

Abstract
Background
Nintedanib is effective for treating idiopathic pulmonary fibrosis (IPF), but some patients may exhibit a suboptimal response and develop on-treatment acute exacerbation (AE-IPF), hepatic injury, or mortality. It remains unclear which patients are at risk for these adverse outcomes.
Methods
We analysed the demographic and clinical data, baseline plasma levels of Krebs von den Lungen-6 (KL-6) and surfactant protein A (SPA), and longitudinal clinical courses of a real-world cohort of IPF patients who received nintedanib ≥ 14 days between March 2017 and December 2020. Cox proportional-hazards regression, subdistribution hazards regression, and sensitivity analyses were performed to investigate the association between baseline predictors and AE-IPF, mortality, and nintedanib-related hepatic injury. The relationship between baseline predictors and pulmonary function decline was determined.
Results
Fifty-seven patients were included, of whom 24 (42%) developed hepatic injury, 20 (35%) had AE-IPF, and 16 (28%) died on-treatment. A baseline plasma KL-6 level ≥ 2.5 ng/mL, and diffusion capacity for carbon monoxide (DLCO) < 55% predicted, were associated with increased risk of hepatic injury (adjusted hazard ratio [aHR] was 3.46; 95% CI 1.13–10.60; p = 0.029 for KL-6, and 6.05; 95% CI 1.89–19.32; p = 0.002 for DLCO). Both factors also predicted severe and recurrent hepatic injury. Patients with baseline KL-6 ≥ 2.5 ng/mL also had a higher risk of AE-IPF (aHR 4.52; 95% CI 1.63–12.55; p = 0.004). For on-treatment mortality, baseline KL-6 ≥ 3.5 ng/mL and SPA ≥ 600 pg/mL were significant predictors (aHR 5.39; 95% CI 1.16–24.97; p = 0.031 for KL-6, and aHR 12.28; 95% CI 2.06–73.05; p = 0.006 for SPA). Results from subdistribution hazard regression and sensitivity analyses supported these findings. Patients with elevated baseline plasma KL-6 levels also exhibited a trend towards faster pulmonary function decline.
Conclusions
For patients with IPF who are receiving nintedanib, we have identified baseline predictors, in particular plasma KL-6 levels, for the risk of adverse outcomes. Patients with these predictors may require close monitoring for unfavourable responses during treatment. Our findings also support the prognostic role of molecular markers like KL-6 and may contribute to future formulation of more individualized therapeutic strategies for IPF.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12890-021-01530-6.
Keywords: Nintedanib, Krebs von den Lungen-6, Surfactant protein A, Diffusion capacity for carbon monoxide, Acute exacerbation, Mortality
Introduction
Idiopathic pulmonary fibrosis (IPF) is a rare but progressive and devastating interstitial lung disease [1–3]. The median survival after diagnosis is around three years, worse than that of many cancers [2–4]. Patients with IPF are also at risk of developing acute exacerbation, which is characterized by extensive alveolar injury and accelerated fibrosis, leading to rapid and severe respiratory deterioration and even death [2, 3, 5, 6]. The pathogenesis of IPF is not fully understood, but much progress has been achieved particularly over the last decade [3]. Two anti-fibrotic agents with proven efficacy, nintedanib and pirfenidone, have recently become available [7, 8]. Nintedanib is a competitive tyrosine kinase inhibitor, interrupting pathways downstream of profibrotic growth factors [3, 9]. Accumulating evidence reveals that nintedanib slows the decline in forced vital capacity and lowers the risk of acute exacerbation [4, 10]. Pooled data from the initial major trials even revealed a possible reduction in the risk of on-treatment mortality [10].
Despite this, in both trials and real-world experience from a range of countries and ethnic groups, the therapeutic efficacy of nintedanib has not been universal: some patients receiving nintedanib might still exhibit a rapid decline in the pulmonary functions or develop acute exacerbation and even died [11–32]. In addition, nintedanib causes adverse reactions, the most common of which are gastrointestinal complaints; hepatotoxicity is also a concern. Hepatic injury during nintedanib therapy has been consistently described in clinical trials [7, 10–14] and post-marketing reports [15–32]. Relatively high incidence rates of hepatitis have been reported particularly from East Asia [19, 20, 26–28]. Nintedanib-related hepatic injury potentially interrupts the therapy and compromises first-pass metabolism, increasing exposure to the drug and thus the risks of other adverse reactions [33–35].
Pulmonary function parameters and radiographic features on computed tomography images are currently important determinants for the diagnosis and severity and prognosis assessment for IPF. However, accumulating evidence shows that blood levels of certain lung-specific macro-molecules, such as Krebs von den Lungen-6 (KL-6, also known as mucin-1, a glycoprotein expressed on the cell membrane of type 2 alveolar cells) and surfactant protein A (SPA, also secreted by type 2 alveolar cells), are also correlated with the severity of fibrosis and may serve as biomarkers for determining the clinical aspects of IPF [36–40]. While pulmonary function parameters are commonly used to assess the efficacy of antifibrotic treatment, the utility of these, as well as clinical and bio-chemical, characteristics for predicting therapeutic response and adverse outcomes during antifibrotic treatment remains to be investigated. In this study, we hypothesized that baseline characteristics, including blood levels of KL-6 and SPA, of patients with IPF could predict the risk of three important adverse outcomes (on-treatment acute exacerbation, mortality, and hepatic injury) during nintedanib treatment.
Methods
Study design and population
This retrospective cohort study involved patients with IPF at National Cheng Kung University Hospital (NCKUH) who had been treated with nintedanib and received regular follow-ups from 1 March, 2017, until 31 December, 2020. The study was approved by the NCKUH institutional review board (B-ER-105-390 and A-ER-107-193). The inclusion criteria for this study were as follows: naïve to anti-fibrotic therapy, aged > 50 years, diagnosed with IPF according to the international guidelines [41, 42] and based on a multi-disciplinary approach, and received uninterrupted nintedanib therapy for ≥ 14 days. We followed the patients until death or 31 December, 2020. Drug compliance was assessed upon each return visit. The electronic medical records of every patient were carefully reviewed and the following data were collected: baseline demographics, comorbidities (which also included the echocardiographic evidence of pulmonary hypertension, and factors necessary for the calculation of the Charlson comorbidity index), serial pulmonary function parameters (recorded at least eight weeks apart), gender-age-physiology (GAP) stages, and pertinent clinical data (including all medications of which the patient took at least three doses concurrently with nintedanib treatment within two weeks before each blood test for hepatic enzymes). We also checked the baseline plasma levels of KL-6 and SPA shortly before nintedanib treatment began, via sandwich enzyme-linked immunosorbent assay (ELISA), using specialized kits from Fine Test (Wuhan Fine Biotech, Wuhan City, China). The protocols for specimen processing and the ELISA are available in Additional file 1: Appendix A.
Important definitions
We focused on three major on-treatment adverse outcomes: acute exacerbation, mortality, and nintedanib-related hepatic injury. The index date was the date when the first dose of nintedanib was taken. “On-treatment” refers to the period between the index date and either day 28 after the last dose of nintedanib [7] (for those who prematurely discontinued the therapy) or 31 December, 2020 (for those who continued the therapy). Patients on the “full dose” took 150 mg of nintedanib twice daily, while patients on the “reduced dose” (for managing drug-related adverse effects) took 100 mg daily, 150 mg daily, or 100 mg twice daily, at the discretion of the treating pulmonologist. Acute exacerbation of IPF (AE-IPF) was defined according to the recent international working group report, which specifically excludes events with identifiable infectious or non-infectious causes of acute deterioration [6]. We defined “mortality” as all-cause mortality. Nintedanib-related hepatitis injury was strictly defined as a blood alanine transaminase (ALT) level above the upper limit of normal (ULN, < 50 IU/L at NCKUH), with or without associated hyperbilirubinemia or symptoms, that could not be attributed to any other aetiology, which occurred while the patient was still receiving nintedanib treatment [26, 27, 43]. Severe hepatic injury was defined as an increase in blood ALT level to ≥ 3 × ULN. Patients were defined as having recurrent hepatic injury if they had already experienced nintedanib-related hepatic injury and recovered after temporary discontinuation or dose reduction of nintedanib, but then again developed unexplained ALT elevation above the ULN upon resumption of nintedanib treatment. Echocardiographic evidence of pulmonary hypertension was defined as an estimated systolic pulmonary arterial pressure (which was derived from tricuspid regurgitation jet velocity) ≥ 35 mmHg [44]. Serial differences in pulmonary function parameters were standardized into 24-week and annual (52-week) rates of change using the following formulae:
24-week rate of change:
Annual (52-week) rate of change:
where FVC represents forced vital capacity and DLCO represents diffusion capacity of the lung for carbon monoxide. The subscript “first” indicates measurements that were closest in time before the initiation of nintedanib treatment, and the subscript “last” indicates measurements that were closest in time before the last dose of nintedanib (for patients who prematurely discontinued the therapy) or 31 December, 2020 (for patients who continued the therapy).
Statistical analysis
Categorical data are presented as counts and percentages, and continuous variables are presented as mean (standard deviation) or median (interquartile range [IQR]) if not normally distributed. No imputation of missing data was made. Variables were compared between patient groups using Fischer’s exact test or the Mann–Whitney U test, whichever was more appropriate. The optimal cut-off value of a continuous variable was determined using receiver operating characteristic (ROC) curve, based on the combined consideration of the area under the curve, accuracy, and Youden’s index. The assumption of proportional hazards was checked using the Shoenfeld test. Cox proportional-hazards regression models were constructed to assess the performance of candidate risk factors in longitudinally predicting the risks of adverse outcomes. When we conducted multi-variable regression analyses, in addition to including the candidate predictors, we routinely adjusted for the baseline GAP stage, Charlson comorbidity index, and duration of nintedanib treatment. For acute exacerbation and hepatic injury, the competing risk of on-treatment mortality was further controlled for by subdistribution hazard regression. Sensitivity analyses were performed to assess the robustness of all the multi-variable models constructed. All tests were two-tailed, and a p-value < 0.05 was considered statistically significant. Statistical analyses were performed using R (Version 3.6.1) and SPSS (Version 22, SPSS, USA). Graphs were plotted using MedCal (Version 16.8.4, MedCal Software, Belgium).
Results
Sixty patients received nintedanib treatment and were followed-up at NCKUH from 1 March, 2017, until 31 December, 2020. Three patients were excluded due to incomplete follow-ups; the remaining 57 were included in this study. Figure 1 shows the inclusion and exclusion flowchart for this study.
Fig. 1.
The inclusion and exclusion flowchart for this study
Table 1 summarizes the baseline characteristics and adverse outcomes of all the included patients. The study cohort was predominantly male and had a mean age of 75.4 ± 9.4 years. Fifteen (26%) patients exhibited sonographic evidence of fatty liver before they began receiving nintedanib, but none had cirrhosis, and all had normal levels of hepatic enzymes at baseline. The median duration of nintedanib treatment was 345 days (IQR, 91–706 days). Overall, 24 (42%) patients developed hepatic injury, manifesting as an asymptomatic elevation in the blood ALT level without concurrent hyperbilirubinemia. Eight patients had severe injury (ALT ≥ 3 × ULN). All the hepatic injury events were nonfatal and spontaneously resolved following transient reduction of the nintedanib dosage or temporary withdrawal of nintedanib, although 14 patients developed recurrent hepatic injury after resumption of nintedanib treatment. Twenty (35%) patients had on-treatment AE-IPF, and 16 (28%) patients died on treatment. Regarding the timing of the first onset of these adverse outcomes, all hepatic injury occurred within one year of the commencement of treatment, and in all cases the onset of AE-IPF occurred within three years of the commencement of treatment. Of the patients who died on-treatment, six (38%) did so within the first year. All the on-treatment mortality events occurred within four years of the commencement of treatment (Additional file 1: Table S1). It is worth noting that in those patients who developed both hepatic injury and AE-IPF, all AE-IPF events occurred after the hepatic injury.
Table 1.
Baseline characteristics and adverse outcomes of the 57 patients
| Baseline characteristics and outcome events | Results |
|---|---|
| Age, years | 75.4 ± 9.4 |
| Sex | |
| Female, n (%) | 9 (16) |
| Male, n (%) | 48 (84) |
| Body height, cm | 162.2 ± 7.7 |
| Body weight, kg | 63.5 ± 10.8 |
| Body mass index, kg/m2 | 24.0 (21.5 to 26.5) |
| Body surface area, m2 | 1.67 ± 0.15 |
| Charlson comorbidity index | 5 (3 to 6) |
| Echocardiographic evidence of pulmonary hypertension, n (%) | 36 (63) |
| Echocardiographic evidence of LV dysfunction, n (%) | 2 (4) |
| Dyslipidaemia, n (%) | 26 (46) |
| Chronic hepatitis B, n (%) | 6 (11) |
| Chronic hepatitis C, n (%) | 5 (9) |
| Other non-viral liver condition, n (%) | 3 (5) |
| Fatty liver on baseline sonography, n (%) | 15 (26) |
| Cigarette smoking status | |
| Never smoker, n (%) | 26 (46) |
| Current smoker, n (%) | 4 (7) |
| Former smoker, n (%) | 27 (47) |
| Baseline plasma KL-6 level, ng/mL | 1.48 (0.55 to 2.82) |
| Baseline plasma SPA level, pg/mL | 283.6 (157.3 to 435.4) |
| Baseline oximetry breathing ambient air, % | 95 (93 to 97) |
| Baseline DLCO, mmol/min/kPa | 2.78 (2.11 to 3.96) |
| Baseline DLCO, % predicted | 55 (38 to 70) |
| Baseline FVC, L | 2.02 ± 0.49 |
| Baseline FVC, % predicted | 67 ± 12 |
| Stages based on the GAP index | |
| Stage 1, n (%) | 14 (25) |
| Stage 2, n (%) | 31 (54) |
| Stage 3, n (%) | 12 (21) |
| Nintedanib-related hepatic injury, n (%) | 24 (42) |
| On-treatment AE-IPF, n (%) | 20 (35) |
| On-treatment mortality, n (%) | 16 (28) |
| Duration of nintedanib therapy, days | 345 (91 to 706) |
| Time to first nintedanib-related hepatic injury, days | 69 (17 to 156) |
| Time to first on-treatment AE-IPF, days | 238 (111 to 431) |
| Time to on-treatment mortality, days | 486 (217 to 811) |
| Time between plasma sampling and nintedanib initiation, days | 6 (0 to 28) |
| Time between baseline pulmonary functions and nintedanib initiation, days | 28 (16 to 51) |
| Time between plasma sampling and baseline pulmonary functions, days | 24 (12 to 81) |
| Annual rate of change in FVC, L/52 weeks | − 0.13 (− 0.26 to + 0.06) |
| Annual rate of change in DLCO, % predicted/52 weeks | − 9 (− 29 to − 2) |
Categorical data are presented as counts and percentages, and continuous variables were presented as mean (± standard deviation) or median (interquartile range) if non-normally distributed. AE-IPF, acute exacerbation of idiopathic pulmonary fibrosis; DLCO, diffusion capacity for carbon monoxide; FVC, forced vital capacity; GAP, gender, age, physiology; KL-6, Krebs von den Lungen-6; LV, left ventricular; SPA, surfactant protein A
Compared to patients without hepatic injury, patients who developed hepatic injury had significantly higher baseline levels of plasma KL-6 (2.72 ng/mL [IQR 1.82–4.05] and 0.94 ng/mL [IQR 0.44–1.63], respectively; p < 0.001) and significantly lower DLCO (42% predicted [IQR 31–54] and 60% predicted [IQR 53–83], respectively; p = 0.001). Patients with hepatic injury also exhibited a borderline-significant pattern of higher plasma SPA, lower pulse oximetry (breathing ambient air), and higher frequency of pulmonary hypertension (Additional file 1: Table S2). No significant difference was identified in the dosing, the time to the first test for hepatic enzymes, or the concurrently used medication (Additional file 1: Table S3) between patients with and without hepatic injury. Patients who had on-treatment AE-IPF also had significantly higher plasma levels of KL-6 (3.11 ng/mL [IQR 1.38–5.07] versus 1.03 ng/mL [IQR 0.48–1.86]; p = 0.001) and SPA (412.6 pg/mL [IQR 181.8–478.5] versus 235.6 pg/mL [IQR 157.3–379.9]; p = 0.042) than those who did not have AE-IPF. They also had a borderline-significant pattern of a higher frequency of pulmonary hypertension than patients without AE-IPF (Additional file 1: Table S4). The 16 patients who died on-treatment had significantly higher baseline plasma levels of KL-6 (3.61 ng/mL [IQR 1.28–8.22] versus 1.31 ng/mL [IQR 0.48–2.20]; p = 0.001) and SPA (447.9 pg/mL [IQR 393.5–697.3] versus 226.0 pg/mL [IQR 140.2–373.7]; p < 0.001) than those who survived. They also had significantly higher frequencies of pulmonary hypertension (88% versus 54%; p = 0.03) and on-treatment AE-IPF (75% versus 20%; p < 0.001) than patients who survived (Additional file 1: Table S5). These variables were thus selected as candidate predictors for further analyses. The results from the ROC analysis for selecting cut-off values for continuous-variable candidate predictors are summarized in Additional file 1: Table S6. It is worth noting that there was no significant difference between patients with and without adverse outcomes in either their dosing regimen or the time intervals between blood sampling, baseline pulmonary functions, or the initiation of nintedanib treatment (Additional file 1: Tables S2, S4, S5).
In the univariate and multi-variable Cox proportional-hazards regression analyses, only two candidate predictors (KL-6 ≥ 2.5 ng/mL and DLCO < 55% predicted; Fig. 2a, b show the distribution of patients with respect to these cut-off values) consistently yielded significantly elevated crude and adjusted hazard ratios for nintedanib-related hepatic injury (the adjusted hazard ratio [aHR] for KL-6 was 3.46 [95% CI 1.13–10.60], p = 0.029; and the aHR for DLCO was 6.05 [95% CI 1.89–19.32], p = 0.002). Pulmonary hypertension yielded a borderline-significant crude hazard ratio, but that failed to reach statistical significance in the multi-variable model (Fig. 3a and Additional file 1: Table S7). A baseline KL-6 ≥ 2.5 ng/mL and DLCO < 55% predicted was also correlated with an increased risk of severe and recurrent hepatic injury during nintedanib treatment. For severe hepatic injury, the adjusted odds ratio (aOR) from the multi-variable ordinal logistic regression analysis was 9.58 (95% CI 1.97–55.67; p = 0.007) for KL-6 and 10.89 (95% CI 2.63–54.25; p = 0.002) for DLCO. For recurrent hepatic injury, the aOR was 26.01 (95% CI 4.19–260.13; p = 0.001) for KL-6 and 60.28 (95% CI 8.30–823.62; p < 0.001) for DLCO (Additional file 1: Table S8). Ikeda et al. reported that patients with BMI < 22 kg/m2 or BSA < 1.58 m2 would have an increased risk for nintedanib-related hepatitis [26, 27]. However, we found no significant differences in BMI or BSA between patients with and without hepatic injury (Additional file 1: Table S2). Univariate regression analyses involving BMI and BSA (using the cut-off values proposed by Ikeda et al.) also found no significant differences (Fig. 3a and Additional file 1: Tables S7 and S8). With the same cut-off value as for hepatic injury (2.5 ng/mL), the baseline plasma KL-6 level was the only significant predictor of on-treatment AE-IPF in both the univariate and the multi-variable Cox proportional-hazards regression (aHR 4.52; 95% CI 1.63–12.55; p = 0.004; Fig. 3b and Additional file 1: Table S9). Figure 2c shows the distribution of patients above and below this cut-off value. For on-treatment mortality, the baseline plasma KL-6 level was also a significant predictor, but with a cut-off value of 3.5 ng/mL (aHR 5.39; 95% CI 1.16–24.97; p = 0.031). Another significant predictor in the multi-variable model for on-treatment mortality was baseline plasma SPA level, with a cut-off value of 600 pg/mL (aHR 12.28; 95% CI 2.06–73.05; p = 0.006; Fig. 2d, e show the distribution of patients above and below these cut-off values, while Fig. 3c shows the results of the Cox proportional-hazards regression analyses). Although the patients who died on-treatment had a significantly higher frequency of pulmonary hypertension at baseline and AE-IPF on treatment, these two predictors were not statistically significant in the regression models (Fig. 3c and Additional file 1: Table S10). To control for the competing risk of on-treatment mortality, we performed a multi-variable subdistribution hazard regression analysis for hepatic injury and for on-treatment AE-IPF, which yielded concordant and supportive results (the lower parts of Fig. 3a, b, and the far-right panels of Additional file 1: Tables S7 and S9). In addition, sensitivity analyses showed that, even in the presence of an unidentified confounder, the above-mentioned multi-variable Cox models and the predictors thereby derived yielded significantly elevated hazard ratios for the corresponding adverse outcomes (Additional file 1: Figures S1a to S1e).
Fig. 2.

Distribution of patients above and below the cut-off values of the major predictors for the three adverse outcomes. Numbers within the bars are the patient counts. a KL-6 level versus nintedanib-related hepatic injury, b DLCO % predicted versus nintedanib-related hepatic injury, c KL-6 level versus on-treatment AE-IPF, d KL-6 level versus on-treatment mortality, and e SPA level versus on-treatment mortality. p-values were derived from Fisher’s exact test. Number of patients with available data: 54 for KL-6, 55 for DLCO % predicted, and 55 for SPA. Abbreviations AE-IPF, acute exacerbation of idiopathic pulmonary fibrosis; DLCO, diffusion capacity for carbon monoxide (in % predicted); KL-6, Krebs von den Lungen-6; SPA, surfactant protein A
Fig. 3.
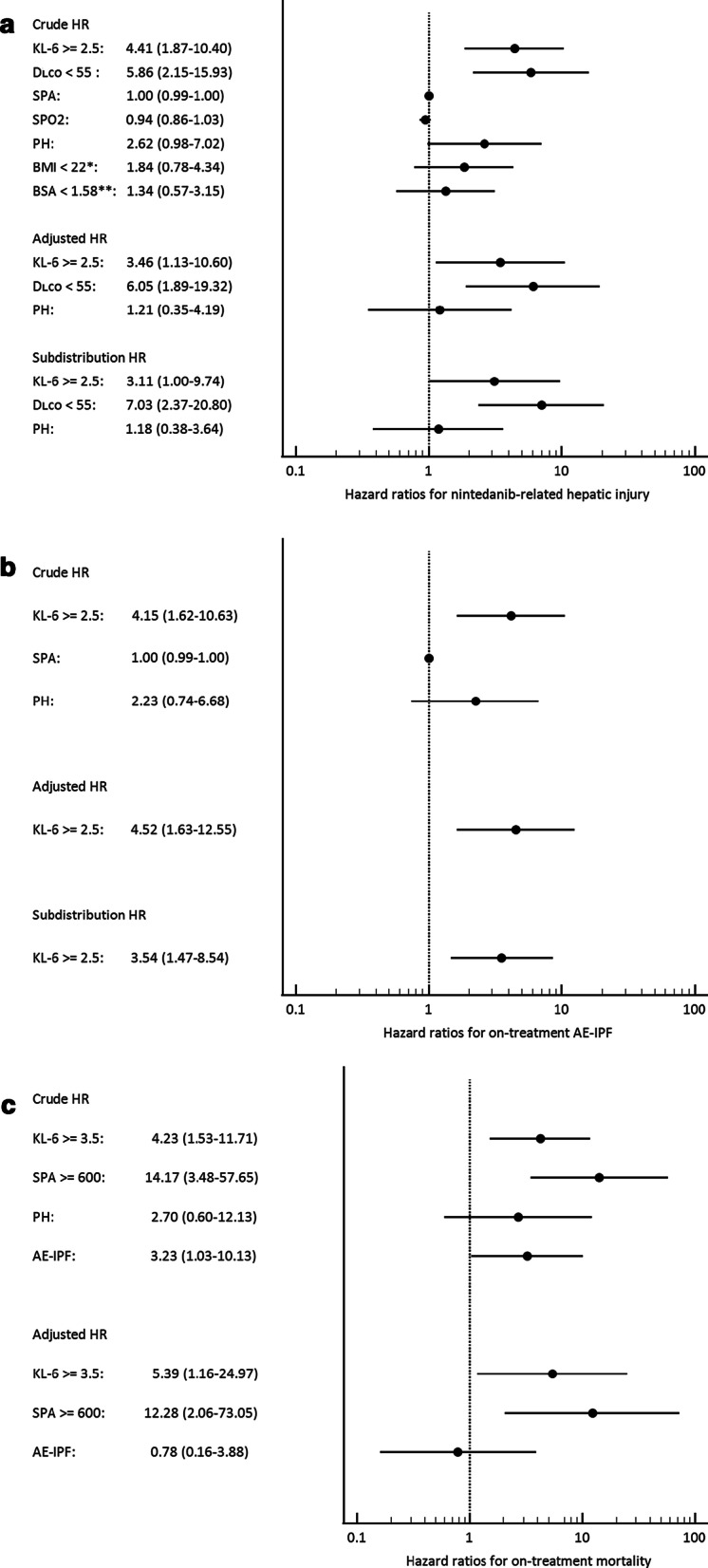
Forest plots showing the results of Cox proportional-hazards regression and subdistribution hazard regression analyses of candidate predictors for a nintedanib-related hepatic injury, b on-treatment acute exacerbation of IPF, and c on-treatment mortality. In addition to the candidate predictors shown, all the multi-variable regression models were also adjusted for gender-age-physiology (GAP) stage, Charlson comorbidity index, and treatment duration. Abbreviation and Notes AE-IPF, acute exacerbation of idiopathic pulmonary fibrosis; BMI, body mass index; BSA, body surface area; DLCO, diffusion capacity for carbon monoxide (in % predicted); HR, hazard ratio; KL-6, Krebs von den Lungen-6; PH, echocardiographic pulmonary hypertension; SPA, surfactant protein A; SPO2, pulse oximetry (while breathing ambient air). *This cut-off value was proposed by Ikeda et al. [26]. **This cut-off value was proposed by Ikeda et al. [27]
Because plasma KL-6 level was the only baseline factor that significantly predicted the risk of all three adverse outcomes, we further explored the relationship between KL-6 level and treatment response in terms of pulmonary function decline. Forty-four and 34 patients had at least one follow-up FVC and DLCO assessment, respectively, at least eight weeks later and while still on-treatment. For FVC, 40 patients had a time interval between the baseline and the last measurements of more than six months, of whom the time interval was more than one year in 25 patients (median interval, 409 days; IQR, 265–712). For DLCO, 32 patients had a time interval between the measurements of more than six months, of whom the time interval was more than one year in 19 patients (median interval, 393 days; IQR, 280–767). The overall annual rate of change in FVC and DLCO in the cohort was − 0.13 L (IQR, − 0.26 to + 0.06) and − 9% (IQR, − 29 to − 2), respectively (Table 1). However, patients with KL-6 ≥ 2.5 ng/mL exhibited a significantly higher annual rate of decline in FVC than patients with KL-6 < 2.5 ng/mL, but a similar annual rate of decline in DLCO (Fig. 4a, b). A significantly higher proportion of patients with KL-6 ≥ 2.5 ng/mL had a decline in FVC of ≥ 5% over 24 weeks than patients with KL-6 < 2.5 ng/mL. For DLCO, a higher proportion of patient with KL-6 ≥ 2.5 ng/mL had a decline ≥ 10% over 24 weeks than patients with KL-6 < 2.5 ng/mL, although this difference was not statistically significant (left half of Fig. 4e, f). Similar patterns of pulmonary function decline were observed with a KL-6 cut-off value of 3.5 ng/mL (Additional file 1: Figures S2a to S2d). Even if we excluded patients with on-treatment AE-IPF from this analysis, we observed similar trends in pulmonary function decline, in addition to a nonsignificant pattern of a faster annual decrease in DLCO, in patients with KL-6 ≥ 2.5 ng/mL (Fig. 4c, d, and the right half of Fig. 4e, f).
Fig. 4.

Comparison between patients with baseline plasma KL-6 < 2.5 ng/mL with patients with KL-6 ≥ 2.5 ng/mL in: a annual rate of FVC decline; b annual rate of DLCO decline; c annual rate of FVC decline after excluding patients with AE-IPF; d annual rate of DLCO decline after excluding patients with AE-IPF; e proportion of patients with ≥ 5% FVC decline over 24 weeks; f proportion of patients with ≥ 10% DLCO decline over 24 weeks. In panels a to d, data are presented as medians with inter-quartile ranges, and p-values were derived from the Mann Whitney U test. In panels e and f, data shown are proportions of patients in each subgroup, and p-values were derived from Fisher’s exact test. Abbreviation AE-IPF, acute exacerbation of idiopathic pulmonary fibrosis; DLCO, diffusion capacity for carbon monoxide; FVC, forced vital capacity; KL-6, Krebs von den Lungen-6
Discussions
In this study, we confirmed our hypothesis and found that for patients with IPF who receive nintedanib treatment, certain baseline markers predict the risk of on-treatment adverse outcomes. Specifically, patients with baseline plasma KL-6 levels ≥ 2.5 ng/mL had a higher risk of nintedanib-related hepatic injury (including severe and recurrent injury) and on-treatment acute-exacerbation of IPF. Patients with baseline plasma KL-6 levels ≥ 3.5 ng/mL had a higher risk of on-treatment mortality. Having a baseline DLCO < 55% predicted and plasma SPA levels ≥ 600 pg/mL was also associated with increased risk of hepatic injury and on-treatment mortality, respectively. In addition, regardless of which cut-off values for KL-6 was used (2.5 or 3.5 ng/mL), a pattern of greater on-treatment decline in FVC and DLCO was observed in patients whose baseline plasma KL-6 levels were above the cut-off value.
The recent introduction of antifibrotic therapy has been a breakthrough in the management of IPF. Although it is not a curative therapy and its efficacy for reducing mortality remains inconclusive, nintedanib has been shown to halt pulmonary function decline and is likely to reduce the risk of acute exacerbation. [13, 16, 19, 20]. However, not all patients receiving nintedanib exhibit the same favourable response. It remains unclear whether and how we can predict who will benefit most from this treatment and who will have a poor response or even develop adverse outcomes. On the other hand, nintedanib has been known to cause drug-induced liver injury ever since its early major trials [7, 10, 24, 45]. Hepatic impairment has also consistently been reported in real-world data from various countries (1.6–9.6% of treated patients) [15, 17, 18, 21–25], and as well as in the recent INPULSIS-ON and INBUILD trials [13, 14]. Late-onset hepatotoxicity has also been described [46]. Data from East Asia have shown relatively high incidence rates of hepatitis, reaching 67.6% [19, 20, 26–29]. In our study cohort, 42% of patients developed hepatic injury.
Functional parameters like FVC and DLCO are important for diagnosing and assessing IPF [16, 20, 47, 48]. Serial decline in pulmonary function parameters is also associated with increasing risk of mortality [49, 50]. However, these parameters may be insensitive to early disease or minor progression, and they are also subject to variation resulting from suboptimal testing procedures and patient-specific factors [51–54]. The blood levels of circulating lung-specific macromolecules such as KL-6 [38, 39, 55–58] and SPA [37, 59] appear to correlate with the severity and prognosis of IPF. Yokoyama et al. retrospectively studied 23 patients with IPF and found that a high baseline serum KL-6 level (≥ 1000 U/mL) was associated with significantly diminished survival [56]. Using the same cut-off value, Wakamatsu et al. also showed that an initially high serum KL-6 level with a subsequent increasing pattern was associated with poor survival and a steep decline in FVC [57]. In addition, Ohshimo et al. proposed that a high baseline serum KL-6 level (≥ 1300 U/mL) predicts a subsequent risk of acute exacerbation [58]. However, none of the patients included in these studies received nintedanib or any other anti-fibrotic treatment. Bergantini et al. followed 23 patients with IPF who received nintedanib for 12 months and found that uninterrupted nintedanib treatment may have stabilized their serum KL-6 levels in serial tests, and that variation in serum KL-6 levels was correlated with serial variations in DLCO [60]. Yoshikawa et al. analysed data from 49 patients receiving anti-fibrotic treatment (including 26 who were receiving nintedanib) and found that the patterns of change in serum SPA, surfactant protein D, and KL-6 levels three and six months into treatment were correlated with the rate of deterioration in FVC and DLCO [61]. Building on these pioneering findings, the present study is novel in showing that the baseline levels of KL-6 in particular, and SPA, have a predictive role in patients with IPF who receive nintedanib treatment, not only for mortality and pulmonary function deterioration, but also for other clinically important outcomes like acute exacerbation and hepatic injury. The findings of our study highlight the heterogeneity of patients with IPF, identify subgroups of patients who may have a poor response and adverse outcomes despite ongoing nintedanib anti-fibrotic treatment, and further support the clinical utility of blood molecular markers (particularly KL-6) for IPF.
We suspect that the mechanism underlying the association between the predictors we have identified and the risk of the three adverse outcomes is related to the severity of lung parenchymal fibrotic destruction and overall physical frailty. The blood levels of KL-6 and SPA probably reveal the degree of injury and dysfunction of type 2 alveolar cells, and the associated fibrotic disruption of the alveolar-endothelial interface [39, 55]. Supporting this rationale is the fact that, when we stratified the cohort according to the level of plasma KL-6 using a cut-off value 2.5 ng/mL (as shown in Additional file 1: Table S11), we found that patients with KL-6 ≥ 2.5 ng/mL indeed had more severe physiological impairment and physical frailty than those with KL-6 < 2.5 ng/mL. This was reflected by differences in relevant clinical indices: significantly lower body weights, body surface area, and DLCO; significantly higher frequencies of pulmonary hypertension; and borderline-significant patterns of lower body mass index and higher frequencies of GAP-stage-3 disease. Interestingly, in the statistical analyses to identify predictors for on-treatment adverse outcomes, these clinical indices individually did not perform as well as KL-6 or, to a lesser extent, SPA. This can probably be explained by the fact that KL-6 and SPA indicate disease severity at the histological-molecular level. They are therefore less susceptible to confounding effects from comorbidities, and variations in function test procedures, than other clinical or functional indices. Considering that pharmacodynamically nintedanib can only slow down and not reverse the fibrogenesis [3, 9], it is plausible that patients with higher levels of KL-6 and SPA (indicating more severe fibrotic destruction) would be less likely to exhibit a strong therapeutic response and more likely to progress towards unfavourable outcomes. This proposed mechanism may also account for the enhanced risk of nintedanib-related hepatic injury. Patients with elevated KL-6 and/or low DLCO probably already had advanced IPF and may have experienced frequent episodes of hypoxemia (either resting or exertion-induced). Hypoxemia can interfere with the oxygen-demanding steps of nintedanib metabolism (such as glucuronidation by UGT-enzymes) in hepatocytes, thereby retarding the clearance of potentially harmful metabolites [35, 62]. The higher risk of hepatic injury appears not to be the direct consequence of increased circulating KL-6 per se, because circulating mucins are cleared mainly by hepatic endothelial cells and Kupffer cells, rather than by hepatocytes [63]. Further research to ascertain whether our proposed mechanisms are valid would be helpful and inspiring.
This study has some limitations. Due to the overall rarity of IPF [2, 3, 64], the size of our study cohort is relatively small. Nevertheless, our findings, which are derived from a detailed real-world database with extended longitudinal follow-up, do provide insights into important clinical issues. In addition, we included only patients of Han Chinese ethnicity who were followed at a single tertiary center in Taiwan. Genetic polymorphisms of KL-6 (such as the rs4072037 single nucleotide polymorphism, which was not checked in the present study) have been described. These may result in different cut-off thresholds in different ethnicities for discriminating between patients with and without interstitial lung diseases (including IPF) [65]. It remains to be determined whether such polymorphisms may also effect the cut-off thresholds for predicting adverse outcomes. Furthermore, we did not include subsequent KL-6 measurements, and therefore could not determine the relationship between longitudinal trends in KL-6 levels and adverse clinical outcomes. Future research involving serial measurements of KL-6 and a larger population with greater diversity, e.g., in both ethnicity and pulmonary spirometry results, or including patients with other progressive fibrosing interstitial lung disease (PF-ILDs) [14, 66], would help to validate the generalizability of our findings and provide more information about the clinical utility of KL-6 levels.
Conclusions
For patients with IPF receiving nintedanib treatment, their baseline plasma level of KL-6 predicted their risk of on-treatment acute exacerbation, mortality, and hepatic injury (including severe and recurrent injury). Patients with elevated baseline plasma KL-6 levels also exhibited a pattern of more rapid pulmonary function decline. Additionally, an initially high plasma SPA level and low DLCO were also associated with adverse on-treatment outcomes. The findings of this study may help to identify patients for whom close monitoring for unfavourable responses during nintedanib treatment would be important. It may also contribute to the future formulation of more individualized therapeutic strategies and support the prognostic roles of blood molecular markers for IPF in real-world clinical practice.
Supplementary Information
Additional file 1. Supplementary figures and tables and the protocols for processing blood specimens and for performing the enzyme-linked immunosorbent assay (ELISA).
Acknowledgements
This study was supported by National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, under the grants NCKUH-10601006 and NCKUH-10804017. We thank the Core Research Laboratory at the Centre of Clinical Medical Research, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, for technical supports. We are thankful to Hsin-Chiao Yang (MS, Division of Thoracic Surgery, Department of Surgery) and Tzu-Hui Yu (MS, Division of Chest Medicine, Department of Internal Medicine), both are senior research assistants at National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, for their technical assistance with the processing of plasma specimens and the ELISA experiment. We are also thankful to Wan-Ni Chen (MS), statistician from the Biostatistics Consulting Centre of National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, for providing statistical consultation and assistance, and to Claire Chang (MA) for the proofreading of this manuscript.
Abbreviations
- AE-IPF
Acute exacerbation of idiopathic pulmonary fibrosis
- aHR
Adjusted hazard ratio
- ALT
Alanine transaminase
- aOR
Adjusted odds ratio
- BMI
Body mass index
- BSA
Body surface area
- DLCO
Diffusion capacity for carbon monoxide
- ELISA
Enzyme-linked immunosorbent assay
- FVC
Forced vital capacity
- IPF
Idiopathic pulmonary fibrosis
- IQR
Inter-quartile range
- KL-6
Krebs von den Lungen-6
- LV
Left ventricular
- ROC
Receiver operating characteristic
- SPO2
Pulse oximetry (while breathing ambient air)
- SPA
Surfactant protein A
- ULN
Upper-limit of normal
- 95% CI
95% confidence interval
Authors' contributions
LSH is the guarantor of this paper and takes responsibility for the integrity of the work as a whole, from inception to published article. HTH and LSH contributed to the conception and design, as well as funding acquisition; HTH, KCW, CCW, TYL contributed to the data collection and curation; HTH, KCW, WCL contributed to the execution of ELISA experiments; HTH, LSH, WCL, KCW, CCW contributed to the data analysis and interpretation; HTH, LSH, KCW, CCW contributed to the drafting of the manuscript; HTH, LSH, WCL, TYL contributed to the critical review of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University (grants numbers NCKUH-10601006 and NCKUH-10804017). The funding source did not involve in the study design, the collection, the analysis, and the interpretation of data, as well as the writing of the report, and the decision to submit the article.
Availability of data and materials
The de-linked datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
This current study was approved by NCKUH Institutional Review Board (B-ER-105-390 and A-ER-107-193). Formal written informed consent for participation was obtained from each patient upon inclusion into this study. We confirmed that all the experiment protocols involving human data were in accordance with the relevant institutional, national, and international regulations and guidelines and the Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
All authors of this manuscript report no conflict of interest. All authors of this manuscript have not received any financial or nonfinancial support from any tobacco or pharmaceutical company.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 2.Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history. Respirology. 2016;21:427–437. doi: 10.1111/resp.12683. [DOI] [PubMed] [Google Scholar]
- 3.Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378:1811–1823. doi: 10.1056/NEJMra1705751. [DOI] [PubMed] [Google Scholar]
- 4.Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010;35:496–504. doi: 10.1183/09031936.00077309. [DOI] [PubMed] [Google Scholar]
- 5.Kim DS. Acute exacerbations in patients with idiopathic pulmonary fibrosis. Respir Res. 2013 doi: 10.1186/1465-9921-14-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, Lee JS, Maher TM, Wells AU, Antoniou KM, Behr J, Brown KK, Cottin V, Flaherty KR, Fukuoka J, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kolb M, Lynch DA, Myers JL, Raghu G, Richeldi L, Taniguchi H, Martinez FJ. Acute exacerbation of idiopathic pulmonary fibrosis—an international working group report. Am J Respir Crit Care Med. 2016;194:265–275. doi: 10.1164/rccm.201604-0801CI. [DOI] [PubMed] [Google Scholar]
- 7.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR, INPULSIS Trial Investigators Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82. doi: 10.1056/NEJMoa1402584. [DOI] [PubMed] [Google Scholar]
- 8.King TE, Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92. doi: 10.1056/NEJMoa1402582. [DOI] [PubMed] [Google Scholar]
- 9.Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, Kolb M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434–1445. doi: 10.1183/09031936.00174914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richeldi L, Cottin V, du Bois RM, Selman M, Kimura T, Bailes Z, Schlenker-Herceg R, Stowasser S, Brown KK. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS trials. Respir Med. 2016;113:74–79. doi: 10.1016/j.rmed.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Costabel U, Inoue Y, Richeldi L, Collard HR, Tschoepe I, Stowasser S, Azuma A. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193:178–185. doi: 10.1164/rccm.201503-0562OC. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi H, Xu Z, Azuma A, Inoue Y, Li H, Fujimoto T, Bailes Z, Schlenker-Herceg R, Kim DS. Subgroup analysis of Asian patients in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Respirology. 2016;21(8):1425–1430. doi: 10.1111/resp.12852. [DOI] [PubMed] [Google Scholar]
- 13.Crestani B, Huggins JT, Kaye M, Costabel U, Glaspole I, Ogura T, Song JW, Stansen W, Quaresma M, Stowasser S, Kreuter M. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study. INPULSIS-ON Lancet Respir Med. 2019;7:60–66. doi: 10.1016/S2213-2600(18)30339-4. [DOI] [PubMed] [Google Scholar]
- 14.Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker-Herceg R, Brown KK, INBUILD Trial Investigators Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2020;382(8):781. doi: 10.1056/NEJMc1917224. [DOI] [PubMed] [Google Scholar]
- 15.Bonella F, Kreuter M, Hagmeyer L, Neurohr C, Keller C, Kohlhaeufl MJ, Müller-Quernheim J, Milger K, Prasse A, German Nintedanib Compassionate Use Consortium Insights from the German compassionate use program of nintedanib for the treatment of idiopathic pulmonary fibrosis. Respiration. 2016;92:98–106. doi: 10.1159/000448288. [DOI] [PubMed] [Google Scholar]
- 16.Harari S, Caminati A, Poletti V, Confalonieri M, Gasparini S, Lacedonia D, Luppi F, Pesci A, Sebastiani A, Spagnolo P, Vancheri C, Balestro E, Bonifazi M, Cerri S, De Giacomi F, Della Porta R, Foschino Barbaro MP, Fui A, Pasquinelli P, Rosso R, Tomassetti S, Specchia C, Rottoli P, for the ILDINET (Interstitial Lung Diseases Italian Network) A real-life multicenter national study on nintedanib in severe idiopathic pulmonary fibrosis. Respiration. 2018;95:433–40. doi: 10.1159/000487711. [DOI] [PubMed] [Google Scholar]
- 17.Toellner H, Hughes G, Beswick W, Crooks MG, Donaldson C, Forrest I, Hart SP, Leonard C, Major M, Simpson AJ, Chaudhuri N. Early clinical experiences with nintedanib in three UK tertiary interstitial lung disease centers. Clin Transl Med. 2017;6:41. doi: 10.1186/s40169-017-0172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galli JA, Pandya A, Vega-Olivo M, Dass C, Zhao H, Criner GJ. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology. 2017;22:1171–1178. doi: 10.1111/resp.13024. [DOI] [PubMed] [Google Scholar]
- 19.Abe M, Tsushima K, Sakayori M, Suzuki K, Ikari J, Terada J, Tatsumi K. Utility of nintedanib for severe idiopathic pulmonary fibrosis: a single-center retrospective study. Drug Des Devel Ther. 2018;12:3369–3375. doi: 10.2147/DDDT.S179427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon HY, Park S, Kim DS, Song JW. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir Res. 2018;19:203. doi: 10.1186/s12931-018-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzouvelekis A, Karampitsakos T, Kontou M, Granitsas A, Malliou I, Anagnostopoulos A, Ntolios P, Tzilas V, Bouros E, Steiropoulos P, Chrysikos S, Dimakou K, Koulouris N, Bouros D. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: a real-life observational study in Greece. Pulm Pharmacol Ther. 2018;49:61–66. doi: 10.1016/j.pupt.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 22.Brunnemer E, Walscher J, Tenenbaum S, Hausmanns J, Schulze K, Seiter M, Heussel CP, Warth A, Herth FJF, Kreuter M. Real-world experience with nintedanib in patients with idiopathic pulmonary fibrosis. Respiration. 2018;95:301–309. doi: 10.1159/000485933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivera-Ortega P, Hayton C, Blaikley J, Leonard C, Chaudhuri N. Nintedanib in the management of idiopathic pulmonary fibrosis: clinical trial evidence and real-world experience. Ther Adv Respir Dis. 2018;12:1–3. doi: 10.1177/1753466618800618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corte T, Bonella F, Crestani B, Demedts MG, Richeldi L, Coeck C, Pelling K, Quaresma M, Lasky JA. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir Res. 2015;16:116. doi: 10.1186/s12931-015-0276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes G, Toellner H, Morris H, Leonard C, Chaudhuri N. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med. 2016;5(9):78. doi: 10.3390/jcm5090078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikeda S, Sekine A, Baba T, Yamakawa H, Morita M, Kitamura H, Ogura T. Hepatotoxicity of nintedanib in patients with idiopathic pulmonary fibrosis: a single-center experience. Respir Investig. 2017;55:51–54. doi: 10.1016/j.resinv.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda S, Sekine A, Baba T, Yamanaka Y, Sadoyama S, Yamakawa H, Oda T, Okuda R, Kitamura H, Okudela K, Iwasawa T, Ohashi K, Takemura T, Ogura T. Low body surface area predicts hepatotoxicity of nintedanib in patients with idiopathic pulmonary fibrosis. Sci Rep. 2017;7(1):10811. doi: 10.1038/s41598-017-11321-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azuma A, Taniguchi H, Inoue Y, Kondoh Y, Ogura T, Homma S, Fujimoto T, Sakamoto W, Sugiyama Y, Nukiwa T. Nintedanib in Japanese patients with idiopathic pulmonary fibrosis: a subgroup analysis of the INPULSIS® randomized trials. Respirology. 2017;22(4):750–757. doi: 10.1111/resp.12960. [DOI] [PubMed] [Google Scholar]
- 29.Xu ZJ, Li HP, Wen FQ, Bai CX, Chen P, Fan F, Hu N, Stowasser S, Kang J. Subgroup analysis for Chinese patients included in the INPULSIS trials on nintedanib in idiopathic pulmonary fibrosis. Adv Ther. 2019;36(3):621–631. doi: 10.1007/s12325-019-0887-1. [DOI] [PubMed] [Google Scholar]
- 30.Bargagli E, Piccioli C, Rosi E, Torricelli E, Turi L, Piccioli E, Pistolesi M, Ferrari K, Voltolini L. Pirfenidone and Nintedanib in idiopathic pulmonary fibrosis: real-life experience in an Italian referral center. Pulmonology. 2019;25:149–153. doi: 10.1016/j.pulmoe.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 31.Noth I, Oelberg D, Kaul M, Conoscenti CS, Raghu G. Safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis in the USA. Eur Respir J. 2018;52(1):1702106. doi: 10.1183/13993003.02106-2017. [DOI] [PubMed] [Google Scholar]
- 32.Fletcher SV, Jones MG, Renzoni EA, Parfrey H, Hoyles RK, Spinks K, Kokosi M, Kwok A, Warburton C, Titmuss V, Thillai M, Simler N, Maher TM, Brereton CJ, Chua F, Wells AU, Richeldi L, Spencer LG. Safety and tolerability of nintedanib for the treatment of idiopathic pulmonary fibrosis in routine UK clinical practice. ERJ Open Res. 2018;4(4):00049-2018. doi: 10.1183/23120541.00049-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaliyaperumal K, Grove JI, Delahay RM, Griffiths WJH, Duckworth A, Aithal GP. Pharmacogenomics of drug-induced liver injury (DILI): molecular biology to clinical applications. J Hepatol. 2018;69:948–957. doi: 10.1016/j.jhep.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Marzin K, Kretschmar G, Luedtke D, Kraemer S, Kuelzer R, Schlenker-Herceg R, Schmid U, Schnell D, Dallinger C. Pharmacokinetics of nintedanib in subjects with hepatic impairment. J Clin Pharmacol. 2018;58:357–363. doi: 10.1002/jcph.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wind S, Schmid U, Freiwald M, Marzin K, Lotz R, Ebner T, Stopfer P, Dallinger C. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin Pharmacokinet. 2019;58:1131–1147. doi: 10.1007/s40262-019-00766-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi H, Sano H, Chiba H, Kuroki Y. Pulmonary surfactant proteins A and D: innate immune functions and biomarkers for lung diseases. Curr Pharm Des. 2006;12:589–598. doi: 10.2174/138161206775474387. [DOI] [PubMed] [Google Scholar]
- 37.Ley B, Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2014;307:L681–L691. doi: 10.1152/ajplung.00014.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papaioannou AI, Kostikas K, Manali ED, Papadaki G, Roussou A, Spathis A, Mazioti A, Tomos I, Papanikolaou I, Loukides S, Chainis K, Karakitsos P, Griese M, Papiris S. Serum levels of surfactant proteins in patients with combined pulmonary fibrosis and emphysema (CPFE) PLoS ONE. 2016;11:e0157789. doi: 10.1371/journal.pone.0157789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.d’Alessandro M, Bergantini L, Cameli P, Vietri L, Lanzarone N, Alonzi V, Pieroni M, Refini RM, Sestini P, Bonella F, Bargagli E. Krebs von den Lungen-6 as a biomarker for disease severity assessment in interstitial lung disease: a comprehensive review. Biomark Med. 2020;14(8):665–674. doi: 10.2217/bmm-2019-0545. [DOI] [PubMed] [Google Scholar]
- 40.King SD, Chen SY. Recent progress on surfactant protein A: cellular function in lung and kidney disease development. Am J Physiol Cell Physiol. 2020;319:C316–C320. doi: 10.1152/ajpcell.00195.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, Behr J, Cottin V, Danoff SK, Morell F, Flaherty KR, Wells A, Martinez FJ, Azuma A, Bice TJ, Bouros D, Brown KK, Collard HR, Duggal A, Galvin L, Inoue Y, Jenkins RG, Johkoh T, Kazerooni EA, Kitaichi M, Knight SL, Mansour G, Nicholson AG, Pipavath SNJ, Buendía-Roldán I, Selman M, Travis WD, Walsh S, Wilson KC, American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society Diagnosis of idiopathic pulmonary fibrosis an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68. doi: 10.1164/rccm.201807-1255ST. [DOI] [PubMed] [Google Scholar]
- 43.Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, Hunt CM, Wilke RA, Avigan M, Kaplowitz N, Bjornsson E, Daly AK. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89:806–815. doi: 10.1038/clpt.2011.58. [DOI] [PubMed] [Google Scholar]
- 44.Mukerjee D, St. George D, Knight C, Davar J, Wells AU, Du Bois RM, Black CM, Coghlan JG. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology. 2004;43(4):461–466. doi: 10.1093/rheumatology/keh067. [DOI] [PubMed] [Google Scholar]
- 45.Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, Brown KK, Flaherty KR, Noble PW, Raghu G, Brun M, Gupta A, Juhel N, Klüglich M, du Bois RM. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–1087. doi: 10.1056/NEJMoa1103690. [DOI] [PubMed] [Google Scholar]
- 46.Olin JL, Woods JA, Garner SJ. Delayed presentation of hepatocellular liver injury after nintedanib administration. Am J Ther. 2017;24:e107–e108. doi: 10.1097/MJT.0000000000000464. [DOI] [PubMed] [Google Scholar]
- 47.Mura M, Porretta MA, Bargagli E, Sergiacomi G, Zompatori M, Sverzellati N, Taglieri A, Mezzasalma F, Rottoli P, Saltini C, Rogliani P. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J. 2012;40:101–109. doi: 10.1183/09031936.00106011. [DOI] [PubMed] [Google Scholar]
- 48.Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, Poletti V, Buccioli M, Elicker BM, Jones KD, King TE, Jr, Collard HR. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–691. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 49.Zappala CJ, Latsi PI, Nicholson AG, Colby TV, Cramer D, Renzoni EA, Hansell DM, du Bois RM, Wells AU. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J. 2010;35:830–835. doi: 10.1183/09031936.00155108. [DOI] [PubMed] [Google Scholar]
- 50.Collard HR, King TE, Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2003;168(5):538–542. doi: 10.1164/rccm.200211-1311OC. [DOI] [PubMed] [Google Scholar]
- 51.Panda A, McHardy G. Diurnal variation in pulmonary diffusing capacity and expiratory volumes. Indian J Physiol Pharmacol. 1980;24(2):112–118. [PubMed] [Google Scholar]
- 52.Schwartz D, Van Fossen D, Davis C, Helmers R, Dayton C, Burmeister L, Hunninghake G. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149:444–449. doi: 10.1164/ajrccm.149.2.8306043. [DOI] [PubMed] [Google Scholar]
- 53.Douglas W, Ryu J, Swensen S, Offord K, Shroeder D, Caron G, DeRemee R. Colchicine versus prednisone in the treatment of idiopathic pulmonary fibrosis: a randomized prospective study. Am J Respir Crit Care Med. 1998;158:220–225. doi: 10.1164/ajrccm.158.1.9709089. [DOI] [PubMed] [Google Scholar]
- 54.Borsboom GJ, van Pelt W, van Houwelingen HC, van Vianen BG, Schouten JP, Quanjer PH. Diurnal variation in lung function in subgroups from two Dutch populations: consequences for longitudinal analysis. Am J Respir Crit Care Med. 1999;159:1163–1171. doi: 10.1164/ajrccm.159.4.9703106. [DOI] [PubMed] [Google Scholar]
- 55.Ishikawa N, Hattori N, Yokoyama A, Kohno N. Utility of KL-6/MUC1 in the clinical management of interstitial lung diseases. Respir Investig. 2012;50:3–13. doi: 10.1016/j.resinv.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M, Bando M, Sugiyama Y, Totani Y, Ishizaki T, Ichiyasu H, Suga M, Hamada H, Kohno N. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. 2006;11(2):164–168. doi: 10.1111/j.1440-1843.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 57.Wakamatsu K, Nagata N, Kumazoe H, Oda K, Ishimoto H, Yoshimi M, Takata S, Hamada M, Koreeda Y, Takakura K, Ishizu M, Hara M, Ise S, Izumi M, Akasaki T, Maki S, Kawabata M, Mukae H, Kawasaki M. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir Investig. 2017;55(1):16–23. doi: 10.1016/j.resinv.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, Kohno N, Bonella F, Guzman J, Costabel U. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med. 2014;108:1031–1039. doi: 10.1016/j.rmed.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 59.Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers: MMP-7 and SP-A predictors of outcome in idiopathic pulmonary fibrosis. Chest. 2013;143:1422–1429. doi: 10.1378/chest.11-2735. [DOI] [PubMed] [Google Scholar]
- 60.Bergantinin L, Bargagli E, Cameli P, Cekorja B, Lanzarone N, Pianigiani L, Vietri L, Bennett D, Sestini P, Rottoli P. Serial KL-6 analysis in patients with idiopathic pulmonary fibrosis treated with nintedanib. Respir Investig. 2019;7(3):290–291. doi: 10.1016/j.resinv.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Yoshikawa T, Otsuka M, Chiba H, Ikeda K, Mori Y, Umeda Y, Nishikiori H, Kuronuma K, Takahashi H. Surfactant protein A as a biomarker of outcomes of anti-fibrotic drug therapy in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. 2020;20(1):27. doi: 10.1186/s12890-020-1060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Angus PW, Morgan DJ, Smallwood RA. Hypoxia and hepatic drug metabolism—clinical implications. Aliment Pharmacol Therap. 1990;4:213–225. doi: 10.1111/j.1365-2036.1990.tb00466.x. [DOI] [PubMed] [Google Scholar]
- 63.Wahrenbrock MG, Varki A. Multiple hepatic receptors cooperate to eliminate secretory mucins aberrantly entering the bloodstream: are circulating cancer mucins the "tip of the iceberg"? Cancer Res. 2006;66(4):2433–41. doi: 10.1158/0008-5472.CAN-05-3851. [DOI] [PubMed] [Google Scholar]
- 64.Lai CC, Wang CY, Lu HM, Chen L, Teng NC, Yan YH, Wang JY, Chang YT, Chao TT, Lin HI, Chen CR, Yu CJ, Wang JD. Idiopathic pulmonary fibrosis in Taiwan—a population-based study. Respir Med. 2012;106:1566–1574. doi: 10.1016/j.rmed.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 65.Horimasu Y, Hattori N, Ishikawa N, Kawase S, Tanaka S, Yoshioka K, Yokoyama A, Kohno N, Bonella F, Guzman J, Ohshimo S, Costabel U. Different MUC1 gene polymorphisms in German and Japanese ethnicities affect serum KL-6 levels. Respir Med. 2012;106:1756–1764. doi: 10.1016/j.rmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 66.Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, Raghu G, Sauter W, Girard M, Alves M, Clerisme-Beaty E, Stowasser S, Tetzlaff K, Kuwana M, Maher TM, SENSCIS Trial Investigators Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med. 2019;380:2518–28. doi: 10.1056/NEJMoa1903076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary figures and tables and the protocols for processing blood specimens and for performing the enzyme-linked immunosorbent assay (ELISA).
Data Availability Statement
The de-linked datasets used and analysed during the current study are available from the corresponding author on reasonable request.