

ABSTRACT
Background
Sufficient fetal fraction (FF) is crucial for quality control of NIPT (Non‐Invasive Prenatal Test) results. Different factors influencing bioinformatic estimation of FF should be considered when implementing NIPT. To what extent the total number of sequencing reads influences FF estimate has been unexplored. In this study, to test the robustness of SeqFF FF estimation and provide additional recommendations for NIPT analysis quality control, we compared the SeqFF FF estimates with two other methods and investigated how the number of sequencing reads and FF level affects the accuracy and precision of FF estimates.
Methods
WGS data of 516 NIPT samples from a prenatal screening program was obtained. Sample data were randomly downsampled by the read count, and FF was calculated by SeqFF software. Then, the outcome was compared with FF estimates from SNP‐ and chrY‐based methods. FF estimated with different read counts and FF levels were compared with FF at 30 M reads as a reference.
Results
SeqFF FF highly correlates with SNP‐ and chrY‐based FF estimates. Raising read count from 2 M to 10 M drastically increased the accuracy of FF estimates. After adding more reads, we saw a further improvement in FF accuracy, reaching a plateau at 20 M reads. Precision of SeqFF FF estimate is independent of FF level in the sample.
Conclusion
SeqFF is a robust method for FF estimation for both genders and for any FF level in range 2–13%. Accuracy of FF estimates highly depends on the read count. We recommend using no less than 10 M reads to achieve accurate FF estimates for NIPT analysis in clinical settings.
Keywords: cell‐free DNA, circulating fetal DNA, fetal fraction, NIPT, prenatal diagnosis
This study tests the robustness of the widely used SeqFF method in determining fetal fraction (FF) using different read counts, and provides recommendations on the minimum number of sequencing reads needed to obtain an accurate FF estimate in NIPT samples. We observed that the SeqFF FF estimates are highly affected by the read count. However, the measurement accuracy was independent of the level of FF in the sample.

1. INTRODUCTION
Non‐Invasive Prenatal Testing (NIPT) is widely used worldwide in prenatal screening settings. Fetal fraction (FF) is the proportion of total cell‐free DNA (cfDNA) present in maternal blood samples that is of fetal origin. The measurement of cell‐free fetal DNA (cffDNA) is a key quality parameter, as a sufficient amount of fetal DNA in the sample is essential to acquire valid NIPT results. Although the minimum threshold varies depending on the assays, the level of FF strongly correlates with the ability to detect aneuploidies in the fetus. Factors known to affect the FF in maternal plasma include gestational age, maternal weight, and placental biology (Hestand et al., 2019; Hou et al., 2019; Kinnings et al., 2015; van Beek et al., 2017). It is crucial to perform an efficient quality control of the NIPT test, considering all the possible factors affecting the outcome, in order to eliminate false negative results in prenatal screening; a failure to do so may result in an incorrect diagnosis or chromosomal aberrations, which are left unnoticed (Cao et al., 2016; Heling et al., 2018; Peng & Jiang, 2017).
Distinguishing between cfDNA of maternal and fetal origin is yet a challenging task. Through the years, a number of methods have been developed to determine the FF. Dedicated tests such as single‐nucleotide polymorphism‐based (SNP‐based) and methylation‐based methods are considered very accurate; however, they require additional lab work. In male pregnancies, relative FF can be estimated with high accuracy by calculating the chromosome Y (chrY) ratio directly from the shallow whole‐genome sequencing (WGS) data. As bioinformatics tools require no additional wet lab preparations, they are preferable resource‐wise, which has led to the development of several software in the field (Kim et al., 2015; Raman et al., 2019; Straver et al., 2016). The most commonly used one is a read‐count‐based multivariate method – SeqFF – enabling FF determination in pregnancies with fetuses of any gender. This open‐source software utilizes the differences in regional read depth data, achieved by shallow single‐end WGS, from autosomes only (Kim et al., 2015; Peng & Jiang, 2017). SeqFF indirectly employs fragmentation differences between maternal and fetal cfDNA. Regions which have more of the shorter cfDNA fragments (i.e., cffDNA) are preferably selected for FF estimation (Kim et al., 2015) Several studies have shown a high overall correlation of SeqFF estimates with chrY‐based ratios, SNP‐based FFs, and trisomy‐based method outcomes (Hestand et al., 2019; Johansen et al., 2016; Kim et al., 2015; van Beek et al., 2017; Wald et al., 2018).
From the introduction of NIPT, the main focus has been on the ability to detect trisomies and estimate FF. Based on their assays, laboratories all over the world use different thresholds for FF levels used in NIPT, keeping 4% FF as the most common lower threshold for quality control. However, there has been very little focus on lab‐to‐lab differences in the number of sequencing reads used for NIPT. Subsequently, it has not yet been described whether the variable number of reads used influence the estimation of FF. The precision of SeqFF software at different levels of FF is also unexplored. This study tests the robustness of the widely used SeqFF method in determining FF in samples with different read counts and FF levels and provides recommendations on the minimum number of reads needed to obtain an accurate FF for NIPT samples.
2. METHODS
2.1. Ethical compliance
This retrospective analysis only used de‐identified patient data, which do not require ethics committee approval at our institution. Consent was considered not required because the data included here are anonymous and de‐identified.
We performed retrospective whole‐genome sequencing (WGS) data analysis of NIPT samples (n = 521), which had been previously taken as part of routine prenatal screening in the Department of Clinical Genetics (Odense University Hospital, Odense, Denmark). On all of the samples, shallow coverage WGS had been performed using ThruPLEX® Plasma‐seq library prep kit (Takara Bio Inc.) and NextSeq 550 (Illumina Inc.) sequencing platform at 1 x 75 base pairs. In this manuscript, we only included samples with the total number of reads of at least 30 million (n = 446).
2.2. SeqFF fetal fraction estimation
Pre‐trained SeqFF models were used to estimate FF in this study. Single‐end sequencing data were aligned to the reference genome GRCh37/ hg19 employing BOWTIE2 (Langmead & Salzberg, 2012). Multimapped reads were removed (mapq>0) using Samtools. The header was removed by Grep from sorted BAM files. After primary data analysis, FF of the original samples and every downsampled data point was estimated by SeqFF software. The mean of the two SeqFF outcomes, elastic net (Enet), and weighted rank selection criterion (WRSC) was used as FF estimate (Kim et al., 2015).
2.3. Determining fetal fraction ratios for male fetuses by chromosome‐Y‐based method
The proportion of reads aligning to chromosome Y out of the total number of reads aligning to the autosomal chromosomes (excluding chromosomes: 13, 18, and 21) was used to achieve chrY‐based FF ratio. The chromosomes 13, 18, and 21 were excluded as these are the most commonly trisomic chromosomes, and, in the case of a trisomy‐positive sample, including them might adversely influence the estimate of FF. ChrY‐based ratio is not a direct FF estimate, but it can be used as a reference for the amount of cffDNA in the sample. This method was only used for samples from pregnancies with male fetuses (n = 244).
Single‐ended BWA (Li & Durbin, 2009) alignment data (reference genome GRCh37/ hg19) were used for this method, which was then sorted, and multi‐mapped reads were removed using Samtools view. Number of reads assigned to chromosome Y and to autosomal chromosomes was collected using Samtools idxstats.
2.4. SNP‐based fetal fraction retrieval
A subset of samples (n = 188) included in this study had previously been analyzed using a commercial SNP‐based method (Clarigo™, Agilent®), and the outcomes were used as a reference in this study. The SNP‐based method is used to screen for aneuploidies of chromosomes 21, 18, and 13 in NIPT samples (Remmerie et al., 2016). Analysis also includes FF and fetal gender. The FF determination technology is based on SNP profiling, where every fragment after PCR contains an SNP. It works by deducting maternal SNP profile from mixed cfDNA SNP profile leaving only DNA fragments, which do not have a pair with loci of maternal origin.
2.5. Statistical data analysis and SeqFF outcome validation
We compared the SeqFF FF outcome with two other well‐established methods (SNP‐based and chrY‐based) for validation of FF estimates in our dataset, proving the robustness of the SeqFF software in determining FF for both genders. The accuracy of FF estimates obtained by SeqFF software with variable number of reads was examined using chrY‐based FF estimates as the reference. For each dataset with different number of reads (from 2 to 30 million), the Pearson correlation coefficient and p‐value (statistically significant when p < 0.05) were calculated and analyzed using R package ggpubr. Residuals were calculated using R function lm.
3. RESULTS AND DISCUSSION
3.1. Strong correlation between SeqFF, chrY‐ and SNP‐based fetal fraction estimates using 30 million reads
Inter‐method comparison between SeqFF outcomes, chrY‐based ratios, and SNP‐based FFs were performed to validate the robustness of SeqFF FF estimates using 30 million reads (SeqFF[30 M]). The original SeqFF validation paper included samples with read number in range of 3.8–34.7 million (Kim et al., 2015). We therefore chose to investigate samples with read count in this range. We expected to obtain the highest accuracy by using 30 million reads. To make numbers comparable, all samples where downsampled to 30 million reads for this comparison, as they originally had different total number of reads. Correlation analysis between SeqFF[30 M] and SNP‐based FF (n = 152), SeqFF[30 M] and chrY‐based (n = 244, male fetuses only), and chrY‐ and SNP‐based (n = 87, male fetuses only) FF outcomes is shown in Figure 1.
FIGURE 1.
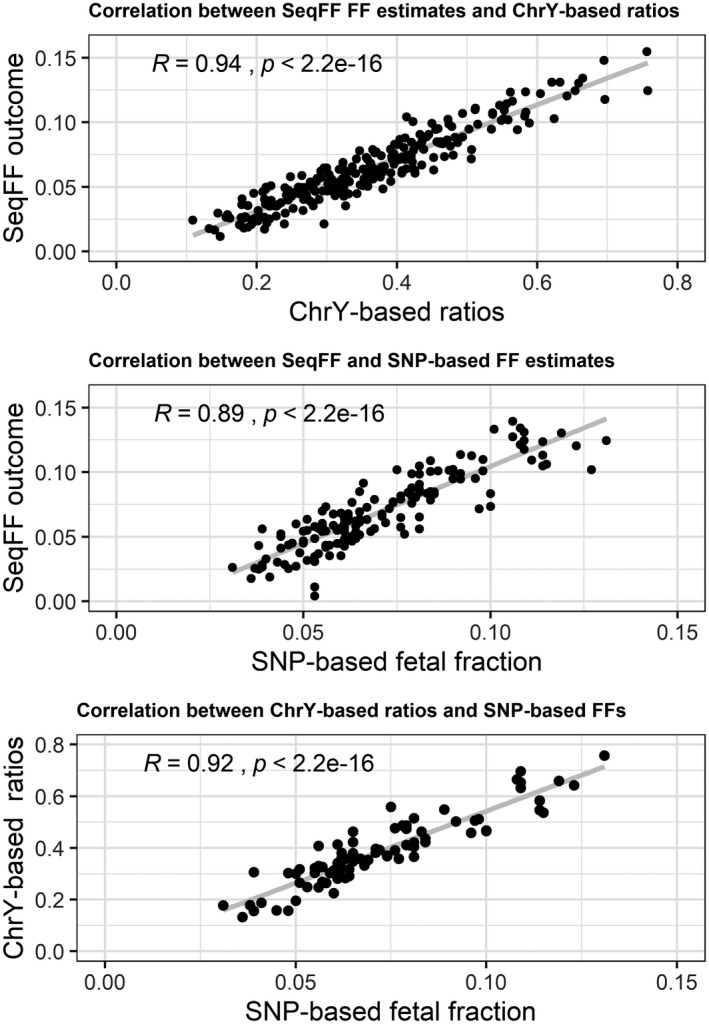
Inter‐method comparison among SeqFF fetal fraction outcomes, chrY‐based ratios and SNP‐based fetal fractions using 30 million reads. From top: in the first and the last graph, where chrYbased ratios are included, only cases with male fetuses are shown. chrY, chromosome Y; FF, fetal fraction; SNP, single nucleotide polymorphism
ChrY‐based methods have previously been shown to provide the most accurate FF estimates in pregnancies with male fetuses (Hestand et al., 2019; van Beek et al., 2017). Thus, it was chosen as reference in this study. As seen in the previous studies (Johansen et al., 2016; Kim et al., 2015; van Beek et al., 2017), showing Pearson correlation coefficient of more than 0.9, our results support that SeqFF outcomes very strongly correlate with FF estimates obtained using chrY‐based method (R = 0.943). As the chrY‐based method only can be used in pregnancies with male fetuses, we also performed an analysis using an SNP‐based method for a subset of samples, which works for both genders. We observed a strong correlation when comparing FFs by SeqFF and SNP‐based methods (R = 0.889), which was close to 0.921 presented by Kim et al. (2015). Comparison of chrY‐ and SNP‐based methods for male fetuses also presented a high correlation (R = 0.917) as seen in the original SeqFF validation article (R = 0.938) (Kim et al., 2015). In summary, our results demonstrate that high accuracy of the SeqFF FF estimates for NIPT samples from pregnancies with both genders can be achieved using 30 million reads. This also adds up to the proof that SeqFF method stays robust across different labs and using different sample preparation kits.
3.2. The total number of reads per sample influences the accuracy of FF estimates
There seems to be no universally accepted cutoff for the number of reads necessary to obtain robust and accurate FFs. Although very variable, the lower cutoff for the number of reads used in NIPT often is <5 million (Balslev‐Harder et al., 2017; Hestand et al., 2019; Hou et al., 2019; Kinnings et al., 2015; Lau et al., 2014; van Beek et al., 2017), and in some cases even samples with as low as 1.5 million reads are used (Du et al., 2020).
To investigate the influence of the number of reads on SeqFF performance, we randomly reduced the read count in each sample. By downsampling, we computationally obtained data simulating WGS performed with lower coverage. Sets of samples were obtained for every million reads in the range 2–30 million.
The highest variation in SeqFF FF estimates using the downsampled data was observed when the total number of reads was <10 million (see Figure 2). The correlation coefficient for FFs increased significantly when the read count was raised from 2 million (R = 0.755) to 10 million (R = 0.9), demonstrating greatly improved accuracy (Figure 4a). There was a further improvement seen when increasing the read count toward 20 million; however, it was a relatively small change. When the read count increased above 20 million, a plateau was reached. These results show a strong positive association between the total number of sequencing reads and the accuracy of FF estimation.
FIGURE 2.
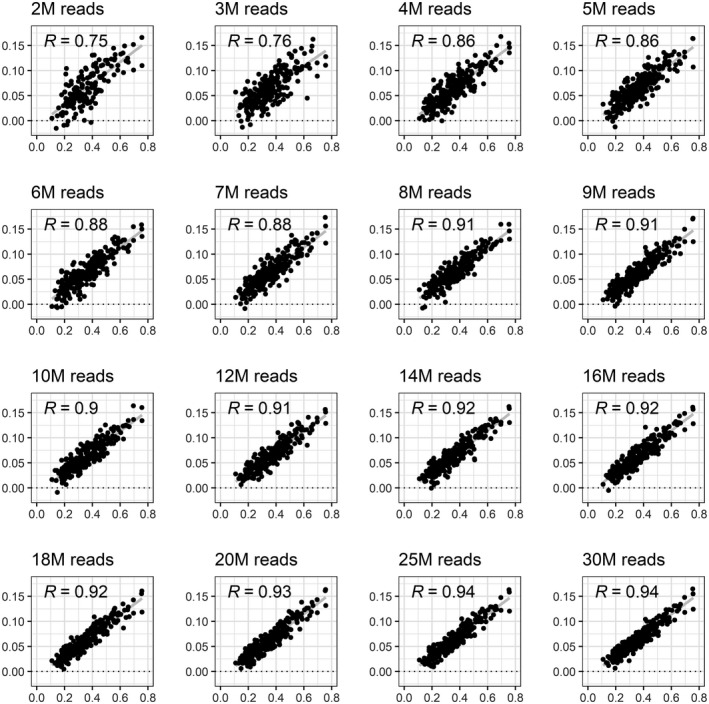
Correlation of SeqFF fetal fraction outcome with chrY‐based ratio when the different number of sequencing reads is used. Only cases with male fetuses are presented here. X axis: chrYbased ratio (reference); Y axis: SeqFF fetal fraction outcome. chrY, chromosome Y; M, million
FIGURE 4.
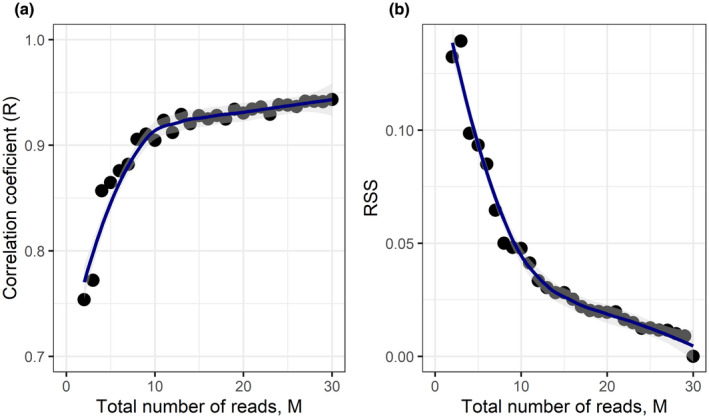
(a) Correlation curve between the correlation coefficient (R) and the total number of reads used for fetal fraction estimation. (b) Correlation curve between the residual sum‐ofsquares (RSS) and the total number of reads. M, million; R, Pearson correlation coefficient; RSS, residual sumof‐squares
The analysis of the residuals of the downsampled set (Figure 3), which were calculated from the SeqFF[30] outcome, confirmed our previously obtained results. The residual sum‐of‐squares (RSS) curve, shown in Figure 4b, decline with the read count increase. Thus, it proves the less erroneous FF estimation by SeqFF method when more sequencing reads are used.
FIGURE 3.
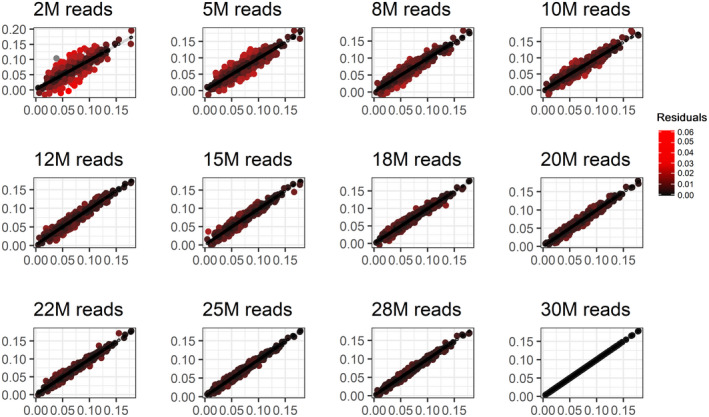
Residuals of SeqFF fetal fraction outcomes from the final SeqFF[30] result in the samples with different read count. Bright red color shows a higher deviation at the respective amount of reads from the SeqFF[30]. M, million; SeqFF[30], the SeqFF fetal fraction estimate at 30 million reads
Our data suggest that to achieve the best accuracy and precision of FF estimates, 20 million or more reads should be used. Additionally, as the biggest improvement in accuracy was seen only by reaching 10 million reads, FF estimated using fewer reads should not be held as a valid quality control for an NIPT sample.
3.3. The accuracy of SeqFF is stable across the full range of fetal fractions
To investigate the robustness of SeqFF FF estimation at different levels of FF, we grouped samples by FF and merged the data, making one huge computational sample for each percentage of FF (in range 2% to 13%). Next, the merged samples were randomly split into subsamples, resulting in sets of 50 subsamples for each million from 2 to 30 million reads. Then, FF was calculated for each subsample in order to study method's accuracy within different levels of cffDNA and how it is influenced by the number of reads. The standard error was calculated for each set of subsamples. We found that the standard error of SeqFF estimates on computational subsamples decreased only when more reads were used (see Figure S1).
To further test if there is any difference between estimates at 2–13% FF for each million reads, we performed the Kruskal–Wallis test (see Table S1). There was no significant change in precision throughout the different levels of FF (Figure 5). Our results indicate that the accuracy of FF estimates is not affected by the actual percentage of cffDNA in the sample, but only by the number of sequencing reads used.
FIGURE 5.
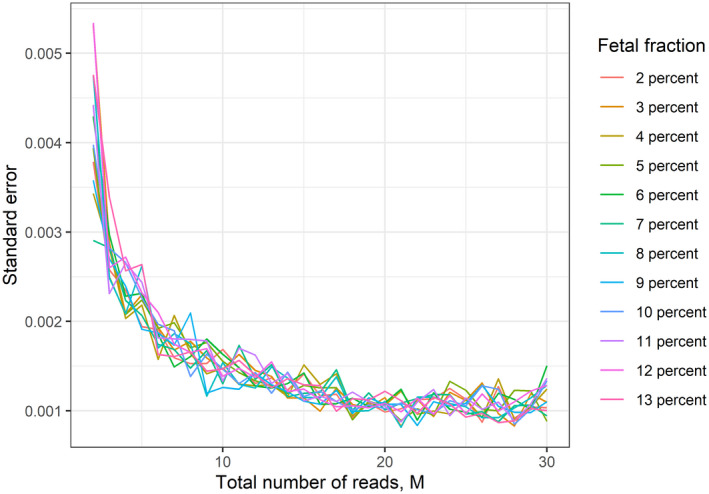
Effect of fetal fraction level and the total read count on the robustness of SeqFF method. M, million
4. CONCLUSIONS
This study adds to the proof that fetal fraction (FF) estimates obtained using SeqFF software strongly correlate with FF from other gold standards, such as SNP‐based method. In pregnancies with male fetuses, SeqFF results show a very high correlation with the ratios obtained by the chrY‐based method. Thus, it is a reliable method for accurate FF estimation for both genders.
When implementing any method for FF estimation, it is important to validate that it can detect FF not only for both genders but also within different levels of cffDNA, as those factors are normally unknown when the NIPT sample is taken. Our results demonstrate that SeqFF outcome is highly affected by the sequencing read count. The highest accuracy of FF is obtained when the read count is 20 million reads or more. However, relatively robust estimates of FF in the range 2% to 15% can be obtained already from 10 million reads. The precision of FF estimates when using this software was not affected by the level of cffDNA in the sample; thus, it worked the same through the whole range of FFs used in this study.
Accurate and precise estimation of FF is crucial for the development of NIPT and the interpretation of the screening test outcome. Our findings using SeqFF software raise the question whether other bioinformatics methods for FF estimation are influenced in the same way. Therefore, further studies including other software are deemed beneficial. To achieve the highest accuracy of FF, at least 20 million reads should be reached. As we are aware that running costs may be an issue in clinical settings, we recommend the read count of at least 10 million reads. Insufficient number of reads will affect the estimation of FF, introducing the possibility of false positive or false negative results in NIPT report.
AUTHORS' CONTRIBUTION
I.M., C.B.A., C.F., and M.J.L. participated in the design of this study. M.J.L. supervised this study. I.M. and M.J.L. performed data analysis and interpretation. I.M. wrote and modified the manuscript. C.F. and C.B.A. added clinical insights for the significance of this study and revised the draft critically for important intellectual content. All authors contributed to reviewing the manuscript. All authors read and approved the final version of manuscript.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interests regarding the publication of this paper.
Supporting information
Figure S1
Table S1
ACKNOWLEDGMENT
This paper is part of the PhD research project, which was supported by grants from the following funds: PhD Scholarship from Faculty of Health Sciences at University of Southern Denmark, Region Syddanmarks PhD Fund (grant no. 18/50642) and Research Fund (grant no. 18/17848), P.A. Messerschmidt og Hustrus Fond (grant no. 028077‐0006), Aase og Ejnar Danielsens Fond (grant no. 19‐10‐0259), A.P. Møller ‐ Fonden til Lægevidenskabens Fremme (grants no. 18‐L‐0459 and no. 19‐L‐0281), and Overlægerådets forskningsfond at Odense University Hospital (grant no. 85‐A4013).
DATA AVAILABILITY STATEMENT
The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Balslev‐Harder, M. , Richter, S. R. , Kjaergaard, S. , & Johansen, P. (2017). Correlation between Z score, fetal fraction, and sequencing reads in non‐invasive prenatal testing. Prenatal Diagnosis, 37(9), 943–945. 10.1002/pd.5116 [DOI] [PubMed] [Google Scholar]
- Cao, Y. , Hoppman, N. L. , Kerr, S. E. , Sattler, C. A. , Borowski, K. S. , Wick, M. J. , Highsmith, W. E. , & Aypar, U. (2016). False Negative cell‐free DNA screening result in a newborn with trisomy 13. Case Reports in Genetics, 2016, 7397405. 10.1155/2016/7397405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Y. , Chen, A. , Yang, R. , Zhou, T. , Zhou, Q. , Yang, L. , Wang, J. , Hong, Y. , Chen, C. , Wan, Q. , Yang, L. , & Chen, Y. (2020). A proof‐of‐concept study on the effects of low total cfDNA content and solutions to increase the NIPT trisomy 21 detection rate. Journal of Clinical Laboratory Analysis, 34(2), e23035. 10.1002/jcla.23035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heling, K. S. , Chaoui, R. , & Floter, M. (2018). False‐negative NIPT result for trisomy 21. Ultraschall in Der Medizin, 39(1), 92. 10.1055/s-0043-120758 [DOI] [PubMed] [Google Scholar]
- Hestand, M. S. , Bessem, M. , van Rijn, P. , de Menezes, R. X. , Sie, D. , Bakker, I. , Boon, E. M. J. , Sistermans, E. A. , & Weiss, M. M. (2019). Fetal fraction evaluation in non‐invasive prenatal screening (NIPS). European Journal of Human Genetics, 27(2), 198–202. 10.1038/s41431-018-0271-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Y. , Yang, J. , Qi, Y. , Guo, F. , Peng, H. , Wang, D. , Wang, Y. , Luo, X. , Li, Y. I. , & Yin, A. (2019). Factors affecting cell‐free DNA fetal fraction: Statistical analysis of 13,661 maternal plasmas for non‐invasive prenatal screening. Human Genomics, 13(1), 62. 10.1186/s40246-019-0244-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen, P. , Richter, S. R. , Balslev‐Harder, M. , Miltoft, C. B. , Tabor, A. , Duno, M. , & Kjaergaard, S. (2016). Open source non‐invasive prenatal testing platform and its performance in a public health laboratory. Prenatal Diagnosis, 36(6), 530–536. 10.1002/pd.4819 [DOI] [PubMed] [Google Scholar]
- Kim, S. K. , Hannum, G. , Geis, J. , Tynan, J. , Hogg, G. , Zhao, C. , Jensen, T. J. , Mazloom, A. R. , Oeth, P. , Ehrich, M. , van den Boom, D. , & Deciu, C. (2015). Determination of fetal DNA fraction from the plasma of pregnant women using sequence read counts. Prenatal Diagnosis, 35(8), 810–815. 10.1002/pd.4615 [DOI] [PubMed] [Google Scholar]
- Kinnings, S. L. , Geis, J. A. , Almasri, E. , Wang, H. , Guan, X. , McCullough, R. M. , Bombard, A. T. , Saldivar, J.‐S. , Oeth, P. , & Deciu, C. (2015). Factors affecting levels of circulating cell‐free fetal DNA in maternal plasma and their implications for noninvasive prenatal testing. Prenatal Diagnosis, 35(8), 816–822. 10.1002/pd.4625 [DOI] [PubMed] [Google Scholar]
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9(4), 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, T. K. , Cheung, S. W. , Lo, P. S. S. , Pursley, A. N. , Chan, M. K. , Jiang, F. , Zhang, H. , Wang, W. , Jong, L. F. J. , Yuen, O. K. C. , Chan, H. Y. C. , Chan, W. S. K. , & Choy, K. W. (2014). Non‐invasive prenatal testing for fetal chromosomal abnormalities by low‐coverage whole‐genome sequencing of maternal plasma DNA: Review of 1982 consecutive cases in a single center. Ultrasound in Obstetrics and Gynecology, 43(3), 254–264. 10.1002/uog.13277 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, X. L. , & Jiang, P. (2017). Bioinformatics approaches for fetal DNA Fraction estimation in noninvasive prenatal testing. International Journal of Molecular Sciences, 18(2), 10.3390/ijms18020453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman, L. , Baetens, M. , De Smet, M. , Dheedene, A. , Van Dorpe, J. , & Menten, B. (2019). PREFACE: In silico pipeline for accurate cell‐free fetal DNA fraction prediction. Prenatal Diagnosis, 39(10), 925–933. 10.1002/pd.5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmerie, N. , Wils, H. , De Schrijver, J. , Vyverman, M. , Crappé, J. , Standaert, L. , Brison, N. , Van Den Bogaert, K. , Vlašín, P. , Kadlecová, J. , Kozubík, K. , Blaháková, I. , Grochová, D. , Vermeesch, J. , Tijsen, J. , Theuns, J. , Vauterin, P. , & Del Favero, J. (2016). Validation of a targeted, multiplex PCR‐based NIPT test in an international multi‐centre study. Belgium: Multiplicon M.V. Retrieved from https://www.thunderbiosci.co.kr/wp‐content/uploads/2017/08/poster_990x1400mm_multi‐centre_study_clarigo_r4_1_0.pdf [Google Scholar]
- Straver, R. , Oudejans, C. B. , Sistermans, E. A. , & Reinders, M. J. (2016). Calculating the fetal fraction for noninvasive prenatal testing based on genome‐wide nucleosome profiles. Prenatal Diagnosis, 36(7), 614–621. 10.1002/pd.4816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beek, D. M. , Straver, R. , Weiss, M. M. , Boon, E. M. J. , Huijsdens‐van Amsterdam, K. , Oudejans, C. B. M. , Reinders, M. J. T. , & Sistermans, E. A. (2017). Comparing methods for fetal fraction determination and quality control of NIPT samples. Prenatal Diagnosis, 37(8), 769–773. 10.1002/pd.5079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald, N. J. , Lau, K. W. , Bestwick, J. P. , Old, R. W. , Huttly, W. J. , & Cheng, R. (2018). Specifying a gold standard for the validation of fetal fraction estimation in prenatal screening. Clinical Chemistry, 64(9), 1394–1399. 10.1373/clinchem.2018.288670 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Data Availability Statement
The data are not publicly available due to privacy or ethical restrictions.