

Abstract
Background
Hyperuricemia, pulmonary hypertension, renal failure, and alkaline intoxication syndrome (HUPRA syndrome) is a rare autosomal recessive mitochondrial disease. SARS2 gene encoding seryl‐tRNA synthetase is the only pathogenic gene of HUPRA syndrome. All the previously reported cases with HUPRA syndrome were detected for homozygous mutation.
Methods
We identified compound heterozygous mutations causing HUPRA syndrome using whole‐exome sequencing, and verifed pathogenicity with ACMG standards. All the previously published cases with SARS2 mutations were reviewed.
Results
SARS2 gene compound heterozygotes variants were detected in this Chinese patient (c.667G>A/c.1205G>A). Bioinformatics studies and protein models predict that a new variant (c.667G>A) is likely to be pathogenic. A total of six patients, five of whom were previously reported with HUPRA syndrome, were analyzed. All of the six had typical clinical manifestations of HUPRA syndrome, except the Chinese girl who had no pulmonary hypertension or alkaline intoxication. The shrunken kidney was more prominent in our proband. The average survival time for previously reported patients was 17 months, and the Chinese girl was 70 months. Three mutation variants were found, including five homozygous mutants, three of which were Palestinian (c.1169A > G), two of which were from a Spanish family (c.1205G> A), and one was a new variant (c.667G>A/c.1205G>A).
Conclusion
We found a new pathogenic form (compound heterozygous mutation) causing HUPRA syndrome, and identified a novel pathogenic site (c.667G>A) of the SARS2 gene, expanding the spectrum of SARS2 pathogenic variants. The mild phenotype in complex heterozygous mutations is described.
Keywords: HUPRA syndrome, SARS2, seryl‐tRNA synthetase
We found a new pathogenic form (compound heterozygous mutation) with a novel pathogenic site (c.667G>A) of the SARS2 gene causing HUPRA syndrome, expanding the spectrum of SARS2 pathogenic variants. Mild phenotype in complex heterozygous mutations was described.

1. INTRODUCTION
HUPRA syndrome (OMIM: #613845) is a rare mitochondrial disease caused by SARS2 mutation. SARS2 is a nuclear gene encoding seryl‐tRNA synthetase, whose primary function is to charge tRNASer with aminoacylated serine (Antonellis & Green, 2008; Wellner et al., 2018). These charged tRNA enters the ribosome during messenger RNA (mRNA) translation, to provide serine needed for mitochondrial protein synthesis. Mitochondrial proteins synthesized by this process participate in forming the mitochondrial respiratory chain complex (Belostotsky et al., 2011). Theoretically speaking, when the SARS2 gene is mutated, it will cause abnormal energy conversion. But given the residual activity of the mutant SARS2, some studies have speculated that it can be maintained above the necessary threshold in most tissues, but not in high‐energy demanding tissues (Belostotsky et al., 2011; Scheper et al., 2007). So, the penetrance after the mutation is influenced by the minimum energy required by tissues and organs to maintain normal function (González‐Serrano et al., 2019). Both the nervous system and kidney, especially the tubules, maintain their function with high energy requirements (Martín‐Hernández et al., 2005; Rahman & Hall, 2013). The previously reported SARS2 gene mutation cases are described as (a) progressive spastic paresis which is a neurological disease in a homozygous splicing mutation in SARS2 (Linnankivi et al., 2016), (b) HUPRA syndrome (Belostotsky et al., 2011; Rivera et al., 2013). HUPRA syndrome has been previously described in homozygous mutations c.1169A > G (p. Asp390Gly) and c.1205G>A (p. Arg402His) in SARS2. We report novel compound heterozygous variants (c. 667G>A p. Val223Met and c. 1205G>A p. Arg402His), which may be the new variants causing HUPRA syndrome.
2. MATERIALS AND METHODS
2.1. Ethical compliance
Ethical approval for this study was gained through the Institutional Review Board, Children's Hospital of Chongqing Medical University (2020‐99). Informed consent was obtained from the patient's parents.
2.2. Patients and pedigree
The proband from a nonconsanguineous Chinese family participated in this study. She was diagnosed, treated, and followed up in the Department of Nephrology, Children's Hospital of Chongqing Medical University, Chongqing, China. Her demographic, clinical, and laboratory data were collated.
2.3. WES and Sanger sequencing
2.3.1. Extraction of DNA
Peripheral blood samples of the proband and her parents were collected, and the genomic DNA was extracted according to the standard procedures using QIAamp DNA Bloodmini kits (Qiagen). Then, Covaris sonicator (Covaris S2, USA) was used to fragment 3 micrograms genomic DNA into 150–300 bp sized fragments.
2.3.2. The construction of the DNA library
The obtuse ends of the purified DNA fragment were repaired and a tail was added. The Illumina paired‐end (PE) adapter was used to connect the fragments overnight, and the combination product was amplified by seven‐cycle polymerase chain reaction (PCR) with an 8‐bp PE primer.
2.3.3. Capture of target region
The purified PCR product containing 3 µg DNA was hybridized with GenCapTM probe solution at 65 c for 22 hr. Then Pre‐activated Dynal Myone Streptavidin C1 magnetic beads (Invitrogen, USA) were taken to fix the products on the rotator for 1 hr at room temperature,and washed it with buffer according to the introduction book. Fifteen‐cycle PCRs were used to amplify the captured DNA libraries, and we purified and eluded them in a 30 µl volume. Finally, the enrichment degree was estimated by Agilent 2100 Bioanalyzer and quantitative PCR.
2.3.4. Sequencing with next‐generation technology
The Illumina HiSeq2500 DNA Sequencer was used to sequence the final captured DNA libraries, providing an average coverage depth for each sample of at least 100‐fold.
2.3.5. Data filtering and analysis
The Illumina pipeline (version 1.3.4) which had the default parameters was used for image analysis, error estimation, and base calls. The different samples in the original data were identified by using index primers. All nonconforming reads (reads contaminated by adapters, more than 10% of nucleotides over‐read length, average mass less than 10, or more than 50% of bases with mass less than 5) are removed. Burrows‐Wheeler comparison tool (BWA‐0.7.12‐r1044) was used to compare the remaining reads with the reference human genome (UCSC HG19). Next, GATK software 3.4–46 was used to identify SNP and indels with the recommended parameters.
2.3.6. Large deletions/duplications analysis
The depths of each genic region in the same sequencing lane which came from different samples were significantly correlated (r > 0.7); so, we used the depth of each capture region to calculate a z‐score equation. A predefined cut‐off point (±3) of derived z‐score was used to identify the large deletions and duplications. We always chose three as the truncation value, for it represents the 99.9th percentile of the normal samples set for one‐tailed region. It will be defined as a deletion (z‐score<−3) or a duplication (z‐score>3).
2.3.7. Functional annotation of genetic variants
These mutations can be classified into nonsense, deletion, splice site, insertion, synonymy, and non‐coding mutations referring to public databases (HapMap database, dbSNP 144, EXAC, ESP6500, 1000 genome variants database, and a local control database). The quality value of single‐base sequencing ≥20 was used to screen the variants, and then the following principles were followed to screen the potential candidate mutations: 1). functional variants, and 2). Variants with an allele frequency lower than 0.01 (refer to any public databases mentioned above).
2.4. Pathogenicity analysis
According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015), we performed pathogenicity analysis to identify the pathogenicity of variants.
Non‐synonymous variants were evaluated using the five algorithms. The SIFT (http://sift.bii.a‐star.edu.sg/); PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/); PROVEAN, (http://provean.jcvi.org/index.php); M‐CAP (http://bejerano.stanford.edu/MCAP/); CADD, (https://cadd.gs.washington.edu). Conservation analysis was performed through multiple sequence alignment across representative species [GenBank (www.ncbi.nlm.nih.gov)]. The pathogenicity of mutations also was assessed using structural domains analysis (https://www.ncbi.nlm.nih.gov/cdd), protein secondary structure (https://npsa‐prabi.ibcp.fr/cgi‐bin/npsa_automat.pl?page=npsa_sopma.html), the spatial structure. The Spatial structure was modeled using PyMOL application.
Reference sequence of SARS2 gene: NCBI Reference Sequence NM_017827.3.
2.5. Literature data review
"SARS2," "HUPRA," "mitochondrial disease," and "seryl‐tRNA synthase" were used as keywords to search in PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) and HGMD (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html) until June 2020. The publication language was limited to English. We also searched Wanfang (http://www.wanfangdata.com.cn/index.html), VIP (http://www.cqvip.com/), and CNKI (https://www.cnki.net/) by using the same keywords in Chinese. All of the data were open and freely accessible.
3. RESULTS
3.1. The identification of Chinese girl with HUPRA syndrome
3.1.1. Clinical manifestations
The child was born prematurely at 36 weeks of gestation (birth weight 2250 g) and was the second child of nonconsanguineous Chinese parents, with a history of thalassemia, whose height and weight always lagged behind peers since she was growing up. At the age of 4 years and 4 months old, she was admitted to the hospital because of growth retardation, anemia, and renal failure. The Proband presented with higher urea levels than creatinine elevation, hyperuricemia, and renal atrophy (right kidney 4.7 × 2.9 × 2.4 cm, left kidney 4.8 × 3.3 × 3.1 cm), without hematuria, proteinuria, and edema. There was no metabolic alkalosis, system, or pulmonary hypertension. The detailed clinical features and laboratory data were outlined in Table 1 (P6). After a total of 1 year and 5 months of follow‐up, the family refused blood purification, and the child died from uremia without manifestations of other systemic diseases or accident at the age of 5 years and 10 months old. At the last follow‐up before her death, the proband had a weight of 13.5 kg and a height of 100 cm, which were lagging behind peers just as before. She showed anemia with the hemoglobin of 98 g/L (110–150), elevated serum urea nitrogen (28.7 mmol/L (2.2–7.14)), serum creatinine (180 μmol/L (14–60)), and serum uric acid (863 μmol/L (100–410)), and decreased estimated glomerular filtration rate (eGFR) (20.83 ml/min 1.73 m−2).
TABLE 1.
Summary of the patient and five previously reported HUPRA Syndrome cases caused by SARS2 mutations.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| SARS2 mutations (NM_017827.3) | c.1169A>G (p. Asp390Gly) | c.1205G>A (p. Arg402His) |
c.667G>A (p. Val223Met) c.1205G>A (p. Arg402His) |
|||
| Reference | Belostotsky et al.,2011 | Rivera et al., 2013 | In this paper | |||
| Gender | M | F | F | F | M | F |
| Initial visit | 4 months | 7 months | 4 months | 5 months | 2 months | 4 years and 4 months |
| Ethnicity | Palestinians | Palestinians | Palestinians | Spanish | Spanish | Chinese |
| Premature | 34 weeks | 27 weeks | 27 weeks | 37 weeks | 36 weeks | 36 weeks |
| Pulmonary hypertension | + | + | + | ND① | + | − |
| Progressive renal failure | + | + | + | + | + | + |
| Metabolic alkalosis | + | + | + | + | + | − |
| Retardation | + | + | + | + | + | + |
| Anemia(g/dl) | 9(N > 10.5) | 4.8(N > 10.5) | ND | 8.4(N > 12) | 7.6(N > 12) | 8.7(N > 11) |
| Diuresis | + | ND | + | ND | ND | + |
| Hypertension | + | + | ND | + | ND | − |
| Hypotonia | + | ND | ND | + | ND | − |
| Hyperuricemia (mg/dl) | 13.8 (2.4–6.4) | 26.8 (2.4–6.4) | 14.1 (2.4–6.4) | 11 (2.2–7) | 9.4 (2.2–7) | 11 (2.4–6.6) |
| Low FeUA | 5.7% (N > 7%) | <5% (N > 7%) | +(DU) | ND | 2–3%(N > 7%) | ND |
| Hyponatremia (mEq/L) | 116 (133–146) | 124 (133–146) | 122 (133–146) | ND | +(DU) | 146.8 (132–145) |
| Hypomagnesemia (mg/dl) | 0.9 (1.58–2.4) | 1.2 (1.58–2.4) | 0.97 (1.58–2.4) | ND | 1.7 (1.5–2.3) | 1.29 (1.29–2.70) |
| High FeMg | 12.80% (N < 5%) | +(DU) | +(DU) | ND | ND | ND |
| Elevated BUN (disproportionate to Scr) | + | + | + | + | + | + |
| Urea(mg/dl) | 44 (5–18) | 87 (5–18) | +(DU) | 159 (20–48) | +(DU) | 129 (15–40) |
| Serum creatinine (mg/dl) | 0.99 (0.2–0.4) | 1.14 (0.2–0.4) | +(DU) | 1.01 (0.35–0.5) | 0.96 (0.35–0.5) | 1.24 (0.17–1.02) |
| Elevated lactic acid(mmol/L) | 10.2 (0.5–2.4) | 5.54 (0.5–2.4) | 8 (0.5–2.4) | − | − | 4.57 (0.7–2.1) |
| Creatinine clearance (ml/min 1.73 m−2) | 29 (39–114) | 22 (39–114) | 31 (39–114) | 42 (>60) | ND | 25 (>90) |
| amino acids and acylcarnitine in plasma(μM) |
Serum alanine 1029 (N < 547) |
ND | ND | − | − | − |
| Proteinuria(mg/mg) | 1.26 (N < 0.5) | ND | ND | − | ND | − |
| Size of kidney | normal | ND | normal | normal | normal | Diminution |
| Survival time | 14 months | 10 months | 1+ year | 26 months | 21 months | 5 years and 10 months |
①, Pulmonary hypertension is unknown, but postmortem reports suggest hypertrophic obstructive cardiomyopathy. P1, P2 are from distant cousins of the same family; the parents of P2 are cousins; P4 and P5 are siblings of the same parents. FeMg, fractional excretion of magnesium; FeUA, fractional excretion of uric acid; ND, not determined; +(DU), it exists but the details are unknown.
We conducted a genealogical survey, and no exact positive family history of HUPRA syndrome was found (Figure 1). Except for the patient's older sister (IV 3), who had retardation of development, dying at the age of 1 year and 10 months old for an unclear reason. Several family members had hyperuricemia without pulmonary hypertension, renal failure, alkaline intoxication, or other symptoms similar to HUPRA syndrome: the brother of the maternal grandmother (Ⅱ7) suffered gout, and the patrilineal grandmother (Ⅱ2) had mild hyperuricemia. The father had transient elevations of serum creatinine and uric acid (Scr 80 μmol/L (14–60), UA 425 μmol/L (100–410)).
FIGURE 1.
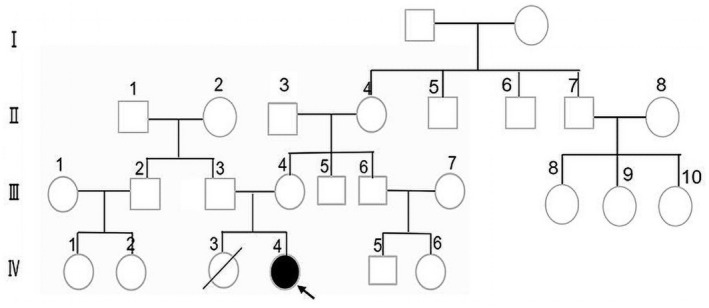
Family pedigree based on clinical presentation (The arrow points to the proband). The brother of maternal grandmother (Ⅱ7) suffered gout, and the patrilineal grandmother (Ⅱ2) had mild hyperuricemia. The father had transient elevated creatinine and uric acid levels.
3.1.2. Molecular genetics
The proband was identified as having compound heterozygous mutations in the SARS2 gene by WES and Sanger sequencing: ①c.667G>A, p. (Val223Met); this variant came from her mother, and it was the first time to be identified. This variant ②c.1205G>A, p. (Arg402His) came from her father, and the homozygous mutation at the same site has been reported in two Spanish children (Rivera et al., 2013) (Figure 2Aa,b)). The parents (I‐1 and I‐2), presented the heterozygosity mutation (Figure 2Ac).
FIGURE 2.
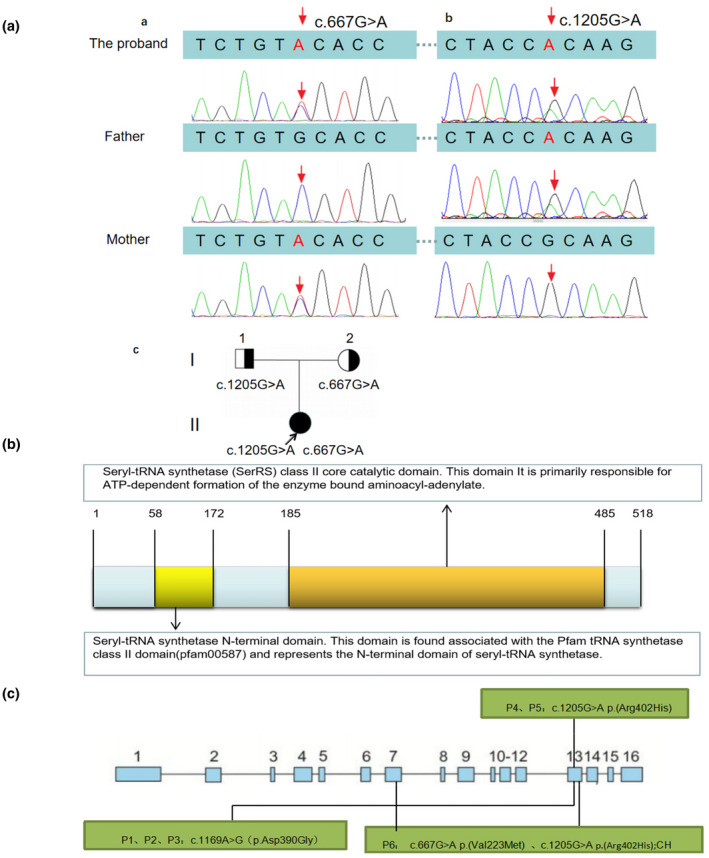
Pathogenic variants in SARS2 gene and the domain of seryl‐tRNA synthetase (NM_017827.3). (A) The proband was compound heterozygotes for two variants in the SARS2 gene. Variant 1 located in chromosome 19 was missense mutation, c.667G>A, p. Val223Met (maternal allele) (a), and Variant 2 were missense mutation too, c.1205G>A, p. Arg402His (paternal allele) (b). Family pedigree based on molecular genetics. (The arrow points to the proband) (c). (B) The domain of seryl‐tRNA synthetase. There are 518 amino acids in seryl‐tRNA synthetase. The 58th to 172nd amino acids form the seryl‐tRNA synthetase N‐terminal domain, the 185th to 485th form the SerRS class II core catalytic domain. (C) Gene structure of SARS2, showing exons 1 to 16 in blue boxes, and its pathogenic variants in patients with HUPRA syndrome reported previously and described in the present study. CH, compound heterozygous.
3.1.3. Pathogenicity analysis
According to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015), we performed a pathogenicity analysis from the following aspects.
3.1.3.1. Analysis in silico prediction
Non‐synonymous variants were evaluated using the five algorithms and the actual results of the electronic version are shown in Table 2.
TABLE 2.
Analysis in silico prediction.
| Nucleotide | SIFT | PROVEAN | PolyPhen−2 | M‐CAP | CADD |
|---|---|---|---|---|---|
| c.667G>A |
0.003 Pathogenic |
−1.84 Pathogenic |
0.796 Possible pathogenic |
0.137 Possible pathogenic |
28 Pathogenic |
| c.1205G>A |
0.001 Pathogenic |
−4.56 Pathogenic |
0.999 Possible pathogenic |
0.530 Possible pathogenic |
31 Pathogenic |
3.1.3.2. Conservation analysis
The SARS2 missense pathogenic variants of the proband are localized in two highly conserved amino acid sequences (the 223rd position and the 402nd position) among representative species (Figure 3A).
FIGURE 3.
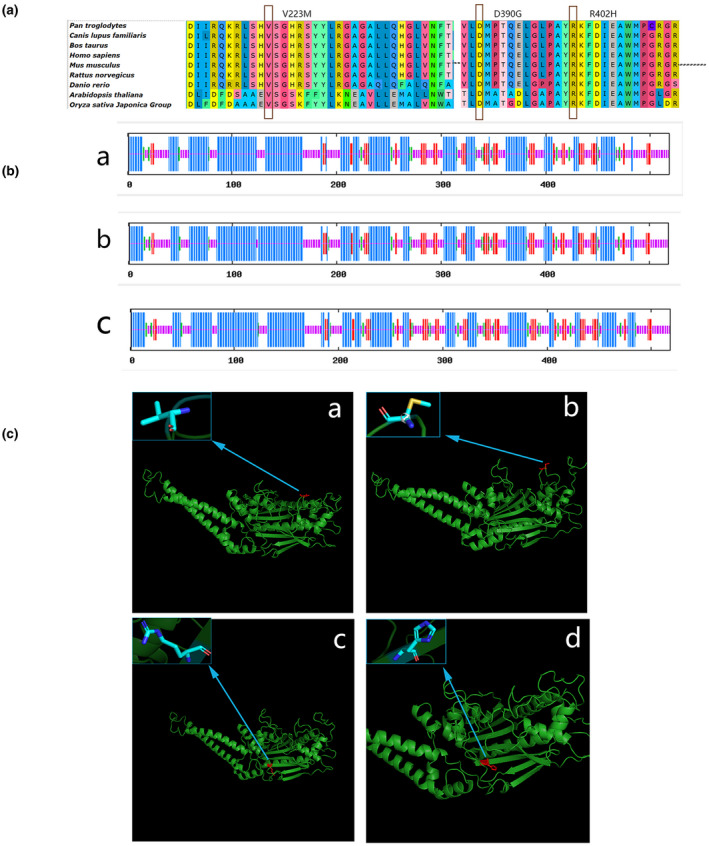
Multiple sequence alignment, secondary and spatial structure of seryl‐tRNA synthetase. (A) Multiple sequence alignment. The SARS2 pathogenic variants (the arrow points) of the proband are localized in two highly conserved amino acid sequences (p. Arg402 and p. Val223) among representative species. (B) Secondary and spatial structure of seryl‐tRNA synthetase. There are four secondary structures: alpha helix(blue), extended strand(red), beta turn(green), and random coil(purple). a: secondary structure of the wild type, b: secondary structure of the 223rd position of mutations Val to Met, c: secondary structure of the 402nd position of mutations Arg to His. C) Spatial structure of seryl‐tRNA synthetase. The predicted wild type and mutated proteins of SARS2(NM_017827.3) through in silico analysis. The mutated sites affected the amino acid side chain; The blue arrow positions represent the 223rd position of mutations Val (a; wild type) to Met (b; mutated), the blue arrow positions represent the 402nd position of mutations Arg (c; wild type) to His (d; mutated). After the mutation from non‐polar amino acid Val (a; wild type) to polar amino acid Met (b; mutated), there is an extra sulfur atom, and the structure ‘‐CH(CH 3)2’ changes to the structure ‘‐CH2CH2SCH3'. After the mutation of Arg (c; wild type) to His (d; mutated), 6 hydrogen atoms and 1 nitrogen atom decreased, and methylene, carbon atoms, amino, and imidogen form a chain structure with a carbon‐nitrogen double bond before mutation, whereas after the mutation, methylene, methyne, carbon atoms, nitrogen atoms, and imidogen form a ring structure (imidazolyl) containing 1 carbon‐carbon double bond and 1 carbon‐nitrogen double bond. Different colors represent different atoms: light blue: C atom; dark blue: N atom; red: O atom; silver: H atom; yellow: S atom.
3.1.3.3. Structural domains
SARS2‐coded protein is a homodimer enzyme, and this protein has two domains. The first one is the seryl‐tRNA synthetase N‐terminal domain: this domain is found to be associated with the Pfam tRNA synthetase class II domain (pfam00587) and represents the N‐terminal domain of seryl‐tRNA synthetase. The second one is the seryl‐tRNA synthetase (SerRS) class II core catalytic domain: this domain is primarily responsible for ATP‐dependent formation of the enzyme‐bound aminoacyl‐adenylate. Both p. Arg402 and p. Val223 are located in the seryl‐tRNA synthetase (SerRS) class II core catalytic domain, participating in the formation of protein functional structure region (Figure 2B).
3.1.3.4. Secondary and spatial structure
Secondary structure: In the wild type, the protein is composed of four secondary structures: alpha‐helix (45.95%), extended strand (12.36%), beta‐turn (5.41%), and random coil (36.29%). Among them, the 223rd position, Val, participates in the random coil and the 402nd position, Arg, contributes to the formation of the alpha helix. When the 223rd position Val mutated to Met, the second structure changed: alpha‐helix (46.33%), extended strand (12.55%), beta‐turn (4.83%), and random coil (36.29%); the 223rd amino acid (Met) participates in the formation of a random coil. When the 402nd position Arg mutated to His, the second structure consists of alpha‐helix (44.98%), extended strand (12.93%), beta‐turn (6.56%), and random coil (35.52%), whereas the 402nd amino acid (His) participates in the formation of the random coil (Figure 3B).
In the normal phenotypic spatial structure of seryl‐tRNA synthetase (SerRS), its class II core catalytic domain is located in the c‐terminal spherical domain which is built around an eight antiparallel β‐sheet, encompassed by three helical bundles (Chimnaronk et al., 2005). The p. Arg402 is positioned at eight antiparallel β‐sheet of the C‐terminal globular domain. The p. Val223 is also located in seryl‐tRNA synthetase (SerRS) class II core catalytic domain, although it does not participate in forming the core β‐sheet nor the surround α‐helix, just form a β‐turn between two α‐helices. We predicted the spatial structures of wild type and mutated proteins of SARS2 through in silico analysis, and the mutated sites affected the amino acid side chain (Figure 3C).
In conclusion, both p. (Val223Met) and p. (Arg402His) are pathogenic mutations in the silico prediction, which are localized in two highly conserved amino acid sequences, participating in the formation of protein structural domains. The mutations lead to change in the secondary structure, affecting the amino acid side chain in the special structure. All of the above is possible to influence the normal expression of the SARS2 gene, leading to mitochondrial disease. The parents were heterozygous carriers, and the mutations were segregated according to a strictly recessive model with full penetrance. The variants are considered likely pathogenic according to ACMG Standards (Richards et al., 2015).
3.2. Literature review
All identified articles were read in full. Five cases with HUPRA syndrome were reported (Belostotsky et al., 2011; Rivera et al., 2013), including the proband we investigated in this study, totally six patients are named P1‐P6 respectively (P6 is the Chinese girl), and the relevant information is summarized in Table 1.
Except for P6, all the other five patients were homozygous mutations. All of the six had retardation of development, a history of preterm delivery, and obvious hyperuricemia. In the five patients with homozygous mutations: all of them had metabolic alkaline intoxication; Four of them had pulmonary hypertension, and postmortem reports of P4 suggest hypertrophic obstructive heart disease. Clinical manifestations of the proband in the present study (P6) were similar to those five previously reported homozygous mutations patients, but without pulmonary hypertension and metabolic alkaline intoxication. Moreover, the shrunken kidney was more prominent in P6. All the cases had a poor prognosis, and the end‐point follow‐up results were death. Among the five homozygous mutations patients, the shortest survival time was 10 months, the longest was 26 months, whereas for the P6, it was 70 months. It was suggested that proband P6 has milder clinical manifestations and longer survival time.
Renal biopsy was performed in three patients, who were characterized by interstitial fibrosis, tubular atrophy, and markedly enlarged mitochondria in tubular epithelial cells without glomerular lesion (Table 3). The pathology suggested that renal failure of HUPRA syndrome is because of tubular interstitial damage.
TABLE 3.
Renal biopsy results of three patients with HUPRA syndrome.
| Patient | Glomerulus | Tubulo‐interstitial | Tubular epithelial cells | Blood vessel | Immunohistochemistry |
|---|---|---|---|---|---|
| 1 | Normal | Dedifferentiated, atrophic tubules with thick basement membrane or tubules that were completely denuded. | Tubular epithelial cells contained markedly enlarged mitochondria with paracrystalline lesions. | Hyperplastic arteriolitis | Negative |
| 4 | Normal | Interstitial fibrosis and tubular atrophy with vacuoles | Enlarged mitochondria | N | N |
| 5 | Normal | Interstitial fibrosis and unspecific tubular damage | Enlarged mitochondria | Normal | N |
N, Not determined.
Three mutation sites were found. Out of the five homozygous mutations cases, three of which were Palestinian (c.1169A>G p. (Asp390Gly)), two of which were from a family in Spain (c.1205G>A p. (Arg402His)), and one compound heterozygous mutations case (c.1205G>A p. (Arg402His), c.667G>A p. (Val223Met)) were identified. (Figure 2C).
4. DISCUSSION
Mitochondrial diseases (MD) are multi‐system diseases caused by mutations in nuclear genes or mitochondrial genes, and nuclear gene mutations account for 75%–95% of mitochondrial diseases (Graham, 2012). The mitochondrial proteome contains about 1,500 proteins, of which only 13 are encoded by mitochondrial genes. HUPRA syndrome is a mitochondrial disease caused by the nuclear gene SARS2 mutation. Nuclear gene SARS2 encodes serine‐tRNA synthase to transport serine in mitochondrial ribosomes. SARS2 gene mutation leads to a decrease in the number of seryl‐tRNA synthase, which fails to participate in mitochondrial protein translation, which leads to interference in mitochondrial oxidative phosphorylation. Belostotsky et al. identified and named the first case of HUPRA syndrome, which is an autosomal recessive disorder caused by homozygous mutations in the SARS2 gene (c.1169A > G (p. Asp390Gly)). Vitro functional verification proved that the mutation damaged the catalytic activity of seryl‐tRNA synthetase. Muscle biopsies showed reduced activity of respiratory chain synthase complex I, complex III, and complex IV in skeletal muscle cells. Combined with conservative analysis and family lineage investigation, c.1169A > G (p. Asp390Gly) was considered a pathogenic mutation (Belostotsky et al., 2011). Also, HUPRA syndrome was reported to be caused by a mutation in c.1205G>A (p. Arg402His), in which skeletal muscle respiratory synthase activity was normal, but cutaneous fibroblast complex I and complex IV were slightly decreased (Rivera et al., 2013).
Our study presented the first HUPRA syndrome of a Chinese patient, which was caused by SARS2 gene mutation. The patient had the following typical clinical manifestations of HUPRA syndrome: development retardation, renal failure, obvious hyperuricemia, and a history of preterm delivery, but had no metabolic alkaline intoxication and pulmonary hypertension. Moreover, the shrunken kidney was more prominent in our proband. Compared with other patients with HUPRA syndrome who died at a very young age, the Chinese girl had a longer life and died at the age of 70 months. And in the previously reported cases, two of whom died of multiorgan failure and three of whom died of pulmonary hypertension with a mean survival age of 17 months. It was suggested this patient with compound heterozygotes variants might have milder clinical manifestations with a longer survival time to progress to renal insufficiency, and finally bilateral renal atrophy due to uremia. Besides, we observed that all HUPRA syndromes manifest mainly as hyperuricemia, whereas the most frequent renal dysfunction of most mitochondrial tubulopathy was hyperuricosuria and hypouricemia resulting from decreased reabsorption (Pérez‐Albert et al., 2018). The reason for hyperuricemia in HUPRA syndrome is probably caused by progressive renal failure. Significant hyperuricemia in HUPRA syndrome is derived from the reduction of uric acid filtration caused by declined glomerular filtration rate, masking the tendency to hypouricemia due to the mitochondrial tubular diseases. Besides this, relative volume depletion due to polyuria may also be responsible for the hyperuricemia with decreased urinary fractional excretion of uric acid. The accepted mechanism underlying hyperuricemia within the context of pulmonary hypertension is increased production of uric acid in ischemic/hypoxic cardiac tissue (Bendayan et al., 2003).
In our study, we detected the first case of HUPRA syndrome with compound heterozygous mutations (c.667G>A/c.1205G>A) in the SARS2 gene. Pathogenicity of mutation c.1205G>A p. Arg402His has been reported (Rivera et al., 2013), and we verified it with ACMG guidelines as follows: (a) The patient fibroblasts have a combined deficiency in complexes of the mitochondrial respiratory chain, which is expected for defects in a gene involved in mitochondrial protein synthesis (Rivera et al., 2013); (b) The mutation is located close to the only previously described mutation in SARS2 associated with HUPRA syndrome, in the class II core catalytic domain of seryl‐tRNA synthetase (SerRS); (c) The mutation is very rare in the population: in ExAC All, the allele frequencies of the variant is 0.00001655, and in ExAC East Asians (EAS) or 1000g2015aug_ALL, it is 0; (d) The mutation segregates with the disease (Rivera et al., 2013); (e) Missense mutation in SARS2 gene is the sole cause of HUPRA syndrome, with a low rate of benign missense variations; (f) The amino acid residue is highly conserved, and the substitution is predicted to have effect in the protein structure. Therefore, according to the ACMG classification criteria (Richards et al., 2015), the mutation c.1205G>A is pathogenic (PS3 + PM1 + PM2 +PP1 + PP2 + PP3). Several findings support the pathogenic role of the novel change (c.667G>A p. Val223Met): (a) The mutation is located in seryl‐tRNA synthetase (SerRS) class II core catalytic domain, participating in the formation of protein functional structure region, which was also the hot spot mutation region of SARS2 gene. (b) This mutation was low‐frequency variation in the database ExAC All (0.000008587), ExAC East Asians (EAS) (0), and 1000g2015aug_ALL (0). (c) Pathogenic variant ((p. (Arg402His)), which has been demonstrated previously, was detected in trans. (d) It was a missense variant of the SARS2 gene that has a low rate of benign missense variations and in which missense variants are a common mechanism of HUPRA syndrome. (e) The amino acid residues p. Val223 and p. Arg402 are highly conserved, and the scores for pathogenicity obtained by in‐silico analysis in different software are probably damaged; besides, p. Val223 and p. Arg402 are involved in protein domain formation. When they mutate, so do the structures of secondary and three‐dimensional. Therefore, according to the ACMG classification criteria (Richards et al., 2015), c.667G>A is likely pathogenic (PM1 + PM2 + PM3 + PP2 + PP3). The evidence showed it is a new pathogenic form with a novel pathogenic site (c.667G>A) of the SARS2 gene causing HUPRA syndrome.
As the present case has a mild clinical phenotype, we attempt to explain the possible reasons for varying severity clinical phenotypes: (a) P. Val223 is less critical than p. Asp390 and p. Arg402. Seryl‐tRNA synthetase core domain is built around eight antiparallel β‐sheet. P. Asp390 is in the second β‐sheet and p. Arg402 in the third. Although p. Val223 is also located in the same domain, it does not participate in forming the core β‐sheet, just forming a β‐turn between two α‐helices. (b) Seryl‐tRNA synthetase may have secondary functions that are critical to the urinary system or other specific tissues, and they have yet to be discovered (Guo & Schimmel, 2013; Kim et al., 2011). These secondary functions are involved in the addition of appended domains (Guo et al., 2010)and protein subtypes produced by alternative splicing (Wakasugi et al., 2002), and function loss may arise due to the impaired secondary functions associated with the pathogenic variants. Based on this, we propose a hypothesis: both the p. Asp390 and the p. Arg402 are associated with the seryl‐tRNA synthetase secondary functional, whereas the p. Val223 is not, and it requires further verification.
In conclusion, we identified the first case of HUPRA syndrome with compound heterozygous mutations (c.667G>A/c.1205G>A) in the SARS2 gene. The present results expanded the spectrum of SARS2 pathogenic variants, describing the differences in clinical manifestations between homozygous and compound heterozygous mutations. But unfortunately, we were unable to obtain pathological reports of the muscle or the kidney, and the study was only based on pathogenicity prediction without functional verification.
CONFLICT OF INTERESTS
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Yi Zhou analyzed the genetic data and wrote the manuscript. Cheng Zhong, Qin Yang collected the clinical data from the patient. Gaofu Zhang, Haiping Yang, Qiu Li conducted literature review. Mo Wang designed and revised the manuscript. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Mygenostics Gene Technology Co., Ltd. (Chongqing, China) for providing whole‐exome and Sanger sequencing technology support for this study.
Yi Zhou made the great contribution to the article and should be regarded as first author.
Funding information
This study was financially supported by“The central government directs special funds for local science and technology development.”
DATA AVAILABILITY STATEMENT
This is an open‐access article under the terms of the Creative Commons Attribution‐NonCommercial‐NoDerivs License, which permits use and distribution in any medium, provided the original work is properly cited, the use is noncommercial and no modifications or adaptations are made.
REFERENCES
- Antonellis, A. , & Green, E. D. (2008). The role of aminoacyl‐tRNA synthetases in genetic diseases. Annual Review of Genomics and Human Genetics, 9, 87–107. 10.1146/annurev.genom.9.081307.164204 [DOI] [PubMed] [Google Scholar]
- Belostotsky, R. , Ben‐Shalom, E. , Rinat, C. , Becker‐Cohen, R. , Feinstein, S. , Zeligson, S. , Segel, R. , Elpeleg, O. , Nassar, S. , & Frishberg, Y. (2011). Mutations in the mitochondrial seryl‐tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. American Journal of Human Genetics, 88(2), 193–200. 10.1016/j.ajhg.2010.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendayan, D. , Shitrit, D. , Ygla, M. , Huerta, M. , Fink, G. , & Kramer, M. R. (2003). Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respiratory Medicine, 97, 130–133. [DOI] [PubMed] [Google Scholar]
- Chimnaronk, S. , Gravers Jeppesen, M. , Suzuki, T. , Nyborg, J. , & Watanabe, K. (2005). Dual‐mode recognition of noncanonical tRNAs(Ser) by seryl‐tRNA synthetase in mammalian mitochondria. The EMBO Journal, 24(19), 3369–3379. 10.1038/sj.emboj.7600811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Serrano, L. E. , Chihade, J. W. , & Sissler, M. (2019). When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl‐tRNA synthetases. Journal of Biological Chemistry, 294(14), 5309–5320. 10.1074/jbc.REV118.002953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, B. H. (2012). Diagnostic challenges of mitochondrial disorders: complexities of two genomes. Methods in Molecular Biology, 837, 35–46. 10.1007/978-1-61779-504-6_3 [DOI] [PubMed] [Google Scholar]
- Guo, M. , & Schimmel, P. (2013). Essential nontranslational functions of tRNA synthetases. Nature Chemical Biology, 9(3), 145–153. 10.1038/nchembio.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Schimmel, P. , & Yang, X. L. (2010). Functional expansion of human tRNA synthetases achieved by structural inventions. FEBS Letters, 584(2), 434–442. 10.1016/j.febslet.2009.11.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , You, S. , & Hwang, D. (2011). Aminoacyl‐tRNA synthetases and tumorigenesis: more than housekeeping. Nature Reviews Cancer, 11(10), 708–718. 10.1038/nrc3124 [DOI] [PubMed] [Google Scholar]
- Linnankivi, T. , Neupane, N. , Richter, U. , Isohanni, P. , & Tyynismaa, H. (2016). Splicing defect in mitochondrial seryl‐tRNA synthetase gene causes progressive spastic paresis instead of HUPRA syndrome. Human Mutation, 37(9), 884–888. 10.1002/humu.23021 [DOI] [PubMed] [Google Scholar]
- Martín‐Hernández, E. , García‐Silva, M. T. , Vara, J. , Campos, Y. , Cabello, A. , Muley, R. , del Hoyo, P. , Martín, M. A. , & Arenas, J. (2005). Renal pathology in children with mitochondrial diseases. Pediatric Nephrology, 20(9), 1299–1305. 10.1007/s00467-005-1948-z [DOI] [PubMed] [Google Scholar]
- Pérez‐Albert, P. , de Lucas Collantes, C. , Fernández‐García, M. Á. , de Rojas, T. , Aparicio López, C. , & Gutiérrez‐Solana, L. (2018). Mitochondrial disease in Children: The nephrologist's perspective. JIMD Reports, 42, 61–70. 10.1007/8904_2017_78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, S. , & Hall, A. M. (2013). Mitochondrial disease–an important cause of end‐stage renal failure. Pediatric Nephrology, 28(3), 357–361. 10.1007/s00467-012-2362-y [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera, H. , Martín‐Hernández, E. , Delmiro, A. , García‐Silva, M. T. , Quijada‐Fraile, P. , Muley, R. , Arenas, J. , Martín, M. A. , & Martínez‐Azorín, F. (2013). A new mutation in the gene encoding mitochondrial seryl‐tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrology, 14, 195. 10.1186/1471-2369-14-195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper, G. C. , van der Klok, T. , van Andel, R. J. , van Berkel, C. G. , Sissler, M. , Smet, J. , Muravina, T. I. , Serkov, S. V. , Uziel, G. , Bugiani, M. , Schiffmann, R. , Krägeloh‐Mann, I. , Smeitink, J. A. M. , Florentz, C. , Van Coster, R. , Pronk, J. C. , & van der Knaap, M. S. (2007). Mitochondrial aspartyl‐tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nature Genetics, 39(4), 534–539. 10.1038/ng2013 [DOI] [PubMed] [Google Scholar]
- Wakasugi, K. , Slike, B. M. , Hood, J. , Otani, A. , Ewalt, K. L. , Friedlander, M. , Cheresh, D. A. , & Schimmel, P. (2002). A human aminoacyl‐tRNA synthetase as a regulator of angiogenesis. Proceedings of the National Academy of Sciences United States of America, 99(1), 173–177. 10.1073/pnas.012602099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner, K. , Betat, H. , & Mörl, M. (2018). A tRNA's fate is decided at its 3’ end: Collaborative actions of CCA‐adding enzyme and RNases involved in tRNA processing and degradation. Biochimica Et Biophysica Acta (BBA) ‐ Gene Regulatory Mechanisms, 1861(4), 433–441. 10.1016/j.bbagrm.2018.01.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is an open‐access article under the terms of the Creative Commons Attribution‐NonCommercial‐NoDerivs License, which permits use and distribution in any medium, provided the original work is properly cited, the use is noncommercial and no modifications or adaptations are made.