

Abstract
Cucurbit downy mildew (DM), caused by the obligate biotroph Pseudoperonospora cubensis, is a destructive disease in cucumber. A valuable source of DM resistance is the Indian cucumber accession PI 197088, which harbours several quantitative trait loci (QTLs) contributing to quantitatively inherited DM resistance. With a combination of fine‐mapping and transcriptomics, we identified Amino Acid Permease 2A (CsAAP2A) as a candidate gene for QTL DM4.1.3. Whole‐genome and Sanger sequencing revealed the insertion of a Cucumis Mu‐like element (CUMULE) transposon in the allele of the resistant near‐isogenic line DM4.1.3. To confirm whether loss of CsAAP2A contributes to partial DM resistance, we performed targeting induced local lesions in genomes on a DM‐susceptible cucumber genotype to identify an additional csaap2a mutant, which indeed was partially DM resistant. In view of the loss of the putative function as amino acid transporter, we measured amino acids in leaves. We found that DM‐inoculated leaves of line DM4.1.3 (with the csaap2a mutation) contained significantly fewer amino acids than wild‐type cucumber. The decreased flow of amino acids towards infected leaves in csaap2a plants compared to the wild type might explain the resistant phenotype of the mutant, as this would limit the available nutrients for the pathogen and thereby its fitness. To examine whether AAP genes play a conserved role as susceptibility factors in plant–oomycete interactions, we made targeted mutations in two AAP genes from tomato and studied the effect on susceptibility to Phytophthora infestans. We conclude that not only CsAAP2A but also SlAAP5A/SlAAP5B are susceptibility genes for oomycete pathogens.
Keywords: cucumber (Cucumis sativus), downy mildew (Pseudoperonospora cubensis), nutrient transport, plant–pathogen interactions, susceptibility gene, transposable element
Loss‐of‐function mutations in amino acid permease genes were found to contribute to resistance to Pseudoperonospora cubensis in cucumber and to Phytophthora infestans in tomato by restricting pathogen feeding.
1. INTRODUCTION
One of the most destructive cucurbit diseases is downy mildew (DM), caused by the obligate biotrophic oomycete Pseudoperonospora cubensis. P. cubensis can infect over 50 different plant species in the family Cucurbitaceae, including several economically important crops such as melon (Cucumis melo), watermelon (Citrullus lanatus), and squash (Cucurbita spp.), and is especially a limiting factor in cucumber (Cucumis sativus) cultivation (Lebeda & Cohen, 2011; Savory et al., 2011).
Cucumber DM is characterized by the appearance of chlorotic lesions on leaves, which appear angular as they are restricted by leaf veins. Brown to black sporulation can be observed on the abaxial side of the lesions. As the disease progresses, lesions become necrotic and multiple lesions will coalesce, leading to wilting and necrosis of the leaf and, eventually, death of the plant (Oerke et al., 2006; Thomas, 1996).
Whereas chemical control by means of fungicide application can play a role in disease management (Urban & Lebeda, 2006), breeding for resistance to DM is the most economic as well as environmentally friendly method to control the disease and prevent DM outbreaks. Modern systematic DM resistance breeding in cucumber started in the 1930s and 1940s, using cvs. Bangalore and Chinese Long as sources of resistance (Jenkins, 1942), eventually leading to the DM‐resistant cultivar Palmetto (Criswell et al., 2010). However, the resistance of Palmetto was broken within 2 years after its commercial release (Epps & Barnes, 1952). Hereafter, a new source of resistance was found in the Indian semi‐wild cucumber accession PI 197087, characterized by small brown (necrotic) lesions instead of larger yellow (chlorotic) lesions (Barnes & Epps, 1954). DM resistance from PI 197087 was found to be inherited as a single recessive gene, named p or dm‐1 (Criswell et al., 2010; van Vliet & Meysing, 1974, 1977), and consequently this gene was introgressed into many cucumber cultivars. DM resistance due to dm‐1 was effective for nearly 50 years (Cohen et al., 2015). Recently, a missense mutation in the gene CsSGR (Staygreen) was found to be causal for dm‐1 resistance (Wang et al., 2019). Since 2004, however, new virulent strains of P. cubensis emerged that have partially overcome dm‐1‐based resistance (Cohen et al., 2015; Holmes et al., 2015), prompting the search for novel sources of DM resistance.
In a large multilocation germplasm screening, wild cucumber accession PI 197088 came up as one of the most resistant genotypes (Call et al., 2012). Interestingly, PI 197088 was originally collected on the same date and location as PI 197087, indicating that the two accessions might be closely related. However, PI 197088 was found to be resistant to post‐2004 strains of P. cubensis that are virulent on PI 197087. In contrast to PI 197087‐based resistance, which is presumably caused by a single recessive gene, several groups discovered that DM resistance in PI 197088 is polygenic, with multiple quantitative trait loci (QTLs) contributing to overall disease resistance. In 2010, Caldwell et al. filed a patent describing three QTLs derived from PI 197088 on chromosomes 2, 4, and 5 in one F3 and two F5 mapping populations with three different susceptible parents (Caldwell et al., 2011). Yoshioka et al. mapped seven QTLs on chromosomes 1, 3, and 5 in a collection of 111 recombinant inbred lines (RILs) derived from a cross between the PI 197088 inbred line CS‐PMR1 and the intermediate susceptible line Santou (Yoshioka et al., 2014). Li et al. mapped five QTLs on chromosomes 1, 3, 4, and 5 in a set of 183 F3 families originating from a cross between PI 197088 and susceptible parent Changchunmici (Li et al., 2018). Wang et al. identified 11 QTLs on chromosomes 1 to 6 in a set of 148 RILs derived from a cross between PI 197088 and Coolgreen (Wang et al., 2018). Whereas these four groups all found that DM resistance in PI 197088 is polygenic, it is striking that there is no consensus at all on the genetic architecture of DM resistance in PI 197088, as each of the groups reached different conclusions on the number of QTLs and the contributions of the different QTLs on overall DM resistance. However, one or more QTLs on chromosome 5 were usually found to have the largest effect on resistance, often followed by a QTL on chromosome 4. Different mapping outcomes are probably caused by differences in experimental set‐up, such as the usage of different susceptible parents, different mapping population structures, different inoculation methods and sources, and different ways of scoring.
Recently, we fine‐mapped a QTL from cucumber accession PI 197088 for DM resistance on chromosome 4 (DM4.1), which was previously shown to explain 13.7%–27% of the phenotypic variance in DM resistance (Li et al., 2018; Wang et al., 2018) and is therefore one of the most promising QTLs for DM resistance in PI 197088. By repeated backcrossing using marker‐assisted selection (MAS) we obtained near‐isogenic lines (NILs), each having small PI 197088‐derived introgressions on chromosome 4 in a susceptible background. To our surprise, we found that instead of a single QTL (DM4.1) three individual loci could be distinguished within a 12 Mb interval, which we named DM4.1.1, DM4.1.2, and DM4.1.3. The individual effects of each of these three QTLs on the disease phenotype is markedly different. The first QTL, DM4.1.1, decreased the amount of disease‐induced necrosis, or “collapsing”, although this effect was not possible to score in the absence of the two other DM resistance QTLs. The second QTL, DM4.1.2, decreased the amount of sporulation of the pathogen. The third QTL, DM4.1.3, had a recessively inherited effect on pathogen‐induced chlorosis (yellowing), especially at early time points after inoculation, indicating that it probably slows the growth of the pathogen. The combined effect of the three loci is markedly stronger than the sum of the separate effects of the individual loci (Berg et al., 2020).
In plants, one of the major limiting mineral nutrients is nitrogen, as it is required to produce numerous compounds, including amino acids and nucleotides as well as several hormones. After inorganic nitrogen (in the form of ammonium or nitrate) is taken up by the roots, it is fixated either directly in the roots or, after transportation of nitrate, in leaves, in an energy‐requiring process producing amino acids. Amino acids are the major transport form of organic nitrogen. As plants require several transmembrane transport steps for amino acids to flow from source to sink tissues, and additionally different types of amino acids (e.g., basic, neutral, and acidic) require different specificities of transporters, it is not surprising that plants encode a plethora of amino acid transporter genes, divided over 10 distinct gene families (Tegeder & Rentsch, 2010). One of the most‐studied amino acid transporter families is the amino acid permease (AAP) family, which generally transport neutral and acidic amino acids, whereas there are also AAP genes transporting basic amino acids (Tegeder & Ward, 2012). The model plant Arabidopsis thaliana has eight AAP paralogs that are involved in several important steps in amino acid transport, that is, root uptake, phloem loading, xylem‐to‐phloem transfer, and seed loading (Tegeder & Ward, 2012). AAP genes were previously found to be transcriptionally up‐regulated in roots of A. thaliana infected with plant‐parasitic nematodes (Hammes et al., 2005), and subsequently it was discovered that mutations in several AAP genes contributed to nematode resistance (Elashry et al., 2013; Marella et al., 2013). Although there are no experimental data to prove it, it was postulated that AAP genes play a role in the transport of amino acids to nematode feeding sites, and hence that mutations in AAP genes limit the availability of amino acids for the parasite, thereby decreasing the nematode's fitness (Marella et al., 2013).
Here, we report that a natural loss‐of‐susceptibility mutation in a cucumber Amino Acid Permease gene (CsAAP2A) within the fine‐mapped DM4.1.3 locus contributes to quantitatively inherited DM resistance in PI 197088. To examine whether AAP genes play a broader role in plant–parasite interactions, we have generated mutant alleles of two tomato AAP genes, and found that these mutants were partially resistant to the hemibiotrophic oomycete Phytophthora infestans. We thereby establish that AAP genes play a conserved role as determinants of susceptibility against multiple parasites.
2. RESULTS
2.1. Fine‐mapping and identification of candidate genes for subQTL DM4.1.3 for reduced chlorosis
We developed a near‐isogenic line (NIL DM4.1.3) with partial DM resistance caused by an introgression on chromosome 4 from PI 197088 in the susceptible HS279 background. Initial QTL mapping results delimited subQTL DM4.1.3 to the c.3.6 Mb interval Chr4: 16,876,817–20,438,834 (Berg et al., 2020). To narrow down this interval, we selected 14 cucumber plants with recombinations within the DM4.1.3 interval from the population used for QTL mapping. These recombinants were selfed to create populations that were tested for DM resistance. Eight of these families were fixed for the resistant allele of subQTL DM4.1.3, characterized by a uniform low level of chlorosis. It should be noted that several of these resistant families were fixed for the susceptible allele at the other subQTLs (DM4.1.1 and DM4.1.3) without consequences on the observed low level of chlorosis, which is in agreement with our previous finding that only subQTL DM4.1.3, and not one of the other subQTLs, is responsible for the antichlorosis phenotype (Berg et al., 2020). Three other families were fixed for the susceptible allele of subQTL DM4.1.3, characterized by a uniform high level of chlorosis. The remaining three families were segregating. Analysis of these recombinant populations allowed fine‐mapping of subQTL DM4.1.3 to the interval Chr4: 18,842,456–19,596,926 (Figure 1), containing 80 predicted genes in the cucumber reference genome (Chinese Long 9930 v. 2).
FIGURE 1.

Fine‐mapping subQTL DM4.1.3. Screening of individuals derived from the mapping population allowed identification of additional informative recombinants within the DM4.1.3 interval. Bars represent genotypes at marker locations. Black bars indicate the PI 197088 allele, white bars indicate the HS279 allele, grey bars represent heterozygosity. Individuals were self‐fertilized to develop populations, which were phenotyped regarding chlorosis
To identify candidate genes for QTL DM4.1.3, we investigated the expression of the 80 predicted genes within the fine‐mapped interval in a previously described RNA‐seq data set (Berg et al., 2020), obtained by sequencing RNA isolated from P. cubensis‐inoculated cucumber leaves of NIL DM4.1.3 and the susceptible recurrent parent HS279. Thirteen genes within the interval were found to be differentially expressed between genotypes NIL DM4.1.3 and HS279 (Data S1). Furthermore, nonsynonymous polymorphisms between the two genotypes in the interval of QTL DM4.1.3 were determined (Data S2).
An Amino Acid Permease gene, CsAAP2A (gene ID: Csa4M573860), was selected as a promising candidate gene based on the finding that this gene was down‐regulated by a factor of 100 in NIL DM4.1.3 compared to HS279, which was highly significant (Benjamini–Hochberg corrected p value = 3.1 × 10−28).
2.2. Lack of CsAAP2A expression is due to the insertion of a CUMULE transposon
To investigate the reason why CsAAP2A was very poorly expressed in NIL DM4.1.3, we resequenced the whole genomes of genotype NIL DM4.1 (an NIL with an introgression spanning subQTL DM4.1.1, DM4.1.2, and DM4.1.3) as well as susceptible parent HS279. The alignment of reads to the reference genome in the genomic locus of AAP (Chr4: 18,882,370–18,886,704) was manually inspected (Data S3). Several indications were found of a structural variation in the fourth exon of CsAAP2A in genotype NIL DM4.1: double coverage of an 11 bp stretch of the exon, alignment problems of reads at either side of this 11 bp sequence, and flanking reads with mate pairs aligning to different regions of the cucumber reference genome (Data S4). These three observations led us to hypothesize that there might be an insertion of a transposable element at this location in the DM4.1.3 allele of CsAAP2A.
To test this hypothesis, PCRs were performed on DNA isolated from resistant donor PI 197088, susceptible recurrent parent HS279, partially DM‐resistant NIL DM4.1, and partially resistant NIL DM4.1.3 (Figure 2a,b) using a primer pair flanking exon four of CsAAP2A (Figure 2c). Cloning and Sanger sequencing of the resulting PCR products revealed the presence of a 7,688 bp insertion in the resistant genotypes compared to the susceptible HS279 (Figure 2c). This insertion had all the hallmarks of an autonomous CUMULE transposon (Figure 2d), that is, it was flanked by a duplication of 11 bp of CsAAP2A (target site duplication, TSD), the first and the last 118 bp of the insert are inverted copies of one another (terminal inverted repeats, TIR) (Data S5), and the insertion contained a 2,206 bp long open reading frame encoding a MuDRA transposase gene, with high sequence homology (90% identical nucleotide sequences) to the MuDRA transposase characterized by van Leeuwen et al. in the melon CUMULE (Cucumis Mutator‐Like Element) transposon (van Leeuwen et al., 2007). Furthermore, we identified an additional gene within the insertion consisting of five exons with a cumulative length of 1,599 bp, with homology to Ubiquitin‐like peptidase genes (Ulp1), which was also identified in the melon CUMULE transposon (van Leeuwen et al., 2007). By realigning RNA‐Seq reads obtained from NIL DM4.1.3 to the 7,688 bp insertion (Data S6), it was found that, whereas there was no evidence of expression of the MuDRA gene, the Ulp1 gene was found to be abundantly expressed.
FIGURE 2.

PCR amplification of AAP alleles from resistant and susceptible cucumber genotypes led to the identification of a CUMULE transposon. (a) Representative downy mildew (DM) phenotypes of cucumber genotypes PI 197088 (resistant donor), HS279 (susceptible recurrent parent), and partially resistant near‐isogenic lines DM4.1 and DM4.1.3 with PI 197088 introgressions in HS279 background, 7 days after spray inoculation with Pseudoperonospora cubensis. (b) Representative DM phenotypes of the four genotypes described in (a), 14 days after spray inoculation with P. cubensis. (c) DNA isolated from the cucumber genotypes described in (a) was used as a template for PCR with AAP‐specific primers. Amplified products were analysed with gel electrophoresis. Whereas the product amplified from HS279 DNA gave a band of the expected size (1.5 kb), products amplified from (partially) resistant genotypes resulted in larger (c.8 kb) fragments. (d) The large fragment amplified in (c) was cloned and sequenced, which revealed the presence of a 7,688 bp insertion with hallmarks of a Cucumis Mu‐like element (CUMULE) in the DM4.1.3 allele of the AAP gene. The CUMULE transposon is schematically represented. Repeat regions (TSD, target site duplication; TIR, terminal inverted repeat) are indicated in black. Additional tandem repeat is indicated in white. Putative coding sequences (short expressed ORF, Ulp1: Ubiquitin‐like protease 1, MudrA: transposase) are indicated in grey. Elements on the negative strand are indicated in opposite direction, below the black line. The full CUMULE sequence plus annotation has been deposited in GenBank (MN062013)
The annotated DNA sequence of the CUMULE transposable element was deposited in the GenBank database (MK936607). Data S8 gives nucleotide sequences of both the wild‐type and the mutant alleles of CsAAP2A on genomic and (predicted) coding sequence level.
A BLASTn search was performed screening nucleotide sequences of the MuDRA transposase gene, the Ulp1 protease gene, and the TIR domains of the newly identified CUMULE transposon as queries against the cucumber reference genome (Chinese Long 9930 v. 2). We identified 67 regions with homology to the MuDRA transposase, 61 regions with homology to the Ulp1, and 94 regions with homology to the TIR domains, scattered over all seven cucumber chromosomes and often in close proximity to one another (Data S9), indicating that CUMULE transposons are rather common in the cucumber genome.
A recently described script (Berg et al., 2015) was used to perform an in silico search for occurrence of the CUMULE‐harbouring allele of CsAAP2A in a resequenced core collection of 115 divergent cucumber accessions (Qi et al., 2013); however, we did not find indications for the presence of this allele in any of these 115 accessions. In a search for other polymorphisms in CsAAP2A in the 115 cucumber genome data, we identified 36 single nucleotide polymorphisms (SNPs), four of which were predicted to cause changes to the CsAAP2A protein. It was found that accessions with the alternative allele of two of these nonhomologous SNPs generally had lower DM disease indices compared to the average (Data S10).
Whereas the insertion of the CUMULE transposon apparently led to abolishment of CsAAP2A expression, we determined the effect of the insertion on the predicted protein sequence (Data S11). We found that the insertion of the transposable element led to truncation of the last 306 amino acid residues out of the total 466, substituting them with 19 other amino acid residues before arriving at an alternative stop codon. This indicates that even residual CsAAP2A expression will presumably not encode a functional AAP protein, and the CUMULE‐containing allele of CsAAP2A is a bona fide loss‐of‐function allele.
2.3. A TILLING mutant in CsAAP2A gains resistance to P. cubensis
To test whether loss‐of‐function mutations in CsAAP2A indeed contributes to DM resistance, a TILLING (targeting induced local lesions in genomes) population of cucumber was screened for mutations in CsAAP2A. In total, seven nonsynonymous mutants were identified (Additional Data S12). The mutant with the most drastic predicted effect on the encoded protein, that is, having a truncation after the first 24 amino acid residues, was chosen for phenotyping. Progeny of a heterozygous plant, segregating for the ethyl methanesulfonate (EMS) mutation in CsAAP2A, was inoculated with P. cubensis, and scored for chlorosis and sporulation at 7 days postinoculation (dpi). It was found that plants homozygous for the mutant csaap2a allele were significantly less chlorotic (Kruskal–Wallis test, p < .01) compared to siblings that were either heterozygous or wild‐type (WT) regarding CsAAP2A (Figure 3 and Data S13). With respect to sporulation, there was a trend for csaap2a mutant plants to sporulate less than WT or heterozygous siblings, although this difference was not significant (Kruskal–Wallis, p = .065) (Data S14). This indicates that loss of CsAAP2A indeed confers partial resistance to P. cubensis, especially regarding chlorosis, which is in agreement with our previous observation that subQTL DM4.1.3 especially has an effect on pathogen‐induced chlorosis and a smaller effect on sporulation (Berg et al., 2020).
FIGURE 3.
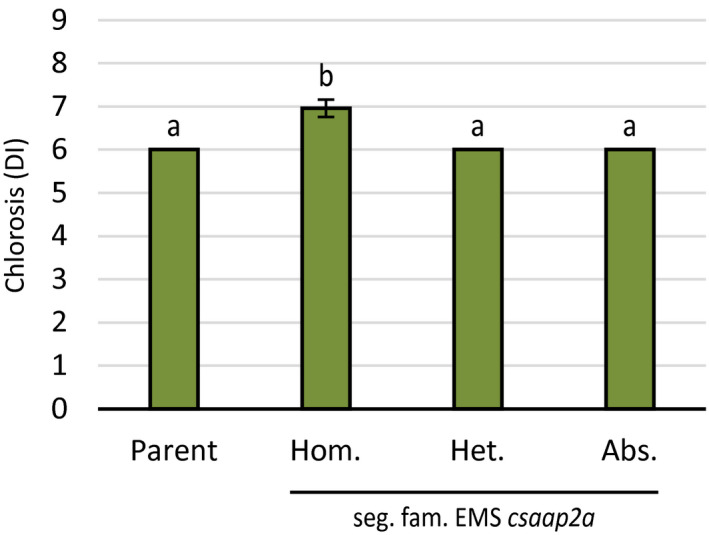
csaap2a TILLING mutant is partially resistant to Pseudoperonospora cubensis. A P. cubensis disease assay was performed on a family segregating for an ethyl methanesulfonate (EMS)‐induced mutation in CsAAP2A leading to an early stop codon (Data S12), with the non‐EMS‐treated parental line as an additional control. Hom., homozygous for the mutant csaap2a allele; Het., heterozygous; Abs., homozygous for the wild‐type CsAAP2A allele. Plants were scored for chlorosis at 7 days postinoculation. Bars represent average disease index (DI) phenotype scores on a 1–9 scale, ranging from susceptible to resistant. Error bars represent standard deviation. Bars with different letters are statistically significant from one another (Kruskal–Wallis, p < .05)
2.4. NIL DM4.1.3 accumulates less amino acids upon P. cubensis inoculation compared to the susceptible parent HS279
As we identified an amino acid transporter gene (CsAAP2A) as a likely susceptibility gene for QTL DM4.1.3, we hypothesized that impairment of this gene may lead to lower amino acid levels in leaves, thereby reducing the feeding of the pathogen. In view of this hypothesis, we extracted total free amino acids from leaves of eight individual plants of NIL DM4.1.3, as well as from eight individuals of susceptible parent HS279, 7 days after inoculation with P. cubensis. For both genotypes, eight mock‐inoculated plants were included as controls. Free amino acid concentrations in leaf extracts were quantified using gas chromatography with flame ionization detection (GC‐FID) (Figure 4).
FIGURE 4.
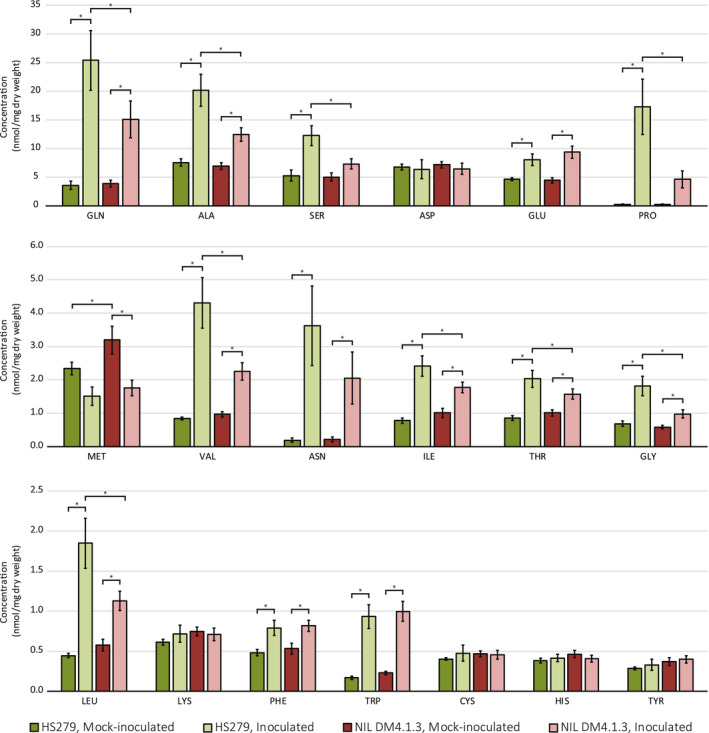
Amino acid profiling of cucumber leaf extracts. Amino acids were extracted from leaves of cucumber genotypes HS279 (downy mildew susceptible) and NIL DM4.1.3 (partially downy mildew resistant), 7 days postinoculation with Pseudoperonospora cubensis or mock treatment. Free amino acid concentrations were quantified using gas chromatography with flame ionization detection (GC‐FID). Coloured bars indicate the average amino acid concentration per amino acid (in nmol per mg dry weight) in each of the genotype × treatment combinations (n = 8 in each group). Error bars indicate standard error of the mean. Differences due to genotype, treatment, and the interaction were all found to be statistically significant (two‐way multivariate analysis of variance, p < .05). Asterisks indicate significant between‐subject differences (p < .05)
Generally, it was found that leaves of both genotypes inoculated with P. cubensis had higher amino acid concentrations compared to mock‐inoculated leaves. However, in P. cubensis‐inoculated leaves of genotype NIL DM4.1.3, the concentration of several amino acids was found to be lower than in inoculated leaves of the susceptible parent, indicating that the QTL partially prevents the increase in amino acids. Differences due to genotype, treatment, and the interaction were all found to be statistically significant (two‐way multivariate analysis of variance [MANOVA], p < .05). A post hoc test for each of the individual amino acids revealed that for nine out of the 19 quantified amino acids (Ala, Gly, Val, Leu, Ile, Thr, Ser, Pro, Gln) there was a significant genotype × treatment interaction (p < .05). For five other amino acids (Asn, Met, Glu, Phe, Trp) there was a significant main effect of the treatment (p < .05) but no significant effect of the genotype (p > .05). For the remaining five amino acids (Asp, Cys, Lys, His, Tyr), no significant differences in concentration were observed (p > .05).
2.5. Phylogenetic analysis of the AAP gene family in several plant species
AAP genes form a gene family with multiple copies in all land plant species characterized so far (Tegeder & Ward, 2012). To study the evolutionary relationship amongst AAP homologs, we constructed a phylogenetic tree using 93 AAP homologs identified in 10 plant species. AAP protein sequences were identified in the genomes of four cucurbit species (cucumber [Cucumis sativus], melon [Cucumis melo], watermelon [Citrullus lanatus], and zucchini [Cucurbita pepo]), as well as the noncucurbit eudicots Arabidopsis thaliana, barrel clover (Medicago trunculata), and tomato (Solanum lycopersicum), the monocot rice (Oryza sativa), and the lower plants Selaginella moellendorffii and Physcomitrella patens. Data S15 gives an overview of the identified AAP proteins.
In the resulting maximum‐likelihood phylogenetic tree (Data S16), the grouping of identified AAP homologs is largely consistent with a previously published phylogeny of AAP genes (Tegeder & Ward, 2012), as four clades can be observed that can be subdivided into two subclades each. According to the nomenclature proposed by Tegeder and Ward (2012), newly identified AAP gene homologs are named based on their positions in the phylogenetic tree. Our candidate gene Csa4M573860 is therefore referred to as CsAAP2A.
Four out of the five cucumber AAP genes, including our candidate gene CsAAP2A, belong phylogenetically to one single subclade, whereas CsAAP6 belongs to another subclade. In the phylogenetic tree (Data S16) one melon (CmAAP2A) and one watermelon (ClAAP2A) ortholog of candidate gene CsAAP2A can be identified, but in other plant species no single CsAAP2A ortholog can be inferred, indicating separate gene/genome duplications and/or gene deletion events in the evolutionary branches leading to these species.
2.6. Transcript abundance profiling of cucumber AAP genes
A previously described RNA‐Seq data set consisting of gene expression data from a variety of cucumber tissues of reference accession Chinese Long 9930 (Berg et al., 2017) was analysed to study tissue‐specific expression patterns of AAP genes in cucumber (Figure 5a). It was found that our candidate gene CsAAP2A, as well as the highly similar homolog CsAAP2B, were abundantly transcribed in stem tissue, at a slightly lower level in root and hypocotyl tissue, and were barely detectable in cotyledons, leaves, flowers, and fruit. In contrast, CsAAP3A was highly expressed in leaf tissue, flowers, fruits, and cotyledon tissue, whereas CsAAP3B was generally highly expressed compared to the other CsAAP genes in all examined tissues and was especially abundant in root tissue. CsAAP6 had relatively high expression values in hypocotyl, cotyledon, and leaf tissue.
FIGURE 5.
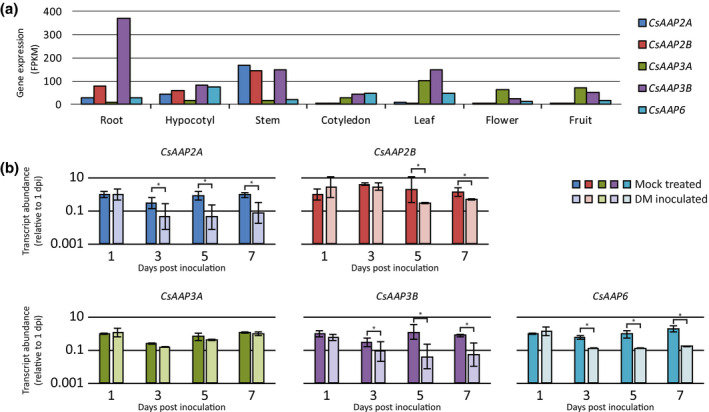
Expression profile of cucumber AAP genes in different tissues and in Pseudoperonospora cubensis‐inoculated leaves. (a) The transcript abundance (in the number of fragments per kilobase of transcript per million mapped reads, FPKM) of cucumber AAP genes in different tissues of inbred line Chinese Long 9930. The amount of independent biological replicates per tissue was either one (hypocotyl, stem, and cotyledon), two (root), or three (leaf and fruit). Data from Berg et al. (2017). (b) Relative expression level of cucumber AAP genes in leaves of downy mildew‐susceptible cucumber genotype HS279 in response to inoculation with P. cubensis or mock treatment. Relative transcript abundances at 1, 3, 5, and 7 days postinoculation (dpi) with P. cubensis were determined using quantitative reverse transcription PCR. Data were normalized relative to reference gene TIP41, and subsequently normalized relative to the average ∆C t value of mock‐treated plants at 1 dpi for each gene. Relative transcript abundances were calculated as 2−∆∆ C t. Each bar shows the relative expression of three biological replicates on a logarithmic scale. Error bars indicate standard deviation. Asterisks indicate significant differences between mock‐treated and inoculated plants (analysis of variance, p < .05)
To determine the effects of DM on AAP gene expression, cucumber leaves of both genotypes HS279 and NIL DM4.1.3 inoculated with P. cubensis or mock‐treated leaves were sampled at 1, 3, 5, and 7 days postinoculation (dpi). RNA was isolated and quantitative reverse transcription PCR (RT‐qPCR) was performed on cDNA samples using primers specific for each of the CsAAP genes (Data S18). In the susceptible genotype HS279, inoculation with P. cubensis significantly reduced the expression of the majority of the AAP genes, including CsAAP2A (Figure 6b). In NIL DM4.1.3, generally the same trends were observed for the majority of the AAP genes (Data S17). However, expression of CsAAP2A was reduced by 100–1,000‐fold compared to HS279 at every time point and in mock‐treated as well as P. cubensis‐inoculated leaves. This is consistent with the RNA‐Seq data. Apparently, the CUMULE insertion reduced the expression considerably.
FIGURE 6.
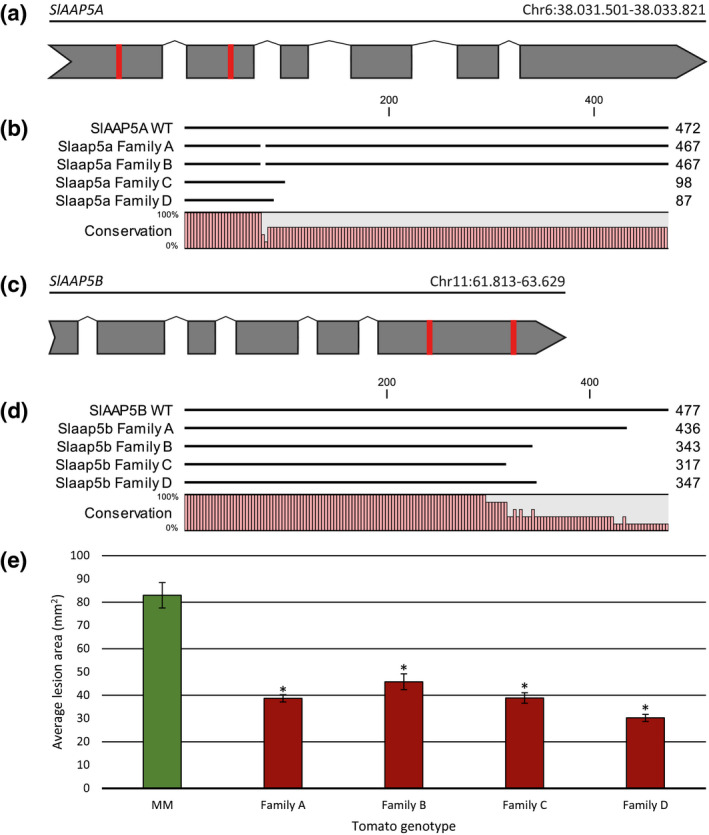
CRISPR‐Cas9 mediated mutagenesis of SlAAP5A and SlAAP5B results in tomato mutants with partial Phytophthora infestans resistance. (a, c) Schematic representations of the gene model of the tomato genes SlAAP5A (a) and SlAAP5B (c). Exons are indicated in grey. 20 bp target sequences for CRISPR‐Cas9 mediated genome editing are indicated in red. (b, d) Multiple sequence alignments of the predicted protein sequences of SlAAP5A (b) and SlAAP5B (d) in four homozygous tomato T2 families with CRISPR‐Cas9‐induced mutations, compared to the reference protein sequence. Data S20 gives the full sequence alignments. (e) Four tomato T2 families with mutations in SlAAP5A and SlAAP5B as indicated in (b) and (d) were tested for susceptibility to P. infestans isolate CA56 compared to wild‐type control cv. Moneymaker (MM). Lesion areas (in mm2) were measured at 4 days postinoculation. Each bar represents the average lesion area of 48–63 biological replicates. Error bars indicate the standard error of the mean. Asterisks indicate significant differences compared to the average lesion area of MM (t test, p < .05)
2.7. CRISPR‐Cas9 generated tomato aap mutants gain partial resistance to P. infestans
To test whether AAP genes in other species also contribute to pathogen susceptibility, we chose to disrupt SlAAP5A and SlAAP5B, the two tomato orthologs of our cucumber candidate gene CsAAP2A (Data S16), by means of CRISPR/Cas9 mutagenesis. We designed four guide RNAs targeting two 20 bp regions in each of the two genes (Figure 6a,b). We developed an expression vector containing a Cas9 gene under control of a constitutive CaMV 35S promoter, and each of the four guide RNAs under U6 promoters, and used this construct to generate stable transformants in tomato cv. Moneymaker (MM) background. A total of 21 primary transformants were obtained and screened for mutations in SlAAP5A and/or SlAAP5B by means of PCR, followed by Sanger sequencing. Three primary transformants, heterozygous for mutations in SlAAP5A and/or SlAAP5B, were selected for selfing to generate T1 families. In segregating T1 families, individuals were screened for homozygous mutations in SlAAP5A and/or SlAAP5B. Four homozygous T1 individuals were selected for selfing in order to generate four homozygous T2 families (families A–D), each with mutations in both SlAAP5A and SlAAP5B (Data S19). Predicted SlAAP5A protein sequences in families A and B had a five amino acid deletion compared to the reference sequence, whereas in families C and D the predicted protein sequence was truncated with 396 amino acids compared to the reference (Figure 6a). Predicted SlAAP5B proteins were truncated in each of the four families, with the last 55 to 180 amino acids truncated compared to the reference sequence (Figure 6b).
Four‐week‐old seedlings of tomato T2 families A–D, and their WT background MM, were inoculated with P. infestans isolate CA65. At 4 dpi, lesion areas were measured. It was found that average lesion sizes in all four slaap5a/b mutant families were significantly smaller compared to WT MM (p < .05), indicating (partial) resistance to P. infestans (Figure 6c).
3. DISCUSSION
3.1. Loss of CsAAP2A contributes to partial DM resistance
We have shown here that a loss‐of‐function allele of the cucumber amino acid transporter CsAAP2A (Csa4M573860) is the most likely candidate gene for the DM4.1.3 locus, contributing to quantitative DM resistance in the Indian cucumber accession PI 197088. The presence of a 7,688 bp insertion corresponding to a Mu‐like transposon in the DM4.1.3 allele of CsAAP2A presumably silences expression of the gene, for example by changing the methylation status of the gene and/or by posttranscriptional breakdown of aberrant transposon‐containing primary transcripts (Lisch & Slotkin, 2011). Even if the DM4.1.3 allele of CsAAP2A were expressed (e.g., in other tissues or other circumstances than the P. cubensis‐inoculated leaves studied here), the integration of a 7,688 bp insertion in the fourth exon of the gene would truncate the protein, removing over two‐thirds of the amino acid sequence, presumably rendering the remaining protein nonfunctional (Data S11).
In a separate line of evidence, we obtained an additional csaap2a mutant in a different genetic background by means of screening a population of cucumber plants derived from EMS‐treated seeds for mutations in CsAAP2A, a process known as TILLING (McCallum et al., 2000). A TILLING mutant with a premature stop codon in CsAAP2A (W25Stop) was indeed partially resistant to P. cubensis, compared to siblings without this mutation, heterozygous plants, and the non‐EMS‐treated background genotype (Figure 3).
Additionally, natural CsAAP2A polymorphisms in a collection of 115 resequenced cucumber accessions were identified (Data S10), two of which occurred in (rather) DM‐resistant cucumber accessions according to recent data by Liu et al. (2020). However, more data (e.g., by screening F2 populations derived from these accessions) would be required to conclude whether or not these polymorphisms contribute to the observed DM resistance in those accessions.
The finding that a loss‐of‐function mutation in the CsAAP2A gene provides (partial) resistance leads to the conclusion that CsAAP2A should be considered to be a susceptibility gene (S‐gene) for downy mildew. This is in agreement with the recessive inheritance of the resistance from the DM4.1.3 locus. To our knowledge, this is the first example of an effect of mutations in amino acid transporter genes on downy mildew resistance.
3.2. SlAAP5A and SlAAP5B contribute to P. infestans susceptibility in tomato
On identifying CsAAP2A as a novel susceptibility gene for DM in cucumber by means of genetic mapping and transcriptomics, we decided to perform functional analysis of the role of AAP genes on oomycete susceptibility in another pathosystem, that is, the interaction between tomato and P. infestans. We found that mutations in the two closest tomato orthologs of CsAAP2A, that is, SlAAP5A and SlAAP5B, led to increased resistance towards P. infestans, indicating that either or both tomato AAP genes are susceptibility factors. While mutant families C and D have truncations with most of the SlAAP5A protein missing, mutant families A and B have only a 15 bp (in‐frame) deletion in SlAAP5A, while showing a similar decrease in average lesion size compared to families C and D, whereas all four families have truncations of SlAAP5B, whereby different parts of the C terminal of the protein are missing (Data S20). Although it is tempting to speculate that the gain in resistance is caused by the mutations in SlAAP5B rather than SlAAP5A, more work is required to study the separate effects of mutations in either gene.
3.3. Amino acid transporters as broadly conserved susceptibility genes
Obligate parasitic organisms are by definition fully dependent on their host to provide them with nutrients. Amino acids, as building blocks of proteins and precursors for many secondary metabolites, are an important category of nutrients. It should therefore not be surprising that amino acid transporters play important roles in plant–pathogen interactions. Pathogens are also able to induce host‐expressed amino acid transporters, and thus increase the availability of amino acids for the pathogen. This is best documented for sedentary plant‐parasitic nematodes, which induce the formation of highly specialized feeding structures (termed giant cells for root‐knot nematodes or syncytia for cyst nematodes) in plant roots from which they acquire nutrients. In roots containing these feeding structures, many host‐encoded amino acid transporter genes are transcriptionally up‐regulated (Elashry et al., 2013; Hammes et al., 2005, 2006). Triggered by these findings two groups studied the interaction of nematodes with Arabidopsis aap mutants and found that reproduction of cyst nematodes is significantly reduced in Ataap1, Ataap2, and Ataap6 T‐DNA knock‐out mutants (Elashry et al., 2013), whereas reproduction of root‐knot nematodes is reduced in Ataap3 and Ataap6 T‐DNA knock‐out mutants (Marella et al., 2013). Another example of an amino acid transporter that plays an important role in plant–parasite interactions is the Arabidopsis LHT1 gene, the expression of which is induced by infection with the bacterium Pseudomonas syringae as well as the fungi Erysiphe cichoracearum (causal agent of powdery mildew) and Colletotrichum higginsianum (causal agent of anthracnose). lht1‐knock‐out mutants were found to be less susceptible to all three diseases (Liu et al., 2010).
Several amino acid transporter genes, most notably in the AAP and LHT families, have now been shown to be susceptibility genes against various pathogens, including nematodes (Elashry et al., 2013; Marella et al., 2013), bacteria, and fungi (Liu et al., 2010) in Arabidopsis as well as the oomycetes P. cubensis in cucumber and P. infestans in tomato (this research), demonstrating the fact that amino acid transport is important for many unrelated pathogens in several unrelated plant species. In the investigated cases so far, loss‐of‐function mutations in single amino acid transporter genes were found to confer partial resistance. As plants have a multitude of amino acid transporter genes (e.g., at least 67 genes in Arabidopsis thaliana; Rentsch et al., 2007) with partially overlapping substrate specificity and expression profiles, it is to be expected that with regards to their role as S‐genes there is functional redundancy between amino acid transporter genes.
3.4. DM resistance in cucumber due to CsAAP2A mutations is caused by perturbing pathogen‐induced amino acid loading of infected leaves
Obligate biotrophic pathogens rely on their host to provide them with amino acids, therefore they are dependent on host‐encoded transporters to transfer amino acids from source tissues to infected cells. Key players in long‐ distance transport of amino acids are members of the AAP gene family. The Arabidopsis gene AAP2, which is expressed along the vasculature, is involved in xylem‐to‐phloem transport of amino acids. Mutations in AAP2 lead to disturbed source‐to‐sink transport of amino acids, and therefore to 26%–52% lower amino acid concentrations in sink tissues such as developing leaves and siliques compared to WT plants (Zhang et al., 2010).
As we study a loss‐of‐function allele of a cucumber ortholog of AAP2 as a candidate gene contributing to downy mildew resistance, we were triggered to examine the effect of this mutation on amino acid concentrations, in the presence as well as in the absence of the pathogen. Our results (Figure 4) suggest that the pathogen manipulates the plant to increase amino acid transport towards infected leaves, thereby creating an artificial sink. In contrast, Liu et al. showed that Arabidopsis plants inoculated with the powdery mildew‐causing fungus Erysiphe cichoracearum had lower concentrations of several amino acids, especially glutamine, compared to mock‐inoculated controls (Liu et al., 2010). Apparently, E. cichoracearum is not able to facilitate amino acid transport towards infected tissue, but rather depletes the existing amino acid pool.
Furthermore, our results showed that CsAAP2A is involved in amino acid loading of infected leaves, and consequently a loss‐of‐function mutation leads to decreased amino acid transport, because in inoculated plants the observed increase in amino acids was generally lower in plants with the mutant CsAAP2A allele compared to WT plants (Figure 4). The spectrum of amino acids for which we found significant differences in concentration between both genotypes after inoculation encompassed nine neutral, nonaromatic amino acids, consistent with the previously found specificity of AAP transporters in Arabidopsis (Rentsch et al., 2007).
4. EXPERIMENTAL PROCEDURES
4.1. Plant materials and growing conditions
Plant introduction line PI 197088, highly resistant to DM caused by P. cubensis (Call et al., 2012), was originally collected in Assam, India on 16 April 1951 and is maintained by the United States National Plant Germplasm System (NPGS). Breeding line HS279 is a pickling type cucumber, susceptible to DM, with good horticultural characteristics. From a cross between these genotypes, F3BC3S3 mapping populations were developed using HS279 as recurrent parent. NILs DM4.1 and DM4.1.3 were derived from F3BC3S3 individuals with PI 197088‐derived introgressions on chromosome 4 corresponding to the interval Chr4: 11,479,953–20,438,834 and Chr4: 18,469,868–23,424,119, respectively. TILLING was performed on a homozygous breeding line, susceptible to P. cubensis, previously described in Berg et al. (2015).
Tomato cv. Moneymaker was used as a susceptible control for experiments with P. infestans, and as background genotype to perform genome editing to create tomato aap mutants, described below.
Plants were grown in climate chambers with temperatures of 22 °C (day) and 17 °C (night), with a 16/8 hr day/night cycle, and a relative humidity of 80%.
4.2. Inoculum maintenance and disease tests
An isolate of P. cubensis obtained from an infected cucumber field in Haelen, the Netherlands, was maintained on fully expanded cucumber leaves, healthy in appearance before inoculation. Detached leaves were kept in closed boxes containing water‐soaked paper towels and inoculated with a spore suspension developed as described below. Boxes containing inoculated cucumber leaves were kept in a climate chamber under 18 °C (day) and 15 °C (night), with a 16/8 hr day/night cycle for 10 days. Heavily infected detached leaves were preserved at −20 °C as inoculum source for <6 months. Spore suspensions were produced by washing spores from frozen infected leaves using tap water and filtering through cheesecloth. The spore concentration was measured using a haemocytometer and adjusted to 104 spores/ml.
Cucumber plants for P. cubensis disease tests were grown in plastic tents, which were closed the day before inoculation to ensure a high relative humidity. Both sides of cucumber leaves were sprayed with spore suspension prepared as described above. After inoculation, plants were left in darkness at 18/15 °C (day/night) for 24 hr in closed plastic tents. Starting from 7 dpi, yellowing (chlorosis), sporulation, and collapsing (necrosis) of leaves were assessed by eye on a 1–9 scale, 9 being fully resistant and 1 being fully susceptible.
P. infestans isolate CA56 was maintained on rye sucrose agar plates. Prior to inoculation, zoospores were washed from plates with sterile Milli‐Q water and filtered through Miracloth. Zoospore concentrations were scored using a haemocytometer and diluted to 105 zoospores/ml with sterile Milli‐Q water. Abaxial sides of tomato leaflets were inoculated with 10 μl droplets of inoculum using a repeater pipette. Plants were placed in closed plastic tents prior to inoculation and during the infection to ensure a high relative humidity. Lesions were measured at 4 dpi using an electronic caliper: the longest diameter of the lesion was measured (d 1), as well as the perpendicular diameter (d 2). Total lesion area was then calculated as ¼ × π × d 1 × d 2.
4.3. Identification and sequencing of CUMULE transposon
DNA was isolated from cucumber leaf samples originating from genotypes PI 197088, HS279, NIL DM4.1, and NIL DM4.1.3 using a CTAB protocol (Healey et al., 2014). PCR was performed on the isolated DNA samples using primers AAP‐clon‐TE‐F and AAP‐clon‐TE‐R as specified in Data S18. In each PCR, 5 μl of template DNA, 2.5 μl of each primer, 1 μl of dNTP, 10 μl of Phusion HF buffer, 28.5 μl of Milli‐Q water, and 0.5 μl of Phusion high‐fidelity polymerase (Thermo Fisher Scientific) were mixed. Cycling conditions were: 30 s initial denaturation at 98 °C, followed by 30 cycles of 10 s denaturation at 98 °C, 20 s annealing at 60 °C, and 3 min 30 s extension at 72 °C, followed by a final incubation of 7 min at 72 °C. PCR products were visualized with GelRed staining and analysed by agarose gel electrophoresis. PCR products were cloned using the Zero Blunt TOPO PCR Cloning Kit (Thermo Fisher Scientific) according to the manufacturer's instructions and transformed to electrocompetent Escherichia coli DH5α cells. The presence of the insert was checked by using primer pairs AAP‐clon‐TE‐F plus AAP‐colPCR‐R1 and AAP‐colPCR‐F2 plus AAP‐colPCR‐R2 (Data S18) on 1 μl of bacterial culture in a 10 μl DreamTaq DNA polymerase (Thermo Fisher Scientific) PCR according to the manufacturer's instructions. Twelve individual E. coli cultures with confirmed AAP‐TE inserts were grown in liquid Luria Bertani broth culture and plasmids were recovered using the Qiaprep spin miniprep kit (Qiagen). Plasmids were sent for Sanger sequencing (GATC Biotech, Germany), first using primers AAP‐clon‐TE‐F and AAP‐clon‐TE‐R, subsequently using primers designed based on the previous sequencing runs. Data S18 gives an overview of the 57 designed sequencing primers. Obtained reads were trimmed and aligned to one another using CLC Genomics Workbench v. 11. A consensus sequence was built based on majority rule.
For annotation, previously described RNA‐Seq reads of NIL DM4.1.3 were aligned to the consensus transposable element sequence using TopHat v. 2.1.1 (Kim et al., 2013). The alignment of RNA‐Seq reads to the transposable element sequence was manually checked using IGV v. 2.3.32 (Robinson et al., 2011). Alignment of repetitive regions and prediction of open reading frames (ORFs) was done using CLC Genomics Workbench v. 11. BLASTn and BLASTp searches of ORFs and translated ORFs were performed at the NCBI BLAST server.
To check for the occurrence of similar transposable elements in the cucumber reference genome, BLASTn searches with annotated regions of the transposable element sequence were performed against the cucumber reference genome, Chinese Long 9930 v. 2.
4.4. Analysis of genome‐wide association study data regarding CsAAP2A
To check for occurrence of the CUMULE insertion at the CsAAP2A locus in cucumber germplasm, reads of the resequencing project of 115 cucumber accessions by Qi et al. (2013) were downloaded from the NCBI short read archive, accession SRA056480. By a simple BASH script, total reads were screened for the presence of 30 bp sequences comprising (a) the last 15 bp of CsAAP2A before the transposable element insertion and the first 15 bp of the transposable element insertion, in forward (5′‐CCAAAAGACATGGGAAATTGACAAAAATAGG‐3′) or reverse (5′‐CCTATTTTTGTCAATTTCCCATGTCTTTTGG‐3′) orientation; (b) the last 15 bp of the transposable element insertion and the first 15 bp of CsAAP2A after the transposable element insertion, in forward (5′‐GGCCTATTTTTGTGAATTCCCCAAAAGACAT‐3′) or reverse (5′‐ATGTCTTTTGGGGAATTCACAAAAATAGGCC‐3′) orientation; and (c) the 30 bp of CsAAP2A surrounding the transposable element insertion site, without transposable element sequence, in forward (5′‐ CCCATTCATGATGTCTTTTGGAGTTGTGGAA‐3′) or reverse (5′‐TTCCACAACTCCAAAAGACATCATGAATGGG‐3′) orientation.
The number of detected reads per accession for each of these six sequences was stored as a tabular file. The genotype of the accessions was determined to be homozygous transposable element‐allele, homozygous WT‐allele, or heterozygous.
Total detected SNPs in the 115 resequenced accessions were obtained from Qi et al. (2013) and manually filtered for the locus containing CsAAP2A. Effects of SNPs on the encoded CsAAP2A protein were predicted using SNPeff software (Cingolani et al., 2012). Data on DM susceptibility of the 115 cucumber accessions were downloaded from Liu et al. (2020).
4.5. Amino acid profiling of P. cubensis inoculated cucumber plants
For amino acid profiling, 16‐day‐old cucumber plants of genotypes NIL DM4.1.3 and HS279 were inoculated with P. cubensis as described above or a mock treatment consisting of spraying the leaves with tap water instead of spore suspension. Inoculated leaves (8 dpi) were sampled and directly frozen in liquid nitrogen. Frozen leaves were ground in liquid nitrogen and c.100–200 mg aliquots were taken for amino acid extraction. Total amino acids were extracted in 750 μl of 80% ethanol at 50 °C in an ultrasonic bath for 30 min. Samples were centrifuged, the supernatants were collected, and a second extraction was performed similarly as described above on the pellet, after which both supernatants per sample were pooled. Solvent was removed by freeze‐drying, after which extracts were resuspended in 100 μl of 80% ethanol.
Free amino acids in amino acid extracts were cleaned and derivatized using the EZ:faast GC/FID kit (Phenomenex). Derivatized samples were injected on an Agilent Technologies 7890A GC system with an FID detector using an Agilent Technologies 7683B series injector. Agilent Chemstation software was used to call peaks in the resulting chromatograms and to determine the peak area per amino acid.
4.6. Phylogenetic analysis
Protein sequences annotated as AAP genes in the A. thaliana genome were obtained from the Arabidopsis Information Resource (TAIR) on www.arabidopsis.org (accessed on 22 February 2019). The eight Arabidopsis AAP proteins (AtAAP1–AtAAP8) were used as queries for BLASTp searches against reference genomes of Medicago trunculata (Mt4.0 v. 1) (Tang et al., 2014), Oryza sativa (v. 7_JGI) (Ouyang et al., 2007), Physcomitrella patens (v. 3.3) (Jenkins et al., 2018), Selaginella moellendorffii (v. 1.0) (Banks et al., 2011), Solanum lycopersicum (ITAG2.40) (Tomato Genome Consortium, 2012), Cucumis sativus (Chinese Long 9930 v. 2) (Huang et al., 2009), Cucumis melo (cv. DHL92 v. 3.5.1) (Garcia‐Mas et al., 2012), Citrullus lanatus subsp. vulgaris (cv. 97103 v. 1.0) (Guo et al., 2013), and Cucurbita pepo subsp. pepo (Zucchini v. 4.1) (Montero‐Pau et al., 2018). Identified BLASTp hits (with e‐value <1E−10) were reciprocally used as queries against the A. thaliana (TAIR10) genome (Berardini et al., 2015) to select genes for which the best hit was an Arabidopsis AAP gene. The resulting list was manually curated to concatenate short predicted genes with subsequent gene identifiers (as they were most likely gene prediction errors), split a very long predicted gene, and remove remaining short (<300 amino acids) genes. Protein sequences were aligned to one another using CLC Genomics Workbench v. 11 using standard parameters. A maximum‐likelihood phylogeny was determined using CLC Genomics Workbench v. 11 using standard settings and 1,000 bootstrap replicates.
4.7. Expression analysis of AAP genes in cucumber using RT‐qPCR
For expression analysis, plants of genotypes HS279 and NIL DM4.1.3 were inoculated with P. cubensis as described above or mock treated. At 1, 3, 5, and 7 dpi, leaf samples were taken of three individual plants per genotype/treatment and immediately frozen in liquid nitrogen. RNA isolation, cDNA synthesis, and RT‐qPCR were performed as previously described (Berg et al., 2015). For quantification of AAP expression, we used the primer sequences as described in Data S18. Primer pairs specific for the cucumber reference gene TIP41, as described by Warzybok and Migocka (2013), were used for normalization of expression. Two technical replicates were taken for each sample/gene combination. C t values per sample were normalized by subtracting the geometric mean of the C t values for TIP41, giving ΔC t. ΔC t values were subsequently normalized by subtracting the average ΔC t value for each gene in mock‐inoculated HS279 plants at 1 dpi, giving ΔΔC t. Averages and standard deviations of ΔΔC t values were calculated over three biological replicates. Normality of ΔΔC t distributions was tested using Shapiro–Wilk tests (p > .05). Differences in ΔΔC t value between time points were analysed with two‐way analysis of variance (ANOVA) tests. Homogeneity of variances were tested using Levene's test. If ANOVA tests showed a significant (p < .05) treatment × time point interaction effect, one‐way ANOVA tests were carried out to test for significance of simple main effects of treatment per time point (p < .05). All statistical analyses were performed using SPSS v. 23 software (IBM). Relative transcript abundances were calculated as 2−ΔΔ C t for plotting.
4.8. Genome editing of tomato by CRISPR‐Cas9
Guide sequences of 20 bp aimed at knocking out SlAAP5A and/or SlAAP5B were designed using the web‐tool Cas‐Designer (Park et al., 2015). Two guide sequences were selected per gene. Forward primers containing the four desired 20 bp target sequences, preceded by BsaI restriction sites at the 5′ end, were ordered at Macrogen Europe B.V. (Data S18). Together with primer gRNA‐R (Data S18), single‐guide RNAs (sgRNAs) containing the desired 20 bp target sequence were amplified by PCR from plasmid pICH86966::AtU6p::sgRNA_PDS. pICH86966::AtU6p::sgRNA_PDS was a gift from Sophien Kamoun (Addgene plasmid # 46,966) and is described in Nekrasov et al. (2013).
Using a Golden Gate cloning strategy employing restriction enzyme BsaI described in Werner et al. (2012), the four sgRNAs were cloned into plasmids pICH47751, pICH47761, pICH47772, or pICH47781, respectively. In a subsequent Golden Gate cloning reaction employing restriction enzyme BpiI, the four sgRNA plasmids were combined with the Cas9 gene in plasmid pICH47742::35Sp::Cas9‐NOST, using pICH47732::Nosp::NPT2‐OCST as in planta selectable marker, and with linker plasmid pICH41822 into binary vector pAGM4723. The abovementioned plasmids belong to the MoClo Toolkit, which was a gift from Sylvestre Marillonnet (Addgene kit # 1,000,000,044) and are described in Werner et al. (2012).
pAGM4723::Cas9::sgRNA_SlAAP5A/B was transformed to electrocompetent cells of Agrobacterium tumefaciens AGL1‐virG by electroporation. Cotyledon explants of tomato cv. Moneymaker were transformed as previously described Berg et al. (2017). A total of 21 tomato transformants were obtained, which were screened for mutations in SlAAP5A by PCR with primers SlAAP5A‐MutScr‐F and SlAAP5A‐MutScr‐R followed by Sanger sequencing using both primers, and similarly for mutations in SlAAP5B with primers SlAAP5B‐MutScr‐F and SlAAP5B‐MutScr‐R (Data S18). Sanger sequencing reads were visually inspected to identify plants with putative (heterozygous and/or biallelic) mutations in either target gene.
Putative mutants were selfed in order to generate segregating T1 families. Individuals in T1 families were again screened for mutations in SlAAP5A and SlAAP5B as described above. Four T1 individuals with homozygous mutations in both target genes, derived from two primary transformants, were selected for selfing to develop fixated T2 families.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
F.W.K.H. and W.H.F. developed plant materials and (fine)mapped the DM 4.1.3 locus. H.A. performed the RT‐qPCR experiment. J.A.B. designed, performed, and analysed the rest of the experiments, with valuable suggestions by H.J.S. and Y.B. J.A.B. drafted the manuscript. W.H.V., R.G.F.V., Y.B., and H.J.S. gave critical feedback on the draft manuscript. All authors read and approved the manuscript.
Supporting information
DATA S1 Differential expression analysis in DM4‐1‐3 interval. RNA‐Seq transcript abundance values (in absolute numbers of uniquely mapped fragments, calculated using HTSeq; Anders et al., 2015) of 95 genes (when applicable with multiple predicted splice isoforms) in the DM4.1.3 locus in genotypes HS279 and NIL DM4.1.3 (three biological replicates per genotype) 3 days post inoculation with Pseudoperonospora cubensis. Gene IDs and annotations according to both reference genomes 9930 v2 (Li et al., 2011) and 9930 v3 (Li et al., 2019) are given. Average numbers of fragments per gene, as well as the average fold change between the two genotypes, are given. Significance of differential expression (BH‐adjusted p value) as determined with R package DEseq2 (Love et al., 2014) is given
DATA S2 Nonsynonymous SNPs in DM4‐1‐3 interval. Nonsynonymous single nucleotide polymorphisms (SNPs) between genotypes HS279 and DM4.1.3 in genes within the DM4.1.3 locus, as determined using the SnpEff (Cingolani et al., 2012) algorithm. Location of the SNP based on the reference genome (Chinese Long 9930 v. 2) is given, the allele in both genotypes, the effect (amino acid substitution) on the encoded protein as well as the Gene ID, and the annotation of the gene in which the SNP occurs
DATA S3 Genomic analysis of the AAP locus. Whole‐genome sequencing (WGS) data (PE reads, 100 bp) of cucumber genotypes NIL DM4.1 and HS279, as well as RNA‐Seq data (PE reads, 100 bp) of genotype HS279 were manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011), layout as described in Additional Data Hernummer 4. Exons of the CsAAP2A gene (Csa4M573860.1) are indicated above as black boxes. A red box indicates the part of the CsAAP2A alignment which is represented in Data S4 in more detail. The RNA‐Seq data of NIL DM4.1 are not shown because of the very low transcript abundance for the CsAAP2A gene
DATA S4 Genomic analysis of AAP locus in NIL DM4.1 reveals a large structural variation. Whole‐genome resequencing data (paired‐end reads, 100 bp) of cucumber genotype NIL DM4.1 were manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011). A scale bar indicates the genomic position on the reference genome (Chinese Long 9930 v. 2). Coverage per bp of the reference genome is indicated by the bar graph. Nucleotides in resequencing reads not identical to the reference genome are indicated with colours (green for A, red for T, blue for C, brown for G), indels are indicated by black horizontal stripes. Reads for which the other mate in the mate pair was mapped on a different chromosome or with deviating insert sizes are indicated by nongrey colours. (a) Local alignment of reads to genomic locus Chr4: 18,885,310–18,885,674 (corresponding to the fourth exon of AAP gene Csa4M573860.1 plus surrounding introns) indicated an 11 bp stretch of the reference genome with double coverage compared to the rest of the reference genome; reads with alignment problems before as well as after this 11 bp stretch, and flanking reads with mate pairs mapping to other genomic loci. (b) Local alignment of reads to genomic locus Chr4: 18,885,439–18,885,547 showing the findings in (a) in more detail
DATA S5 Alignment of CUMULE TIR domains. Nucleotide sequence alignment of the first 118 bp of the CUMULE transposon sequence identified in the DM4.1.3 allele of the CsAAP2A gene with the reverse complement of the last 118 bp of the transposon sequence, indicating a near‐perfect terminal inverted repeat
DATA S6 Alignment of RNA‐Seq data NIL DM4‐1‐3 to CUMULE transposon. Alignment of RNA‐Seq data (paired‐end reads, 100 bp) of cucumber genotype NIL DM4.1.3 to the newly identified CUMULE transposon sequence was manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011), graphical representation as described in Data S4. Reads for which the other mate in the mate pair could not be mapped on the transposon are highlighted with red outlines. Below the alignment, a schematic representation of the annotated CUMULE transposon (as in Figure 2b) is drawn to scale
DATA S7 Alignment of CUMULE tandem repeat region. Multiple nucleotide sequence alignment of the 10 satellites in the tandem repeat region identified in the CUMULE transposon in CsAAP2A. A bar graph indicates the percentage conservation between the 10 repeats per bp
DATA S8 Nucleotide sequences CsAAP2A. Wild‐type and mutant alleles of CsAAP2A on genomic and coding sequence level
DATA S9 BLAST output of CUMULE elements in reference genome. A BLASTn search was performed using the 118 bp (5′) TIR domain, the 2,205 bp predicted MudrA gene, and the 1,599 bp predicted Ulp1 gene from the CUMULE transposon as queries against the cucumber reference genome (Chinese Long 9930 v. 2). Tabular blast output (standard BLAST output format 6) is given
DATA S10 Natural CsAAP2A polymorphisms in 115 resequenced cucumber accessions. Single nucleotide polymorphisms (SNPs) detected by Qi et al. (2013) in a collection of 115 cucumber accessions were manually filtered to select polymorphisms in CsAAP2A (i.e. filtered on genomic position Chr4: 18,882,371–18,886,704) and annotated using the SnpEff algorithm (Cingolani et al., 2012). Data on DM susceptibility of these accessions was obtained from Liu et al. (2020)
DATA S11 Alignment of predicted AAP protein sequences with/without TE insertion. Multiple amino acid sequence alignment of the predicted sequence of the wild‐type CsAAP2A gene with the allele encoded by the TE‐allele of the CsAAP2A gene identified in the DM4.1.3 locus
DATA S12 Identification of EMS mutant alleles CsAAP2A. A TILLING library of cucumber ethyl methanesulfonate (EMS) mutants was screened for mutant alleles of CsAAP2A. Seven mutants were identified. Single nucleotide polymorphisms (SNPs) and their effects on the encoded AAP protein are indicated
DATA S13 Phenotype of EMS mutant csaap2a in Pseudoperonospora cubensis disease assay. Representative photographs of plants from the bioassay described in Figure 3, 7 days postinoculation with P. cubensis. Left: a plant homozygous for the ethyl methanesulfonate (EMS)‐induced mutation in CsAAP2A. Right: a plant homozygous for the wild‐type allele of CsAAP2A
DATA S14 csaap2a TILLING mutant is partially resistant to Pseudoperonospora cubensis. A cucumber TILLING population was screened for mutants in CsAAP2A (Data S12). A mutant with an early stop codon—W25Stop—was selected for phenotyping. The disease assay described in Figure 3 was also scored for sporulation at 7 days postinoculation. Bars represent average phenotype scores on a 1–9 scale, ranging from susceptible to resistant. Error bars represent standard deviation. Whereas plants with the ethyl methanesulfonate (EMS)‐induced mutation in CsAAP2A appeared to sporulate slightly less compared to other plants, this effect was not statistically significant (Kruskal–Wallis, p > .05)
DATA S15 Identified AAP homologs in 10 plant species. AAP genes identified in the genomes of Arabidopsis thaliana, Medicago trunculata, Oryza sativa, Physcomitrella, Selaginella moellendorffii, Solanum lycopersicum, Cucumis sativus, Cucumis, Citrullus lanatus subsp. Vulgaris, and Cucurbita pepo subsp. pepo. Gene name abbreviations as used in Data S16 are given, the gene accession codes of the respective genome annotations, the clades to which the genes belong according to the phylogenetic analysis described in Data S16, and the sizes of the predicted protein (in number of amino acids)
DATA S16 Phylogenetic analysis of AAP genes in 10 plant species. A multiple protein sequence alignment was made of putative AAP proteins identified in the genomes of 10 plant species. A maximum‐likelihood tree was constructed using CLC Genomics Workbench v. 11, represented here as a cladogram. Numbers at nodes represent bootstrap values in percentages (1,000 bootstrap replications). Nodes with bootstrap support values <50% were collapsed. Candidate gene CsAAP2A (Csa4M573860.1) is highlighted in green. Accession numbers of sequences in the phylogenetic tree are available in Data S11
DATA S17 Expression profile of cucumber AAP genes in Pseudoperonospora cubensis inoculated leaves of genotype NIL DM4.1.3. Relative expression level of cucumber AAP genes in leaves of partial downy mildew resistant cucumber genotype NIL DM4.1.3 in response to inoculation with P. cubensis or a mock treatment. Relative transcript abundances at 1, 3, 5, and 7 days postinoculation (dpi) with P. cubensis were determined using quantitative reverse transcription PCR (RT‐qPCR). Data were normalized relative to reference gene TIP41 and subsequently normalized relative to the average ∆C t value of mock‐treated HS279 plants (Figure 5b) at 1 dpi for each gene. Relative transcript abundances were calculated as 2−∆∆ C t. Each bar shows the relative expression of three biological replicates on a logarithmic scale. Note that the y axis for CsAAP2A is different compared to the other genes, as the gene was barely detectable by RT‐qPCR in this genotype. Error bars indicate standard deviation. Asterisks indicate significant differences between mock‐treated and inoculated plants (analysis of variance, p < .05)
DATA S18 List of used primers. A list of primers used in this manuscript. Names as used in this manuscript are given, plus the nucleotide sequences of the primers, in 5′ to 3′ orientation
DATA S19 cDNA sequence alignment of SlAAP5A and SlAAP5B alleles in CRISPR‐Cas9 mediated mutants. Multiple sequence alignment of cDNA from four tomato mutants compared to the reference allele
DATA S20 Protein sequence alignment of SlAAP5A and SlAAP5B alleles in CRISPR‐Cas9 mediated mutants. Multiple sequence alignment of predicted protein sequences from four tomato mutants compared to the reference allele
ACKNOWLEDGEMENTS
We gratefully acknowledge TKI Starting Materials for funding this research, project number EZ‐2012‐08. Furthermore, we are thankful to Gerard Bijsterbosch for assistance during sequencing of the CUMULE transposon as well as assistance in performing the P. infestans assay, and to Annemarie Dechesne for assistance and recommendations during gas chromatography experiments.
Berg JA, Hermans FWK, Beenders F, et al. The amino acid permease (AAP) genes CsAAP2A and SlAAP5A/B are required for oomycete susceptibility in cucumber and tomato. Mol Plant Pathol. 2021;22:658–672. 10.1111/mpp.13052
Funding information
This project was partially funded by TKI Starting Materials, the Netherlands, project number EZ‐2012‐08, and partially funded by Nunhems BV, the Netherlands
DATA AVAILABILITY STATEMENT
The RNA‐Seq data set analysed in this manuscript was submitted to the NCBI Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra under accession number PRJNA544259. All other data generated or analysed during this study are included in this published article and its additional data files.
REFERENCES
- Anders, S. , Pyl, P.T. & Huber, W. (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks, J.A. , Nishiyama, T. , Hasebe, M. , Bowman, J.L. , Gribskov, M. , dePamphilis, C. et al. (2011) The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science, 332, 960–963. 10.1126/science.1203810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, W. & Epps, W. (1954) An unreported type of resistance to cucumber downy mildew. Plant Disease Reporter, 38, 620. [Google Scholar]
- Berardini, T.Z. , Reiser, L. , Li, D. , Mezheritsky, Y. , Muller, R. , Strait, E. et al. (2015) The Arabidopsis information resource: Making and mining the “gold standard” annotated reference plant genome. Genesis, 53, 474–485. 10.1002/dvg.22877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J.A. , Appiano, M. , Bijsterbosch, G. , Visser, R.G.F. , Schouten, H.J. & Bai, Y. (2017) Functional characterization of cucumber (Cucumis sativus L.) Clade V MLO genes. BMC Plant Biology, 17, 80. 10.1186/s12870-017-1029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J.A. , Appiano, M. , Santillán Martínez, M. , Hermans, F.W.K. , Vriezen, W.H. , Visser, R.G.F. et al. (2015) A transposable element insertion in the susceptibility gene CsaMLO8 results in hypocotyl resistance to powdery mildew in cucumber. BMC Plant Biology, 15, 243. 10.1186/s12870-015-0635-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, J.A. , Hermans, F.W. , Beenders, F. , Lou, L. , Vriezen, W.H. , Visser, R.G. et al. (2020) Analysis of QTL DM4.1 for downy mildew resistance in cucumber reveals multiple subQTL: A novel RLK as candidate gene for the most important subQTL. Frontiers in Plant Science, 11, 569876. 10.3389/fpls.2020.569876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell, D. , Chan, E. , de Vries, J. , Joobeur, T. , King, J. , Reina, A. , et al. (2011). Methods and compositions for identifying downy mildew resistant cucumber plants. Patent EP‐2491147‐A1. Filed 22 October 2010. [Google Scholar]
- Call, A.D. , Criswell, A.D. , Wehner, T.C. , Klosinska, U. & Kozik, E.U. (2012) Screening cucumber for resistance to downy mildew caused by Pseudoperonospora cubensis (Berk. and Curt.) Rostov. Crop Science, 52, 577–592. 10.2135/cropsci2011.06.0296 [DOI] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L.L. , Coon, M. , Nguyen, T. , Wang, L. et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6, 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, Y. , Van den Langenberg, K.M. , Wehner, T.C. , Ojiambo, P.S. , Hausbeck, M. , Quesada‐Ocampo, L.M. et al. (2015) Resurgence of Pseudoperonospora cubensis: The causal agent of cucurbit downy mildew. Phytopathology, 105, 998–1012. 10.1094/PHYTO-11-14-0334-FI [DOI] [PubMed] [Google Scholar]
- Criswell, A.D. , Call, A.D. & Wehner, T.C. (2010) Genetic control of downy mildew resistance in cucumber—a review. Cucurbit Genetics Cooperative, 33–34, 13–16. [Google Scholar]
- Elashry, A. , Okumoto, S. , Siddique, S. , Koch, W. , Kreil, D.P. & Bohlmann, H. (2013) The AAP gene family for amino acid permeases contributes to development of the cyst nematode Heterodera schachtii in roots of Arabidopsis . Plant Physiology and Biochemistry, 70, 379–386. 10.1016/j.plaphy.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epps, W. & Barnes, W. (1952) The increased susceptibility of the palmetto cucumber to downy mildew in South Carolina. Plant Disease Reporter, 36, 14–15. [Google Scholar]
- Garcia‐Mas, J. , Benjak, A. , Sanseverino, W. , Bourgeois, M. , Mir, G. , Gonzalez, V.M. et al. (2012). The genome of melon (Cucumis melo L.).) Proceedings of the National Academy of Sciences of the United States of America, 109, 11872–11877. 10.1073/pnas.1205415109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, S. , Zhang, J. , Sun, H. , Salse, J. , Lucas, W.J. , Zhang, H. et al. (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nature Genetics, 45, 51–58. 10.1038/ng.2470 [DOI] [PubMed] [Google Scholar]
- Hammes, U.Z. , Nielsen, E. , Honaas, L.A. , Taylor, C.G. & Schachtman, D.P. (2006) AtCAT6, a sink‐tissue‐localized transporter for essential amino acids in Arabidopsis . The Plant Journal, 48, 414–426. 10.1111/j.1365-313X.2006.02880.x [DOI] [PubMed] [Google Scholar]
- Hammes, U.Z. , Schachtman, D.P. , Berg, R.H. , Nielsen, E. , Koch, W. , McIntyre, L.M. et al. (2005) Nematode‐induced changes of transporter gene expression in Arabidopsis roots. Molecular Plant‐Microbe Interactions, 18, 1247–1257. 10.1094/MPMI-18-1247 [DOI] [PubMed] [Google Scholar]
- Healey, A. , Furtado, A. , Cooper, T. & Henry, R.J. (2014) Protocol: A simple method for extracting next‐generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods, 10, 21.– 10.1186/1746-4811-10-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, G.J. , Ojiambo, P.S. , Hausbeck, M.K. , Quesada‐Ocampo, L. & Keinath, A.P. (2015) Resurgence of cucurbit downy mildew in the United States: A watershed event for research and extension. Plant Disease, 99, 428–441. 10.1094/PDIS-09-14-0990-FE [DOI] [PubMed] [Google Scholar]
- Huang, S. , Li, R. , Zhang, Z. , Li, L.I. , Gu, X. , Fan, W. et al. (2009) The genome of the cucumber, Cucumis sativus L. Nature Genetics, 41, 1275–1281. 10.1038/ng.475 [DOI] [PubMed] [Google Scholar]
- Jenkins, J.M. (1942) Downy mildew resistance in cucumbers. Journal of Heredity, 33, 35–38. 10.1093/oxfordjournals.jhered.a105122 [DOI] [PubMed] [Google Scholar]
- Jenkins, J. , Haas, F.B. , Schmutz, J. & Rensing, S.A. (2018) The Physcomitrella patens chromosome‐scale assembly reveals moss genome structure and evolution. The Plant Journal, 93, 515–533. 10.1111/tpj.13801 [DOI] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. & Salzberg, S.L. (2013) TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeda, A. & Cohen, Y. (2011) Cucurbit downy mildew (Pseudoperonospora cubensis)–biology, ecology, epidemiology, host–pathogen interaction and control. European Journal of Plant Pathology, 129, 157–192. 10.1007/s10658-010-9658-1 [DOI] [Google Scholar]
- van Leeuwen, H. , Monfort, A. & Puigdomenech, P. (2007) Mutator‐like elements identified in melon, Arabidopsis and rice contain ULP1 protease domains. Molecular Genetics and Genomics, 277, 357–364. 10.1007/s00438-006-0194-9 [DOI] [PubMed] [Google Scholar]
- Li, L. , He, H. , Zou, Z. , Li, Y. , Horticulture, C. & Northwest, A. (2018) QTL analysis for downy mildew resistance in cucumber inbred line PI 197088. Plant Disease, 102, 1240–1245. 10.1094/PDIS-04-17-0491-RE [DOI] [PubMed] [Google Scholar]
- Li, Q. , Li, H. , Huang, W.U. , Xu, Y. , Zhou, Q. , Wang, S. et al. (2019) A chromosome‐scale genome assembly of cucumber (Cucumis sativus L.). Gigascience, 8, giz072. 10.1093/gigascience/giz072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Zhang, Z. , Yan, P. , Huang, S. , Fei, Z. & Lin, K. (2011) RNA‐Seq improves annotation of protein‐coding genes in the cucumber genome. BMC Genomics, 12, 540. 10.1186/1471-2164-12-540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisch, D. & Slotkin, R.K. (2011) Strategies for silencing and escape: The ancient struggle between transposable elements and their hosts. International Review of Cell and Molecular Biology, 292, 119–152. 10.1016/B978-0-12-386033-0.00003-7 [DOI] [PubMed] [Google Scholar]
- Liu, G. , Ji, Y. , Bhuiyan, N.H. , Pilot, G. , Zou, J. , Wei, Y. et al. (2010) Amino acid homeostasis modulates salicylic acid‐associated redox status and defense responses in Arabidopsis . The Plant Cell, 22, 3845–3863. 10.1105/tpc.110.079392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Lu, H. , Liu, P. , Miao, H. , Bai, Y. , Gu, X. et al. (2020) Identification of novel loci and candidate genes for cucumber downy mildew resistance using GWAS. Plants, 9, 1659. 10.3390/plants9121659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. & Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella, H.H. , Nielsen, E. , Schachtman, D.P. & Taylor, C.G. (2013) The amino acid permeases AAP3 and AAP6 are involved in root‐knot nematode parasitism of Arabidopsis . Molecular Plant‐Microbe Interactions, 26, 44–54. 10.1094/MPMI-05-12-0123-FI [DOI] [PubMed] [Google Scholar]
- McCallum, C.M. , Comai, L. , Greene, E.A. & Henikoff, S. (2000) Targeted screening for induced mutations. Nature Biotechnology, 18, 455–457. 10.1038/74542 [DOI] [PubMed] [Google Scholar]
- Montero‐Pau, J. , Blanca, J. , Bombarely, A. , Ziarsolo, P. , Esteras, C. , Martí‐Gómez, C. et al. (2018) De novo assembly of the zucchini genome reveals a whole‐genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnology Journal, 16, 1161–1171. 10.1111/pbi.12860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov, V. , Staskawicz, B. , Weigel, D. , Jones, J.D.G. & Kamoun, S. (2013) Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA‐guided endonuclease. Nature Biotechnology, 31, 691–693. 10.1038/nbt.2655 [DOI] [PubMed] [Google Scholar]
- Oerke, E. , Steiner, U. , Dehne, H. & Lindenthal, M. (2006) Thermal imaging of cucumber leaves affected by downy mildew and environmental conditions. Journal of Experimental Botany, 57, 2121–2132. 10.1093/jxb/erj170 [DOI] [PubMed] [Google Scholar]
- Ouyang, S. , Zhu, W. , Hamilton, J. , Lin, H. , Campbell, M. , Childs, K. et al. (2007) The TIGR rice genome annotation resource: Improvements and new features. Nucleic Acids Research, 35, 8–11. 10.1093/nar/gkl976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. , Bae, S. & Kim, J. (2015) Cas‐designer: A web‐based tool for choice of CRISPR‐Cas9 target sites. Bioinformatics, 31, 4014–4016. 10.1093/bioinformatics/btv537 [DOI] [PubMed] [Google Scholar]
- Qi, J. , Liu, X. , Shen, D.I. , Miao, H. , Xie, B. , Li, X. et al. (2013) A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nature Genetics, 45, 1510–1515. 10.1038/ng.2801 [DOI] [PubMed] [Google Scholar]
- Rentsch, D. , Schmidt, S. & Tegeder, M. (2007) Transporters for uptake and allocation of organic nitrogen compounds in plants. FEBS Letters, 581, 2281–2289. 10.1016/j.febslet.2007.04.013 [DOI] [PubMed] [Google Scholar]
- Robinson, J.T. , Thorvaldsdóttir, H. , Winckler, W. , Guttman, M. , Lander, E.S. , Getz, G. et al. (2011) Integrative genomics viewer. Nature Biotechnology, 29, 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory, E.A. , Granke, L.L. , Quesada‐Ocampo, L.M. , Varbanova, M. , Hausbeck, M.K. & Day, B. (2011) The cucurbit downy mildew pathogen Pseudoperonospora cubensis . Molecular Plant Pathology, 12, 217–226. 10.1111/j.1364-3703.2010.00670.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Krishnakumar, V. , Bidwell, S. , Rosen, B. , Chan, A. , Zhou, S. et al. (2014) An improved genome release (version Mt4.0) for the model legume Medicago truncatula . BMC Genomics, 15, 312. 10.1186/1471-2164-15-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder, M. & Rentsch, D. (2010) Uptake and partitioning of amino acids and peptides. Molecular Plant, 3, 997–1011. 10.1093/mp/ssq047 [DOI] [PubMed] [Google Scholar]
- Tegeder, M. & Ward, J.M. (2012) Molecular evolution of plant AAP and LHT amino acid transporters. Frontiers in Plant Science, 3, 21. 10.3389/fpls.2012.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. (1996) Downy mildew. In: Zitter, T.A. (Ed.) Compendium of Cucurbit Diseases. St Paul, MN, USA: APS Press, pp. 25–27. [Google Scholar]
- Tomato Genome Consortium (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature, 485, 635–641. 10.1038/nature11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban, J. & Lebeda, A. (2006) Fungicide resistance in cucurbit downy mildew—Methodological, biological and population aspects. Annals of Applied Biology, 149, 63–75. 10.1111/j.1744-7348.2006.00070.x [DOI] [Google Scholar]
- van Vliet, G.J.A. & Meijsing, W.D. (1974) Inheritance of resistance to Pseudoperonospora cubensis Rost. in cucumber (Cucumis sativus L.). Euphytica, 23, 251–255. 10.1007/BF00035865 [DOI] [Google Scholar]
- van Vliet, G.J.A. & Meijsing, W.D. (1977) Relation in the inheritance of resistance to Pseudoperonospora cubensis Rost. and Sphaerotheca fuliginea Poll. in cucumber (Cucumis sativus L.). Euphytica, 26, 793–796. [Google Scholar]
- Wang, Y. , Tan, J. , Wu, Z. , VandenLangenberg, K. , Wehner, T.C. , Wen, C. et al. (2019) STAYGREEN, STAY HEALTHY: A loss‐of‐susceptibility mutation in the STAYGREEN gene provides durable, broad‐spectrum disease resistances for over 50 years of US cucumber production. New Phytologist, 221, 415–430. 10.1111/nph.15353 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , VandenLangenberg, K. , Wen, C. , Wehner, T.C. & Weng, Y. (2018) QTL mapping of downy and powdery mildew resistances in PI 197088 cucumber with genotyping‐by‐sequencing in RIL population. Theoretical and Applied Genetics, 131, 597–611. 10.1007/s00122-017-3022-1 [DOI] [PubMed] [Google Scholar]
- Warzybok, A. & Migocka, M. (2013) Reliable reference genes for normalization of gene expression in cucumber grown under different nitrogen nutrition. PLoS One, 8, e72887. 10.1371/journal.pone.0072887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, S. , Engler, C. , Weber, E. , Gruetzner, R. & Marillonnet, S. (2012) Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system and the MoClo system. Bioengineering, 3, 38–43. 10.4161/bbug.3.1.18223 [DOI] [PubMed] [Google Scholar]
- Yoshioka, Y. , Sakata, Y. , Sugiyama, M. & Fukino, N. (2014) Identification of quantitative trait loci for downy mildew resistance in cucumber (Cucumis sativus L.). Euphytica, 198, 265–276. 10.1007/s10681-014-1102-8 [DOI] [Google Scholar]
- Zhang, L. , Tan, Q. , Lee, R. , Trethewy, A. , Lee, Y. & Tegeder, M. (2010) Altered xylem‐phloem transfer of amino acids affects metabolism and leads to increased seed yield and oil content in Arabidopsis. The Plant Cell, 22, 3603–3620. 10.1105/tpc.110.073833 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1 Differential expression analysis in DM4‐1‐3 interval. RNA‐Seq transcript abundance values (in absolute numbers of uniquely mapped fragments, calculated using HTSeq; Anders et al., 2015) of 95 genes (when applicable with multiple predicted splice isoforms) in the DM4.1.3 locus in genotypes HS279 and NIL DM4.1.3 (three biological replicates per genotype) 3 days post inoculation with Pseudoperonospora cubensis. Gene IDs and annotations according to both reference genomes 9930 v2 (Li et al., 2011) and 9930 v3 (Li et al., 2019) are given. Average numbers of fragments per gene, as well as the average fold change between the two genotypes, are given. Significance of differential expression (BH‐adjusted p value) as determined with R package DEseq2 (Love et al., 2014) is given
DATA S2 Nonsynonymous SNPs in DM4‐1‐3 interval. Nonsynonymous single nucleotide polymorphisms (SNPs) between genotypes HS279 and DM4.1.3 in genes within the DM4.1.3 locus, as determined using the SnpEff (Cingolani et al., 2012) algorithm. Location of the SNP based on the reference genome (Chinese Long 9930 v. 2) is given, the allele in both genotypes, the effect (amino acid substitution) on the encoded protein as well as the Gene ID, and the annotation of the gene in which the SNP occurs
DATA S3 Genomic analysis of the AAP locus. Whole‐genome sequencing (WGS) data (PE reads, 100 bp) of cucumber genotypes NIL DM4.1 and HS279, as well as RNA‐Seq data (PE reads, 100 bp) of genotype HS279 were manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011), layout as described in Additional Data Hernummer 4. Exons of the CsAAP2A gene (Csa4M573860.1) are indicated above as black boxes. A red box indicates the part of the CsAAP2A alignment which is represented in Data S4 in more detail. The RNA‐Seq data of NIL DM4.1 are not shown because of the very low transcript abundance for the CsAAP2A gene
DATA S4 Genomic analysis of AAP locus in NIL DM4.1 reveals a large structural variation. Whole‐genome resequencing data (paired‐end reads, 100 bp) of cucumber genotype NIL DM4.1 were manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011). A scale bar indicates the genomic position on the reference genome (Chinese Long 9930 v. 2). Coverage per bp of the reference genome is indicated by the bar graph. Nucleotides in resequencing reads not identical to the reference genome are indicated with colours (green for A, red for T, blue for C, brown for G), indels are indicated by black horizontal stripes. Reads for which the other mate in the mate pair was mapped on a different chromosome or with deviating insert sizes are indicated by nongrey colours. (a) Local alignment of reads to genomic locus Chr4: 18,885,310–18,885,674 (corresponding to the fourth exon of AAP gene Csa4M573860.1 plus surrounding introns) indicated an 11 bp stretch of the reference genome with double coverage compared to the rest of the reference genome; reads with alignment problems before as well as after this 11 bp stretch, and flanking reads with mate pairs mapping to other genomic loci. (b) Local alignment of reads to genomic locus Chr4: 18,885,439–18,885,547 showing the findings in (a) in more detail
DATA S5 Alignment of CUMULE TIR domains. Nucleotide sequence alignment of the first 118 bp of the CUMULE transposon sequence identified in the DM4.1.3 allele of the CsAAP2A gene with the reverse complement of the last 118 bp of the transposon sequence, indicating a near‐perfect terminal inverted repeat
DATA S6 Alignment of RNA‐Seq data NIL DM4‐1‐3 to CUMULE transposon. Alignment of RNA‐Seq data (paired‐end reads, 100 bp) of cucumber genotype NIL DM4.1.3 to the newly identified CUMULE transposon sequence was manually inspected using Integrative Genomics Viewer software (Robinson et al., 2011), graphical representation as described in Data S4. Reads for which the other mate in the mate pair could not be mapped on the transposon are highlighted with red outlines. Below the alignment, a schematic representation of the annotated CUMULE transposon (as in Figure 2b) is drawn to scale
DATA S7 Alignment of CUMULE tandem repeat region. Multiple nucleotide sequence alignment of the 10 satellites in the tandem repeat region identified in the CUMULE transposon in CsAAP2A. A bar graph indicates the percentage conservation between the 10 repeats per bp
DATA S8 Nucleotide sequences CsAAP2A. Wild‐type and mutant alleles of CsAAP2A on genomic and coding sequence level
DATA S9 BLAST output of CUMULE elements in reference genome. A BLASTn search was performed using the 118 bp (5′) TIR domain, the 2,205 bp predicted MudrA gene, and the 1,599 bp predicted Ulp1 gene from the CUMULE transposon as queries against the cucumber reference genome (Chinese Long 9930 v. 2). Tabular blast output (standard BLAST output format 6) is given
DATA S10 Natural CsAAP2A polymorphisms in 115 resequenced cucumber accessions. Single nucleotide polymorphisms (SNPs) detected by Qi et al. (2013) in a collection of 115 cucumber accessions were manually filtered to select polymorphisms in CsAAP2A (i.e. filtered on genomic position Chr4: 18,882,371–18,886,704) and annotated using the SnpEff algorithm (Cingolani et al., 2012). Data on DM susceptibility of these accessions was obtained from Liu et al. (2020)
DATA S11 Alignment of predicted AAP protein sequences with/without TE insertion. Multiple amino acid sequence alignment of the predicted sequence of the wild‐type CsAAP2A gene with the allele encoded by the TE‐allele of the CsAAP2A gene identified in the DM4.1.3 locus
DATA S12 Identification of EMS mutant alleles CsAAP2A. A TILLING library of cucumber ethyl methanesulfonate (EMS) mutants was screened for mutant alleles of CsAAP2A. Seven mutants were identified. Single nucleotide polymorphisms (SNPs) and their effects on the encoded AAP protein are indicated
DATA S13 Phenotype of EMS mutant csaap2a in Pseudoperonospora cubensis disease assay. Representative photographs of plants from the bioassay described in Figure 3, 7 days postinoculation with P. cubensis. Left: a plant homozygous for the ethyl methanesulfonate (EMS)‐induced mutation in CsAAP2A. Right: a plant homozygous for the wild‐type allele of CsAAP2A
DATA S14 csaap2a TILLING mutant is partially resistant to Pseudoperonospora cubensis. A cucumber TILLING population was screened for mutants in CsAAP2A (Data S12). A mutant with an early stop codon—W25Stop—was selected for phenotyping. The disease assay described in Figure 3 was also scored for sporulation at 7 days postinoculation. Bars represent average phenotype scores on a 1–9 scale, ranging from susceptible to resistant. Error bars represent standard deviation. Whereas plants with the ethyl methanesulfonate (EMS)‐induced mutation in CsAAP2A appeared to sporulate slightly less compared to other plants, this effect was not statistically significant (Kruskal–Wallis, p > .05)
DATA S15 Identified AAP homologs in 10 plant species. AAP genes identified in the genomes of Arabidopsis thaliana, Medicago trunculata, Oryza sativa, Physcomitrella, Selaginella moellendorffii, Solanum lycopersicum, Cucumis sativus, Cucumis, Citrullus lanatus subsp. Vulgaris, and Cucurbita pepo subsp. pepo. Gene name abbreviations as used in Data S16 are given, the gene accession codes of the respective genome annotations, the clades to which the genes belong according to the phylogenetic analysis described in Data S16, and the sizes of the predicted protein (in number of amino acids)
DATA S16 Phylogenetic analysis of AAP genes in 10 plant species. A multiple protein sequence alignment was made of putative AAP proteins identified in the genomes of 10 plant species. A maximum‐likelihood tree was constructed using CLC Genomics Workbench v. 11, represented here as a cladogram. Numbers at nodes represent bootstrap values in percentages (1,000 bootstrap replications). Nodes with bootstrap support values <50% were collapsed. Candidate gene CsAAP2A (Csa4M573860.1) is highlighted in green. Accession numbers of sequences in the phylogenetic tree are available in Data S11
DATA S17 Expression profile of cucumber AAP genes in Pseudoperonospora cubensis inoculated leaves of genotype NIL DM4.1.3. Relative expression level of cucumber AAP genes in leaves of partial downy mildew resistant cucumber genotype NIL DM4.1.3 in response to inoculation with P. cubensis or a mock treatment. Relative transcript abundances at 1, 3, 5, and 7 days postinoculation (dpi) with P. cubensis were determined using quantitative reverse transcription PCR (RT‐qPCR). Data were normalized relative to reference gene TIP41 and subsequently normalized relative to the average ∆C t value of mock‐treated HS279 plants (Figure 5b) at 1 dpi for each gene. Relative transcript abundances were calculated as 2−∆∆ C t. Each bar shows the relative expression of three biological replicates on a logarithmic scale. Note that the y axis for CsAAP2A is different compared to the other genes, as the gene was barely detectable by RT‐qPCR in this genotype. Error bars indicate standard deviation. Asterisks indicate significant differences between mock‐treated and inoculated plants (analysis of variance, p < .05)
DATA S18 List of used primers. A list of primers used in this manuscript. Names as used in this manuscript are given, plus the nucleotide sequences of the primers, in 5′ to 3′ orientation
DATA S19 cDNA sequence alignment of SlAAP5A and SlAAP5B alleles in CRISPR‐Cas9 mediated mutants. Multiple sequence alignment of cDNA from four tomato mutants compared to the reference allele
DATA S20 Protein sequence alignment of SlAAP5A and SlAAP5B alleles in CRISPR‐Cas9 mediated mutants. Multiple sequence alignment of predicted protein sequences from four tomato mutants compared to the reference allele
Data Availability Statement
The RNA‐Seq data set analysed in this manuscript was submitted to the NCBI Sequence Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra under accession number PRJNA544259. All other data generated or analysed during this study are included in this published article and its additional data files.