics, 102, 1143–1157. 10.1016/j.ajhg.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, T. C. , Lidral, A. C. , McCoy, J. C. , Liu, H. , Cox, L. L. , Zhu, Y. , Anderson, R. D. , Moreno Uribe, L. M. , Anand, D. , Deng, M. , Richter, C. T. , Nidey, N. L. , Standley, J. M. , Blue, E. E. , Chong, J. X. , Smith, J. D. , Kirk, E. P. , Venselaar, H. , Krahn, K. N. , … Roscioli, T. (2019). Mutations in GDF11 and the extracellular antagonist, Follistatin, as a likely cause of Mendelian forms of orofacial clefting in humans. Human Mutation, 40(10), 1813–1825. 10.1002/humu.23793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullup, T. , Kho, A. L. , Dionisi‐Vici, C. , Brandmeier, B. , Smith, F. , Urry, Z. , Simpson, M. A. , Yau, S. , Bertini, E. , McClelland, V. , Al‐Owain, M. , Koelker, S. , Koerner, C. , Hoffmann, G. F. , Wijburg, F. A. , Hoedt, A. E. T. , Rogers, R. C. , Manchester, D. , Miyata, R. , … Jungbluth, H. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics, 45, 83–87. 10.1038/ng.2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delplancq, G. , Tarris, G. , Vitobello, A. , Nambot, S. , Sorlin, A. , Philippe, C. , Carmignac, V. , Duffourd, Y. , Denis, C. , Eicher, J. C. et al (2020). Cardiomyopathy due to PRDM16 mutation: First description of a fetal presentation, with possible modifier genes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 184(1), 129–135. 10.1002/ajmg.c.31766 [DOI] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, M. J. , Marazita, M. L. , Beaty, T. H. , & Murray, J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nature Reviews Genetics, 12(30), 167–178. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, X. , Wang, Y. , Fan, Y. , Du, H. , Luo, N. , Zhang, S. , & Chen, X. (2019). TCOF1 pathogenic variants identified by Whole‐exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet Journal of Rare Diseases, 14(1), 178. 10.1186/s13023-019-1136-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans, L. , Adeyemo, W. L. , Eshete, M. , Mossey, P. A. , Busch, T. , Aregbesola, B. , Donkor, P. , Arthur, F. , Bello, S. A. , Martinez, A. , Li, M. , Augustine‐Akpan, E. A. , Deressa, W. , Twumasi, P. , Olutayo, J. , Deribew, M. , Agbenorku, P. , Oti, A. A. , Braimah, R. , … Butali, A. (2016). Association studies and direct DNA sequencing implicate genetic susceptibility loci in the etiology of nonsyndromic orofacial clefts in sub‐saharan african populations. Journal of Dental Research, 95(11), 1245–1256. 10.1177/0022034516657003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans, L. J. J. , Busch, T. D. , Mossey, P. A. , Eshete, M. A. , Adeyemo, W. L. , Aregbesola, B. , Donkor, P. , Arthur, F. K. N. , Agbenorku, P. , Olutayo, J. , & Twumasi, P. (2017). The prevalence, penetrance, and expressivity of etiologic IRF6 variants in orofacial clefts patients from sub‐Saharan Africa. Molecular Genetics & Genomic Medicine, 5(2), 164–171. 10.1002/mgg3.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowans, L. J. J. , Oseni, G. , Mossey, P. A. , Adeyemo, W. L. , Eshete, M. A. , Busch, T. D. , Donkor, P. , Obiri‐Yeboah, S. , Plange‐Rhule, G. , Oti, A. A. et al (2018). Novel GREM1 variations in sub‐saharan african patients with cleft lip and/or cleft palate. The Cleft Palate‐Craniofacial Journal, 55(5), 736–742. 10.1177/1055665618754948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueneau, L. , Fish, R. J. , Shamseldin, H. E. , Voisin, N. , Tran Mau‐Them, F. , Preiksaitiene, E. , Monroe, G. R. , Lai, A. , Putoux, A. , Allias, F. , Ambusaidi, Q. , Ambrozaityte, L. , Cimbalistienė, L. , Delafontaine, J. , Guex, N. , Hashem, M. , Kurdi, W. , Jamuar, S. S. , Ying, L. J. , … Reymond, A. (2018). KIAA1109 Variants Are Associated with a Severe Disorder of Brain Development and Arthrogryposis. American Journal of Human Genetics, 102(1), 116–132. 10.1016/j.ajhg.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S. , Zhang, Y. , Zhou, T. , Wang, D. , Weng, Y. , Chen, Q. , Ma, J. , Li, Y. P. , & Wang, L. (2018). GATA4 as a novel regulator involved in the development of the neural crest and craniofacial skeleton via Barx1. Cell Death and Differentiation, 25(11), 1996–2009. 10.1038/s41418-018-0083-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, J. G. (2014). Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. European Journal of Medical Genetics, 57, 464–472. 10.1016/j.ejmg.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Huang, L. , Jia, Z. , Shi, Y. I. , Du, Q. , Shi, J. , Wang, Z. , Mou, Y. , Wang, Q. , Zhang, B. , Wang, Q. , Ma, S. , Lin, H. E. , Duan, S. , Yin, B. , Lin, Y. , Wang, Y. , Jiang, D. , Hao, F. , Zhang, L. , … Yang, Z. (2019). Genetic factors define CPO and CLO subtypes of nonsyndromic orofacial cleft. PLoS Genetics, 15(10), e1008357. 10.1371/journal.pgen.1008357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsanis, S. H. , & Jabs, E. W. (2018). Treacher collins syndrome. 2004 Jul 20 [Updated 2018 Sep 27]. In: Adam M. P., Ardinger H. H., & Pagon R. A. et al, GeneReviews® [Internet] (1993–2020). University of Washington. [Google Scholar]
- Kondo, S. , Schutte, B. C. , Richardson, R. J. , Bjork, B. C. , Knight, A. S. , Watanabe, Y. , Howard, E. , Ferreira de Lima, R. L. L. , Daack‐Hirsch, S. , Sander, A. , McDonald‐McGinn, D. M. , Zackai, E. H. , Lammer, E. J. , Aylsworth, A. S. , Ardinger, H. H. , Lidral, A. C. , Pober, B. R. , Moreno, L. , Arcos‐Burgos, M. , … Murray, J. C. (2002). Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nature Genetics, 32(2), 285–289. 10.1038/ng985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivicka‐Uzkurele, B. , Pilmane, M. , & Akota, I. (2008). Barx1, growth factors and apoptosis in facial tissue of children with clefts. Stomatologija, 10(2), 62–66. [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4, 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Labno, A. , Warkocki, Z. , Kulinski, T. , Krawczyk, P. S. , Bijata, K. , Tomecki, R. , & Dziembowski, A. (2016). Perlman syndrome nuclease DIS3L2 controls cytoplasmic non‐coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Research, 44, 21. 10.1093/nar/gkw649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , O’Donnell‐Luria, A. H. , Ware, J. S. , Hill, A. J. , Cummings, B. B. , Tukiainen, T. , Birnbaum, D. P. , Kosmicki, J. A. , Duncan, L. E. , Estrada, K. , Zhao, F. , Zou, J. , Pierce‐Hoffman, E. , Berghout, J. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536, 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie, E. J. , Taub, M. A. , Liu, H. , Steinberg, K. M. , Koboldt, D. C. , Zhang, Q. , Carlson, J. C. , Hetmanski, J. B. , Wang, H. , Larson, D. E. , Fulton, R. S. , Kousa, Y. A. , Fakhouri, W. D. , Naji, A. , Ruczinski, I. , Begum, F. , Parker, M. M. , Busch, T. , Standley, J. , … Murray, J. C. (2015). Identification of functional variants for cleft lip with or without cleft palate in or near PAX7, FGFR2, and NOG by targeted sequencing of GWAS loci. American Journal of Human Genetics, 96(3), 397–411. 10.1016/j.ajhg.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. D. , Wu, J. , Zhao, Y. , Wang, X. , Jiang, F. , Hou, Q. , Chen, D. N. , Zheng, R. , Yu, R. , Zhou, W. , & Men, M. (2020). Phenotypic spectrum of idiopathic hypogonadotropic hypogonadism patients with CHD7 variants from a large Chinese cohort. Journal of Clinical Endocrinology and Metabolism, 105(5), dgz182. 10.1210/clinem/dgz1822 [DOI] [PubMed] [Google Scholar]
- Li, Y. I. , Zhao, W. , Li, D. , Tao, X. , Xiong, Z. , Liu, J. , Zhang, W. , Ji, A. , Tang, K. , Liu, F. , & Li, C. (2019). EDAR, LYPLAL1, PRDM16, PAX3, DKK1, TNFSF12, CACNA2D3, and SUPT3H gene variants influence facial morphology in a Eurasian population. Human Genetics, 138(6), 681–689. 10.1007/s00439-019-02023-7 [DOI] [PubMed] [Google Scholar]
- Luan, S. , Luo, J. , Liu, H. , & Li, Z. (2019). Regulation of RNA decay and cellular function by 3′‐5′ exoribonuclease DIS3L2. RNA Biology, 16(2), 160–165. 10.1080/15476286.2018.1564466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midro, A. T. , Stasiewicz‐Jarocka, B. , Borys, J. , Hubert, E. , Skotnicka, B. , Hassmann‐Poznańska, E. , Sierpińska, T. , Panasiuk, B. , Schanze, D. , & Zenker, M. (2020). Two unrelated families with variable expression of Fraser syndrome due to the same pathogenic variant in the FRAS1 gene. American Journal of Medical Genetics. Part A, 182(4), 773–779. 10.1002/ajmg.a.61495. [DOI] [PubMed] [Google Scholar]
- Mossey, P. A. , & Modell, B. (2012). Epidemiology of oral clefts 2012: an international perspective. Frontiers in Oral Biology, 16, 1–18. 10.1159/000337464 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, N. , Bishop, M. , Mortillo, M. , Chopra, P. , Hetmanski, J. B. , Taub, M. A. , Moreno, L. M. , Valencia‐Ramirez, L. C. , Restrepo, C. , Wehby, G. L. , Hecht, J. T. , Deleyiannis, F. , Butali, A. , Weinberg, S. M. , Beaty, T. H. , Murray, J. C. , Leslie, E. J. , Feingold, E. , & Marazita, M. L. (2020). Whole genome sequencing of orofacial cleft trios from the Gabriella Miller Kids First Pediatric Research Consortium identifies a new locus on chromosome 21. Human Genetics, 139(2), 215–226. 10.1007/s00439-019-02099-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, S. S. , Salian, S. , Shukla, A. , Mathew, M. , & Girisha, K. M. (2017). Variable presentation of Fraser syndrome in two fetuses and a novel mutation in FRAS1. Congenit Anom (Kyoto), 57(3), 83–85. 10.1111/cga.12188 [DOI] [PubMed] [Google Scholar]
- Niceta, M. , Barresi, S. , Pantaleoni, F. , Capolino, R. , Dentici, M. L. , Ciolfi, A. , Pizzi, S. , Bartuli, A. , Dallapiccola, B. , Tartaglia, M. , & Digilio, M. C. (2019). TARP syndrome: Long‐term survival, anatomic patterns of congenital heart defects, differential diagnosis and pathogenetic considerations. European Journal of Medical Genetics, 62(6), 103534. 10.1016/j.ejmg.2018.09.001 [DOI] [PubMed] [Google Scholar]
- O’Shea, R. M. , & Sabatini, C. S. (2016). What is new in idiopathic clubfoot? Curr Rev Musculoskelet Med, 9, 470–477. 10.1007/s12178-016-9375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlakis, E. , Chiotaki, R. , & Chalepakis, G. (2011). The role of Fras1/Frem proteins in the structure and function of basement membrane. International Journal of Biochemistry & Cell Biology, 43(4), 487–495. 10.1016/j.biocel.2010.12.016 [DOI] [PubMed] [Google Scholar]
- Pavone, V. , Chisari, E. , Vescio, A. , Lucenti, L. , Sessa, G. , & Testa, G. (2018). The etiology of idiopathic congenital talipes equinovarus: a systematic review. Journal of Orthopaedic Surgery and Research, 13, 206. 10.1186/s13018-018-0913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilmane, M. , Sidhoma, E. , Akota, I. , & Kazoka, D. (2019). Characterization of cytokines and proliferation marker Ki67 in cleft affected lip tissue. Medicina (Kaunas), 55(9), 518. 10.3390/medicina55090518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch, P. , Witten, D. , Cooper, G. M. , Shendure, J. , & Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittler, M. , López‐Camelo, J. S. , Castilla, E. E. , Bermejo, E. , Cocchi, G. , Correa, A. , Csaky‐Szunyogh, M. , Danderfer, R. , De Vigan, C. , De Walle, H. , da Graça Dutra, M. , Hirahara, F. , Martínez‐Frías, M. L. , Merlob, P. , Mutchinick, O. , Ritvanen, A. , Robert‐Gnansia, E. , Scarano, G. , Siffel, C. , … Mastroiacovo, P. (2008). Preferential associations between oral clefts and other major congenital anomalies. The Cleft Palate‐Craniofacial Journal, 45(5), 525–532. 10.1597/06-250.1 [DOI] [PubMed] [Google Scholar]
- Sadler, B. , Gurnett, C. A. , & Dobbs, M. B. (2019). The genetics of isolated and syndromic clubfoot. Journal of Children's Orthopaedics, 13, 238–244. 10.1302/1863-2548.13.190063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura, T. , Kawakami, M. , Tatsumi, K. , Tanaka, T. , Morita‐Takemura, S. , Kirita, T. , & Wanaka, A. (2019). The role of the Wnt signaling pathway in upper jaw development of chick embryo. Acta Histochemica Et Cytochemica, 52(1), 19–26. 10.1267/ahc.18038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siavrienė, E. , Petraitytė, G. , Mikštienė, V. , Rančelis, T. , Maldžienė, Ž. , Morkūnienė, A. , Byčkova, J. , Utkus, A. , Kučinskas, V. , & Preikšaitienė, E. (2019). A novel CHD7 variant disrupting acceptor splice site in a patient with mild features of CHARGE syndrome: a case report. BMC Medical Genetics, 20(1), 127. 10.1186/s12881-019-0859-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavotinek, A. M. , & Tifft, A. J. (2002). Fraser syndrome and cryptophthalmos: review of the diagnostic criteria and evidence for phenotypic modules in complex malformation syndromes. Journal of Medical Genetics, 39, 623–633. 10.1136/jmg.39.9.623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36, 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soma, N. , Higashimoto, K. , Imamura, M. , Saitoh, A. , Soejima, H. , & Nagasaki, K. (2017). Long term survival of a patient with Perlman syndrome due to novel compound heterozygous missense mutations in RNB domain of DIS3L2. American Journal of Medical Genetics, 173, 1077–1081. 10.1002/ajmg.a.38111 [DOI] [PubMed] [Google Scholar]
- Umair, M. , Khan, A. , Hayat, A. , Abbas, S. , Asiri, A. , Younus, M. , Amin, W. , Nawaz, S. , Khan, S. , Malik, E. , Alfadhel, M. , & Ahmad, F. (2019). Biallelic missense mutation in the ECEL1 underlies distal arthrogryposis type 5 (DA5D). Frontiers in Pediatrics, 7, 343. 10.3389/fped.2019.00343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gijn, D. R. , Tucker, A. S. , & Cobourne, M. T. (2013). Craniofacial development: current concepts in the molecular basis of Treacher Collins syndrome. British Journal of Oral and Maxillofacial Surgery, 51(5), 384–388. 10.1016/j.bjoms.2012.09.008 [DOI] [PubMed] [Google Scholar]
- van Haelst, M. M. , Maiburg, M. , Baujat, G. , Jadeja, S. , Monti, E. , Bland, E. , Pearce, K. , Hennekam, R. C. , & Scambler, P. J. (2008). Molecular study of 33 families with Fraser syndrome: new data and mutation review. American Journal of Medical Genetics, 146A, 2252–2257. 10.1002/ajmg.a.32440 [DOI] [PubMed] [Google Scholar]
- van Rooij, I. A. L. M. , Ludwig, K. U. , Welzenbach, J. , Ishorst, N. , Thonissen, M. , Galesloot, T. E. , Ongkosuwito, E. , Bergé, S. J. , Aldhorae, K. , Rojas‐Martinez, A. , Kiemeney, L. A. L. M. , Vermeesch, J. R. , Brunner, H. , Roeleveld, N. , Devriendt, K. , Dormaar, T. , Hens, G. , Knapp, M. , Carels, C. , & Mangold, E. (2019). Non‐syndromic cleft lip with or without cleft palate: Genome‐wide association study in Europeans identifies a suggestive risk locus at 16p12.1 and supports SH3PXD2A as a clefting susceptibility gene. Genes, 10, 1023. 10.3390/genes10121023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venselaar, H. , Te Beek, T. A. , Kuipers, R. K. , Hekkelman, M. L. , & Vriend, G. (2010). Protein structure analysis of variants causing inheritable diseases. An eScience approach with life scientist friendly interfaces. BMC Bioinformatics, 11, 548. 10.1186/1471-2105-11-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, M. , Geneviève, D. , Ostertag, A. , Marlin, S. , Lacombe, D. , Martin‐Coignard, D. , Coubes, C. , David, A. , Lyonnet, S. , Vilain, C. , Dieux‐Coeslier, A. , Manouvrier, S. , Isidor, B. , Jacquemont, M.‐L. , Julia, S. , Layet, V. , Naudion, S. , Odent, S. , Pasquier, L. , … Collet, C. (2016). Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genetics in Medicine, 18(1), 49–56. 10.1038/gim.2015.29 [DOI] [PubMed] [Google Scholar]
- Vojcek, E. , Keszthelyi, T. M. , Jávorszky, E. , Balogh, L. , & Tory, K. (2020). EPG5 c.1007A > G mutation in a sibling pair with rapidly progressing Vici syndrome. Annals of Human Genetics, 84(1), 80–86. 10.1111/ahg.12337 [DOI] [PubMed] [Google Scholar]
- Waryah, A. M. , Shahzad, M. , Shaikh, H. , Sheikh, S. A. , Channa, N. A. , Hufnagel, R. B. , Makhdoom, A. , Riazuddin, S. , & Ahmed, Z. M. (2016). A novel CHST3 allele associated with spondyloepiphyseal dysplasia and hearing loss in Pakistani kindred. Clinical Genetics, 90(1), 90–95. 10.1111/cge.12694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. , Zheng, Z. , Cai, H. , Li, H. , Ye, X. , Zhang, X. , Wang, Z. , & Fu, Q. (2016). Three novel missense mutations in the filamin B gene are associated with isolated congenital talipes equinovarus. Human Genetics, 135(10), 1181–1189. 10.1007/s00439-016-1701-7 [DOI] [PubMed] [Google Scholar]
- Yin, B. , Shi, B. , & Jia, Z. L. (2018). Associations among PRDM16 polymorphisms, environmental exposure factors during mother's pregnancy, and nonsyndromic cleft lip with or without cleft palate. Hua Xi Kou Qiang Yi Xue Za Zhi, 36(5), 503–507. 10.7518/hxkq.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, B. , Shi, B. , & Jia, Z. L. (2019). Pathogenic genes and clinical therapeutic strategies for Treacher Collins syndrome. Hua Xi Kou Qiang Yi Xue Za Zhi, 37(3), 330–335. 10.7518/hxkq.2019.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. X. , Haller, G. , Lin, P. , Alvarado, D. M. , Hecht, J. T. , Blanton, S. H. , Stephens Richards, B. , Rice, J. P. , Dobbs, M. B. , & Gurnett, C. A. (2014). Genome‐wide association study identifies new disease loci for isolated clubfoot. Journal of Medical Genetics, 51(5), 334–339. 10.1136/jmedgenet-2014-102303 [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Wojcik, A. , Sanders, V. R. , Rahmani, B. , & Kurup, S. P. (2018). Ocular findings in a patient with oculofaciocardiodental (OFCD) syndrome and a novel BCOR pathogenic variant. International Ophthalmology, 38(6), 2677–2682. 10.1007/s10792-017-0754-5 [DOI] [PubMed] [Google Scholar]
- Zucchero, T. M. , Cooper, M. E. , Maher, B. S. , Daack‐Hirsch, S. , Nepomuceno, B. , Ribeiro, L. , Caprau, D. , Christensen, K. , Suzuki, Y. , Machida, J. , Natsume, N. (2004). Interferon Regulatory Factor 6 (IRF6) Gene Variants and the Risk of Isolated Cleft Lip or Palate. New England Journal of Medicine, 351(8), 769–780. 10.1056/NEJMoa032909 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.