

Abstract
THE PRION DISEASES POSE UNIQUE SCIENTIFIC, medical, veterinary and regulatory challenges. Here, we summarize current information bearing on the natural history, pathobiology and epidemiology of these disorders and public policy responses to the potential threats to public health posed, particularly, by bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease (vCJD). Six years after the first case reports of vCJD, there is still no clear indication of the magnitude of the primary epidemic, or of the likelihood of lateral transmission of this untreatable disease by iatrogenic means, particularly by blood and blood products. However, the unsettling nature of the available evidence warrants prudence regarding public health policy and regulation, as well as a forward-looking approach to research.
As a novel group of infectious diseases, prion diseases (also known as transmissible spongiform encephalopathies, or TSEs) present the clinician, the scientist and the public health decision-maker with a range of distinctive phenomena and challenges. These diseases have been recognized for decades,1 or even centuries,2 as rapidly progressing, fatal, potentially transmissible neurological syndromes found in both humans and animals. Examples of prion diseases that affect different mammalian species include Creutzfeldt–Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome, fatal familial insomnia, variant Creutzfeldt–Jakob disease (vCJD) and kuru in humans; scrapie in sheep and goats; mad cow disease (bovine spongiform encephalopathy, BSE) in cattle; chronic wasting disease in deer and elk; transmissible mink encephalopathy in mink and feline spongiform encephalopathy in domestic and captive cats.3,4,5
The accumulated evidence now strongly suggests that the sporadically, iatrogenically and familially occurring forms of human prion disease, collectively termed “classic Creutzfeldt–Jakob disease” (cCJD), do not currently constitute a serious threat to the safety of the blood supply, despite strong evidence that this subset of diseases can under certain circumstances be transmitted between individuals by iatrogenic routes.6,7 This rationale led to a policy decision by Health Canada in 1998 that recalls and withdrawals of blood products were not indicated by virtue of those products' association with donors found later to have developed cCJD.8 However, the recent appearance in the United Kingdom of vCJD and BSE as novel prion diseases has led to unprecedented challenges on several levels. Here, we review key features of current scientific knowledge that has informed a different set of public policy decisions9,10,11 concerning blood safety in relation to vCJD. We present an argument that can be summarized as follows: first, due to wide exposure of human populations to an infective agent that causes a prion disease in cattle and is the likely cause of vCJD, we face the possibility of a significant international epidemic of vCJD in future decades; second, because vCJD, unlike cCJD, is a new human prion disease, experience with it is still inadequate to conclude that it poses no threat to the biological safety of blood products; and, third, although limited, the data so far available concerning the distinctive pathobiology of vCJD are sobering and point to a need to consider the possibility of its being transmissible by blood.
Basic epidemiology, diagnosis and causes
The most common form of human TSE, sporadic CJD, has been estimated to occur endemically at a rate of 0.5–1 case per million population per year.12 The clinical course of sporadic CJD entails rapidly progressing dementia, with death usually ensuing within weeks or months of onset; in only about 10% of patients does survival exceed a year. The median age of onset is in the seventh decade, with cases in individuals aged less than 50 years being rare. No effective treatment is available. In about one-third of sporadic CJD cases, the initial presentation is “cerebellar,” with ataxia, dysarthria and coordination defects. With disease progression, there may be pyramidal involvement, basal ganglionic dysfunction, spinal cord involvement with motor neuron degeneration (indicated by muscle atrophy and fasciculations) and, frequently, myoclonus.
Definitive diagnosis of prion disease requires brain biopsy or necropsy and neurohistopathological analysis. There is no central nervous system inflammation of any conventional type associated with prion disease. However, in addition to more readily observable alterations (especially spongiform change, often accompanied by gliosis), a critical observation in all prion diseases is the accumulation in brain tissue of PrPSc, an abnormal, insoluble, protease-resistant amyloid form of a normal cellular protein named PrPC (Fig. 1). (The term PrPSc, originally named for its association with scrapie, is now most often used generically to refer to native misfolded PrP; this is the sense in which we employ it. Another term, PrPres, is used to refer sometimes to the native misfolded form of PrP and sometimes to the residual protease-resistant core of PrPSc, also known as PrP 27–30. Here, we use PrP 27–30 to denote this protein fragment.) The pattern of accumulation of PrPSc is also variable, appearing as distinct accretions or plaques in a minority of sporadic cases and in some inherited prion disease (e.g., Gerstmann-Sträussler-Scheinker syndrome), or distributed diffusely, even to the point of being invisible by direct immunostaining in situ and detectable only by Western immunoblotting of brain extracts. The protease resistance of PrPSc may contribute to its accumulation in TSE-affected brain, as well as to the ability of TSE infectivity to resist complete inactivation by many physical, chemical and enzymatic treatments that would normally be sterilizing for a conventional virus or bacterium.13,14 This impressive resistance has contributed to the current concern over the potential for transmission of prion disease by blood, tissues, organs and medical procedures.
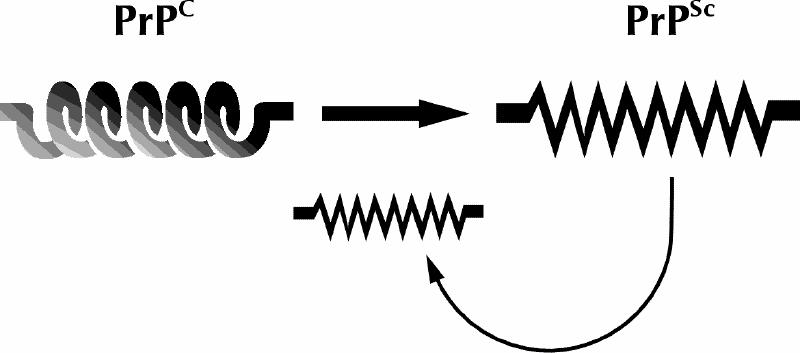
Fig. 1: Schematic depiction of the prion hypothesis. A normally folded cellular form of the prion protein called PrPCC, which consists largely of α helix, for unknown and perhaps diverse reasons adopts an alternative conformation (PrPSc) that is rich in β-sheet structure, has a tendency to aggregate and possesses additional properties that lead directly or indirectly to CNS toxicity. Once formed, molecules in the misfolded conformation have a tendency to stimulate the formation of additional PrPSc by post-translational recruitment of PrPC.
The term TSE refers partly to the distinctive pattern of vacuolating neuronal degeneration and loss found in the brains of affected individuals. It also denotes the fact that, although these diseases occur both sporadically (with no traceable original cause) and in familial clusters (which are linked to genetic mutations), they can also be directly transmitted from one host individual to another within the same species or even between widely diverged species. Such infectious transmission of TSEs was first demonstrated experimentally with laboratory animals in the 1960s.15 However, it has also occurred accidentally, both in humans (in artificial or unusual situations like neurosurgery, transplantation, parenteral injection of hormones derived from cadavers and — for kuru — ritual cannibalism)16 and in animals, where TSE contamination of vaccines or feed has been implicated. Spontaneous natural transmission (vertical and horizontal) has also been inferred for some animal TSEs such as scrapie, BSE and chronic wasting disease, although such claims remain controversial and the potential mechanisms are poorly understood.4 Convincing evidence for spontaneous horizontal transmission of prion disease between humans has never been adduced, although some intriguing observations would seem to offer hints to the contrary.17
The current leading biological explanation for the cause of TSEs invokes a novel mechanism that not only implicates PrPSc as a pathognomonic marker and a pivotal component of pathogenesis in these diseases, but also postulates that this molecule acts as a nonliving infective agent composed solely of protein: a proteinaceous infectious particle, or prion.18 This claim is the basis of the prion hypothesis, for which Dr. Stanley Prusiner was awarded the Nobel Prize for Physiology or Medicine in 1997.5 It has not yet been demonstrated to the satisfaction of all biologists that the infectivity and other biological behaviour of TSEs are independent of a nucleic acid genome like that found in conventional infective agents.19,20,21,22 However, many now do believe that, in some fashion that has yet to be completely elucidated, inoculation of a susceptible host even peripherally with a minute quantity of PrPSc can be sufficient to initiate a systemic cascade of recruitment of the host's endogenous PrPC into the misfolded form. PrPSc then accumulates in neural tissues to a degree and in a fashion, again that is not well understood, that ultimately causes neurotoxicity, neurodegeneration and death. The estimated 10%–15% of human prion disease cases that are associated with dominant mutations in the autosomal gene, PRNP,23 that encodes PrPC are thought to result from subtle alterations in PrPC structure or stability, or both. Such alterations, in turn, presumably have a marked effect on the lifetime risk of a spontaneous occurrence of widespread pathogenic misfolding into PrPSc and consequent pathology, leading to a clear pattern of highly penetrating inherited disease. In sporadic cases of CJD, the point of origin for such a cascade of PrP misfolding and pathogenesis is far from clear. However, the prion hypothesis would be consistent with initiation by a mechanism originating with random somatic events of protein misfolding, or perhaps somatic mutation predisposing to such misfolding.
BSE and vCJD: coemergence of 2 novel prion diseases
BSE
In December 1986, the Central Veterinary Laboratory in Weybridge, England, made the first formal diagnosis of a novel prion disease in cattle, namely, BSE.24 A rapid rise in the incidence of BSE ensued in the UK over the following months. Although the precise origins of BSE in British cattle remain uncertain,25,26 the epidemic trend was inferred to be the result of infectious oral transmission to cattle via feed supplements containing rendered bovine protein into which tissues from BSE-affected cattle had been accidentally incorporated.27,28 Legislative measures to curb the transmission of BSE were initiated by the government of the UK in summer 1988, with the imposition of a ruminant-to-ruminant feeding ban, institution of compulsory disease notification and a program to destroy affected animals, as well as the herds to which they belonged. Additional measures imposed later included the following: first, an extension of the ruminant-to-ruminant feeding ban to include prohibition of the use of mammalian protein in any ruminant feedstuffs and prohibition of the use of mammalian meat-and-bone meal in feed destined for any farmed animal species; second, the Specified Bovine Offals ban of November 1989 — later extended to other potentially hazardous materials (Specified Risk Materials) — which prohibited the use of certain bovine tissues and organs for human consumption that, by analogy with sheep scrapie, posed the largest risk of harbouring TSE infectivity, although they were otherwise suitable for use in human food; and, third, the implementation in May 1996 of the Over Thirty Month Scheme, according to which no bovine animal over the age of 30 months would be permitted to enter the human food chain.
Human exposure to BSE
These legislative actions were designed both to bring the BSE epidemic under control and to minimize the risks to human health posed by possible interspecies transmission of the infection. Nevertheless, the incidence of BSE in the UK continued to rise steadily until 1992, when it reached a yearly peak at 36 680 confirmed cases, and after which it has continuously declined to a total of 2254 cases in 1999 and a nearly complete total in 2000 (1308 as of Feb. 2, 2001) that is consistent with a continuing decline.29 Although this trend is very encouraging, and it is hoped that BSE will have been eradicated from the UK by some time early in the first decade of the 21st century, between December 1986 and Feb. 2, 2001, a total of 177 706 cases of BSE had been formally diagnosed in that country.28 Furthermore, because of the protracted asymptomatic incubation period of BSE (estimated to be 4–5 years on average), in excess of 700 000 infected animals are estimated to have been slaughtered and unknowingly consumed, with perhaps 40 000 of these being in an advanced state of disease development.30
Another sobering consideration is that, because of the typically broad distribution of foodstuffs containing bovine products, according to one recent estimate31 one infected animal could expose up to 500 000 consumers to BSE. To complicate matters further, very recent evidence32 suggests that prion infectivity can propagate in experimental inoculations across species boundaries, in the absence of any clinical signs in the recipient animals. If such phenomena have occurred in species other than cattle (e.g., sheep, pigs and poultry) that were also exposed to BSE through contaminated feed, these species could also have contributed to the total level of human exposure. This suggests that a large and unknown portion of the British population has been exposed at some level to the infectious agent of BSE. In addition, cases of BSE from outside the UK reported up until Feb. 16, 2001, total 3291 in 15 different countries,33 including one case that occurred in Canada in a bull imported from the UK.34 Because of these BSE cases from outside the UK, as well as broad interactions between the UK and other countries (e.g., tourism, trade, migration), the risk to humans currently residing outside the UK of having had primary (e.g., foodborne) exposure to this TSE agent cannot be discounted.
vCJD
Concerns over the possibility of transmission of BSE to humans led to the re-establishment in 1990 of a comprehensive system of national surveillance for CJD in the UK.35 Because it was not known a priori in what form human prion disease resulting from exposure to BSE might appear clinically, this surveillance was directed not necessarily toward the identification of a novel clinical syndrome, but toward detection of any rise in the incidence of human prion disease against a comprehensively enumerated and characterized background of endemic disease. In fact, it was against such a well-characterized, relatively constant background incidence of cCJD cases (including those with identifiable genetic and iatrogenic causes) that a series of 10 cases of a novel clinical form of CJD was first observed and reported.36 This syndrome — initially named “new variant CJD” (nvCJD) but now simply “variant CJD” (vCJD) or Will–Ironside syndrome — presented a clinical picture that, when considered in all of its aspects, contrasted strikingly with that of all forms of cCJD. Since the onset of the first reported case in 1995 and up to Feb. 5, 2001, a total of 94 definite and probable cases of vCJD have been referred to the UK CJD Surveillance Unit in Edinburgh,37 with an additional 2 confirmed cases and 1 probable case in France and 1 in the Republic of Ireland.
Perhaps the most striking clinical difference between classic and variant CJD is the downward shift in average age for individuals affected by the latter disease. The median age at death of the 52 definite and probable cases of vCJD identified in the UK up to the end of January 2000 was 29 years (range 14–53 years), as compared with a median age at death of 65 years for classic sporadic CJD diagnosed under the same surveillance system.38 The rate of progression of vCJD is marginally slower than that of cCJD (mean duration 14 months, range 7–38 months for the 52 cases mentioned above, compared with a median duration of less than 1 year for cCJD). Certain clinical features, which are very rare in cCJD, are common in vCJD:39,40,41 involuntary movements are virtually universal, and sensory symptoms (often painful) occur in about 50% of affected individuals. “Psychiatric” presentation of the disease (depression and anxiety) is common, confounding clinical suspicion of vCJD. Paraclinical diagnostic data also differ between the 2 syndromes: on electroencephalography the classic periodic triphasic wave complexes of cCJD are not observed in vCJD, and MRI brain signal hyperintensities differ in frequency and distribution.42 vCJD also appears to be a distinct neuropathological entity, characterized by marked brain accumulation of PrPSc, some of which is in plaques surrounded by prominent spongiosis (“florid” or “daisy” plaques).36,43
Evidence for a causal link between BSE and vCJD
Obviously, a direct demonstration of transmission of BSE to humans is ethically inconceivable, and given that no specific exogenous nucleic acid genome has yet been associated with TSEs, we cannot apply the powerful methods of “molecular epidemiology” whereby microbial genetic markers are used to trace transmission pathways for infectious disease.44 Nevertheless, the total evidence available so far (Table 1) strongly implicates exposure to BSE, most probably through contaminated food or other bovine-derived products, as the key risk factor for vCJD and very probably its cause through interspecies transmission of an infective agent.
Table 1

Estimation of risk for secondary transmission of vCJD
Pathobiology and potential transmissibility
Awareness of the potential for transmission of vCJD via blood or peripheral tissues was heightened recently by the publication of 3 sets of findings in the UK. The first of these was the immunocytochemical demonstration of PrPSc deposits in lymphoid tissue in the excised appendix of a 48-year-old man who underwent an appendectomy 8 months before he exhibited clinical signs of vCJD.70 PrPSc deposits had been demonstrated by similar means in the tonsils of patients with diagnoses of vCJD.71 These observations indicated clearly that the pathological hallmark of prion disease (and, thus, by extrapolation, TSE infectivity) could be found months before the appearance of neurological symptoms in peripheral lymphoid tissue (such as tonsils, the appendix, lymph nodes or the spleen) of patients with vCJD.
The second set of results72 indicated that PrPSc deposition in peripheral lymphoid tissues of patients with vCJD was not an isolated finding but, rather, a highly reproducible pattern. In their study, Collinge's laboratory found that such deposits could be detected readily in tissue from 40 lymph nodes, 68 tonsils and 64 spleens of a series of patients with diagnoses of definite or probable vCJD, but not in any of a series of corresponding tissue samples taken from control individuals who were either healthy or had diagnoses of cCJD or other neurological disease. One interpretation of these data is that, in association with its putative peripheral route of entry and early propagation in lymphoid tissues before the stage of neuroinvasion, vCJD infectivity accumulates to much higher levels in peripheral tissues than does cCJD infectivity. Combined with the demonstrated presence of PrPC on the surfaces of diverse types of circulating blood cells,73 which may provide a biochemical substrate for agent propagation or transport in blood, or both, this would suggest in turn that presymptomatic individuals infected with vCJD may present a very different set of risks for secondary transmission through donated blood or via other routes than do individuals in the asymptomatic phase of conventional CJD.
The third, very recently reported finding is that whole blood transfused from a single sheep that had been experimentally infected with BSE by the oral route was apparently capable of transmitting the same disease to a recipient sheep, as early as midway through the asymptomatic incubation period of the donor animal.74 This report has been criticized as premature, given the single observation and the currently incomplete status of the experiments,75 however, because of the positive match in glycoform patterns between the donor and recipient PrP 27-30s, it seems probable that these early results will be borne out by the full experiment. This work has provided the first experimental evidence of bloodborne transmission of BSE-derived prion infectivity in any species — including sheep-to-sheep transmission of scrapie — and raises the question whether this finding can be extrapolated to vCJD in humans. The findings also complement those of ongoing work in rodent models, where the presence of low levels of blood infectivity has been confirmed in advanced preclinical stages following intracerebral inoculation.6
One careful quantitative assessment of secondary vCJD disease risk per infected vCJD blood donation76 used conservative, empirically supported assumptions (e.g., concentration of infectivity in donated blood; degree to which current harvesting and manufacturing processes dilute, remove or inactivate the vCJD agent and distribute it among different fractions; age distribution and expected lifespan of recipients of infectious materials; incubation periods from exposure to clinical disease, and so on) to construct a statistical model and a “base case” scenario for bloodborne transmission of vCJD in the UK. The author admitted that the assumed values of many of the variables incorporated remain highly uncertain, so that the estimated values of output parameters of the model may in reality diverge widely in either direction from those of the base case. To offset this weakness, which is inherent in complex statistical models of this type, “sensitivity analyses” were also carried out by systematically changing the key input variables over a range of plausible values. For example, in the base case, one important output parameter was that approximately 0.8 new cases of vCJD could be expected per infected blood donation.76 However, this estimate was strongly influenced by the input estimate of vCJD infectivity levels in blood. In the base case, this input value was derived from those found in the blood of experimental rodents infected with cCJD (i.e., approximately 1 ID50 per mL of whole blood). If the level of infectivity in vCJD blood was assumed for instance to be 10 times higher, the number of new cases of vCJD expected per infected donation estimated by the model rose to 3.6. With an infectivity level of 100 ID50 per mL, this figure rose still further to 35.4. Assuming a lower level of blood infectivity had the opposite but proportionally smaller effect. As another example, changing the median incubation period from the basal value of 15 years (log-normal distribution, 99% range 5–40 years) to 5 years or 30 years altered the model output much less markedly, by raising the estimate to approximately 1.2 and lowering it to approximately 0.5 new cases per infected donation respectively.
Size of the primary epidemic of vCJD
Taken together with the epidemiological features of BSE itself and the magnitude of the resulting human exposure (see previous section), current evidence for the zoonotic origin of vCJD indicates that a human epidemic — on some as-yet-undetermined scale ranging from hundreds of individuals to tens or even hundreds of thousands — of primary (BSE-related) vCJD cases is expected to continue in the UK and Europe over the next few years to a decade or more. Clearly, any reliable estimate of the number of cases in the UK could provide an indication of what other, less-exposed countries, such as Canada, can expect. So far, however, such an estimate has proven elusive.
Computer simulation studies have shown that, even when restricting assumptions are placed on epidemiological scenarios, for example, that only individuals homozygous for a common polymorphic allele encoding methionine at codon 129 in PRNP are susceptible to infection by BSE, it remains difficult to estimate usefully the total magnitude of the human vCJD epidemic by extrapolation from the current surveillance data.77,78,79 The most recent report derived by this approach79 suggests that the absence of a marked increase in numbers of vCJD cases in 1999 compared with 1998 is consistent with a total epidemic of most probably fewer than 6000 cases, unless one assumes that the mean incubation period approaches the mean lifespan, in which case the projected total rises to 136 000. The authors go farther and postulate that, if the total annual incidence of vCJD in the UK remains at or below 15 cases during 2000–2002, even with such a protracted mean incubation period the total falls to 20 000.79
Relevant to this point is a very recently published statistical analysis of the UK CJD surveillance data from 1994 to June 30, 2000, which shows that, when analyzed for the presence of significant trends in the annual incidence of vCJD onsets and deaths, the data are consistent with an average 23% annual increase in incidence of onsets and a 33% annual increase in death rate over that period.80 In fact, as of Feb. 5, 2001, the total number of deaths in the UK definitely caused by vCJD stands at 26 for the year 2000, with an additional 2 probable cases awaiting postmortem examination,36 indicating that the total number of deaths for last calendar year is twice the 1999 total of 14. This observation would seem to imply that the most optimistic scenarios developed by Ghani and colleagues79 are not particularly likely to apply, especially if we relax the somewhat restrictive assumption that only methionine/ methionine homozygotes are susceptible.
The uncertainty inherent in such extrapolations is compounded by the possibility that the epidemic curve may be “multiphasic,” reflecting incidence of disease onset in various population subgroups that may vary in susceptibility or exposure factors such as genotype, age at exposure, or intensity, level or route of exposure. For example, all confirmed definite or probable vCJD cases sequenced by the UK CJD Surveillance System have been homozygous for the methionine-encoding allele at codon 129 of PRNP,38 despite a frequency in the general white population for this genotype of about 35%. Genotype at this polymorphic genetic site has a well-known correlation with susceptibility to sporadic CJD,16,81 and the human subpopulations carrying the other 2 known genotypes at this site, encoding methionine/valine and valine/valine, could be differently susceptible to vCJD or express its symptoms differently, or both. This effectively means that, even if the shape of the current incidence curve begins to become apparent in the next few years, it is by no means certain that the course of the entire epidemic will then be predictable. Because of the possibility that genotype could modify disease expression, it is also conceivable that more than one variant CJD syndrome may emerge from the same BSE-related human epidemic in the UK, thus presenting additional challenges for diagnosis and surveillance.
Epidemiological study of the first-identified peripherally acquired human prion disease, kuru, has also yielded some intriguing results that are relevant to attempts to predict the future course of the vCJD epidemic. First, despite the abrupt decline and likely termination of exposure to this infectious illness among the Fore people of Papua New Guinea around 1960, due to cessation of traditional cannibalism, clinical cases continued to appear at a low rate well into the 1990s.2 This indicates that, in the case of transmission of prion disease among humans via a peripheral (oral?) route, the incubation period from exposure to clinical disease can be at least as long as 30 years, and perhaps longer. If this pattern applies to vCJD (for which the statistical dispersion of incubation periods may be conjectured to be even broader, because of the relative inefficiency of interspecies transmission), then the total duration of the present epidemic may be comparable to that of kuru. In fact, in a similar vein, Collinge82 has recently proposed that it is perhaps more plausible that the present cohort of patients with vCJD has resulted from exposure of individuals early in the course of the BSE epidemic than to suppose that these exposures occurred during its peak. Second, in a study83 of the distribution of the PRNP methionine/valine polymorphism among 92 patients with kuru, it was found that, despite similarity in clinical presentation, homozygotes at this codon position showed a reduced average age of onset and duration of illness compared with methionine/valine heterozygotes. This observation is consistent with the suggestion that the vCJD incidence curve may be genetically “multiphasic,” which again would lead to serious consideration of the possibility of an epidemic of protracted duration.
The results of a direct approach to the estimation of vCJD prevalence, using immunohistochemical staining of archived tissues from 3170 appendectomies and tonsillectomies to search for diagnostic deposits of PrPSc, proved completely negative.84 Despite the fact that these results are encouraging, caveats must be applied, because fixed tissues may on average yield lower sensitivities for such examination. Partly for this reason, a prospective tissue-based prevalence study is currently being planned. However, even if the technical obstacles are overcome, it may prove difficult on the basis of a possibly very low rate of positives to estimate population prevalence with useful statistical precision.85
Conclusion
What this discussion highlights is that, aside from the difficulties inherent in detecting trends in data consisting of small numbers of observations, it is difficult to move with any precision, with a novel, complex and slowly progressing infectious disease such as vCJD, from the distribution of dates of death per se to the shape, size and dynamics of a possibly much larger epidemic curve. We also lack critical basic knowledge of the infection process and its dynamics, which could help to estimate the risk of secondary transmission, assuming infection in a blood donor. Despite a comprehensive CJD surveillance system86 that has been in full operation by Health Canada since 1998, there has not yet been a case of vCJD identified in Canada, and the probability of this occurring cannot presently be reliably estimated. In the final analysis, it may never be possible to predict precisely the quantitative features of the vCJD epidemic in the UK, and a policy of waiting until the pattern becomes clear from data from existing cases may be tantamount to ensuring that the best opportunities for early intervention are missed. Because countries other than the UK (including Canada) may be significantly affected by vCJD, it would seem prudent to treat the threat to Canadian public health from bloodborne transmission of vCJD as not purely theoretical, but as real and large enough to warrant intensified efforts in prion disease surveillance, prevention and research. Arguably, one of the highest priorities here is the development of a screening test for prion infectivity in blood, for which candidate technological platforms (so far based on ultrasensitive fluorescence detection of PrPSc) are beginning to appear.87,88 This novel and difficult task will require international cooperation and long-term validation studies to succeed.89
Footnotes
This article has been peer reviewed.
Competing interests: None declared.
Correspondence to: Dr. Michael B. Coulthart, National Laboratory for Host Genetics and Prion Diseases, Canadian Science Centre for Human and Animal Health, Health Canada, 1015 Arlington St., Winnipeg MB R3E 3R2; fax 204 789-5021; mike_coulthart@hc-sc.gc.ca and to Dr. Neil R. Cashman, Centre for Research in Neurodegenerative Diseases, University of Toronto, 6 Queen's Park Cres. W, Toronto ON M5S 1A8; fax 416 978-1878; neil.cashman@utoronto.ca
References
- 1.Creutzfeldt HG. Über eine eigenartige herdförmige Erkrankung des Zentralnervensystems. Zeitschrift für die gestamte Neurologie und Psychiatrie 1920;57:1-18.
- 2.Brown P, Bradley R. 1755 and all that: a historical primer of transmissible spongiform encephalopathy. BMJ 1998;317:19-26. [DOI] [PMC free article] [PubMed]
- 3.Cashman NR. A prion primer. CMAJ 1997;157(10):1381-85. Available: www.cma.ca/cmaj/vol-157/issue-10/1381.htm [PMC free article] [PubMed]
- 4.Ridley RM, Baker HF. Fatal protein. Oxford: Oxford University Press; 1998.
- 5.Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998;95:13363-83. [DOI] [PMC free article] [PubMed]
- 6.Brown P. The risk of blood-borne Creutzfeldt-Jakob disease. Dev Biol Stand 2000; 102:53-9. [PubMed]
- 7.Wilson K, Code C, Ricketts MN. Risk of acquiring Creutzfeldt-Jakob disease from blood transfusions: systematic review of case-control studies. BMJ 2000;321:17-9. [DOI] [PMC free article] [PubMed]
- 8.Health Canada. Health Canada advises blood agencies on disposition of blood products [news release]. Ottawa: Health Canada; 1998 Dec 24. Available: www.hc-sc.gc.ca/english/archives/releases/1998/98_106e.htm (accessed 2001 June 1).
- 9.Therapeutic Products Programme, Health Canada. Directive 99-01: donor exclusion to address theoretical risk of transmission of variant CJD through the blood supply. Ottawa: Health Canada; 1999 Aug 17. Available: www.hc-sc.gc.ca/hpb-dgps/therapeut/htmleng/btox.html (accessed 2001 June 1).
- 10.Therapeutic Products Programme, Health Canada. Directive 99-02: donor exclusion to address theoretical risk of transmission of variant CJD through the use of commercial blood products. Ottawa: Health Canada; 1999 Aug 17. Available: www.hc-sc.gc.ca/hpb-dgps/therapeut/htmleng/btox.html (accessed 2001 June 1).
- 11.Therapeutic Products Programme, Health Canada. Directive D2000-01: donor exclusion to address theoretical risk of transmission of variant CJD through the blood supply. Ottawa: Health Canada; 2000 Aug 30. Available: www.hc-sc.gc.ca/hpb-dgps/therapeut/htmleng/btox.html (accessed 2001 June 1).
- 12.Masters CL, Harris JO, Gajdusek DC, Gibbs CJ, Bernoulli C, Asher DM. Creutzfeldt-Jakob disease: patterns of worldwide occurrence and the significance of familial and sporadic clustering. Ann Neurol 1979;5:177-88. [DOI] [PubMed]
- 13.Rohwer RG. Virus-like sensitivity of the scrapie agent to heat inactivation. Science 1984;223:600-2. [DOI] [PubMed]
- 14.Rohwer RG. Scrapie infectious agent is virus-like in size and susceptibility to inactivation. Nature 1984;308:658-62. [DOI] [PubMed]
- 15.Gajdusek DC, Gibbs CJ, Alpers MP. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 1966;209:794-6. [DOI] [PubMed]
- 16.Brown P, Preece M, Brandel JP, Sato T, McShane L, Zerr I, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000;55:1075-81. [DOI] [PubMed]
- 17.Brown P, Cervenakova L, McShane L, Goldfarb LG, Bishop K, Bastian F, et al. Creutzfeldt-Jakob disease in a husband and wife. Neurology 1998;50(3):684-8. [DOI] [PubMed]
- 18.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136-44. [DOI] [PubMed]
- 19.Rohwer RG. The scrapie agent: “a virus by any other name”. Curr Top Microbiol Immunol 1991;172:195-232. [PubMed]
- 20.Chesebro B. Human TSE disease — viral or protein only? Nat Med 1997; 3:491-2. [DOI] [PubMed]
- 21.Chesebro B, Caughey B. Prion protein and the transmissible spongiform encephalopathies. Trends Cell Biol 1997;7:56-62. [DOI] [PubMed]
- 22.Chesebro B. BSE and prions: uncertainties about the agent. Science 1998;279: 42-3. [DOI] [PubMed]
- 23.Windl O, Dempster M, Estibeiro JP, Lathe R, de Silva R, Esmonde T, et al. Genetic basis of Creutzfeldt-Jakob disease in the United Kingdom: a systematic analysis of predisposing mutations and allelic variation in the PRNP gene. Hum Genet 1996;98:259-64. [DOI] [PubMed]
- 24.Wells GAH, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, et al. A novel progressive spongiform encephalopathy in cattle. Vet Rec 1987; 121: 419-20. [DOI] [PubMed]
- 25.Taylor DM, Fernie K, McConnell I, Ferguson CE, Steele PI. Solvent extraction as an adjunct to rendering: the effect on BSE and scrapie agents of hot solvents followed by dry heat and steam. Vet Rec 1998;143:6-9. [DOI] [PubMed]
- 26.Brown P. On the origins of BSE. Lancet 1998;352:252-3. [DOI] [PubMed]
- 27.Wilesmith JW, Wells GAH, Cranwell MP, Ryan JBM. Bovine spongiform encephalopathy: epidemiological studies. Vet Rec 1988;123:638-44. [PubMed]
- 28.Wilesmith JW. The epidemiology of bovine spongiform encephalopathy. Sem Virol 1991;2:239-45.
- 29.Ministry of Agriculture, Fisheries and Food, United Kingdom. MAFF BSE information: weekly cumulative statistics. Updated 2001 May 11. London (UK): The Ministry; 2001. Available: www.maff.gov.uk/animalh/bse/index.html (accessed 2001 June 4).
- 30.Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, Udy HJ, et al. Transmission dynamics and epidemiology of BSE in British cattle. Nature 1996;382:779-88. [DOI] [PubMed]
- 31.European Union Scientific Steering Committee. Opinion of the scientific steering committee on the human exposure risk (HER) via food with respect to BSE (adopted 1999 Dec 10). Brussels: The Committee; 1999. Available: //europa.eu.int/comm/food/fs/sc/ssc/out67_en.html (accessed 2001 June 4).
- 32.Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant species. Proc Natl Acad Sci U S A 2000;97(18):10248-53. [DOI] [PMC free article] [PubMed]
- 33.Office International des Epizooties. Number of reported cases of BSE worldwide (excluding the United Kingdom). Paris: The Office; updated 2001 June 1. Available: www.oie.int/eng/info/en_esbmonde.htm (accessed 2001 June 4).
- 34.Canadian Food Inspection Agency. Fact sheet: bovine spongiform encephalopathy. Ottawa: The Agency; updated 2001 May 25. Available: //inspection .gc.ca/english /anima/heasan/disemala/bseesbe.shtml (accessed 2001 June 4).
- 35.Southwood Committee. Report of the working party on bovine spongiform encephalopathy. London (UK): Department of Health and Ministry of Agriculture, Fisheries and Food; 1989.
- 36.Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovich A et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996;347:921-5. [DOI] [PubMed]
- 37.UK Department of Health. Monthly Creutzfeldt-Jakob disease statistics, February 5 2001. London: The Department; 2001. Available: www.doh.gov.uk/cjd/stats/feb01.htm (accessed 2001 June 4).
- 38.UK National CJD Surveillance Unit Creutzfeldt-Jakob Disease Surveillance. 8th Annual Report, 1999. Edinburgh: The Unit; 1999. Available: www.cjd.ed.ac.uk/jan.html (accessed June 4).
- 39.Zeidler M, Stewart GE, Barraclough CR, Bateman DE, Bates D, Burn DJ, et al. New variant Creutzfeldt-Jakob disease: neurological features and diagnostic tests. Lancet 1997;350:903-7. [DOI] [PubMed]
- 40.Zeidler M, Johnstone EC, Bamber RW, Dickens CM, Fisher CJ, Francis AF, et al. New variant Creutzfeldt-Jakob disease: psychiatric features. Lancet, 1997; 350:908-10. [DOI] [PubMed]
- 41.Will RG, Zeidler M, Stewart GE, Macleod MA, Ironside JW, Cousens SN, et al. Diagnosis of new variant Creutzfeldt-Jakob disease. Ann Neurol 2000;47: 575-82. [PubMed]
- 42.Zeidler M, Sellar RJ, Collie DA, Knight R, Stewart G, Macleod MA, et al. The pulvinar sign on magnetic resonance imaging in variant Creutzfeldt-Jakob disease. Lancet 2000;355:1412-8. [DOI] [PubMed]
- 43.Ironside JW, Head MW, Bell JE, McCardle L, Will RG. Laboratory diagnosis of variant Creutzfeldt-Jakob disease. Histopathology 2000;37:1-9. [DOI] [PubMed]
- 44.Hahn BH, Shaw GM, De Cock KM, Sharp PM. AIDS as a zoonosis: scientific and public health implications. Science 2000;287:607-14. [DOI] [PubMed]
- 45.Chazot G, Broussolle E, Lapras Cl, Blattler T, Aguzzi A, Kopp N. New variant of Creutzfeldt-Jakob disease in a 26-year-old French man. Lancet 1996; 347: 1181. [DOI] [PubMed]
- 46.Oppenheim C, Brandel JP, Jauw JJ, Deslys JP, Fontaine B. MRI and the second French case of vCJD. Lancet 2000;356:253-4. [DOI] [PubMed]
- 47.Tiwana H, Wilson C, Pirt J, Cartmell W, Ebringer A. Autoantibodies to brain components and antibodies to Acinetobacter calcoaceticus are present in bovine spongiform encephalopathy. Infect Immun 1999;67:6591-5. [DOI] [PMC free article] [PubMed]
- 48.Purdey M. The UK epidemic of BSE: slow virus or chronic pesticide-initiated modification of the prion protein? Med Hypotheses 1996;46:429-54. [DOI] [PubMed]
- 49.Purdey M. Ecosystems supporting clusters of sporadic TSEs demonstrate excesses of the radical-generating divalent cation manganese and deficiencies of antioxidant cofactors Cu, Se, Fe, Zn: Does a foreign cation substitution at prion protein's Cu domain initiate TSE? Med Hypotheses 2000;54:278-306. [DOI] [PubMed]
- 50.Wyatt JM, Pearson GR, Smerdon TN, Gruffydd-Jones TJ, Wells GAH, Wilesmith JW. Naturally occurring scrapie-like encephalopathy in five domestic cats. Vet Rec 1991;129:233-36. [DOI] [PubMed]
- 51.Bradley R, Matthews D. Sub-acute, transmissible spongiform encephalopathies: current concepts and future needs. Rev Sci Tech 1992;11:605-32. [DOI] [PubMed]
- 52.UK Ministry of Agriculture, Fisheries and Food. MAFF BSE information: other TSEs. London: The Ministry; updated 2001 April 1. Available: www.maff.gov.uk/animalh/bse/bse-statistics/level-3-tsestat.html (accessed 2001 June 4).
- 53.Bons N, Mestre-Frances N, Charnay Y, Tagliavini F. Spontaneous spongiform encephalopathy in a young adult rhesus monkey. Lancet 1996;348(9019):55. [DOI] [PubMed]
- 54.Bons N, Mestre-Frances N, Guiraud I, Charnay Y. Prion immunoreactivity in brain, tonsil, gastrointestinal epithelial cells, and blood and lymph vessels in lemurian zoo primates with spongiform encephalopathy. C R Acad Sci III 1997; 320:971-9. [DOI] [PubMed]
- 55.Bons N, Mestre-Frances N, Belli P, Cathala F, Gajdusek DC, Brown P. Natural and experimental oral infection of nunhuman primates by bovine spongiform encephalopathy agents. Proc Natl Acad Sci U S A 1999;96:4046-51. [DOI] [PMC free article] [PubMed]
- 56.Will RG, Ironside JW. Oral infection by the bovine spongiform encephalopathy prion. Proc Natl Acad Sci U S A 1999;96:4738-9. [DOI] [PMC free article] [PubMed]
- 57.Wilesmith JW, Ryan JBM, Hueston WD. Bovine spongiform encephalopathy: case-control studies of calf feeding practices and meat and bone meal inclusion in proprietary concentrates. Res Vet Sci 1991;52:325-31. [DOI] [PubMed]
- 58.Ryder SJ, Hawkins SA, Dawson M, Wells GA. The neuropathology of experimental bovine spongiform encephalopathy in the pig. J Comp Pathol 2000; 122: 131-43. [DOI] [PubMed]
- 59.Foster JD, Hope J, Fraser H. Transmission of bovine spongiform encephalopathy to sheep and goats. Vet Rec 1993;133:339-41. [DOI] [PubMed]
- 60.Baker HF, Ridley RM, Wells GAH. Experimental transmission of BSE and scrapie to the common marmoset. Vet Rec 1993;132:403-6. [DOI] [PubMed]
- 61.Fraser H, Foster JD. Transmission to mice, sheep and goats and bioassay of bovine tissues. In: Bradley R, Marchant B, editors. Transmissible spongiform encephalopathies. Proceedings of a consultation on BSE with the Scientific Veterinary Committee of the Commission of the European Communities; 1993 Sept 14–15; Brussels: The Commission; 1994. p. 145-59.
- 62.Wells GA, Hawkins SA, Green RB, Austin AR, Dexter I, Spencer YI, et al. Preliminary observations on the pathogenesis of experimental bovine spongiform encephalopathy (BSE): an update. Vet Rec 1998;142:103-6 [DOI] [PubMed]
- 63.Wells GA, Hawkins SA, Green RB, Spencer YI, Dexter I, Dawson M. Limited detection of sternal bone marrow infectivity in the clinical phase of experimental bovine spongiform encephalopathy (BSE). Vet Rec 1999;144:292-4. [DOI] [PubMed]
- 64.UK Institute for Food Science and Technology. Position statement on bovine spongiform encephalopathy (BSE). London: The Institute; 1999 June 16. Available: www.ifst.org/hottop5.htm (accessed 2001 June 4).
- 65.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997;389(6650):498-501. [DOI] [PubMed]
- 66.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and aetiology of ‘new variant’ CJD. Nature 1996;383:685-90. [DOI] [PubMed]
- 67.Hill AF, Desbrulais M, Joiner S, Sidle KCL, Gowland I, Collinge J, et al. The same prion strain causes vCJD and BSE. Nature 1997;389:448-50. [DOI] [PubMed]
- 68.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994;68:7859-68. [DOI] [PMC free article] [PubMed]
- 69.Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci U S A 1999;96:15137-42. [DOI] [PMC free article] [PubMed]
- 70.Hilton DA, Fathers E, Edwards P, Ironside JW, Zajicek J. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet 1998;352:703-4 [DOI] [PubMed]
- 71.Hill AF, Zeidler M, Ironside J, Collinge J. Diagnosis of new variant Creutzfeldt-Jakob disease by tonsil biopsy. Lancet 1997;349:99-100. [DOI] [PubMed]
- 72.Hill AF, Butterworth RJ, Joiner S, Jackson G, Rossor MN, Thomas DJ, et al. Investigation of variant Creutzfeldt-Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999;353(9148):183-9. [DOI] [PubMed]
- 73.Dodelet VC, Cashman NR. Prion protein expression in human leukocyte differentiation. Blood 1998;91:1556-61. [PubMed]
- 74.Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet 2000;356:999-1000. [DOI] [PubMed]
- 75.Brown P. BSE and transmission through blood. Lancet 2000;356:955-6. [DOI] [PubMed]
- 76.Comer PJ. Assessment of the risk of exposure to vCJD infectivity in blood and blood products: final report for the Spongiform Encephalopathy Advisory Committee and the Department of Health. London: Det Norske Veritas; 1999.
- 77.Ghani AC, Ferguson NM, Donnelly CA, Hagenaars TJ, Anderson RM. Epidemiological determinants of the pattern and magnitude of the vCJD epidemic in Great Britain. Proc R Soc Lond B Biol Sci 1998;265:2443-52. [DOI] [PMC free article] [PubMed]
- 78.Ghani AC, Ferguson NM, Donnelly CA, Hagenaars TJ, Anderson RM. Estimation of the number of people incubating variant CJD. Lancet 1998;352:1353-4. [DOI] [PubMed]
- 79.Ghani AC, Ferguson NM, Donnelly CA, Anderson RM. Predicted vCJD mortality in Great Britain. Nature 2000;406:583-4. [DOI] [PubMed]
- 80.Andrews NJ, Farrington CP, Cousens SN, Smith PG, Ward H, Knight RSG, et al. Incidence of variant Creutzfeldt-Jakob disease in the UK. Lancet 2000;356:481-2. [DOI] [PubMed]
- 81.Windl O, Giese A, Schulz-Schaeffer W, Zerr I, Skworc K, Arendt S, et al. Molecular genetics of human prion diseases in Germany. Hum Genet 1999; 105:244-52. [DOI] [PubMed]
- 82.Collinge J. Variant Creutzfeldt-Jakob disease. Lancet 1999;354:317-23. [DOI] [PubMed]
- 83.Cervenakova L, Goldfarb LG, Garruto R, Lee HS, Gajdusek DC, Brown P. Phenotype-genotype studies in kuru: implications for new variant Creutzfeldt-Jakob disease. Proc Natl Acad Sci U S A 1998;95(22):13239-41. [DOI] [PMC free article] [PubMed]
- 84.Ironside JW, Hilton DA, Ghani A, Johnston NJ, Conyers L, McCardle LM, et al. Retrospective study of prion-protein accumulation in tonsil and appendix tissues. Lancet 2000;355:1693-4. [DOI] [PubMed]
- 85.Ghani AC, Donnelly CA, Ferguson NM, Anderson RM. Assessment of the prevalence of vCJD through testing tonsils and appendices for abnormal prion protein. Proc R Soc Lond B Biol Sci 2000;267:23-9. [DOI] [PMC free article] [PubMed]
- 86.Division of Blood-borne Pathogens, Health Canada. Surveillance for Creutzfeldt-Jakob disease in Canada. Ottawa: Health Canad; 1999 Sept. Available: www.hc-sc.gc.ca/hpb/lcdc/bid/bbp/cjd/cjd0999e.html (accessed 20001 June 4).
- 87.Schmerr MJ, Jenny AL, Bulgin MS, Miller JM, Hamir AN, Cutlip RC, et al. Use of capillary electrophoresis and fluorescent labeled peptides to detect the abnormal prion protein in the blood of animals that are infected with a transmissible spongiform encephalopathy. J Chromatogr A 1999;853:207-14. [DOI] [PubMed]
- 88.MacGregor I, Hope J, Barnard G, Kirby L, Drummond O, Pepper D, et al. Application of a time-resolved fluoroimmunoassay for the analysis of normal prion protein in human blood and its components. Vox Sang 1999;77:88-96. [DOI] [PubMed]
- 89.Bonn D. Future uncertain for reliable vCJD screening tests. Lancet 2000; 356:228. [DOI] [PubMed]