

Abstract
Receptor Tyrosine Kinases (RTKs) are transmembrane receptors of great clinical interest due to their role in disease. Historically, therapeutics targeting RTKs have been identified using in vitro kinase assays. Due to frequent development of drug resistance, however, there is a need to identify more diverse compounds that inhibit mutated but not wild-type RTKs. Here, we describe MaMTH-DS (Mammalian Membrane Two-Hybrid Drug Screening), a live-cell platform for high-throughput identification of small-molecules targeting functional protein-protein interactions of RTKs. We applied MaMTH-DS to an oncogenic Epidermal Growth Factor Receptor (EGFR) mutant resistant to the latest generation of clinically approved tyrosine kinase inhibitors (TKIs). We identified four mutant-specific compounds, including two that would not have been detected by conventional in vitro kinase assays. One of these targets mutant EGFR via a novel mechanism of action, distinct from classical TKI inhibition. Our results demonstrate how MaMTH-DS is a powerful complement to traditional drug screening approaches.
INTRODUCTION
Receptor tyrosine kinases (RTKs) are an important class of integral membrane cell surface receptors, responsible for triggering diverse intracellular signalling cascades in response to external stimuli and playing a major role in regulating many cellular processes1. They all share a similar tripartite architecture, with an extracellular, ligand-binding region, a single alpha-helical membrane spanning domain, and a cytoplasmic region containing a tyrosine kinase domain alongside C-terminal and juxtamembrane regulatory elements1,2. Binding of ligand to RTK monomers induces dimerization and conformational changes, leading to activation of the intracellular tyrosine kinase domains, trans-autophosphorylation of tyrosine residues, and recruitment/activation of intracellular signalling proteins. Due to their central importance, RTK dysfunction is causally associated with a variety of diseases (including many cancers) making them targets of therapeutic importance1.
To date, small molecules targeting RTK activity have been primarily identified using in vitro kinase assays, powerful enzymatic methods suited for high-throughput screening of tens to hundreds of thousands of compounds in parallel. By necessity, these assays are performed outside of the natural cellular environment, using purified kinase domain instead of full-length protein, introducing a number of limitations3. One significant limitation is the inability to detect compounds that affect RTK function independent of direct inhibition of kinase activity, or whose action depends upon additional protein domains or cellular factors, potentially leading to molecules of therapeutic value being missed. Moreover, in vitro kinase assays do not assess cellular toxicity or permeability of molecules, leading to the identification of numerous candidates found to be impractical for use in follow-up testing. Other common assays, such as cell viability approaches, offer the natural environmental advantages of working in live cells, but only detect compounds affecting cell growth/metabolism, and do not allow screening using specific disease-associated protein targets, making them less selective.
We previously reported the development of the Mammalian Membrane Two-Hybrid (MaMTH)4, a split-ubiquitin-based technology adapted from our well established Membrane Yeast Two-Hybrid (MYTH)5–7. MaMTH is designed to detect protein-protein interactions (PPIs) involving full-length integral membrane proteins directly in their natural membrane context in live mammalian cells4 (Supplementary Fig. 1). Notably, MaMTH is highly sensitive and able to detect subtle, dynamic alterations in PPIs in response to mutation state and environmental changes5,8,9.
In this study, we exploit these key features of MaMTH to convert it into a high-throughput, small-molecule screening platform to detect compounds that specifically target RTK functional interactions. This new platform, called MaMTH-DS (for MaMTH Drug Screening) is highly sensitive, easy to setup up, readily scalable, and combines the specificity of in vitro kinase assays with the advantages of working in live cells. In this way, MaMTH-DS allows identification of compounds inhibiting specific interactions of interest, but with the advantages that this inhibition can be mediated by diverse mechanisms, and that detected compounds are already cell permeable and have their toxicity assessed.
As a proof of principle, we used MaMTH-DS to screen an osimertinib-resistant EGFR mutant important in non-small-cell lung cancer (NSCLC) against a pilot library of 2,960 small molecules and, in conjunction with follow-up assays, identified four mutant specific compounds. We demonstrate that two of these compounds would not have been identified as specific mutant EGFR-targeting agents by classical in vitro kinase or cell-based assays, and that one of these compounds displays a completely novel mechanism of action with respect to its effect on mutant EGFR signalling. Overall, our results illustrate key advantages of MaMTH-DS, including its ability to identify diverse types of compounds, and show how it can serve as a powerful complement to traditional drug screening approaches. Additionally, several of the molecules identified in this study are promising candidates for treatment of mutant EGFR-associated, drug-resistant NSCLC for which therapeutic options are currently lacking.
RESULTS
Development of MaMTH-DS platform
To develop MaMTH-DS we introduced several significant modifications to traditional MaMTH. The first was a transition from transiently transfected to stably expressed baits, a necessary step to minimize variability/noise and allow for sensitive detection of small-molecule activity in a large-scale multi-well format. To improve ease of stable generation, we made reporter cell lines and MaMTH bait vector compatible with the Flp-IN TREx system (Thermo Fisher), a Flp recombinase-based method for rapid generation of isogenic stables in as little as two weeks. Expression of bait in this format is also under the control of a tetracycline-inducible promoter, allowing controlled bait induction during screening. The MaMTH bait vector was also adapted for use with Gateway cloning technology (Thermo Fisher) to facilitate rapid generation of constructs. A schematic diagram of the MaMTH-DS bait vector is given in Supplementary Fig 2a. Furthermore, to reduce cell loss and make the system compatible with automated handling steps, we greatly enhanced the adherence of our reporter cells to tissue culture plastic by genomic integration and overexpression of the macrophage scavenger receptor (MSR1)10 (Supplementary Fig 2b). We also changed our reporter from Firefly to Gaussia princeps luciferase, a significantly more stable enzyme that has the advantage of being secreted from cells into the growth media. This eliminates the cell lysis step of the original MaMTH, reducing handling steps and associated variability. Additionally, G. princeps luciferase produces significantly higher signal than Firefly luciferase, allowing for more sensitive detection, particularly in a 384-well format (Supplementary Fig 2c). Collectively these changes make the assay easy to setup, improve sensitivity and signal strength, and greatly reduce variability, permitting its use in an automated, high-throughput, multi-well small-molecule screening system.
Testing and validation of MaMTH-DS
To test MaMTH-DS sensitivity and suitability for use in drug screening, we selected several RTKs whose dysfunction is associated with cancer, and prepared stable MaMTH-DS RTK baits in our reporter cell lines. In our assays, we chose Shc1 as a ‘prey’ since it is well documented to interact with a wide variety of activated RTKs in a spatial-temporal manner and is important for their signalling function11,12 (though other RTK interactors could also be used). We then performed MaMTH-DS assays in the presence of small-molecule TKIs, including control molecules and compounds known to specifically target the function of the corresponding RTKs. First, we examined the response of the RTK MET to the TKIs crizotinib and erlotinib. As expected, the interaction was strongly reduced in a dose-dependent manner when exposed to crizotinib, consistent with crizotinib’s reported activity against MET13, but not erlotinib, which does not target MET (Supplementary Fig. 3a, Left Panel). The response to crizotinib was not due to a loss in cell viability (Supplementary Fig. 3a, Middle Panel), although some reduction in bait expression was observed (Supplementary Fig. 3a, Right Panel), suggesting crizotinib reduces MET stability (and consequently interaction with Shc1). We next tested the response of FGFR4 bait to BLU9931, a compound reported to target this receptor14. Similar to our results with MET, reporter activity was strongly reduced in the presence of BLU9931, but not in the presence of erlotinib control (Supplementary Fig. 3b, Left Panel). Once again BLU9931 had no effect on reporter cell viability (Supplementary Fig. 3b, Middle Panel) but some effect on FGFR4 bait expression was observed (Supplementary Fig. 3b, Right Panel), although this was less pronounced than that observed with MET. We then proceeded to examine the response of two additional RTKs, AXL and ALK, to the compounds foretinib and brigatinib, previously shown to target these receptors, respectively15–17. Once again, AXL and ALK activities were strongly reduced, in a dose-dependent manner, in the presence of targeting compound, but not erlotinib control (Supplementary Fig. 3c and d, Left Panels), while cell viability was unaffected (Supplementary Fig. 3c and d, Middle Panels). Unlike with MET and FGFR4, however, AXL and ALK expression level was not altered by compound (Supplementary Fig. 3c and d, Right Panels), suggesting that the effect of these TKIs on bait interaction with Shc1 is not due to a reduction in receptor protein amount/stability.
Finally, we looked at the more subtle case of whether the reported differential effects of two therapeutic TKIs (erlotinib and osimertinib) on EGFR mutants important in NSCLC could be detected in MaMTH-DS. These included EGFR L858R (responsive to the first generation TKI erlotinib18), EGFR L858R/T790M (which confers resistance to erlotinib, but is sensitive to the third generation TKI osimertinib19), and the recently reported EGFR L858R/T790M/C797S (which is resistant to all current clinically available therapeutic TKIs, including osimertinib)20. In agreement with clinical results, erlotinib inhibited interaction of Shc1 with L858R mutant, but not with WT or either drug-resistant mutant, whereas osimertinib affected both L858R and L858R/T790M mutants, but not WT and the C797S triple mutants (Supplementary Fig. 4a). As with the other RTKs tested, these effects were not due to a reduction in reporter cell viability (Supplementary Fig. 4b) and, as with AXL and ALK, no effect of TKIs on bait expression was observed (Supplementary Fig. 4c).
MaMTH-DS screen of drug-resistant EGFR triple mutant
Based on the success of our preliminary assays, we assembled and optimized a high-throughput, automated MaMTH-DS screening workflow for rapid detection of small-molecules targeting RTK functional interactions (Fig. 1a). To test this new workflow, we performed screening of the clinically relevant, osimertinib-resistant EGFR L858R/T790M/C797S mutant20 against a pilot library of 2,960 diverse small-molecules (Supplementary Fig. 5a, Supplementary Dataset 1). Screening was carried out twice (two independent experiments) to test for reproducibility and was performed using robotics for cell seeding, sample transfection and small-molecule addition. All screen data were subject to Box-Cox power transformation to improve data distribution symmetry and normality prior to further analysis (Supplementary Fig. 5b). Z’ values across all ten screened plates exceeded 0.5 in the first round of screening (average 0.68 overall). All ten plates exceeded 0.4 (with seven plates exceeding 0.5) in the second round (average 0.56 overall), supporting excellent assay quality21 (Supplementary Fig. 5c). Data normalization was performed on a per plate basis, using controls-based (Normalized Percent Inhibition, NPI) and sample-based (BScore22) approaches, to correct for plate variation and positional effects (Supplementary Fig. 6a). NPI and BScore correlated well, and inhibitory hits were scored using a combined cut-off of greater than 70% NPI and a BScore of −3 or less (Supplementary Fig. 6b). Overall, we detected a total of 49 candidates from Round 1 and 45 candidates from Round 2, with an overlap of 34 shared hits between both rounds (Supplementary Fig. 6c) and a relatively even distribution of hits across all plates (Supplementary Fig. 7). All raw data for these screens are provided in Supplementary Datasets 2 and 3.
Figure 1.
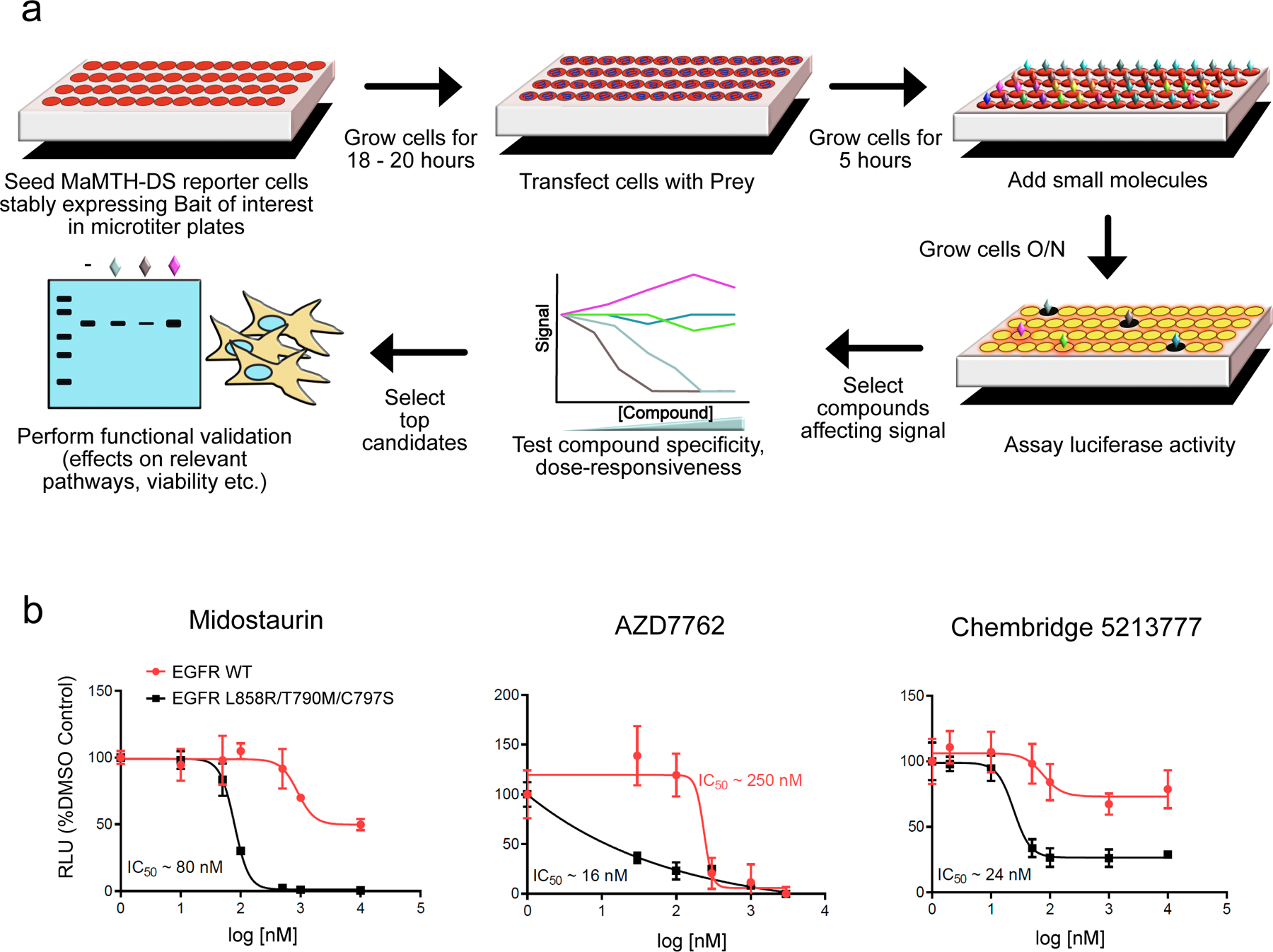
(a) Schematic representation of the MaMTH-DS platform workflow. (b) Dose response curves for the top three candidate EGFR L858R/T790M/C797S inhibitors (midostaurin, AZD7762 and Chembridge 5213777) showing robust, dose-responsive and mutant specific inhibition. Results are shown as the average ± SD for three independent experiments.
To determine reproducibility, and eliminate compounds displaying significant activity against EGFR WT and/or general toxicity, all 34 shared hits were retested in triplicate using MaMTH-DS, against both EGFR L858R/T790M/C797S and EGFR WT (Supplementary Table 1). Compounds were selected for further consideration only if they inhibited EGFR L858R/T790M/C797S (but not EGFR WT) by more than 50% and if the difference in their inhibition of mutant vs. WT was both statistically significant and at least two-fold (Supplementary Table 1). The five compounds satisfying these criteria were subjected to dose-response testing from which three (the TKIs midostaurin and AZD7762, and ChemBridge compound 5213777) displayed robust, dose-dependent inhibition of EGFR L858R/T790M/C797S (Fig. 1b), making them candidates for further study.
Functional validation of TKI Hits
Midostaurin (Rydapt™) is a multi-kinase inhibitor previously investigated for use against mutant EGFR23, but its activity has never been shown against EGFR C797S triple mutant. Significantly, the compound has been recently FDA approved for treatment of FLT3-mutant acute myeloid leukemia (AML)24. Although the kinase inhibitor library used in our collection contains other known, highly-specific FLT3 inhibitors (including crenolanib25, sorafenib26 and quizartinib27), none were found to target the EGFR C797S triple mutant in our screen (Supplementary Datasets 2 and 3). However, gilteritinib, another next generation TKI recently approved by the FDA for treatment of FLT3-mutant AML28, was absent from our library. Strikingly, secondary testing of this compound in MaMTH-DS found that, despite considerable structural differences with midostaurin (Supplementary Fig. 8a), it is also a potent and highly specific inhibitor of the EGFR triple mutant (Supplementary Fig. 8b). We therefore decided to include gilteritinib in our validation studies.
We first examined the activity of midostaurin and gilteritinib using in vitro kinase assays with recombinant kinase domain from EGFR C797S triple mutants. These included the EGFR L858R/T790M/C797S mutant used in our screen and the other commonly occurring, drug-resistant, NSCLC mutant ex19del/T790M/C797S. Both midostaurin and gilteritinib directly inhibited the kinase domain of EGFR C797S triple mutants with IC50 values in the low nanomolar range (at 100 µM ATP), displaying significantly more potent activity towards the mutants than WT EGFR (Fig 2a, Supplementary Table 2). We also assessed the effect of midostaurin and gilteritinib on recombinant oncogenic EGFR single and double mutants using in vitro kinase assays, and found that both compounds were less effective against L858R and ex19del single mutants, but displayed moderate to strong activity against the ex19del/T790M and L858R/T790M double mutants (with slightly greater potency observed for midostaurin), suggesting their activity is not restricted to the triple oncogenic mutants alone (Supplementary Table 2).
Figure 2.
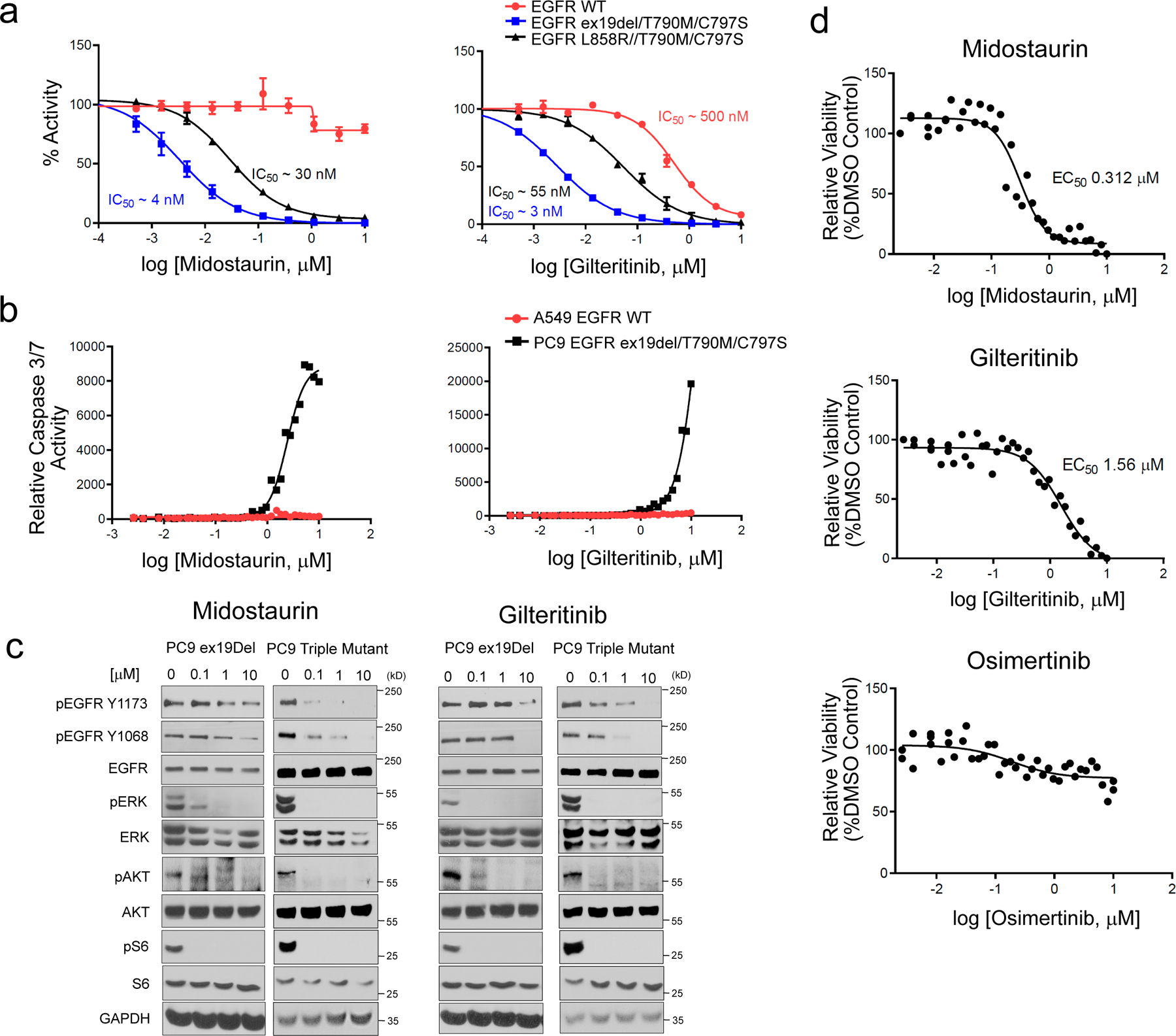
Validation of midostaurin and gilteritinib as EGFR ex19del/T790M/C797S and EGFR L858R/T790M/C797S activating mutant inhibitors. (a) in vitro kinase assay of recombinant kinase domain (residues 696–1022) of indicated mutant or WT EGFR in the presence of midostaurin (left panel) or gilteritinib (right panel). Results are shown as the average ± SD for two independent experiments. (b) Effect of midostaurin and gilteritinib on caspase 3/7 activity in PC9 EGFR ex19del/T790M/C797S and A549 EGFR WT cells. Results are shown as single 36-point dose response experiments. (c) Effects of midostaurin (left panels) and gilteritinib (right panels) on EGFR activation and downstream signalling in PC9 EGFR ex19del and EGFR ex19del/T790M/C797S cells after 2 hours treatment (see Supplementary Figs 21–24 for source blot images). Results are representative of at least two independent experiments. (d) Midostaurin and gilteritinib mediated reduction of PC9 EGFR ex19del/T790M/C797S organoid viability. Osimertinib control, which does not target triple mutant EGFR activity, has no effect. Results are shown as single 36-point dose response experiments.
We next looked at the effects of midostaurin and gilteritinib in cancer cell lines. Both compounds induced caspase 3 and 7 activity in PC9 adenocarcinoma cells carrying EGFR ex19del/T790M/C797S triple mutant, but not in A549 epithelial cells carrying EGFR WT (Fig. 2b). Midostaurin and gilteritinib also reduced EGFR phosphorylation/activation in PC9 ex19del/T790M/C797S triple mutant cells more strongly than in PC9 ex19del single mutant background cells (Fig. 2c). Phosphorylation of downstream signalling partners was also affected, although more subtly than EGFR phosphorylation, with midostaurin more potently reducing pAKT and pERK levels and Gilteritinib more potently reducing pAKT levels in triple mutant cells (Fig. 2c). This more subtle effect is likely due to midostaurin and gilteritinib having modest activity towards single mutant EGFR (Supplementary Table 2) and possibly targeting additional signalling pathway kinases. Midostaurin and gilteritinib also potently affected the growth of PC9 triple mutant organoids, in contrast to osimertinib control which doesn’t target the activity of EGFR ex19del/T790M/C797S (Fig. 2d).
The remaining TKI identified in our screen, AZD7762, is a CHK1/2 kinase inhibitor that has not been reported as active against EGFR NSCLC mutants, but has been the subject of numerous studies, including a Phase 1 clinical trial for treatment of solid tumors; however work was discontinued due to cardiac toxicity29,30. Analysis of activity by in vitro kinase assay using recombinant EGFR-WT, EGFR ex19del/T790M/C797S and EGFR L858R/T790M/C797S proteins revealed that AZD7762 effectively inhibited the kinase activity of all three forms of EGFR, and was most potent against the ex19del triple mutant, with an IC50 of 10 nM at 100 µM ATP (Supplementary Fig 9). However, AZD7762 activity towards WT and L858R triple mutants was comparable (with IC50 values of 240 nM and 280 nM, respectively) in contrast to the differential effect on the interaction of Shc1 with EGFR WT and mutant observed in MaMTH-DS (IC50 values of 250 nM and 16 nM, respectively). While these data are interesting and suggest involvement of additional cellular factors in the mutant-specificity observed in MaMTH-DS, the compound still displays substantial activity towards WT. Based on this, as well as AZD7762’s previously described toxicity and detailed coverage in the literature, we decided not to pursue further validation of this compound.
Functional validation of ChemBridge 5213777 (EMI1)
Our final hit, ChemBridge 5213777 (3-(1,3-benzoxazol-2-yl)-7-(diethylamino)-2H-chromen-2-one), which we have renamed EMI1 (EGFR MaMTH Inhibitor) (1) (Fig. 1a), is a coumarin derivative. Although published information on EMI1 is limited, previous studies have shown it displays anti-proliferative activity against various cancer cell lines and potential inhibitory effects on microtubule dynamics31. Interestingly, EMI1, while potently reducing the interaction of EGFR triple mutant with Shc1 in our MaMTH-DS assay, did not behave as a TKI and displayed no inhibition of the kinase activity of EGFR triple-mutant protein in vitro (Fig. 3b). EMI1 did, however, more strongly inhibit the viability and increase the caspase 3/7 activity of PC9 EGFR ex19del/T790M/C797S triple mutant cells than non-cancerous human bronchial epithelial (HBE) cells (Fig. 3c,d), as well as potently reduce PC9 EGFR ex19del/T790M/C797S organoid viability (Fig. 3e). Notably, EMI7 (2), a structurally similar analog lacking the diethyl amino group, obtained as part of our effort to identify molecular regions important in compound activity and eliminate functional groups associated with potential in vivo toxicity (Supplementary Fig. 10a), had no effect on the interaction of EGFR triple mutant with Shc1 (Supplementary Fig. 10b) or on PC9 triple mutant cell or organoid viability (Supplementary Fig. 10c,d). This suggests an important role for the diethyl amino group in EMI1 activity. We decided to use EMI7 as a control in our functional studies of EMI1.
Figure 3.
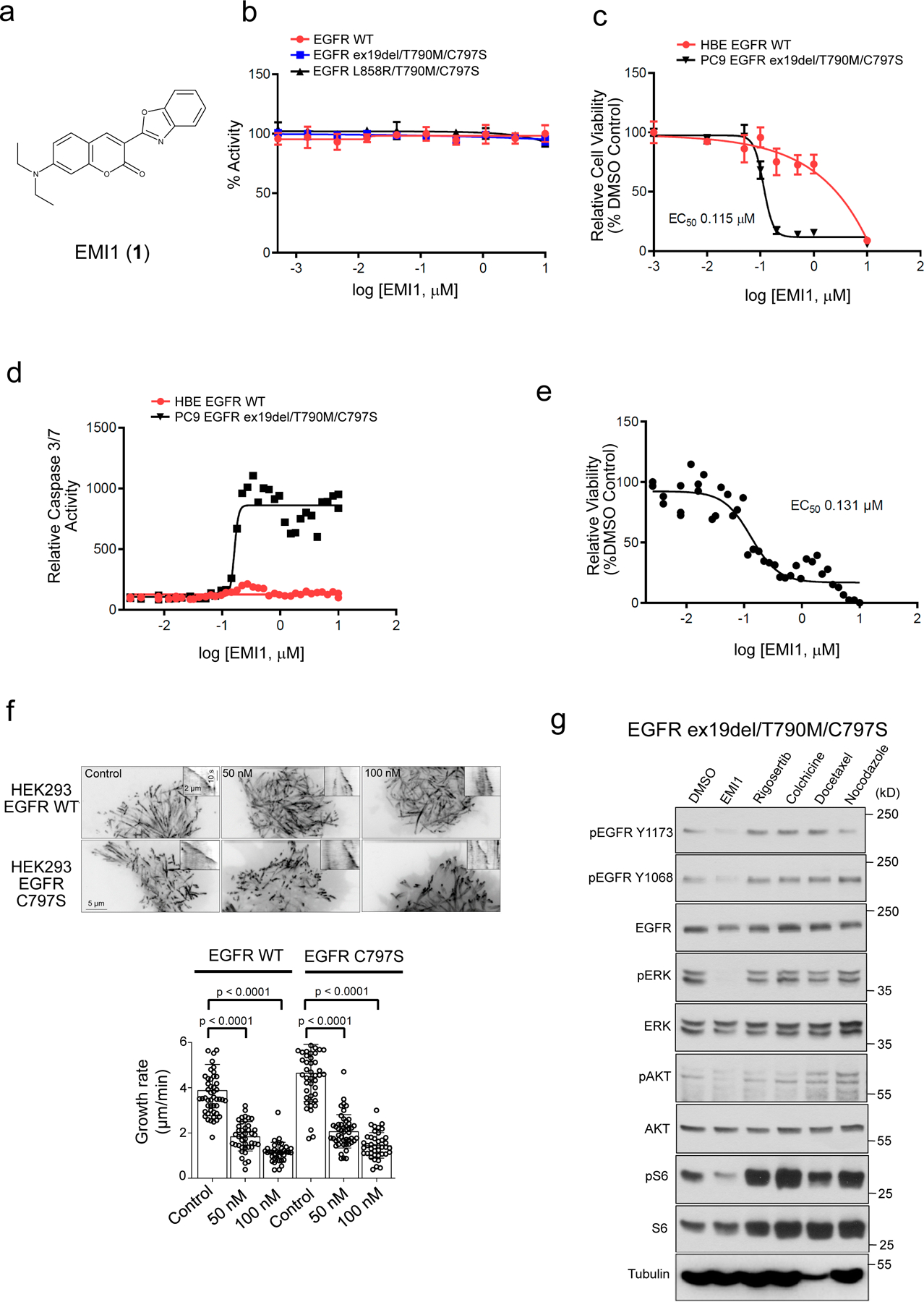
Validation of EMI1 as an EGFR ex19del/T790M/C797S and EGFR L858R/T790M/C797S activating mutant inhibitor. (a) Chemical structure for EMI1. (b) in vitro kinase assay of recombinant kinase domain (residues 696–1022) of indicated mutant or WT EGFR in the presence of EMI1. Results are shown as the average ± SD for two independent experiments. (c) Effect of EMI1 on the viability of PC9 EGFR ex19del/T790M/C797S and HBE bronchial epithelial lung EGFR WT control cells. Results are shown as the average ± SD for three independent experiments (d) Effect of EMI1 on caspase 3/7 activity in PC9 EGFR ex19del/T790M/C797S and HBE EGFR WT cells. Results are shown as single 36-point dose response experiments. (e) Viability assay measuring effect of EMI1 on PC9 EGFR ex19del/T790M/C797S organoid growth. Results are shown as single 36-point dose response experiments. (f) Maximum intensity projections (stream acquisition/exposure time 500 ms/100 frames) showing the effect of EMI1 on microtubule dynamics in HEK293 MaMTH reporter cells stably expressing EGFR WT or EGFR L858R/T790M/C797S transfected with EB3-TagRFP as a microtubule plus end marker. The contrast is inverted. Graph shows quantification of microtubule plus end velocity in HEK293 reporter cells for EMI1. n = 51, 41, 36 for HEK293 EGFR WT, control, 50 and 100 nM. n = 49, 47, 41 for HEK293 EGFR C797S control, 50 and 100 nM. Significant p-values are displayed and were calculated using the Mann-Whitney test. (g) Western blot analysis showing activity of EMI1 and other microtubule targeting compounds after 2 hours treatment on EGFR ex19del/T790M/C797S activation and downstream signalling in PC9 triple mutant cells (see Supplementary Fig. 25 for source blot images). Results are representative of at least two independent experiments.
To investigate EMI1 mechanism of action we first examined its reported activity as a microtubule-targeting agent. Using HEK293 MaMTH reporter EGFR stable cells expressing fluorescently tagged microtubule plus-end binding protein EB3, we showed that EMI1 had a similar inhibitory effect on microtubule plus end growth in both EGFR WT and EGFR-C797S triple mutant cells at 50–100 nM concentration (Fig. 3f). At 1 µM concentration, EMI1 strongly depolymerized interphase microtubules, perturbed spindle formation and induced strong mitotic block in PC9 EGFR ex19del/T790M/C797S cells after 20 hours of treatment (Supplementary Fig 11). Furthermore, in vitro microtubule dynamics assays using purified tubulin and mCherry-EB3 showed that EMI1 reduced microtubule growth rate and time, while increasing catastrophe frequency (Supplementary Fig 12). The inactive EMI7 derivative had no effect on microtubule dynamics (Supplementary Fig 12). Together these results show that EMI1 directly targets microtubules. Interestingly, however, EMI1 displayed an effect on EGFR activation and signalling through ERK and AKT in PC9 EGFR ex19del/T790M/C797S cells (at a 20 µM concentration) that was not evident when using other microtubule destabilizing compounds including Rigosertib, Nocodazole and Colchicine, or the microtubule-stabilizer Docetaxel. Thus, EMI1 appears to have a unique mutant EGFR-specific polypharmacology (Fig. 3g).
We next used MaMTH-DS to determine whether EMI1 activity towards EGFR is due to direct and specific inhibition of its physical interaction with Shc1. To this end, we examined interaction of EGFR L858R/T790M/C797S bait with two other functionally important EGFR-binding partners CrkII and Hsp90. EMI1 inhibited interaction of both proteins with EGFR at a level similar to that observed with Shc1 (Supplementary Fig. 13, suggesting it is not a specific inhibitor of the EGFR-Shc1 PPI interface. Rather, the loss of interaction mediated by EMI1 appears to be due to a more general alteration in EGFR activity.
Next, we assessed the effect of EMI1 on additional NSCLC cells including PC9 cells expressing either EGFR ex19del or EGFR ex19del/T790M and A549 EGFR WT cells. EMI1 induced EGFR degradation, and inhibited the activation of EGFR, ERK, AKT and S6 in PC9-ex19del and PC9-ex19del/T790M cells (Supplementary Fig 14a). Interestingly, EGFR activation was not affected in A549 cells, but there was a modest decrease in total EGFR levels, and attenuation of downstream signalling components ERK, AKT and S6 (Supplementary Fig. 14b). These data indicate that there is a differential response to EMI1 with respect to EGFR activation between wild-type and mutant EGFR, where mutant EGFR activation is decreased.
It is well established that changes in receptor internalization, recycling and degradation affect the signaling response and can contribute to tumorigenesis32. In addition, the concentration of active (phosphorylated) RTK within early endosomes and the number of early endosomes containing phosphorylated RTK regulate signaling amplitude and duration33. Molecules targeting EGFR may thus result in alterations of endosomal trafficking which could in turn feed-back on its activity. We therefore assessed whether EMI1, as well as midostaurin and osimertinib for comparison, affect EGFR endosomal trafficking, in terms of kinetics and endosomal distribution, using an automated microscopy approach. HEK293 MaMTH reporter EGFR WT and EGFR L858R/T790M/C797S cells were incubated with fluorescently labelled EGF for 30 minutes plus or minus 1 µM compound, fixed and stained with antibodies against the early endosomal marker EEA1 to label early endosomes, and anti-EGFR pY1068 to label phosphorylated pEGFR, as previously described33. Representative images for trafficking assays are provided in Supplementary Figures 15 and 16. We found that EMI1, like midostaurin, significantly reduced EGF accumulation in EEA1-positive early endosomes in EGFR L858R/T790M/C797S cells, in contrast to osimertinib which had no effect (Fig. 4a, red). A similar effect was observed for EGFR WT cells but to a lesser degree (Fig. 4a, black). EMI1 and midostaurin, but not osimertinib, also significantly reduced the overall amount of EGFR pY1068 localized to EEA1-positive endosomes in EGFR L858R/T790M/C797S, but not in EGFR WT, cells (Fig. 4b). These results indicate that EMI1 and midostaurin exert an inhibitory effect on the uptake and distribution of activated, mutant EGFR receptor in early endosomes.
Figure 4.
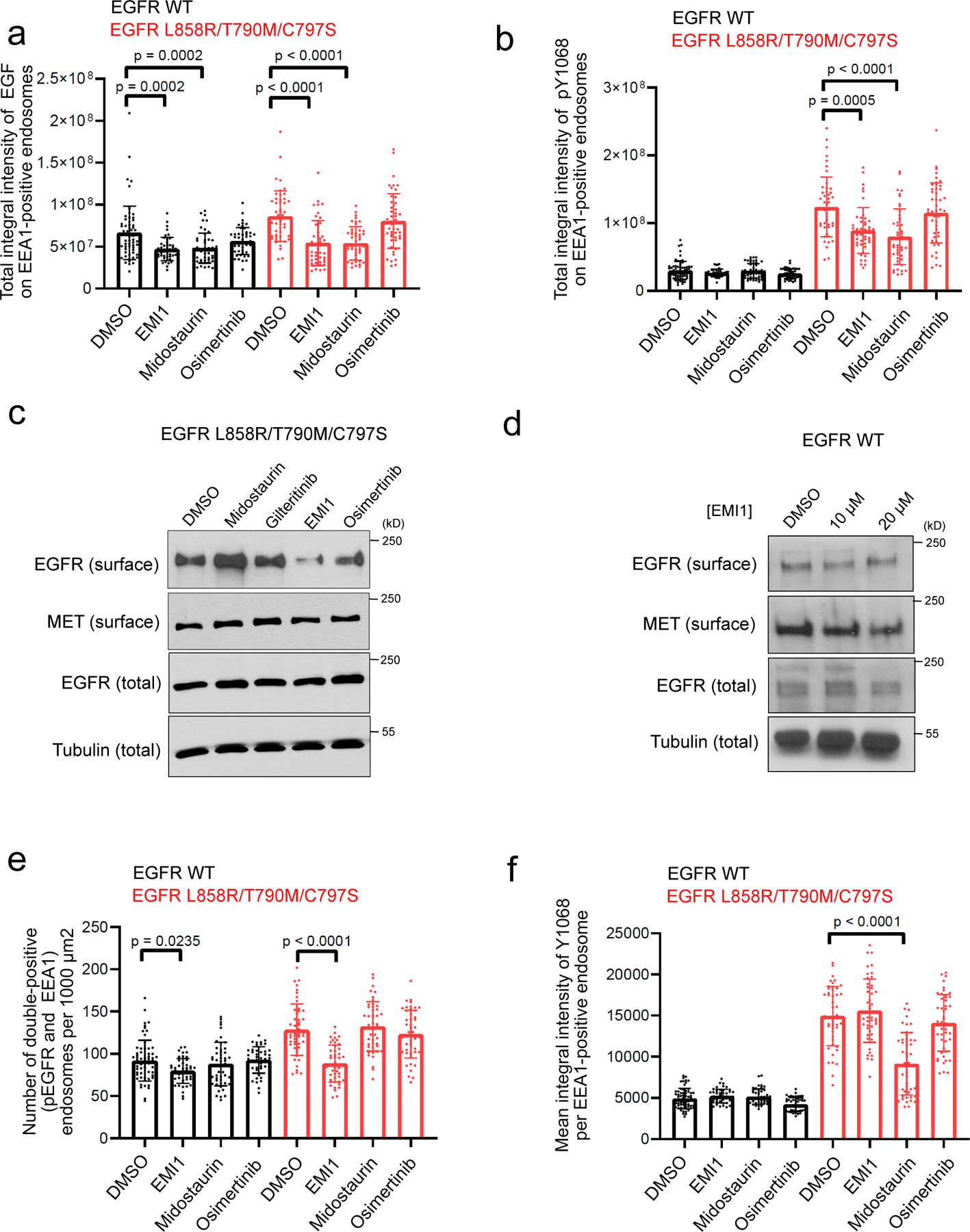
Investigating effect of EMI1 on activated EGFR L858R/T790M/C797S endosomal trafficking. (a) Total integral intensity of EGF on EEA1-positive endosomes normalized on cytoplasm area after 30 minutes of EGF stimulation upon 1 µM compound treatment in HEK293 EGFR WT cells or EGFR L858R/T790M/C797S cells. (b) Total integral intensity of pY1068 on EEA1-positive endosomes normalized on cytoplasm area after 30 minutes of EGF stimulation with 1 µM compound treatment in HEK293 EGFR WT cells or EGFR L858R/T790M/C797S cells. Results are shown as dot plots representing the average ± SD. For EGFR WT cells, n = 63, 46, 48 and 48 images were analyzed for DMSO, EMI1, midostaurin and osimertinib treatment, respectively. For EGFR-C797S cells, n = 48, 46, 46, 48 images were analyzed for DMSO, EMI1, midostaurin and osimertinib treatment, respectively. Significant p-values are displayed and were calculated using the Dunn’s multiple comparison test. (c) Cell surface biotinylation assay assessing surface levels of EGFR L858R/T790M/C797S after treatment with 5 µM compound for 2 hours. Results are representative of at least two independent experiments. (d) Cell surface biotinylation assay assessing surface levels of EGFR WT after treatment with EMI1 for 2 hours. Results are representative of at least two independent experiments (see Supplementary Fig. 26 for source blot images). (e) Number of pEGFR and EEA1 double-positive endosomes per 1000μm2 after 30 min stimulation by EGF. (f) Mean integral intensity of pEGFR on double-positive (pEGFR and EEA1) endosomes after 30 min stimulation by EGF. Results are shown as dot plots representing the average ± SD. For EGFR WT cells, n = 63, 46, 48 and 48 images were analyzed for DMSO, EMI1, midostaurin and osimertinib treatment, respectively. For EGFR-C797S cells, n = 48, 46, 46, 48 images were analyzed for DMSO, EMI1, midostaurin and osimertinib treatment, respectively. Significant p-values are displayed and were calculated using the Dunn’s multiple comparison test.
The inhibition of EGF uptake may be due to lower cell surface levels of EGFR. Indeed, EMI1 treatment for 2 hours led to a marked decrease in surface levels of EGFR in mutant cells (Fig. 4c), an effect not observed upon midostaurin or osimertinib treatment, nor in cells carrying wild-type EGFR (Fig. 4d). Total levels of EGFR did not change in the mutant or WT EGFR expressing cells. Also, no changes occurred in the surface level of another RTK, c-MET. These results argue that EMI1 and midostaurin exert their effects on uptake of by different mechanisms.
EMI1 treatment also decreased the number of early endosomes carrying activated EGFR L858R/T790M/C797S to levels similar to wild-type cells (Fig. 4e) but did not affect the average amount of activated EGFR per endosome (Fig. 4f). Conversely, midostaurin reduced the amount of activated EGFR per endosome (Fig. 4f) but had no effect on total endosome number (Fig. 4e), consistent with its activity as a direct inhibitor of mutant EGFR kinase activity (and therefore endosomal internalization). These observations have important consequences for the modulatory activity of EMI1 on the signalling of the EGFR L858R/T790M/C797S mutant.
Medicinal chemistry analysis of EMI1
To improve potency of EMI1 and explore the chemical groups involved in its function, we performed medicinal chemistry to modify the structure of the compound. Our strategy involved breaking the molecule into three groups: the diethyl-amino substituent, the coumarin backbone and the benzoxazole substituent. We generated 17 derivatives (Supplementary Fig. 17) and tested each for their ability to reduce PC9 EGFR ex19del/T790M/C797S cell viability (Supplementary Fig. 18) and affect EGFR triple mutant activation and downstream signalling (at a 10 µM dose) in PC9 EGFR ex19del/T790M/C797S cells (Supplementary Fig. 19). We identified two derivatives, EMI48 (7) and EMI56 (15), displaying greater potency towards mutant EGFR than EMI1. These contain an additional methyl group on either the benzoxazole ring (EMI48) or coumarin backbone (EMI56). Dose response experiments showed EMI48 more strongly inhibited total EGFR levels, activation and downstream signalling than EMI1, with effects observed at a 5 µM concentration, compared to the 20 µM required for EMI1 (Fig 5a and b). EMI56 also displayed an enhanced effect, but at the 10 µM level (Fig 5c). Interestingly, unlike EMI1, EMI48 and EMI56 did not affect interphase microtubules, nor have an effect on spindle formation in PC9 EGFR ex19del/T790M/C797S cells (Supplementary Fig 20), suggesting that the microtubule and EGFR-targeting activity of EMI1 are distinct. Both analogs warrant further investigation to better understand the mechanism involved in their activity and increased potency.
Figure 5.
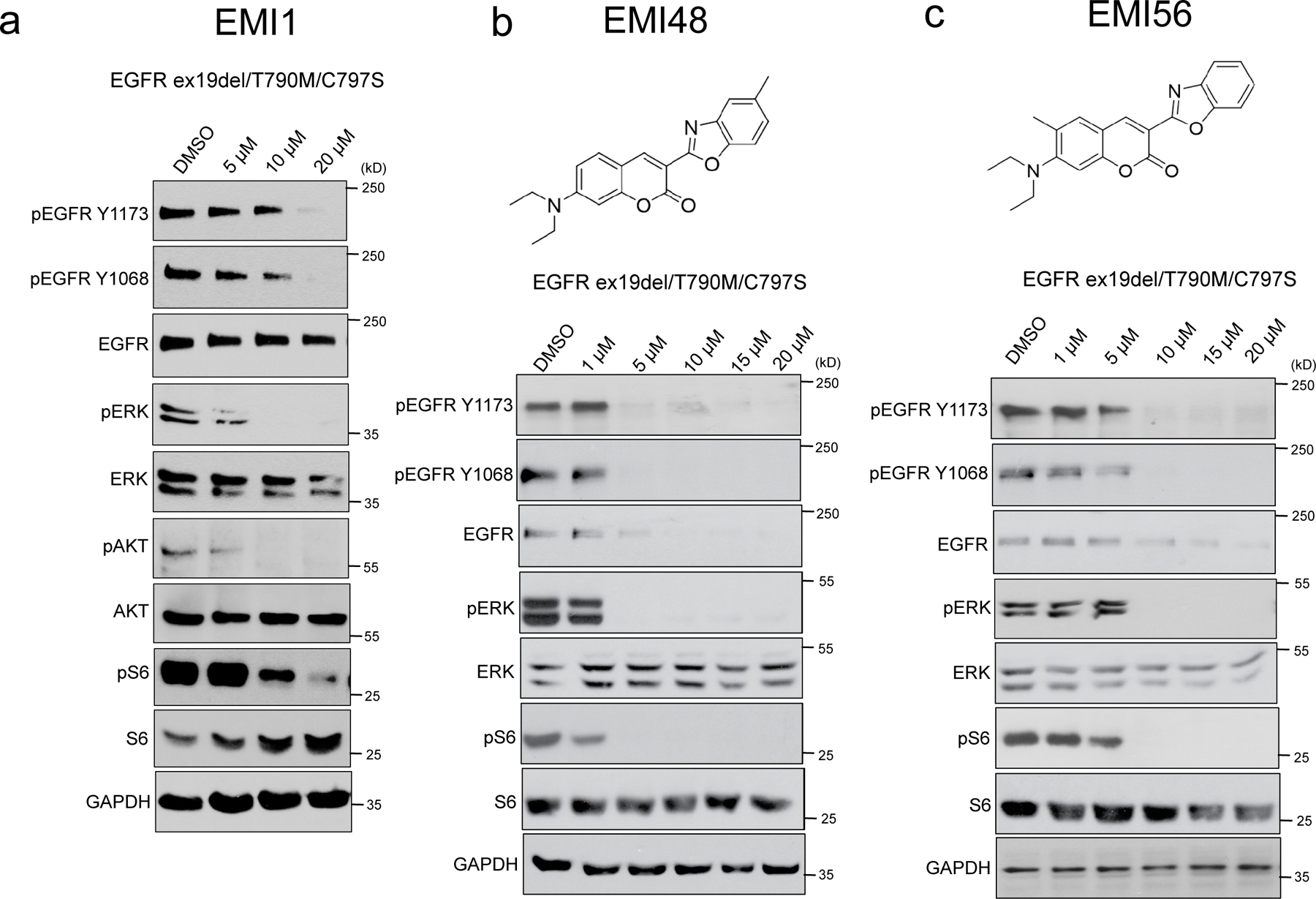
Generation and testing of EMI1 chemical analogs. (b,c,d) Western blot analysis showing effects of EMI1 (b) EMI48 (c) and EMI56 (d) on total EGFR levels, activation and downstream signalling after two hours treatment in PC9 EGFR ex19del/T790M/C797S cells (see Supplementary Figs 27 and 28 for source blot images). Results are representative of at least two independent experiments.
DISCUSSION
We developed MaMTH-DS, a powerful drug-discovery platform for rapid, high-throughput identification of small-molecules modulating interactions between full-length RTKs and functional partner proteins such as Shc1 or other relevant EGFR phospho-binding proteins, directly in living cells, with the benefit that identified small molecule candidates have already passed cell permeability and toxicity tests. We applied this method to examine and identify specific modulators of disease-associated RTKs. Preliminary testing of MaMTH-DS using various RTK baits associated with cancer, including MET, FGFR4, AXL, ALK and EGFR, revealed highly specific, dose-dependent reduction in reporter activity in response to known targeting compounds in all cases. Results closely mirrored those observed in a clinical setting, demonstrating the robustness of MaMTH-DS as a drug-discovery tool. Notably, our results suggested that compound effects on the functional interaction of the RTK baits with adapter protein was mediated by distinct mechanisms, with MET and FGFR4 interactions modulated at least in part by changes in protein level.
Full proof-of-principle MaMTH-DS screening of a pilot collection of 2,960 small-molecules identified three new compounds (midostaurin, AZD7762 and EMI1) specifically targeting the EGFR L858R/T790M/C797S triple mutant associated with drug-resistant NSCLC. Additionally, based upon functional similarities with midostaurin, we identified the TKI gilteritinib as a fourth EGFR triple mutant-targeting compound. The potency and specificity of these molecules were successfully validated in follow-up studies using the both biochemical assays and the NSCLC PC9 EGFR ex19del/T790M/C797S lung adenocarcinoma cell line and associated organoid model, suggesting that all may be of value in further research and therapeutic development. Midostaurin and gilteritinib, in particular, both of which were recently FDA-approved for use in the treatment of FLT3-mutant acute myeloid leukemia, could potentially be repurposed for treatment of patients suffering from EGFR L858R/T790M/C797S and EGFR ex19del/T790M/C797S associated NSCLC and, as such, are currently the subject of clinical studies.
Significantly, neither AZD7762 nor EMI1 would have been detected as mutant specific inhibitors of our target bait by in vitro kinase assay; AZD7762 because (although a TKI) its specificity towards mutant EGFR appears to depend on additional factors present in the live-cell format, and EMI1 because it employs a novel mechanism of action distinct from direct kinase inhibition. Our discovery of both of these compounds highlights the potential advantages of using MaMTH-DS alongside conventional assays.
The unique mode of action of EMI1 will be the focus of future studies. Thus far we identified two distinct activities for EMI1 – a direct effect on microtubule polymerization and an indirect effect on mutant EGFR signalling and trafficking. Chemical separation of these activities will be essential for follow-up studies and potential therapeutic applications. Analysis of the effect of EMI1 on the endosomal distribution of EGFR has provided important first clues about its mutant EGFR-specific activity. Investigating the nature of this specific activity should be invaluable in identifying new mechanisms for targeting oncogenic EGFR and potentially other RTK receptors. Further work will focus on in-depth monitoring of mutant movement throughout the endosomal pathway, examining potential changes in late endosomal representation and lysosomal degradation in response to EMI1 treatment. Additionally, the dual role of EMI1 as a microtubule destabilizer and modulator of mutant EGFR endosomal distribution will be explored to better understand how EMI1 mediates both effects, taking into account the impact of drug dosages and treatment times. We will also expand our medicinal chemistry studies, performing more thorough characterization of our current compound set, as well as preparing additional analogs. The generation of two derivatives, EMI48 and EMI56, which displayed greater potency towards mutant EGFR signalling than EMI1 and a reduced microtubule-depolymerization activity represents a useful step in this direction. These and future analogs will also be studied in further depth in animal models to better understand their mode of action and therapeutic potential.
Collectively our data illustrate the ability of the live-cell MaMTH-DS assay to sensitively detect the loss of functional interactions in response to diverse, but specific, effects of drug action, via mechanisms that may not be detectable by conventional in vitro kinase screening approaches. Specifically, MaMTH-DS can theoretically identify several classes of RTK inhibitors that lead to reduced binding of a functional protein partner including (i) direct inhibitors of RTK enzymatic activity (such as midostaurin and gilteritinib); (ii) direct physical inhibitors of PPIs; (iii) inhibitors that affect RTK dimerization; (iv) allosteric inhibitors of RTK enzymatic activity and (v) inhibitors that affect EGFR trafficking. In this way, our platform represents a potent new drug discovery tool that can complement conventional in vitro biochemical assays for drug screening of RTKs such as EGFR. The highly sensitive and flexible nature of the technology also make it amenable for use with other integral membrane protein targets, potentially expanding its use beyond RTKs. The MaMTH-DS platform represents a technical advance in the area of drug screening research that should greatly facilitate the identification of valuable new therapeutic molecules.
ONLINE METHODS
MaMTH assays.
Cells stably expressing bait of interest (EGFR, MET) were seeded into 96-well TC-treated plates and grown at 37˚C/5% CO2 overnight in DMEM/10%FBS/1%PS to ~ 50–60% confluency. Cells were transfected with 50 ng/well of Nub-Shc1 ‘prey’ protein by calcium phosphate precipitation. Five hours after transfection, media was aspirated out and cells were treated with 100 μL of fresh media containing specific compound and 0.5 μg/ml of tetracycline to induce bait expression. After 24 hours, luciferase activity was measured by chemiluminescence.
Western analysis of bait and downstream signalling molecule expression and phosphorylation.
Cells grown under the specified conditions were washed with ice cold PBS before addition of the cell lysis buffer (Cell Lysis Buffer 10X, Cell Signalling Technology, #9803) supplemented with protease inhibitors. Lysates were transferred to 1.5 mL microtubes, and centrifuged for 16,000 x g for 10 min. The supernatants were mixed with Laemmli sample buffer, and boiled at 95°C for 5 min. Protein quantification was performed using the BCA Protein Assay Reagent (Pierce) according to the manufacturer’s protocol prior to addition of sample buffer. Western blot analyses were performed after separation by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then blocked with 2% BSA in Tris-buffered saline/Tween 20 (TBS-T). Antibodies used for Western blot analysis were: phospho-EGFR antibody (Tyr1173; Santa Cruz, sc101668, 1:10,000), total EGFR (Cell signalling Technology, #4267, 1:10,000), phospho-AKT (Ser473; Cell Signalling Technology, #4060, 1:10,000), total AKT (Cell signalling Technology, #4691, 1:10,000), phospho-ERK (Thr202/Tyr204; Cell Signalling Technology, #9101, 1:10,000), total ERK1/2 (Cell Signalling Technology, #9102, 1:10,000), phospho-S6 (Ser240/244, Cell Signalling Technology, #5364, 1:10,000), total S6 (Cell Signalling Technology, #5364, 1:10,000), anti-GAPDH (Santa Cruz, 1:10,000), anti-tubulin (Santa Cruz, 1:10,000) or anti-V5 (Cell Signalling Technology, 1:10,000).
Cell viability assays.
MaMTH stable bait cells, PC9 or HBE cells were seeded into 96 well plates at 10,000 cells per well. For MaMTH stable cells, the cells were treated the next day with each inhibitor in a dose-dependent manner in addition to 0.5 μg/mL tetracycline to induce bait expression. For PC9 and HBE cells, the cells were treated with each compound the same day as seeding. After 72 hours of drug treatment, cell viability was measured using the CellTiter-Glo assay (Promega).
EGFR localization and trafficking analyses.
The experiment was performed in 384 well CellCarrier imaging plates. Cells were treated with 100 ng/ml of EGF-biotin/streptavidin-Alexa-647 complex, Invitrogen, E35351, equivalent to ~10ng/ml of EGF for 30 minutes. Each condition (EGF stimulation time, treatment and mutation) was repeated in at least 6 wells. 11 images (276x234μm) were collected from each well by automated confocal microscope CV7000 (Yokogawa) with a 60x water immersion objective (NA = 1.2), with a total of 1063±251 (mean±SD) imaged cells per well. Images were analyzed by MotionTracking software (http://motiontracking.mpi-cbg.de)34,35 and 147±80 (mean±SD) EEA1-positive endosomes per cell were found. All statistics were calculated per image, then averaged between images in the well, and, finally, averaged between wells of equal conditions. The SEM was calculated from the last averaging step.
Generation of adherent HEK293 cells.
Flp-In 293 TREx cells (Thermo Fisher) were grown at 37˚C/5% CO2 in DMEM/10%FBS/1%PS media in 6-well TC-treated plates to ~ 50–60% confluency. Cells were then transfected with pcDNA3.1 plasmid, expressing the gene for human Macrophage Scavenger Receptor 1 (MSR1) transcript variant A alongside G418 resistance cassette, using PolyJet transfection reagent (SignaGen), as per manufacturer instructions. Cells were grown overnight and then split into 10 cm plate containing 10 mL of DMEM/10%FBS/1%PS/800 μg/mL G418 and grown at 37˚C/5% CO2 until distinct foci appeared. Individual foci were expanded, and screened for enhanced adherence using methylene blue staining and stringent washing in a 96-well plate format as previously described10. The most highly adherent cell line displaying robust growth in media and appropriate Flp-In 293 TREx resistance to Zeocin and Blasticidin was selected for use in the generation of MaMTH reporter cells.
Generation of stable MaMTH reporter cells.
Reporter vector was generated in a pcDNA3.1(−) backbone using ORFs expressing Gaussia princeps luciferase (New England Biolabs) under the control of a 5xGAL4 UAS and puromycin resistance marker under the control of a constitutive PGK promoter, via Gibson assembly36. Adherent FLP-compatible HEK293 cells (prepared above) were grown at 37˚C/5% CO2 in DMEM/10%FBS/1%PS media in 6-well TC-treated plates to ~ 50–60% confluency. Cells were transfected with 1000 ng reporter vector using X-tremeGENE 9 DNA transfection reagent (Roche) as per manufacturer instructions. After 5 hours, media containing transfection reagent was removed and replaced with fresh DMEM/10%FBS/1%PS. Cells were grown for 48 hours and then split 1 in 2 into new 6-well plates using DMEM/10%FBS/1%PS + 0.5 μg/mL puromycin and grown until individual foci appeared. Individual foci were expanded, and monoclonal populations isolated by sorting of individual cells into 96-well plates using a FACS Aria II Flow Cytometer (BD Biosciences), followed by further expansion. Expanded cell populations were screened individually and a cell line displaying strong MaMTH-responsive reporter activity and minimal background was selected for further use in MaMTH-DS.
Generation of Flp-In TREx compatible MaMTH bait vectors.
Gateway-cloning cassette followed by Cub-GAL4/RelA TF sequence was PCR-amplified from our previously reported MaMTH bait vector4 using KAPA 2X HiFi DNA Polymerase (Kapa Biosystems). The amplified fragment was combined with EcoRV-digested Flp-compatible pcDNA5/FRT/TO vector (Thermo Fisher) via Gibson Assembly36. Generated constructs were fully sequence verified, and construct containing all of the elements necessary for Gateway cloning, tetracycline-induction, MaMTH bait C-tagging and use in generation of isogenic stables via the Flp-In TREx system, was isolated. This final bait vector construct was designated A1160 (Supplementary Note 1).
Generation of Flp-In TREx compatible MaMTH bait constructs.
All bait and prey constructs were generated using the Gateway cloning technology (Thermo Fisher) and destination vectors A1160 (MaMTH bait) or A1245 (MaMTH prey, Supplementary Note 1). ShcI ORF in entry clone format was obtained from the Human ORFeome Collection V8.137. EGFR-WT and single L858R and double L858R/T790M mutant entry clones were generated as described previously4. EGFR triple mutant containing the C797S mutation was generated via site-directed mutagenesis of EGFR double mutant using primers 5’-atgcccttcggcagcctcctggact-3’ and 5’- agtccaggaggctgccgaagggcat-3’. MET entry clone was obtained from OpenFreezer (V9936). All final bait and prey constructs were fully sequence verified.
Generation of double stable MaMTH bait cell lines.
Isogenic MaMTH reporter cell lines stably expressing baits of interest were generated using the Flp-In TREx system (Thermo Fisher). Briefly, MaMTH reporter cells were grown at 37˚C/5% CO2 in DMEM/10%FBS/1%PS media in 6-well TC-treated plates to ~ 50–60% confluency. Cells were transfected with 900 ng pOG44 and 100 ng of bait construct in A1160 using X-tremeGENE 9 DNA transfection reagent (Roche) as per manufacturer instructions. After 5 hours, media containing transfection reagent was removed and replaced with fresh DMEM/10%FBS/1%PS. Cells were grown for 48 hours and then split 1 in 2 into new 6-well plates using DMEM/10%FBS/1%PS + 100 μg/mL hygromycin and grown until individual foci appeared. Foci were expanded and proper, tetracycline-induced bait expression was verified by Western blotting.
MaMTH-DS high-throughput screening workflow.
MaMTH reporter cells stably expressing EGFR L858R/T790M/C797S bait or EGFR L858R/T790M control bait were seeded into 384-well plates (5000 cells/well) in DMEM/10%FBS/1%PS media using a MultiDrop Combi (Thermo) fitted with a standard cassette. Plates were covered with MicroClime Environmental Lids (Labcyte; hydrated with ~10 mL ddH2O) and grown at 37°C/5% CO2 overnight. The next day cells were transfected with 25 ng of MaMTH ShcI prey DNA using X- tremeGENE 9 DNA transfection reagent (Roche) as per manufacturer instructions. Transfection mix (5 μL total volume/well) was added to 384-well plates containing cells using a Bravo Automated Liquid Handling Platform (Agilent) fitted with a 96ST pipette head. Plates were once again covered with MicroClime Lids and grown at 37°C/5% CO2 for 5 hours. Media was then removed from plates using a BioTek 405 Select microplate washer, and a fresh 50 μL of DMEM/10%FBS/1%PS media containing 0.5 μg/mL Tetracycline was added to each well using a MultiDrop Combi. 50 nL of DMSO, Osimertinib (70 μM) or library compound (10 mM for ChemBridge/Maybridge compounds, 1 mM for OICR TKIs) were then added to individual wells using an ECHO 550 (Labcyte). Plates were covered with MicroClime lids and grown for an additional 17–18 hours at 37°C/5% CO2. Cells were then subjected to luciferase assay using 20 μL of 20 μM coelenterazine per well. Luminescence was measured in an injector-equipped SynergyNeo microplate reader, using linear shaking for 2 seconds after substrate addition. All reads were performed from the top using a Gain of 100 and a 1 second integration time.
Data analysis of MaMTH-DS screening results.
All data analysis was performed in an automated fashion using in house-software developed in the R programming language38. Raw data from screens were subjected to Box-Cox transformation as previously described39 in order to improve data distribution symmetry and normality. Z’ values were calculated on a per plate basis using EGFR L858R/T790M and EGFR L858R/T790M/C797S in the presence of Osimertinib as positive and negative controls, respectively (with the exception of ShcI Round 2 Plate 10, where, due to a technical issue, EGFR L858R/T790M/C797S in the presence of DMSO was used as a negative control instead). Prior to Z’ calculations, the single most extreme value from each control dataset was excluded if it was classified as an outlier based on a cut-off of 1.5 times the IQR. Data normalization was performed using both controls-based Normalized Percent Inhibition (NPI) and sample-based (controls independent) BScore. NPI was calculated as (Negative Control Signal – Sample Signal)/(Negative Control Signal – Positive Control Signal)*100. B-Score was calculated using the cellHTS2 package40. NPI was plotted against BScore and hits were scored using a combined cut-off of 70% NPI and a BScore of −3 or less.
In vitro kinase assays.
Kinase assays were performed using recombinant proteins of the kinase domain of wild-type EGFR, EGFR-C797S/T790M/L858R, EGFR-C797S/T790M/ex19del, and EGFR-C797S (Reaction Biology Corporation). Compounds (Midostaurin, AZD7762, Chembridge 5213777 and Gilteritinib) were tested in a 10-dose IC50 duplicate mode with 3-fold serial dilution starting at 10 µM. Reactions were carried out at 10µM ATP.
Caspase 3 and 7 assays.
PC9-C797S, A549 or HBE cells were seeded into 384 well plates at 5,000 cells per well. After 72 hours of drug treatment, caspase 3 and 7 activity were measured using the Caspase-Glo 3/7 assay (Promega).
PC9 EGFR ex19del/T790M/C797S organoid viability assays.
PC9 EGFR ex19del/T790M/C797s cells were adapted to grow in Matrigel conditions, to generate organoid cultures. In viability assays, organoids were dissociated to single cells and seeded on top of a thin layer of Matrigel in culture medium. Drugs were added after 3 days of culturing and grown in the absence or presence of drug for the indicated time period. Organoid viability was determined by ATP quantification using the CellTiter-Glo 3D luminescence-based assay (Promega). Organoid generation and screening were performed in Princess Margaret Living Biobank Facility (https://www.livingbiobank.ca/).
Surface biotinylation assays.
HEK293 EGFR L858R/T790M/C797S cells were seeded at density of 200,000 cells/well in a 6 well plate. Cells were serum-starved for 18 hours and then treated with 10 µM compound for 2 hours at 37°C. Cells were then washed three times with ice-cold PBS and biotinylated using 2 mg/ml biotin for 30 minutes at 4°C. Cells were washed three times with 100 mM glycine and lysed in lysis buffer (Cell Lysis Buffer 10X, Cell Signalling Technology, #9803) supplemented with protease inhibitors. Lysates were transferred to 1.5 mL microtubes, and centrifuged for 16,000 x g for 10 min. Supernatants were transferred to fresh tubes, with 20% of the supernatant being kept as an input control. The remainder of the supernatant was used for pull-down using streptavidin beads. Pull-down of the beads and supernatants was carried out overnight at 4°C on a rotating shaker. Beads were washed, resuspended in Laemmli sample buffer and heated at 95°C for 5 minutes. Samples were run on a 12% SDS-PAGE gel and western blotting was performed.
EMI1 medicinal chemistry.
EMI1 was purchased from InterBioScreen Ltd., with a purity greater than 95%. EMI7 was purchased from Life Chemicals Inc., with a purity greater than 99%. EMI1 medicinal chemistry synthesis schemes along with corresponding mass spectrometry, NMR and HPLC data for each compound is provided in Supplementary Note 2.
Tracking of EB3-positive microtubule plus ends in cells.
MaMTH reporter HEK293 cells stably expressing EGFR WT or EGFR L858R/T790M/C797S were transfected with EB3-TagRFP. Cell were stimulated with 0.5 µg/ml tetracycline to induce EGFR expression 18 hours prior to imaging. Cells were incubated with EMI for 30 min prior to imaging. Inverted research microscope Nikon Eclipse Ti-E (Nikon) supplemented with the perfect focus system (PFS) (Nikon) and equipped with Nikon CFI Apo TIRF 100x 1.49 N.A. oil objective (Nikon), Photometrics Evolve 512 EMCCD (Roper Scientific) controlled with MetaMorph 7.7 software (Molecular Devices) was used to perform the live cell imaging. Images were acquired in a stream mode with an exposure time of 500 ms. Kymographs were generated using ImageJ plugin KymoResliceWide (https://github.com/ekatrukha/KymoResliceWide). Parameters of microtubule dynamics were analyzed as described previously41,42.
Microtubule imaging.
PC-9 EGFR ex19del/T790M/C797S cells were incubated with 1 μM of EMI-1 for 20 hours. Cells were fixed and immunostained for tubulin (1°, rat-anti-α-tubulin (YL1/2) (MA1–80017, Pierce Antibodies, MA) antibody and 2°, Alexa-568 conjugated goat antibodies against rat IgG (Molecular Probes, Eugene, OR)). Nucleus was stained using DAPI. Imaging was performed with a Nikon Eclipse 80i upright fluorescence microscope equipped with Plan Apo VC N.A. 1.40 oil 100x
In vitro microtubule polymerization assays.
To monitor the direct effects of EMI1 on microtubule dynamics, in vitro assays were performed as described previously using purified pig brain tubulin and mCherry-EB343.
Supplementary Material
ACKNOWLEDGEMENTS
We thank L. Riley for valuable discussions and editing of this manuscript. We also thank P.A. Jänne1 and M.J. Eck1 for providing us with the PC9-T790M and PC9-C797S cells used in our study, and the Center for Information Services and High Performance Computing (ZIH) of the TU Dresden for providing support and computational resources.
This research was supported by funding from Consortium Québécois sur la Découverte du Médicament (CQDM) (Explore) and OCE (#23929). In addition, work in the Stagljar lab is supported by the Canadian Cancer Society Research Institute (#703889), Genome Canada via Ontario Genomics (#9427 & #9428), Ontario Research fund (ORF/DIG-501411 & RE08–009), CQDM (Quantum Leap) and Brain Canada (Quantum Leap), and Cancer Research Society (#23235). J.H.P.’s work in the laboratory of Mark Lemmon at Yale was supported by NIH grants R01-CA198164 and R35-GM122485.
GP would like to acknowledge the support of the Ontario Institute for Cancer Research, and its funding from the Government of Ontario.
Footnotes
Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, USA.
COMPETING FINANCIAL INTERESTS
I.S., P.S. and J.S. (in conjunction with the University of Toronto) are listed as inventors on a patent (publication number 20190091205) for the use of EMI1 (and structurally related analogues), midostaurin, gilteritinib and AZD7762 (and structurally related analogues) in the treatment of mutant EGFR-mediated non-small-cell lung cancer.
CODE AVAILABILITY STATEMENT
All R code used in the analysis of the presented drug screening data is available from the authors upon request.
DATA AVAILABILITY STATEMENT
The authors declare that all data supporting the findings presented in this study are available within the paper and its supplementary information files.
REFERENCES
- 1.Lemmon MA & Schlessinger J Cell Signaling by Receptor Tyrosine Kinases. Cell 141, 1117–1134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volinsky N & Kholodenko BN Complexity of Receptor Tyrosine Kinase Signal Processing. Cold Spring Harb. Perspect. Biol 5, a009043–a009043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jia Y, Quinn CM, Kwak S & Talanian RV Current in vitro kinase assay technologies: the quest for a universal format. Curr. Drug Discov. Technol 5, 59–69 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Petschnigg J et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 11, 585–92 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Sokolina K et al. Systematic protein–protein interaction mapping for clinically relevant human GPCRs. Mol. Syst. Biol 13, 918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snider J et al. Mapping the functional yeast ABC transporter interactome. Nat. Chem. Biol 9, 565–72 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stagljar I, Korostensky C, Johnsson N & te Heesen S A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. USA 95, 5187–92 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao Z et al. A Global Analysis of the Receptor Tyrosine Kinase-Protein Phosphatase Interactome. Mol. Cell 65, 347–360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petschnigg J et al. Systematic Identification of Oncogenic EGFR Interaction Partners. J. Mol. Biol 429, 280–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins AK & Horlick RA Macrophage scavenger receptor confers an adherent phenotype to cells in culture. Biotechniques 25, 240–244 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Zheng Y et al. Temporal regulation of EGF signalling networks by the scaffold protein Shc1. Nature 499, 166–171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saucier C et al. The Shc adaptor protein is critical for VEGF induction by Met/HGF and ErbB2 receptors and for early onset of tumor angiogenesis. Proc. Natl. Acad. Sci 101, 2345–2350 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goździk-Spychalska J et al. C-MET inhibitors in the treatment of lung cancer. Curr. Treat. Options Oncol 15, 670–82 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Hagel M et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov 5, 424–437 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Eder JP et al. A Phase I Study of Foretinib, a Multi-Targeted Inhibitor of c-Met and Vascular Endothelial Growth Factor Receptor 2. Clin. Cancer Res 16, 3507–3516 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Qian F et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res 69, 8009–8016 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Zhang S et al. The Potent ALK Inhibitor Brigatinib (AP26113) Overcomes Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in Preclinical Models. Clin. Cancer Res 22, 5527–5538 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Walter AO et al. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLC. Cancer Discov 3, 1404–1415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang T & Zhou C Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients with EGFR inhibitor-resistant non-small cell lung cancer. Transl. lung cancer Res 3, 370–2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thress KS et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med 21, 560–2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J-H, Chung & Oldenburg. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 4, 67–73 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Brideau C, Gunter B, Pikounis B & Liaw A Improved Statistical Methods for Hit Selection in High-Throughput Screening. J. Biomol. Screen 8, 634–647 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Lee HJ et al. Noncovalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov 3, 168–181 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.M., L. Midostaurin approved for FLT3-mutated AML. Blood 129, 3403–3406 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Galanis A et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 123, 94–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antar A et al. Inhibition of FLT3 in AML: a focus on sorafenib. Bone Marrow Transplant 52, 344–351 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Cortes J et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol 19, 889–903 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perl AE et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol 18, 1061–1075 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zabludoff SD et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther 7, 2955–2966 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Sausville E et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol 73, 539–549 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SN et al. 7-Diethylamino-3(2′-benzoxazolyl)-coumarin is a novel microtubule inhibitor with antimitotic activity in multidrug resistant cancer cells. Biochem. Pharmacol 77, 1773–1779 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Mellman I & Yarden Y Endocytosis and cancer. Cold Spring Harb. Perspect. Biol 5, a016949 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villaseñor R, Nonaka H, Del Conte-Zerial P, Kalaidzidis Y & Zerial M Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. Elife 4, e06156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-ONLY REFERENCES
- 34.Collinet C et al. Systems survey of endocytosis by multiparametric image analysis. Nature 464, 243–9 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Rink J, Ghigo E, Kalaidzidis Y & Zerial M Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Yang X et al. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.R-Core-Team. R: A language and environment for statistical computing (2017).
- 39.Box GEP and Cox DR An analysis of transformations. R. Stat. Soc 26, 211–252 (1964). [Google Scholar]
- 40.Boutros M, Brás LP & Huber W Analysis of cell-based RNAi screens. Genome Biol 7, (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aher A et al. CLASP Suppresses Microtubule Catastrophes through a Single TOG Domain. Dev. Cell 46, 40–58.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mohan R et al. End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc. Natl. Acad. Sci 110, 8900–8905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jost M et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 68, 210–223.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings presented in this study are available within the paper and its supplementary information files.