

Abstract
Fus3/Kss1, also known as Pmk1 in several pathogenic fungi, is a component of the mitogen‐activated protein kinase (MAPK) signalling pathway that functions as a regulator in fungal development, stress response, mating, and pathogenicity. Cytospora chrysosperma, a notorious woody plant‐pathogenic fungus, causes canker disease in many species, and its Pmk1 homolog, CcPmk1, is required for fungal development and pathogenicity. However, the global regulation network of CcPmk1 is still unclear. In this study, we compared transcriptional analysis between a CcPmk1 deletion mutant and the wild type during the simulated infection process. A subset of transcription factor genes and putative effector genes were significantly down‐regulated in the CcPmk1 deletion mutant, which might be important for fungal pathogenicity. Additionally, many tandem genes were found to be regulated by CcPmk1. Eleven out of 68 core secondary metabolism biosynthesis genes and several gene clusters were significantly down‐regulated in the CcPmk1 deletion mutant. GO annotation of down‐regulated genes showed that the ribosome biosynthesis‐related processes were over‐represented in the CcPmk1 deletion mutant. Comparison of the CcPmk1‐regulated genes with the Pmk1‐regulated genes from Magnaporthe oryzae revealed only a few overlapping regulated genes in both CcPmk1 and Pmk1, while the enrichment GO terms in the ribosome biosynthesis‐related processes were also found. Subsequently, we calculated that in vitro feeding artificial small interference RNAs of CcPmk1 could silence the target gene, resulting in inhibited fungal growth. Furthermore, silencing of BcPmk1 in Botrytis cinerea with conserved CcPmk1 and BcPmk1 fragments could significantly compromise fungal virulence using the virus‐induced gene silencing system in Nicotiana benthamiana. These results suggest that CcPmk1 functions as a regulator of pathogenicity and can potentially be designed as a target for broad‐spectrum disease control, but unintended effects on nonpathogenic fungi need to be avoided.
Keywords: CcPmk1, Cytospora chrysosperma, gene silencing, pathogenicity, transcriptional analysis
The pathogenicity‐related mitogen‐activated protein kinase Pmk1, acting as a global regulator, can be used as a potential target for broad‐spectrum plant disease resistance.

1. INTRODUCTION
Cytospora chrysosperma, the causal agent of plant twig and stem canker diseases in more than 80 woody plant species all over the world, causes serious economic and ecological losses annually (Adams et al., 2005). In China, C. chrysosperma is a major and notorious pathogenic fungus on poplar, especially in northern China, where various poplar species are widely planted for landscape, industry, and shelter purposes (Adams et al., 2006; Wang et al., 2015). In addition, poor living conditions and weak tree vigour in the planted lands can help this disease spread, leading to a high incidence of poplar canker. It is worth mentioning that C. chrysosperma is regarded as a necrotrophic fungus similar to the Cryphonectria parasitica and Valsa mali, which tend to quickly kill their host cells (Adams et al., 2006; Biggs et al., 1983; Rigling & Prospero, 2018; Yin et al., 2015). The fungus overwinters with mycelia and conidia, which act as primary infection sources of the disease cycle and infect the host through microwounds (Biggs et al., 1983). Moreover, the disease has a long latent period. Once the tree vigour becomes weak, C. chrysosperma will quickly propagate, colonizing the host plants, which will exhibit canker symptoms, including the collapse of rotting stems, the formation of the ring spot, and even the death of the whole tree (Fan et al., 2020). To date, studies on C. chrysosperma and this canker disease have mainly focused on biological characteristics of pathogens, disease epidemiology, taxonomy, and disease control by pesticides, while the molecular mechanism underlying the pathogenicity of poplar canker disease remains enigmatic and scarcely reported (Fan et al., 2015; Madar et al., 2004). Clarification of the pathogenic mechanism is a prerequisite for developing useful and efficient pathogen control methods. Recently, Wang and Wang (2020) described the functions of three oxalic acid biosynthesis and catabolism genes that contribute to fungal pathogenicity in C. chrysosperma. Additionally, we characterized the functions of the mitogen‐activated protein kinase gene, CcPmk1, and found that CcPmk1 regulates fungal growth, conidiation, cell wall integrity, and pathogenicity in C. chrysosperma (Yu et al., 2019). Research on these conserved and important pathogenicity‐related components prompts us to better understand the molecular mechanism of C. chrysosperma pathogenicity.
Mitogen‐activated protein kinase (MAPK) signalling cascades are important for the transmission, integration, and amplification of signals and are key evolutionarily conserved components involved in diverse cellular processes in eukaryotes. In plant‐pathogenic fungi, MAPK pathways are crucial regulators in fungi and host plant interactions, which promote the infection process (Jiang et al., 2018). The MAPK cascades in plants have been shown to contribute to the activation of plant immunity (Hamel et al., 2012). The central module of each MAPK cascade generally consists of three interlinked protein kinases: a MAP kinase kinase kinase (MAPKKK), a MAP kinases kinase (MAPKK), and a MAP kinase (MAPK). The signal is relayed and amplified through the cascades via phosphorylation of the next protein by the upstream protein kinase (Turrà et al., 2014). The final kinase regulates downstream elements such as transcription factors and metabolic enzymes, leading to specific output responses (Manfiolli et al., 2019). MAPK signalling pathways have been well characterized in yeast and filamentous fungi, and their functions are mainly assigned to pheromone responses and filamentous growth, cell wall integrity, and high osmolarity stress (Hamel et al., 2012). Importantly, the Fus3/Kss1 MAPK signal pathway has been found to play a vital role in pathogenicity in several plant‐pathogenic fungi. In Saccharomyces cerevisiae, the Fus3/Kss1 pathway contains these elements: the central complex of MAPKKK Ste11, MAPKK Ste7, and MAPK Fus3/Kss1, which are assembled on the scaffold protein Ste5 (Bayram et al., 2012). Importantly, the Fus3/Kss1 orthologs are one of the well‐characterized MAP kinases in plant‐pathogenic fungi, which are essential for fungal pathogenicity and are denominated as “pathogenic MAPKs” (Pmk1) (Jiang et al., 2018).
Previous investigations on Fus3/Kss1 orthologs in several phytopathogenic fungi have shown their conserved and important roles in plant infection and various developmental processes. In appressorium‐forming pathogens such as Magnaporthe oryzae, Colletotrichum sp., and Ustilago maydis, Pmk1 homologs are essential for infection structure differentiation and invasive growth (He et al., 2017; Mayorga & Gold, 1999; Müller et al., 1999; Wei et al., 2016; Xu & Hamer, 1996). In nonclassical appressorium‐forming plant‐pathogenic fungi such as Botrytis cinerea, Verticillium dahliae, and V. mali, Pmk1 homologs are also indispensable to infection morphology and pathogenicity (Di Pietro et al., 2001; Jenczmionka et al., 2003; Rauyaree et al., 2005; Wu et al., 2017; Zheng et al., 2000). Nevertheless, distinct functions in fungal growth, morphology, and conidiation are found among Pmk1 homologs from different fungi. In Cochliobolus heterostrophus, the deletion of the Pmk1 homolog CHK1 causes significant defects in aerial hyphae development and conidiation (Lev et al., 1999), whereas the Pmk1 homolog Ubc3/Kpp2 is not required for fungal growth or morphology in U. maydis (Mayorga & Gold, 1999; Müller et al., 1999). Additionally, the MAP kinase Pmk1 homologs are also required for secondary metabolism in several fungal species (Bayram et al., 2012; He et al., 2017; Priegnitz et al., 2015; Zhang et al., 2011). Taken together, the MAP kinase Pmk1 acts as a conserved regulator in various biological processes in phytopathogenic fungi.
Recently, an increasing number of studies have focused on downstream components controlled by Pmk1, which have identified various elements regulated by the Fus3/Kss1 homologs. Among them, there are many reports on Ste12, a C2H2 transcription factor, which is involved in invasive growth and pathogenicity as well as Pmk1. However, it is not required for several other functions of Pmk1, such as the host adhesion and secretion of pectinolytic enzymes in Fusarium oxysporum, indicating that these Pmk1‐dependent functions are mediated by other downstream MAPK targets (Rispail & Di Pietro, 2009). Similar results were described for Mst12, a Ste12 homolog in M. oryzae, which is essential for virulence as Pmk1, but deletion of Mst12 can form typical normal‐appearance appressoria while Pmk1 deletion mutants cannot form appressoria. Interestingly, the normal‐appearance appressoria produced by the Mst12 deletion mutant cannot penetrate the host cells (Park et al., 2002). In addition, several other transcription factors may also be the downstream targets of Fus3/Kss1 or Pmk1, for example the bZIP transcription factor Atf1, which is required for cell integrity (Jin et al., 2013; Takada et al., 2007). Importantly, Pmk1 is essential for cell‐to‐cell invasion in the rice blast fungus and the secretion of a subset of effector genes that can suppress plant immunity and promote infection (Sakulkoo et al., 2018). Overall, Pmk1 can regulate the expression of a subset of genes belonging to different functionally categorized groups (Jin et al., 2013; Sakulkoo et al., 2018; Soanes et al., 2012).
RNA interference (RNAi) is a conserved regulatory mechanism that is involved in many kinds of biological processes by affecting gene expression in eukaryotic organisms (Baulcombe, 2005). RNAi has been developed as a powerful technique to analyse gene functions because of its significant advantages, such as short operation period, simple process, and relatively low cost. Tobacco rattle virus (TRV)‐based virus‐induced gene silencing (VIGS) is widely used in various plant species and can induce posttranscriptional gene silencing (PTGS) of target plant genes (Senthil‐Kumar & Mysore, 2011). As bidirectional cross‐kingdom trafficking of small RNA is commonly reported during plant–pathogen interactions (Cai et al., 2018; Hou et al., 2019; Hua et al., 2018; Wang et al., 2016), TRV‐based VIGS can also be used to target pathogen genes. Therefore, RNAi can potentially be developed as an efficient genetic strategy against pathogens for pesticide‐free disease control in plants by targeting crucial pathogenicity‐inducing genes of phytopathogens, which is known as host‐induced gene silencing (HIGS) (Nowara et al., 2010; Song & Thomma, 2018). Recently, attempts at HIGS in plant diseases control have emerged across many kinds of phytopathogens, such as Puccinia spp., V. dahliae, M. oryzae, Blumeria graminis, Phytophthora infestans, and Fusarium graminearum (Guo et al., 2019; Panwar et al., 2018; Qi et al., ,2018, 2019; Sanju et al., 2015; Song & Thomma, 2018; Xu et al., 2018; Zhang et al., 2016). Remarkably, the Pmk1 homology in P. triticina has been used as a target for leaf rust disease control; transgenic wheat harbouring a Pmk1‐RNAi construct shows significant resistance to the disease (Panwar et al., 2018).
Although the basic functions of CcPmk1 have been characterized in C. chrysosperma, its regulation network is still unclear. In this study, we performed a transcriptional analysis between wild type and CcPmk1 deletion mutants (ΔCcPmk1) and revealed several conserved biological processes that were regulated by CcPmk1, such as the RNA process, ribonucleoprotein biosynthesis, and secondary metabolism. Furthermore, we silenced CcPmk1 in vitro by adding artificial small interfering RNAs (asiRNAs) and caused a similar defect in fungal growth as the CcPmk1 deletion mutant. The expression of B. cinerea BcPmk1 can be silenced with CcPmk1 and BcPmk1 homologous fragments using the TRV‐based VIGS system in Nicotiana benthamiana, which would compromise the virulence of B. cinerea. The results indicate that Pmk1 homologs can be used as potential targets for disease control.
2. RESULTS
2.1. Pmk1 homologs show high sequence identity in fungi
We previously found that CcPmk1 is involved in fungal growth and pathogenicity in C. chrysosperma (Yu et al., 2019). Interestingly, the conserved functions in pathogenicity have been described in Pmk1 homologs from other plant‐pathogenic fungi (Table 1). In addition, we analysed the sequences of Pmk1 homologs in pathogenic fungi and nonpathogenic fungi, including saprophytes, endophytes, and symbionts, and revealed high similarity among them, especially the protein kinase domain (PF00069) (Figures 1 and S1, Table S1). However, sequence identities of Pmk1 homologs from phytopathogens and nonpathogens belonging to Ascomycota (over 90%) were higher than those from plant‐pathogenic oomycetes and basidiomycetes. Importantly, a conserved “TEY” phosphorylation site was found in almost all selected Pmk1 homologs from phytopathogens as well as in nonpathogenic fungi. These results indicate a conserved function of Pmk1 homologs from different eukaryotic microbes.
TABLE 1.
The reported functions of Fus3/Kss1/Pmk1 in several plant pathogens
| Gene name | Sequence identity to CcPmk1 (sequence identity of domain regions) a | Plant pathogen | Functions | References |
|---|---|---|---|---|
| CcPmk1 | 100% (100%) | Cytospora chrysosperma | Pathogenicity, growth, cell wall integrity | Yu et al. (2019) |
| VmPmk1 | 100% (100%) | Valsa mali | Pathogenicity, growth, cell wall integrity | Wu et al. (2017) |
| Pmk1 | 98.5% (100%) | Magnaporthe oryzae | Pathogenicity, appressorium formation, mating | Xu and Hamer (1996) |
| VMK1 | 98% (99.3%) | Verticillium dahliae | Pathogenicity, microsclerotia formation, conidiation | Rauyaree et al. (2005) |
| PsMAPK1 | 63.9% (77.8%) | Puccinia striiformis f. sp. tritici | Pathogenicity | Guo et al. (2011) |
| BMP1 | 94.3% (97.9%) | Botrytis cinerea | Pathogenicity, growth | Zheng et al. (2000) |
| Ubc3/Kpp2 | 72.8% (78.6%) | Ustilago maydis | Pathogenicity, mating | Müller et al. (1999), Mayorga and Gold (1999) |
| FMK1 | 98.3% (99.3%) | Fusarium oxysporum | Pathogenicity, growth surface hydrophobicity | Di Pietro et al. (2001) |
| Gpmk1 | 98.5% (99.3%) | Fusarium graminearum | Pathogenicity, conidiation, mating | Jenczmionka et al. (2003) |
| FvMK1 | 98.3% (99.3%) | Fusarium verticillioides | Pathogenicity, conidiation, toxin production | Zhang et al. (2011) |
| ChMK1 | 98.8% (99.6%) | Colletotrichum higginsianum | Pathogenicity, appressorium formation, cell wall integrity | Wei et al. (2016) |
| CgMK1 | 98.8% (99.6%) | Colletotrichum gloeosporioides | Pathogenicity, appressorium formation, melanin biosynthesis | He et al. (2017) |
| CpMK2 | 96% (97%) | Cryphonectria parasitica | Pathogenicity, pigmentation, development | Choi et al. (2005) |
| Smk1 | 95% (97%) | Sclerotinia sclerotiorum | Pathogenicity, asexual development | Chen et al. (2004) |
Numbers in brackets represent the sequence identity of MAPK domains from different plant‐pathogenic fungi compared to the MAPK domain of CcPmk1.
FIGURE 1.

Multiple sequence alignment of Fus3/Kss1/Pmk1 from different species. The protein kinase domain (PF00069) is shadowed in blue. The red box represents the conserved phosphorylation site. The detailed information of the Fus3/Kss1/Pmk1 orthologs are listed in Tables 1 and S1
2.2. Overview of transcriptome analysis between the wild type and the CcPmk1 deletion mutant
To identify the putative genes regulated by CcPmk1 in C. chrysosperma, we conducted transcriptome analyses of the wild type and CcPmk1 deletion mutants during the mimetic infection process. Three biological replicates were established for each treatment, and six data sets were generated. More than 300 million 150 bp paired‐end reads were generated, and the majority of the reads were mapped to the draft genome of C. chrysosperma (NCBI accession number JAEQMF000000000). Raw reads were submitted to the NCBI SRA database with accession numbers SRR12262932, SRR12262933, SRR12262934 (wild‐type‐1–3), and SRR12262935, SRR12262936, SRR12262937 (ΔCcPmk1‐1–3). Principal component analysis (PCA) of read counts of the wild type and CcPmk1 deletion mutants revealed a clear separation of the two tested conditions and the proximity of biological replicates (Figure S2).
Gene expression analysis showed a total of 8,923/10,454 (85.4%) and 8,832/10,454 (84.5%) predicted genes detected in the wild type and CcPmk1 deletion mutants, respectively (Figure 2a), and comparable expression patterns were observed between the wild type and CcPmk1 deletion mutants (Figure S3). A total of 8,746 predicted genes were detected in both the wild type and CcPmk1 deletion mutants, and 177 and 86 predicted genes were specifically expressed in the wild type and CcPmk1 deletion mutants (Figure 2a). Differential expression analysis revealed that 523 predicted genes were significantly down‐regulated and 254 predicted genes were significantly up‐regulated in the CcPmk1 deletion mutant compared with the wild type (Figure 2b).
FIGURE 2.

Gene expression data between the wild type (WT) and CcPmk1 deletion mutant. (a) The global view of expressed genes in WT and CcPmk1 deletion mutant. (b) Differentially expressed genes in the CcPmk1 deletion mutant compared with WT with adjusted p < .05
Gene ontology (GO) annotation showed that the significantly down‐regulated genes were involved in many biological processes, such as ribosome biogenesis (GO: 0042254), carbohydrate metabolic process (GO: 0005975), RNA processing (GO: 0006396), transmembrane transport (GO: 0055085), and protein phosphorylation (GO: 0006468). The cellular component analysis revealed that they were mainly related to the cellular membrane (GO: 0016021, GO: 0043231) and the nucleus (GO: 0005634). The molecular functions of the significantly down‐regulated genes were mainly involved in cofactor binding (GO: 0048037), hydrolase activity (GO: 0004553), oxidoreductase activity (GO: 0016705), and ATP binding (GO: 0005524). Among them, four biological processes were significantly enriched, including ribosome biogenesis (GO: 0042254), ribonucleoprotein complex biogenesis (GO: 0022613), rRNA processing (GO: 0006364), and rRNA metabolic processing (GO: 0016072). Ten molecular functions were significantly enriched, such as flavin adenine dinucleotide binding (GO: 0050660), cofactor binding (GO: 0048037), oxidoreductase activity (GO: 0016705), and O‐methyltransferase activity (GO: 0008171) (Figure 3). Similarly, the significantly up‐regulated genes were enriched in carbohydrate metabolic process (GO: 0005975), acetyltransferase activity (GO: 0016407), transferase activity (GO: 0016747), hydrolase activity (GO: 0016798), and carbohydrate binding (GO: 0030246). Significantly enriched KEGG pathway of ribosome biogenesis in eukaryotes (KEGGID: bfu03008) was found in the differentially down‐regulated genes, while no significant enrichment KEGG pathway was detected in the differentially up‐regulated genes.
FIGURE 3.
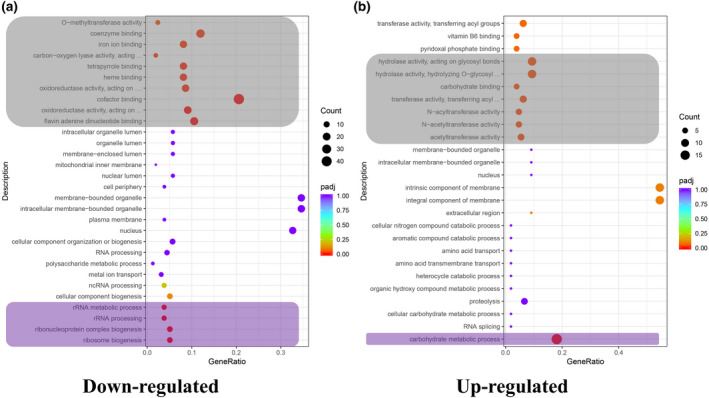
Gene ontology (GO) annotation of differentially expressed genes. (a) GO annotation of significantly down‐regulated genes in the CcPmk1 deletion mutant compared with wild type. (b) GO annotation of significantly up‐regulated genes in CcPmk1 deletion mutant compared with wild type. The grey and purple boxes represent the significantly enriched GO terms
Differentially expressed genes were visualized in a heat map, created using log2(FPKM + 1) and normalized by Z‐score treatment. Genes showing similar patterns of expression were grouped by hierarchical clustering (Figure 4a). Three wild‐type and CcPmk1 deletion mutant repeats were clustered together, indicating the high quality of sequencing samples. Fuzzy clustering analysis of differentially expressed genes generated three clusters. The major differentially expressed genes belonged to cluster 2 (511 genes) and cluster 3 (254 genes) with mild up‐ or down‐regulation, while cluster 1 contained 11 genes that showed dramatically reduced expression levels in the CcPmk1 deletion mutant compared with the wild type (Figure 4b). Interestingly, annotation of these 11 genes revealed that all of them contained known Pfam domains belonging to different gene families, including FAD binding, adh_short, p450, GFO_IDH_MocA, 2OG‐FeII_Oxy, and MFS_1 (Table 2).
FIGURE 4.

Gene expression pattern of differentially expressed genes. (a) The heatmap shows the expression level in the CcPmk1 deletion mutant and the wild type (WT). Levels of expression are represented as normalized by Z‐score treatment. Genes showing similar patterns of gene expression are clustered. (b) Gene expression analysis according to the expression level. The number of genes in each cluster is bracketed
TABLE 2.
List of differentially expressed genes in cluster 1
| Gene ID | Gene length (bp) | Gene description | Pfam_family |
|---|---|---|---|
| GME3437_g | 2,202 | PF01565: FAD binding domain; PF11578: protein of unknown function (DUF3237) | FAD_binding_4 |
| GME5853_g | 747 | PF00106: short chain dehydrogenase | adh_short |
| GME3442_g | 1,599 | PF00067: cytochrome P450 | p450 |
| GME3443_g | 1,362 | PF00891: O‐methyltransferase | NA |
| GME3441_g | 1,686 | PF01565: FAD binding domain; PF08031: berberine and berberine‐like | FAD_binding_4 |
| GME3438_g | 1,227 | PF13450: NAD(P)‐binding Rossmann‐like domain; PF01494: FAD binding domain | FAD_binding_3 |
| GME3434_g | 1,230 | PF01408: oxidoreductase family, NAD‐binding Rossmann fold | GFO_IDH_MocA |
| GME3439_g | 1,116 | PF03171: 2OG‐Fe(II) oxygenase superfamily; PF14226: nonhaem dioxygenase in morphine synthesis N‐terminal | 2OG‐FeII_Oxy |
| GME3446_g | 1,773 | PF08031: berberine and berberine‐like; PF01565: FAD binding domain | FAD_binding_4 |
| GME3445_g | 1,680 | PF07690: major facilitator superfamily | MFS_1 |
| GME3436_g | 882 | PF00106: short chain dehydrogenase | adh_short |
Cluster 1 represents genes that are dramatically down‐regulated in the CcPmk1 deletion mutant compared with that in the wild type, which are clustered together according to their expression patterns as shown in Figure 4b.
2.3. CcPmk1 regulated the expression of secondary metabolism gene clusters
According to the 11 genes listed in Table 2, we found that 10 were in tandem arrangement (GME3434_g to GME3446_g, lacking GME3435_g, GME3440_g, and GME3444_g) (Table 2). Further investigation revealed that GME3435_g, GME3440_g, and GME3444_g, belonging to cluster 2, were also significantly down‐regulated in the CcPmk1 deletion mutant, which meant that the whole gene cluster from GME3434_g to GME3446_g was significantly down‐regulated in the CcPmk1 deletion mutant. Additionally, we found that many other tandem genes were also significantly down‐regulated in the CcPmk1 deletion mutant, such as GME9191_g to GME9200_g, GME3317_g to GME3326_g, GME8084_g to GME8090_g (data not shown). Generally, genes involved in the synthesis of secondary metabolites are tandem arrangements. To clarify whether the tandem genes mentioned above were involved in secondary metabolism, we systematically identified the secondary metabolite gene clusters in the C. chrysosperma genome using antiSMASH. A total of 68 secondary metabolite core genes were identified, including 15 PKS types, 18 NRPS and NRPS‐like types, 15 terpene types, four hybrid types, and two other types (Table 3). Among them, 11 secondary metabolite core biosynthesis genes were significantly down‐regulated in the CcPmk1 deletion mutant, while only one secondary metabolite core biosynthesis gene was significantly up‐regulated in the CcPmk1 deletion mutant. Several predicted gene clusters showed consistent gene expression patterns. For example, genes in the predicted terpene type gene cluster from GME3317_g to GME3324_g were all significantly down‐regulated in the CcPmk1 deletion mutant, and the adjacent genes around the clusters were also significantly down‐regulated in the CcPmk1 deletion mutant (Figure 5a). Similar results were obtained in the hybrid‐NRPS and T1PKS type gene clusters from GME3437_g to GME3444_g as mentioned above, and the genes from GME3434_g to GME3448_g were all significantly down‐regulated in the CcPmk1 deletion mutant (Figure 5b). Remarkably, this hybrid‐NRPS and T1PKS type gene cluster shared 38% sequence identity with the curvupallide‐B biosynthetic gene cluster from Cochliobolus pallescens. However, not all genes in the secondary metabolite gene clusters shared similar gene expression patterns. Several secondary metabolite core biosynthesis genes were significantly down‐regulated in the CcPmk1 deletion mutant, but the adjacent genes showed comparable gene expression between the wild type and the CcPmk1 deletion mutants. Alternatively, the secondary metabolite core biosynthesis genes were not differentially expressed in the CcPmk1 deletion mutant, but the adjacent genes were significantly down‐regulated in the CcPmk1 deletion mutant (Table S2).
TABLE 3.
The core genes involved in the biosynthesis of secondary metabolites in Cytospora chrysosperma and differentially expressed genes in CcPmk1 deletion mutant compared to the wild type
| PKS | NRPS and NRPS‐like | Hybrid | Terpene | Others | Total | |
|---|---|---|---|---|---|---|
| Classification | 29 | 18 | 4 | 15 | 2 | 68 |
| Down‐regulated in CcPmk1 deletion mutant | 4 | 2 | 1 | 4 | 0 | 11 |
| Up‐regulated in CcPmk1 deletion mutant | 1 | 0 | 0 | 0 | 0 | 1 |
Genes with an adjusted p value < .05 found by DESeq2 were assigned as differentially expressed.
FIGURE 5.

Two secondary metabolism gene clusters were regulated by CcPmk1. (a) The predicted secondary metabolism gene cluster was significantly down‐regulated in CcPmk1 from GME_3317 to GME_3324. (b) The predicted secondary metabolism gene cluster was significantly down‐regulated in CcPmk1 from GME_3434 to GME_3444. The blue curves indicate read coverage for the samples from the RNA‐Seq. The green box represents the predicted gene clusters and the red box represents the adjacent genes were also regulated by CcPmk1
2.4. CcPmk1 regulated the expression of transcription factor genes and putative effector genes
Transcription factors (TFs) act as the downstream targets of mitogen‐activated protein kinases, which are important for signal transduction and carrying out various functions, such as fungal growth and pathogenicity. Here, we found 34 significantly down‐regulated TF genes and three significantly up‐regulated TF genes in the CcPmk1 deletion mutant, including 15 Zn2Cys6 TFs (13 down‐regulated and two up‐regulated), seven winged helix repressor DNA‐binding TFs, six C2H2 zinc finger TFs, four bZIP TFs (three down‐regulated and one up‐regulated), three homeobox/homeodomain‐like TFs, one MADS‐box TF, and one p53‐like TF (Figure 6a). Subsequently, we searched through the PHI‐database with the default parameters (http://www.phi‐base.org/) and found that 20 out of the 34 down‐regulated TF homologs and one out of the three up‐regulated TF homologs have been functionally characterized in fungal species (Table S3). Over half of these down‐regulated TF homologs are required for pathogenicity, especially the winged helix repressor DNA‐binding TF homologs, homeobox/homeodomain‐like TF homologs, and the MADS‐box TF homolog. Deletion of these TF homologs resulted in reduced virulence or loss of pathogenicity (Table S3), indicating a virulence‐related function of CcPmk1 in C. chrysosperma.
FIGURE 6.

Putative downstream genes regulated by CcPmk1. (a) Categories of differentially expressed transcription factors in the CcPmk1 deletion mutant. (b) Putative secreted proteins and effector genes regulated by CcPmk1
Traditional effectors are usually small in size, cysteine‐rich, no GPI anchor site, no additional transmembrane, or containing a signal peptide, which can disturb the plant immunity and promote pathogenic processes (Lo Presti et al., 2015). Previous studies have shown that Pmk1 can regulate the expression of effectors (Sakulkoo et al., 2018). Here, we systematically analysed the secreted proteins regulated by CcPmk1 and found that 87 differentially down‐regulated genes and 46 differentially up‐regulated genes contained a signal peptide (Figure 6b). Further analysis revealed that 12 down‐regulated genes and 7 up‐regulated genes met the basic criteria of traditional effectors (Table S3). Among the down‐regulated putative effectors, a cysteine‐rich secretory protein family gene, GME7477_g, is important for C. chrysosperma pathogenicity and can disturb the plant immunity (Han et al., 2021), while a necrosis‐inducing protein (NPP1) was found in the up‐regulated putative effectors (Table S3).
2.5. Comparative transcriptome analysis between CcPmk1 and MoPmk1 regulated genes
The functions of Pmk1 orthologs have been described in more than 20 pathogenic fungi. However, genome‐wide transcriptional profiling of Pmk1 orthologs is rare. Pmk1 from M. oryzae (named MoPmk1 in this study) was regarded as a global regulator of fungal pathogenicity. RNA sequencing has revealed 715 significantly up‐regulated genes and 742 significantly down‐regulated genes in the MoPmk1 inhibition mutant compared with the wild type, including a subset of effector genes that can suppress plant immunity suppression (Sakulkoo et al., 2018).
To identify the putative genes that were regulated by both MoPmk1 and CcPmk1, differentially expressed genes in the CcPmk1 deletion mutant and MoPmk1 inhibition mutant were analysed. First, we identified the homolog genes of 523 differentially down‐regulated genes and 254 differentially up‐regulated genes in M. oryzae. A total of 379 homolog genes (corresponding 346 M. oryzae homolog genes) and 204 homolog genes (corresponding 197 M. oryzae homolog genes) of 523 differentially down‐regulated genes and 254 differentially up‐regulated genes were found in M. oryzae, respectively (Table 4). However, 346 M. oryzae homologs of 523 differentially down‐regulated genes in the CcPmk1 deletion mutant shared only 45 genes with the 742 significantly down‐regulated genes in the MoPmk1 inhibition mutant. In contrast, 197 M. oryzae homologs of 254 differentially up‐regulated genes in CcPmk1 deletion mutant shared only 28 genes with the 715 significantly up‐regulated genes in the MoPmk1 inhibition mutant (Table 4).
TABLE 4.
Comparative transcriptomics analysis between CcPmk1 and MoPmk1 regulated genes
| CcPmk1 vs Cytospora chrysosperma wild type | Number of homolog genes found in Magnaporthe oryzae a | MoPmk1 vs M. oryzae wild type | Overlapping regulated genes between CcPmk1 and MoPmk1 | |
|---|---|---|---|---|
| Down‐regulated | 523 | 379 (346) | 742 | 45 |
| Up‐regulated | 254 | 204 (197) | 715 | 28 |
| Total | 777 | 583 (543) | 1,457 | 73 |
The number in parentheses represents the unique genes.
GO annotation of the overlapping regulated genes between CcPmk1 and MoPmk1 revealed that RNA processing and ribosome biogenesis were enriched, similar to the GO annotation of the 523 differentially down‐regulated genes in the CcPmk1 deletion mutant, indicating the conserved regulation mechanism in RNA processing or ribosome biogenesis‐related pathways between CcPmk1 and MoPmk1 (Figure S4). In the overlapping 45 down‐regulated genes, almost half of them were annotated as rRNA processing related proteins, such as ribosome assembly protein rrb1 (MGG_14350), rRNA biogenesis protein rrp5 (MGG_05260), rRNA methyltransferase nop1 (MGG_0919), and small nucleolar ribonucleoprotein snu13 (MGG_02467). Two translation initiation factors (MGG_06261 and MGG_04026) were significantly down‐regulated in the Pmk1 mutant. The results suggest that CcPmk1 and MoPmk1 may be essential for the translation process. In the overlapping up‐regulated genes, an elicitor protein (MGG_08454) and its homolog (GME2273_g) were found in the MoPmk1 inhibition mutant and CcPmk1 deletion mutant, respectively.
2.6. The asiRNAs targeting CcPmk1 effectively suppressed the fungal growth
According to previous studies and the results mentioned above, we concluded that Pmk1 is a conserved regulator of pathogenicity. Therefore, we regarded it as a target for disease control by gene silencing. We designed and added the asiRNAs of CcPmk1 in an axenic culture (Figure 7a), and the results showed that in vitro treatment with CcPmk1 asiRNAs significantly reduced the expression level of CcPmk1 (over 60% reduction) (Figure 7b). Fluorescence microscopy revealed that asiRNAs binding with FAM could be observed in the intracellular hyphae, indicating that the reduced expression of CcPmk1 resulted from the treatment with CcPmk1 asiRNAs (Figure S5). CcPmk1‐silenced mutants showed obvious defects in fungal growth compared with the control sample (Figure 7c,d), which were similar to the phenotype of the CcPmk1 deletion mutants as described previously (Yu et al., 2019).
FIGURE 7.
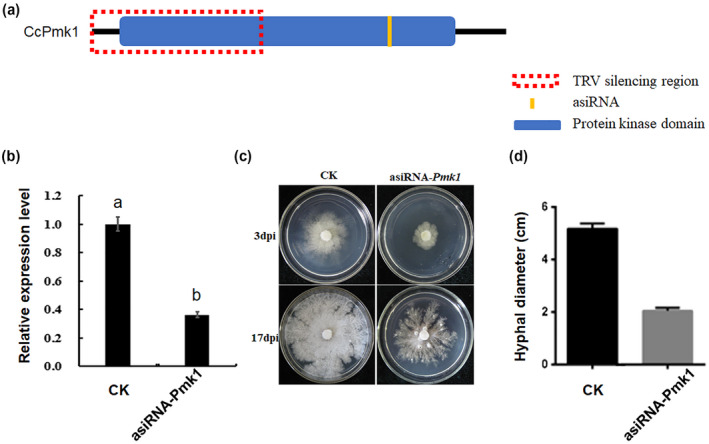
Gene silencing of CcPmk1 by in vitro artificial small interfering RNA. (a) CcPmk1 gene structure and location of designed interfering RNA. (b) Relative expression level of CcPmk1 after in vitro addition of artificial small interfering (asi) RNA. The relative expression levels of CcPmk1 were calculated using the 2−ΔΔ C t method with the actin gene (CcActin) as an internal reference. The different letters represent significant differences at p = .05. (c) The growth of CcPmk1‐silenced mutant on potato dextrose agar (PDA) plates. (d) Calculation of the hyphal diameter of CcPmk1‐silenced mutant and control on PDA plates 3 days after inoculation. CK represents the control treatments without the artificial small interfering RNA. All the experiments were repeated three times with similar results
2.7. TRV‐based gene silencing of BcPmk1 compromised the virulence of B. cinerea
To further determine whether Pmk1 could be targeted for pathogen control, the TRV‐based VIGS system on N. benthamiana was used. Here, we used B. cinerea to calculate the results because C. chrysosperma cannot infect N. benthamiana. First, we chose and amplified a conserved fragment of CcPmk1 and BcPmk1 (approximately 500 bp, shown in Figure S6), a fragment of the phytoene desaturase (PDS) gene (positive control), and a fragment of green fluorescent protein (GFP) (negative control), and successfully fused them into pTRV2, creating TRV2‐CcPmk1, TRV2‐BcPmk1, TRV2‐PDS, and TRV2‐GFP (Figure 8a). Then a 1:1 mixture of Agrobacterium tumefaciens cultures carrying TRV1 and TRV2‐CcPmk1, TRV2‐BcPmk1, TRV2‐GFP, and TRV2‐PDS were infiltrated into leaves of N. benthamiana. Ten days after infiltration, the N. benthamiana leaves were challenged with B. cinerea and inspected for symptoms. The N. benthamiana leaves infiltrated with A. tumefaciens cultures carrying TRV1 and TRV2‐PDS displayed an obvious photobleaching phenotype, indicating the normal state of the TRV‐based VIGS system (Figure 8b). Subsequently, we calculated the lesion areas of the leaves caused by B. cinerea, and the results showed that the control leaves and TRV2‐GFP‐treated leaves showed comparable lesion areas and larger lesion areas than the leaves treated with TRV2‐CcPmk1 and TRV2‐BcPmk1 (Figure 8c,d). Remarkably, the leaves treated with TRV2‐CcPmk1 and TRV2‐BcPmk1 showed similar lesion areas after infection with B. cinerea (Figure 8c,d). We then extracted the total RNA of B. cinerea isolated from the infected N. benthamiana leaves and determined the expression level of BcPmk1. As expected, the expression levels of BcPmk1 were significantly reduced after TRV2‐CcPmk1 and TRV2‐BcPmk1 treatment compared with that in the TRV2 control and TRV2‐GFP samples (Figure 8e). This finding suggests that BcPmk1 expression in B. cinerea is indeed compromised as a result of TRV‐induced VIGS in N. benthamiana, and therefore retarded fungal virulence.
FIGURE 8.
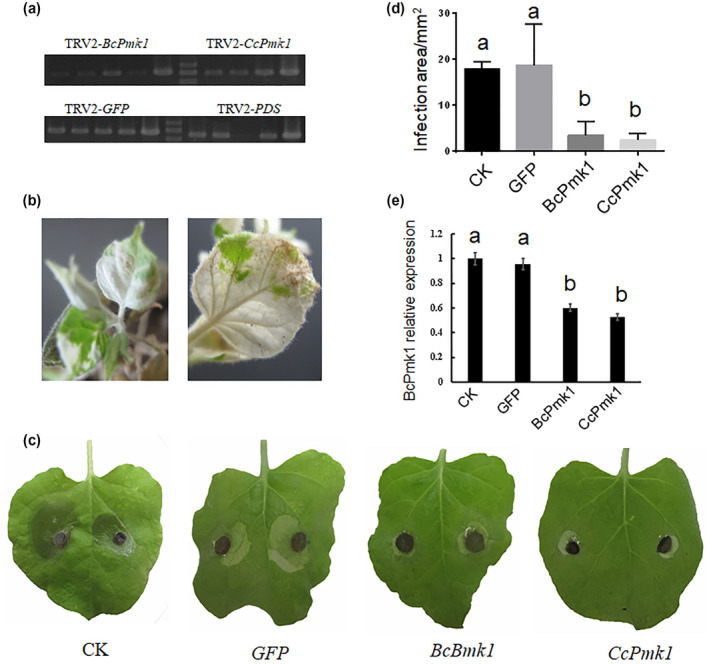
Virus‐induced gene silencing of BcPmk1 in Botrytis cinerea. (a) Construction of the expression cascades of TRV2‐GFP, TRV2‐CcPmk1, and TRV2‐BcPmk1. (b) Silencing the NbPDS gene in Nicotiana benthamiana resulted in the photobleaching. (c) Pathogenicity assay after inoculation of N. benthamiana with the Agrobacterium‐carrying TRV1 and TRV2 constructs. (d) The infection areas of different inoculated N. benthamiana leaves. (e) The relative expression level of BcPmk1
3. DISCUSSION
In this study, we conducted a transcriptional analysis between the wild type and CcPmk1 deletion mutants and found that 777 genes were differentially expressed in the CcPmk1 deletion mutant compared with the wild type, including 523 significantly down‐regulated genes and 254 significantly up‐regulated genes. Functional annotation revealed that CcPmk1 was required for the expression of genes involved in many biological processes, such as ribosome biogenesis, rRNA processing, secondary metabolism, and transcriptional regulation. Silencing BcPmk1 in B. cinerea with the conserved BcPmk1 and CcPmk1 sequences using the TRV‐based VIGS system significantly compromised the virulence of B. cinerea to N. benthamiana. Overall, the results suggest that CcPmk1 is a regulator of pathogenicity in C. chrysoperma, which can potentially be used as a target for disease control.
Fus3/Kss1‐type MAPK is required for virulence in several plant‐pathogenic fungi, and this crucial function is also proven for CcPmk1 of C. chrysosperma. Here, we observed that CcPmk1 shared more than 90% sequence identity with Pmk1 orthologs from phytopathogens belonging to Ascomycota, which were higher than those from plant‐pathogenic oomycetes and basidiomycetes. For the Pmk1 orthologs from nonplant pathogens, several also showed high sequence identities with CcPmk1 (over 90%), such as Microdochium bolleyi, Trichoderma reesei, and Cenococcum geophilum, while Pmk1 orthologs from S. cerevisiae, Laccaria bicolor, and Exidia glandulosa showed low sequence identities with CcPmk1 (below 75%). Pathogenesis is a very complex process involving multiple signalling pathways and various gene regulatory relationships as well as other processes. It is reasonable that the Pmk1 homologs in pathogenic and nonpathogenic fungi exhibit distinct functions in pathogenicity even if their sequences are highly similar. Previous reports from phytopathogens reveal conserved regulatory functions of Pmk1 orthologs in addition to pathogenicity, such as mating, fungal growth, and conidiation (Hu et al., 2007; Mayorga & Gold, 1999; Müller et al., 1999; Xu & Hamer, 1996). These functions may be conservative and important in nonpathogenic fungi as well. Additionally, a conserved TEY phosphorylation site was found among almost all the Pmk1 orthologs from selected fungi and plant‐pathogenic oomycetes, including pathogenic and nonpathogenic microorganisms, indicating that this kinase is evolutionarily conserved. Remarkably, MAPK cascades use protein phosphorylation/dephosphorylation cycles to channel information. Therefore, we considered that the phosphorylation site TEY may play a key and conserved role in Pmk1 kinase and further research on the function of the phosphorylation site should be conducted to reveal the specific mechanism of Pmk1.
Moreover, phosphorylation signals are important in many cellular processes, hence kinases and phosphatases are among the most extensively studied enzymes (Ghosh et al., 2014). Reversible protein phosphorylation on serine, threonine, tyrosine, or histidine residues plays a critical role in the regulation of physiological processes in the cells of microorganisms (Ravikumar et al., 2014). Protein phosphorylation in nonpathogenic fungi is reported to affect an estimated 30% of the proteome and is a major regulatory mechanism that controls many basic cellular processes (Manning et al., 2002; Ptacek et al., 2005). Studies on plant fungal pathogens have demonstrated that protein phosphorylation is also required for various regulatory functions (Bohnert et al., 2019). For example, in M. oryzae, protein phosphorylation regulates infection‐related morphogenesis and pathogenicity via signalling pathways, that is, the MAPK and cyclic adenosine monophosphate (cAMP)‐dependent protein kinase (protein kinase A [PKA]) (Xu & Hamer, 1996). Our results showed that many of the genes involved in protein phosphorylation were significantly down‐regulated in the CcPmk1 deletion mutant, such as GME6673_g and GME8763_g, which were predicted to be serine/threonine protein kinases, and GME7613_g, predicted as a CAMK protein kinase. Therefore, we speculate that CcPmk1 may affect the expression levels of other kinases to control signal transduction and phosphorylation processes, resulting in diverse phenotypes that distinguish the mutant from the wild type.
The downstream elements of Pmk1 homologs in different fungi may be diverse. For instance, Ste12, the downstream C2H2 transcription factor, is involved in the regulation of the mating process in S. cerevisiae as well as Fus3, while the Ste12 homolog Mst12 in M. oryzae is involved in fungal pathogenicity but is dispensable for appressorium formation and mating unlike Pmk1 (Park et al., 2002). In addition, the regulatory functions of Pmk1 in downstream components may vary considerably among different species, and even within a species, because of the life stages and environments of the species. For example, Soanes et al. (2012) identified 481 genes that were significantly down‐regulated in the Pmk1 deletion mutant compared with the wild type in the appressorium development stage. GO functional annotation revealed that the responses to cAMP (GO: 0051591), organic substance (GO: 0010033), chemical stimulus (GO: 0042221), and stimulus (GO: 0050896) were over‐represented, and were involved in appressorium formation (Soanes et al., 2012). However, transcriptional analysis of M. oryzae Pmk1 inhibition mutants during the infection process revealed that thousands of genes were differentially expressed compared with the wild type, including 742 significantly down‐regulated genes (Sakulkoo et al., 2018). The expression of a subset of well‐characterized effectors was Pmk1‐dependent, such as Bas2, Bas3, and Bas4. However, only 36 overlapping regulated genes without well‐characterized effectors were found between the appressorium formation stage and the infection process, indicating the various regulated subset genes during different processes (Sakulkoo et al., 2018; Soanes et al., 2012). Additionally, the gene expression analysis of the Pmk1 deletion mutant by array hybridization showed that protein biosynthesis and processes such as electron transport and oxidative phosphorylation and carbohydrate metabolism were significantly affected during the mycelial growth stage (Jin et al., 2013). In this study, a few overlapping Pmk1‐regulated genes (45 down‐regulated and 28 up‐regulated) were found between the CcPmk1‐regulated genes and the MoPmk1‐regulated genes in M. oryzae during the infection process. Unexpectedly, GO annotation revealed that several similar regulation functions of Pmk1 might be conserved in different fungal species. For example, the ribosome biosynthesis‐related processes were significantly enriched in 45 overlapping down‐regulated genes in the CcPmk1 and MoPmk1 deletion mutants. Similarly, the ribosome biosynthesis KEGG pathway was significantly enriched in the CcPmk1 deletion mutant. Ribosomes are the cellular factories that translate the genetic code in mRNA and make proteins in all cells, and hundreds of proteins are required for ribosome assembly (Granneman & Baserga, 2004). In the absence of these proteins, ribosome biogenesis is stalled and cell growth is terminated even under optimal growth conditions. Here, a subset of genes involved in this process was significantly down‐regulated in the CcPmk1 deletion mutant, which would affect protein biosynthesis and therefore cause multiple defects in phenotypes. The results may suggest the conserved functions of Pmk1 homologs in ribosome biogenesis in different fungal species.
Importantly, the conserved and critical function of Pmk1 homologs from different plant‐pathogenic microorganisms in pathogenicity is well known. In M. oryzae, Pmk1 is essential for appressorium formation and invasive growth (Xu & Hamer, 1996). Recently, taking advantage of living cell imaging, Pmk1 inhibition mutants have been shown to fail to invade plant cells (Sakulkoo et al., 2018). In the soilborne pathogenic fungus F. oxysporum, Fmk1 regulates some pathogenicity‐regulated processes such as invasive growth, reduced expression of the pectate lyase encoding gene, and root attachment (Di Pietro et al., 2001). In addition, Fmk1 is also involved in surface hydrophobicity and reactive oxygen species (ROS) homeostasis (Di Pietro et al., 2001; Segorbe et al., 2017). Deletion of VmPmk1 from V. mali, a necrotrophic fungus that harms the twig and stem of woody plants, shows obvious defects in oxidative stress response, cell wall integrity, and fungal pathogenicity (Wu et al., 2017). Similarly, our previous studies also found that CcPmk1 is required for fungal pathogenicity, growth, cell wall integrity, and expression of effectors (Yu et al., 2019). In this study, we conducted a transcription analysis of CcPmk1 deletion mutants during the infection process, and a subset of genes was differentially expressed, including many secondary metabolism gene clusters, transcription factors, and putative effectors that might be important for fungal pathogenicity. For example, GME2992_g, a putative MADS‐box transcription factor, was significantly down‐regulated in the CcPmk1 deletion mutant and its homolog Mig1 (MGG_01204) from M. oryzae is required for pathogenicity (Mehrabi et al., 2008). Moreover, GME557_g, named CcOah, was down‐regulated in the CcPmk1 deletion mutant, which contributes to oxalic acid biogenesis and virulence (Wang & Wang, 2020). In addition, GME7477_g, a small cysteine‐rich secretory protein belonging to the CAP superfamily, can inhibit PAMP INF1‐triggered cell death and plays a vital role in fungal pathogenicity to the host poplar (Han et al., 2021). Hence, we believe that CcPmk1 may participate in regulating the expression of a subset of downstream components to promote virulence.
Pathogenic fungi have evolved sophisticated ways to infect their hosts, mainly by adapting to the host environment and producing pathogenesis‐related products such as toxic secondary metabolites, effectors, and/or extracellular enzymes (Jonkers et al., 2012). Toxins produced by plant‐pathogenic fungi are often crucial determinants of plant disease, as toxins can lead to partial disease symptoms and nontoxic mutants are nonpathogenic or show reduced virulence (Richard, 2007). The Pmk1 homologs in Aspergillus fumigatus contributes to the positive regulation of secondary metabolite production, which is similar to the findings observed in both Aspergillus nidulans (Bayram et al., 2012; Frawley et al., 2018) and Aspergillus flavus (Frawley et al., 2020). In plant pathogens, the Pmk1 orthologs in F. graminearum and F. verticillioides are important for deoxynivalenol production and fumonisin biosynthesis, which are important for fungal virulence (Turrà et al., 2014). In this study, 11 secondary metabolite core biosynthesis genes were significantly down‐regulated in the CcPmk1 deletion mutant. Importantly, the whole terpene type gene cluster (GME3317_g to GME3324_g) and hybrid‐NRPS and T1PKS type gene cluster (GME3437_g to GME3444_g) were all significantly down‐regulated in ΔCcPmk1. These results suggest that Pmk1 orthologs may share a conserved function in secondary metabolism.
Moreover, we showed that the virulence of B. cinerea to N. benthamiana was retarded using the TRV‐based VIGS system with homologous fragments from CcPmk1 and BcPmk1. Importantly, the fragment of CcPmk1 can be used to cross‐species silence BcPmk1 in B. cinerea. Taken together, the Fus3/Kss1/Pmk1 homologs from different plant‐pathogenic microorganisms play a conserved regulatory role in pathogenicity; therefore, they can be potentially used as a target for the control of broad‐spectrum plant diseases. Recently, the Pmk1 homolog in P. triticina has been used as a target for leaf rust disease control, and transgenic wheat harbouring a Pmk1‐RNAi construct shows significant resistance to the disease (Panwar et al., 2018). Whether the transgenic plants containing the Pmk1‐RNAi could reduce the severity of other diseases was not examined. Although there are off‐target risks of adopting HIGS, such as silencing unintended genes in the host plants as well as in beneficial plant‐associated organisms, this could also be used as a promising approach to control plant diseases by carefully designing it.
In conclusion, we performed a transcriptional analysis between the wild type and CcPmk1 deletion mutants to explore the regulation network of CcPmk1. Our results revealed several key biological processes regulated by CcPmk1. In addition, we confirmed that CcPmk1 can be used as a potential target for disease control. These findings improve our understanding toward the functions of the MAPK signalling pathway in C. chrysosperma. In‐depth studies should augment the understanding of the molecular mechanisms of CcPmk1 downstream elements to analyse the molecular pathogenic mechanism of C. chrysosperma, which, in turn, could be used to develop novel approaches for poplar canker disease control in the future.
4. EXPERIMENTAL PROCEDURES
4.1. Fungi, plants, and their growth conditions
The C. chrysosperma strain (CFCC 89981), isolated from poplar, was used as the wild‐type strain in this study. The B. cinerea was kindly provided by Dr Tingting Dai from Nanjing Forestry University. The strains were regularly cultured on potato dextrose agar (PDA) at 25 °C or potato dextrose broth (PDB) with shaking at 150 rpm and 25 °C. B. cinerea was also grown on malt extract agar (MEA). N. benthamiana was grown in a glasshouse at 22 °C with 16 hr/8 hr light/dark photoperiods, with a relative humidity of approximately 60%.
4.2. Sequence and phylogenetic analysis of CcPmk1 and its homologs
The sequence of CcPmk1 was acquired from the draft genome sequence of C. chrysosperma, which had been sequenced by our laboratory (NCBI GenBank accession number JAEQMF000000000). The other Fus3/Pmk1/Kss1 homologs from other microorganisms were acquired from the JGI database, NCBI, and reported references (Hamel et al., 2012; Jiang et al., 2018). The amino acid sequence alignments were performed using ClustalX v. 2.0 and visualized with Jalview. The phylogenetic tree was constructed with MEGA 10.0 using the neighbour‐joining method with the bootstrap test replicated 1,000 times.
4.3. RNA isolation and quantitative reverse transcription PCR analysis
To prepare the transcription sequencing samples, the wild‐type C. chrysosperma and CcPmk1 deletion mutants were grown in PDB supplemented with sterilized poplar twigs to mimic the states of infection, and then incubated at 25 °C with shaking at 150 rpm for 2 days. The cultures were harvested with Miracloth (Calbiochem), then flash‐frozen in liquid nitrogen and ground to powder for RNA isolation. The experiments were repeated three times.
To determine the expression level of CcPmk1, asiRNAs‐treated C. chrysosperma hyphae were collected for RNA isolation. To identify the expression of the BcPmk1 gene after A. tumefaciens infiltration and B. cinerea inoculation, B. cinerea was isolated from the infected N. benthamiana leaves and then grown in PDA for 2 days, flash‐frozen in liquid nitrogen, and ground to powder for RNA isolation. All RNA samples were isolated with TRIzol reagent (Invitrogen) and purified with a PureLink RNA Mini Kit (Invitrogen) in accordance with the manufacturer's instructions.
For quantitative reverse transcription PCR (RT‐qPCR) assays, cDNA was synthesized using oligo(dT)18 primer and SuperScript IV reverse transcriptase (Invitrogen). Quantitative PCR was performed using SuperReal PreMix Plus (Tiangen) with the Applied Biosystems 7,500 Real‐Time PCR system (Applied Biosystems). The actin gene (CcActin and BcActin) was used as an internal reference for all RT‐qPCR analyses. Relative expression levels were calculated using 2−ΔΔ C t method. All primers used in the present study are listed in Table S5.
4.4. Transcription sequencing and bioinformatic analysis
After calculating the quality and quantity of RNA, the sequencing libraries were generated using the NEBNext Ultra RNA Library Prep Kit for Illumina (NEB) according to the manufacturer's recommendations. The libraries were sequenced on an Illumina HiSeq platform and 150 bp paired‐end reads were generated. The paired‐end clean reads were aligned to the reference genome using Hisat2 v. 2.0.5.
Differential expression analysis was performed using the DESeq2 R package v. 1.16.1. The resulting p values were adjusted using Benjamini and Hochberg's approach for controlling the false discovery rate. Genes with an adjusted p < .05 found by DESeq2 were assigned as differentially expressed. GO and KEGG enrichment analyses of differentially expressed genes were performed using the clusterProfiler R package. GO terms with corrected p < .05 were considered significantly enriched by differentially expressed genes.
The secondary metabolism gene clusters in C. chrysosperma were predicted by the antiSMASH (Blin et al., 2019). The RNA‐Seq coverage data of secondary metabolism gene clusters were visualized using Integrative Genomics Viewer (Thorvaldsdóttir et al., 2013). Signal peptides were predicted using SignalP v. 5.0 (http://www.cbs.dtu.dk/services/SignalP/) and TMHMM v. 2.0 was used to predict the transmembrane helices in proteins (http://www.cbs.dtu.dk/services/TMHMM/). Effector candidates in C. chrysosperma were predicted based on a query of the genome sequence with the following criteria: small in length (≤300 amino acids), cysteine‐rich (≥4 cysteine residues), containing a signal peptide, and no GPI anchor.
4.5. Design and synthesis of asiRNA
Nineteen nucleotide sequences among the conserved MAPK domain were designed as the asiRNA candidates according to the nucleotide sequence of CcPmk1. To avoid off‐targets, the asiRNA candidate sequences were BLAST searched against the C. chrysosperma genomic sequence. We then complemented asiRNA candidate sequences and generated double‐stranded asiRNAs. Double T nucleotides were added to the C‐terminus to stabilize the asiRNAs, and the FAM fluorescent mark was added to observe the entrance of asiRNAs into the hyphae. The designed double‐stranded asiRNAs were synthesized by Shanghai Genepharma. Co., Ltd. The asiRNAs were added to PDB at a final concentration of 0.125 nM and incubated with shaking at 150 rpm and 25 °C for 1 day. The cultures were then harvested and grown in PDA at 25 °C.
4.6. Plasmid construction and TRV treatment
The fragments of CcPmk1, BcPmk1, PDS, and GFP were PCR amplified with specific primer pairs. The TRV1 and TRV2 plasmids were kindly given by Tingli Liu from the Jiangsu Academy of Agricultural Sciences. The PDS gene is involved in carotenoid biosynthesis and silencing it in plants leads to a photobleaching phenotype, therefore it was used as a positive control in this study (Senthil‐Kumar & Mysore, 2014). The resulting fragments were fused into SmaI‐ and EcoRⅠ‐digested TRV2 plasmids. Subsequently, the constructs TRV2‐CcPmk1, TRV2‐BcPmk1, TRV2‐GFP, TRV2‐Empty, and TRV2‐PDS were transformed into A. tumefaciens GV3101. The A. tumefaciens‐carrying TRV1 and TRV2 vectors were coinfiltrated into N. benthamiana leaves as described previously (Senthil‐Kumar & Mysore, 2014).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
FIGURE S1 Phylogenetic analysis of Fus3/Kss1/Pmk1 from different species. The detailed information of the Fus3/Kss1/Pmk1 orthologs is listed in Tables 1 and S1
FIGURE S2 Principal component analysis of read counts of the wild‐type Cytospora chrysosperma and CcPmk1 deletion mutant revealed the clear separation of the two tested conditions and the proximity of biological replicates
FIGURE S3 Global view of gene expression level in CcPmk1 deletion mutant and wild type
FIGURE S4 GO annotation of overlapped down‐regulated genes in both CcPmk1 deletion mutant and MoPmk1 inhibition mutant in Magnaporthe oryzae. The yellow dotted box represents the significant enrichment GO terms. BP, biological process; CC, cellular component; MF, molecular function
FIGURE S5 Fluorescence microscopy of the hyphae treated with the artificial small interfering RNA, indicating that the small interfering RNA was successfully taken up by the hyphae
FIGURE S6 Sequence alignment of BcPmk1 and CcPmk1 fragments used for virus‐induced gene silencing. The same residues are shadowed in black
TABLE S1 The percentage of sequence identity between CcPmk1 and Pmk1 homologues in nonpathogenic fungi
TABLE S2 The secondary metabolism gene clusters in Cytospora chrysosperma predicted by antiSMASH
TABLE S3 The list of transcription factors regulated by CcPmk1
TABLE S4 The list of putative effector genes regulated by CcPmk1
TABLE S5 Primers used in this study
ACKNOWLEDGEMENTS
The authors thank Dr Fanli Meng at Beijing Forestry University for helpful suggestions on this manuscript. This work was supported by funding from the National Key Research and Development Program (2017YFD0600100) and the National Natural Science Foundation of China (31800540).
Xiong D, Yu L, Shan H, Tian C. CcPmk1 is a regulator of pathogenicity in Cytospora chrysosperma and can be used as a potential target for disease control. Mol Plant Pathol. 2021;22:710–726. 10.1111/mpp.13059
Dianguang Xiong and Lu Yu contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- Adams, G.C. , Roux, J. & Wingfield, M.J. (2006) Cytospora species (Ascomycota, Diaporthales, Valsaceae): Introduced and native pathogens of trees in South Africa. Australasian Plant Pathology, 35, 521–548. 10.1071/AP06058 [DOI] [Google Scholar]
- Adams, G.C. , Wingfield, M.J. , Common, R. & Roux, J. (2005) Phylogenetic relationships and morphology of Cytospora species and related teleomorphs (Ascomycota, Diaporthales, Valsaceae) from Eucalyptus . Studies in Mycology, 52, 1–144. [Google Scholar]
- Baulcombe, D. (2005) RNA silencing. Trends in Biochemical Sciences, 30, 290–293. 10.1016/j.tibs.2005.04.012 [DOI] [PubMed] [Google Scholar]
- Bayram, Ö. , Bayram, Ö.S. , Ahmed, Y.L. , Maruyama, J.‐I. , Valerius, O. , Rizzoli, S.O. et al. (2012) The Aspergillus nidulans MAPK module AnSte11‐Ste50‐Ste7‐Fus3 controls development and secondary metabolism. PLoS Genetics, 8, e1002816. 10.1371/journal.pgen.1002816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs, A.R. , Davis, D.D. & Merrill, W. (1983) Histopathology of cankers on Populus caused by Cytospora chrysosperma . Canadian Journal of Botany, 61, 563–574. [Google Scholar]
- Blin, K. , Shaw, S. , Steinke, K. , Villebro, R. , Ziemert, N. , Lee, S.Y. et al. (2019) antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research, 47, W81–W87. 10.1093/nar/gkz310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnert, S. , Heck, L. , Gruber, C. , Neumann, H. , Distler, U. , Tenzer, S. et al. (2019) Fungicide resistance toward fludioxonil conferred by overexpression of the phosphatase gene MoPTP2 in Magnaporthe oryzae . Molecular Microbiology, 111, 662–677. 10.1111/mmi.14179 [DOI] [PubMed] [Google Scholar]
- Cai, Q. , He, B. , Kogel, K.H. & Jin, H. (2018) Cross‐kingdom RNA trafficking and environmental RNAi – nature’s blueprint for modern crop protection strategies. Current Opinion in Microbiology, 46, 58–64. 10.1016/j.mib.2018.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Harel, A. , Gorovoits, R. , Yarden, O. & Dickman, M.B. (2004) MAPK regulation of sclerotial development in Sclerotinia sclerotiorum is linked with pH and cAMP sensing. Molecular Plant‐Microbe Interactions, 17, 404–413. 10.1094/MPMI.2004.17.4.404 [DOI] [PubMed] [Google Scholar]
- Choi, E.‐S. , Chung, H.‐J. , Kim, M.‐J. , Park, S.‐M. , Cha, B.‐J. , Yang, M.‐S. et al. (2005) Characterization of the ERK homologue CpMK2 from the chestnut blight fungus Cryphonectria parasitica . Microbiology, 151, 1349–1358. 10.1099/mic.0.27796-0 [DOI] [PubMed] [Google Scholar]
- Di Pietro, A. , Garcia‐MacEira, F.I. , Meglecz, E. & Roncero, M.I. (2001) A MAP kinase of the vascular wilt fungus Fusarium oxysporum is essential for root penetration and pathogenesis. Molecular Microbiology, 39, 1140–1152. 10.1111/j.1365-2958.2001.02307.x [DOI] [PubMed] [Google Scholar]
- Fan, X.L. , Bezerra, J.D.P. , Tian, C.M. & Crous, P.W. (2020) Cytospora (Diaporthales) in China. Persoonia, 45, 1–45. 10.3767/persoonia.2020.45.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, X. , Hyde, K.D. , Liu, M. , Liang, Y. & Tian, C. (2015) Cytospora species associated with walnut canker disease in China, with description of a new species C. gigalocus . Fungal Biology, 119, 310–319. 10.1016/j.funbio.2014.12.011 [DOI] [PubMed] [Google Scholar]
- Frawley, D. , Greco, C. , Oakley, B. , Alhussain, M.M. , Fleming, A.B. , Keller, N.P. et al. (2020) The tetrameric pheromone module SteC‐MkkB‐MpkB‐SteD regulates asexual sporulation, sclerotia formation and aflatoxin production in Aspergillus flavus . Cellular Microbiology, 22, e13192. 10.1111/cmi.13192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frawley, D. , Karahoda, B. , Sarikaya Bayram, Ö. & Bayram, Ö. (2018) The HamE scaffold positively regulates MpkB phosphorylation to promote development and secondary metabolism in Aspergillus nidulans . Scientific Reports, 8, 16588. 10.1038/s41598-018-34895-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, A. , Servin, J.A. , Park, G. & Borkovich, K.A. (2014) Global analysis of serine/threonine and tyrosine protein phosphatase catalytic subunit genes in Neurospora crassa reveals interplay between phosphatases and the p38 mitogen‐activated protein kinase. G3 Genes, Genomes, Genetics, 4, 349–365. 10.1534/g3.113.008813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman, S. & Baserga, S.J. (2004) Ribosome biogenesis: of knobs and RNA processing. Experimental Cell Research, 296, 43–50. 10.1016/j.yexcr.2004.03.016 [DOI] [PubMed] [Google Scholar]
- Guo, J. , Dai, X. , Xu, J.R. , Wang, Y. , Bai, P. , Liu, F. et al. (2011) Molecular characterization of a Fus3/Kss1 type MAPK from Puccinia striiformis f. sp. tritici, PsMAPK1. PLoS One, 6, e21895. 10.1371/journal.pone.0021895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, X.‐Y. , Li, Y. , Fan, J. , Xiong, H. , Xu, F.‐X. , Shi, J. et al. (2019) Host‐induced gene silencing of moap1 confers broad‐spectrum resistance to Magnaporthe oryzae . Frontiers in Plant Science, 10, 433. 10.3389/fpls.2019.00433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel, L.P. , Nicole, M.C. , Duplessis, S. & Ellis, B.E. (2012) Mitogen‐activated protein kinase signaling in plant‐interacting fungi: distinct messages from conserved messengers. The Plant Cell, 24, 1327–1351. 10.1105/tpc.112.096156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, Z. , Xiong, D. , Xu, Z. , Liu, T. & Tian, C. (2021) The Cytospora chrysosperma virulence effector CcCAP1 mainly localizes to the plant nucleus to suppress plant immune responses. mSphere, 6, e883–e920. 10.1128/mSphere.00883-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, P. , Wang, Y. , Wang, X. , Zhang, X. & Tian, C. (2017) The mitogen‐activated protein kinase CgMK1 governs appressorium formation, melanin synthesis, and plant infection of Colletotrichum gloeosporioides . Frontiers in Microbiology, 8, 2216. 10.3389/fmicb.2017.02216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Y. , Zhai, Y.I. , Feng, L.I. , Karimi, H.Z. , Rutter, B.D. , Zeng, L. et al. (2019) A Phytophthora effector suppresses trans‐kingdom RNAi to promote disease susceptibility. Cell Host & Microbe, 25, 153–165.e5. 10.1016/j.chom.2018.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, G. , Kamp, A. , Linning, R. , Naik, S. & Bakkeren, G. (2007) Complementation of Ustilago maydis MAPK mutants by a wheat leaf rust, Puccinia triticina homolog: potential for functional analyses of rust genes. Molecular Plant‐Microbe Interactions, 20, 637–647. 10.1094/MPMI-20-6-0637 [DOI] [PubMed] [Google Scholar]
- Hua, C. , Zhao, J.H. & Guo, H.S. (2018) Trans‐kingdom RNA silencing in plant–fungal pathogen interactions. Molecular Plant, 11, 235–244. 10.1016/j.molp.2017.12.001 [DOI] [PubMed] [Google Scholar]
- Jenczmionka, N.J. , Maier, F.J. , Lösch, A.P. & Schäfer, W. (2003) Mating, conidiation and pathogenicity of Fusarium graminearum, the main causal agent of the head‐blight disease of wheat, are regulated by the MAP kinase gpmk1. Current Genetics, 43, 87–95. 10.1007/s00294-003-0379-2 [DOI] [PubMed] [Google Scholar]
- Jiang, C. , Zhang, X. , Liu, H. & Xu, J.R. (2018) Mitogen‐activated protein kinase signaling in plant pathogenic fungi. PLoS Pathogens, 14, e1006875. 10.1371/journal.ppat.1006875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Q. , Li, C. , Li, Y. , Shang, J. , Li, D. , Chen, B. et al. (2013) Complexity of roles and regulation of the PMK1‐MAPK pathway in mycelium development, conidiation and appressorium formation in Magnaporthe oryzae . Gene Expression Patterns, 13, 133–141. 10.1016/j.gep.2013.02.003 [DOI] [PubMed] [Google Scholar]
- Jonkers, W. , Dong, Y. , Broz, K. & Kistler, H.C. (2012) The Wor1‐like protein Fgp1 regulates pathogenicity, toxin synthesis and reproduction in the phytopathogenic fungus Fusarium graminearum . PLoS Pathogens, 8, e1002724. 10.1371/journal.ppat.1002724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev, S. , Sharon, A. , Hadar, R. , Ma, H. & Horwitz, B.A. (1999) A mitogen‐activated protein kinase of the corn leaf pathogen Cochliobolus heterostrophus is involved in conidiation, appressorium formation, and pathogenicity: diverse roles for mitogen‐activated protein kinase homologs in foliar pathogens. Proceedings of the National Academy of Sciences of the United States of America, 96, 13542–13547. 10.1073/pnas.96.23.13542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti, L. , Lanver, D. , Schweizer, G. , Tanaka, S. , Liang, L. , Tollot, M. et al. (2015) Fungal effectors and plant susceptibility. Annual Review of Plant Biology, 66, 513–545. 10.1146/annurev-arplant-043014-114623 [DOI] [PubMed] [Google Scholar]
- Madar, Z. , Solel, Z. & Kimchi, M. (2004) First report of cytospora canker caused by Cytospora chrysosperma on white poplar in Israel. Plant Disease, 88, 220. 10.1094/PDIS.2004.88.2.220C [DOI] [PubMed] [Google Scholar]
- Manfiolli, A.O. , Siqueira, F.S. , Dos Reis, T.F. , Van Dijck, P. , Schrevens, S. , Hoefgen, S. et al. (2019) Mitogen‐activated protein kinase cross‐talk interaction modulates the production of melanins in Aspergillus fumigatus . mBio, 10, e215–e219. https://doi.org10.1128/mBio.00215‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, G. , Plowman, G.D. , Hunter, T. & Sudarsanam, S. (2002) Evolution of protein kinase signaling from yeast to man. Trends in Biochemical Sciences, 27, 514–520. 10.1016/S0968-0004(02)02179-5 [DOI] [PubMed] [Google Scholar]
- Mayorga, M.E. & Gold, S.E. (1999) A MAP kinase encoded by the ubc3 gene of Ustilago maydis is required for filamentous growth and full virulence. Molecular Microbiology, 34, 485–497. 10.1046/j.1365-2958.1999.01610.x [DOI] [PubMed] [Google Scholar]
- Mehrabi, R. , Ding, S. & Xu, J.R. (2008) MADS‐box transcription factor mig1 is required for infectious growth in Magnaporthe grisea . Eukaryotic Cell, 7, 791–799. 10.1128/EC.00009-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, P. , Aichinger, C. , Feldbrügge, M. & Kahmann, R. (1999) The MAP kinase kpp2 regulates mating and pathogenic development in Ustilago maydis . Molecular Microbiology, 34, 1007–1017. 10.1046/j.1365-2958.1999.01661.x [DOI] [PubMed] [Google Scholar]
- Nowara, D. , Gay, A. , Lacomme, C. , Shaw, J. , Ridout, C. , Douchkov, D. et al. (2010) HIGS: host‐induced gene silencing in the obligate biotrophic fungal pathogen Blumeria graminis . The Plant Cell, 22, 3130–3141. 10.1105/tpc.110.077040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panwar, V. , Jordan, M. , McCallum, B. & Bakkeren, G. (2018) Host‐induced silencing of essential genes in Puccinia triticina through transgenic expression of RNAi sequences reduces severity of leaf rust infection in wheat. Plant Biotechnology Journal, 16, 1013–1023. 10.1111/pbi.12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, G. , Xue, C. , Zheng, L. , Lam, S. & Xu, J.R. (2002) MST12 regulates infectious growth but not appressorium formation in the rice blast fungus Magnaporthe grisea . Molecular Plant‐Microbe Interactions, 15, 183–192. 10.1094/MPMI.2002.15.3.183 [DOI] [PubMed] [Google Scholar]
- Priegnitz, B.E. , Brandt, U. , Pahirulzaman, K.A. , Dickschat, J.S. & Fleißner, A. (2015) The AngFus3 mitogen‐activated protein kinase controls hyphal differentiation and secondary metabolism in Aspergillus niger . Eukaryotic Cell, 14, 602–615. 10.1128/EC.00018-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptacek, J. , Devgan, G. , Michaud, G. , Zhu, H. , Zhu, X. , Fasolo, J. et al. (2005) Global analysis of protein phosphorylation in yeast. Nature, 438, 679–684. 10.1038/nature04187 [DOI] [PubMed] [Google Scholar]
- Qi, T. , Guo, J. , Peng, H. , Liu, P. , Kang, Z. & Guo, J. (2019) Host‐induced gene silencing: a powerful strategy to control diseases of wheat and barley. International Journal of Molecular Sciences, 20, 206.– 10.3390/ijms20010206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, T. , Zhu, X. , Tan, C. , Liu, P. , Guo, J. , Kang, Z. et al. (2018) Host‐induced gene silencing of an important pathogenicity factor PsCPK1 in Puccinia striiformis f. sp. tritici enhances resistance of wheat to stripe rust. Plant Biotechnology Journal, 16, 797–807. 10.1111/pbi.12829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauyaree, P. , Ospina‐Giraldo, M.D. , Kang, S. , Bhat, R.G. , Subbarao, K.V. , Grant, S.J. et al. (2005) Mutations in VMK1, a mitogen‐activated protein kinase gene, affect microsclerotia formation and pathogenicity in Verticillium dahliae . Current Genetics, 48, 109–116. 10.1007/s00294-005-0586-0 [DOI] [PubMed] [Google Scholar]
- Ravikumar, V. , Shi, L. , Krug, K. , Derouiche, A. , Jers, C. , Cousin, C. et al. (2014) Quantitative phosphoproteome analysis of Bacillus subtilis reveals novel substrates of the kinase PrkC and phosphatase PrpC. Molecular and Cellular Proteomics, 13, 1965–1978. 10.1074/mcp.M113.035949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard, J.L. (2007) Some major mycotoxins and their mycotoxicoses—an overview. International Journal of Food Microbiology, 119, 3–10. 10.1016/j.ijfoodmicro.2007.07.019 [DOI] [PubMed] [Google Scholar]
- Rigling, D. & Prospero, S. (2018) Cryphonectria parasitica, the causal agent of chestnut blight: Invasion history, population biology and disease control. Molecular Plant Pathology, 19, 7–20. 10.1111/mpp.12542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rispail, N. & Di Pietro, A. (2009) Fusarium oxysporum Ste12 controls invasive growth and virulence downstream of the Fmk1 MAPK cascade. Molecular Plant‐Microbe Interactions, 22, 830–839. 10.1094/MPMI-22-7-0830 [DOI] [PubMed] [Google Scholar]
- Sakulkoo, W. , Osés‐Ruiz, M. , Oliveira Garcia, E. , Soanes, D.M. , Littlejohn, G.R. , Hacker, C. et al. (2018) A single fungal MAP kinase controls plant cell‐to‐cell invasion by the rice blast fungus. Science, 359, 1399–1403. 10.1126/science.aaq0892 [DOI] [PubMed] [Google Scholar]
- Sanju, S. , Siddappa, S. , Thakur, A. , Shukla, P.K. , Srivastava, N. , Pattanayak, D. et al. (2015) Host‐mediated gene silencing of a single effector gene from the potato pathogen Phytophthora infestans imparts partial resistance to late blight disease. Functional & Integrative Genomics, 15, 697–706. 10.1007/s10142-015-0446-z [DOI] [PubMed] [Google Scholar]
- Segorbe, D. , Di Pietro, A. , Perez‐Nadales, E. & Turrà, D. (2017) Three Fusarium oxysporum mitogen‐activated protein kinases (MAPKs) have distinct and complementary roles in stress adaptation and cross‐kingdom pathogenicity. Molecular Plant Pathology, 18, 912–924. 10.1111/mpp.12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senthil‐Kumar, M. & Mysore, K.S. (2011) New dimensions for VIGS in plant functional genomics. Trends in Plant Science, 16, 656–665. 10.1016/j.tplants.2011.08.006 [DOI] [PubMed] [Google Scholar]
- Senthil‐Kumar, M. & Mysore, K.S. (2014) Tobacco rattle virus‐based virus‐induced gene silencing in Nicotiana benthamiana . Nature Protocols, 9, 1549–1562. 10.1038/nprot.2014.092 [DOI] [PubMed] [Google Scholar]
- Soanes, D.M. , Chakrabarti, A. , Paszkiewicz, K.H. , Dawe, A.L. & Talbot, N.J. (2012) Genome‐wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae . PLoS Pathogens, 8, e1002514. 10.1371/journal.ppat.1002514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y. & Thomma, B. (2018) Host‐induced gene silencing compromises Verticillium wilt in tomato and Arabidopsis . Molecular Plant Pathology, 19, 77–89. 10.1111/mpp.12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, H. , Nishimura, M. , Asayama, Y. , Mannse, Y. , Ishiwata, S. , Kita, A. et al. (2007) Atf1 is a target of the mitogen‐activated protein kinase Pmk1 and regulates cell integrity in fission yeast. Molecular Biology of the Cell, 18, 4794–4802. 10.1091/mbc.e07-03-0282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir, H. , Robinson, J.T. & Mesirov, J.P. (2013) Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14, 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrà, D. , Segorbe, D. & Di Pietro, A. (2014) Protein kinases in plant‐pathogenic fungi: conserved regulators of infection. Annual Review of Phytopathology, 52, 267–288. 10.1146/annurev-phyto-102313-050143 [DOI] [PubMed] [Google Scholar]
- Wang, M. , Weiberg, A. , Lin, F.M. , Thomma, B.P. , Huang, H.D. & Jin, H. (2016) Bidirectional cross‐kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nature Plants, 2, 16151. 10.1038/nplants.2016.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.L. , Lu, Q. , Decock, C. , Li, Y.X. & Zhang, X.Y. (2015) Cytospora species from Populus and Salix in China with C. davidiana sp. nov. Fungal Biology, 119, 420–432. 10.1016/j.funbio.2015.01.005 [DOI] [PubMed] [Google Scholar]
- Wang, Y. & Wang, Y. (2020) Oxalic acid metabolism contributes to full virulence and pycnidial development in the poplar canker fungus Cytospora chrysosperma . Phytopathology, 110, 1319–1325. 10.1094/PHYTO-10-19-0381-R [DOI] [PubMed] [Google Scholar]
- Wei, W. , Xiong, Y. , Zhu, W. , Wang, N. , Yang, G. & Peng, F. (2016) Colletotrichum higginsianum mitogen‐activated protein kinase ChMK1: role in growth, cell wall integrity, colony melanization, and pathogenicity. Frontiers in Microbiology, 7, 1212. 10.3389/fmicb.2016.01212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y. , Xu, L. , Liu, J. , Yin, Z. , Gao, X. , Feng, H. et al. (2017) A mitogen‐activated protein kinase gene (VmPmk1) regulates virulence and cell wall degrading enzyme expression in Valsa mali . Microbial Pathogenesis, 111, 298–306. 10.1016/j.micpath.2017.09.003 [DOI] [PubMed] [Google Scholar]
- Xu, J.R. & Hamer, J.E. (1996) MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea . Genes & Development, 10, 2696–2706. 10.1101/gad.10.21.2696 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Wang, X. , Li, Y. , Zeng, J. , Wang, G. , Deng, C. et al. (2018) Host‐induced gene silencing of a regulator of G protein signalling gene (VdRGS1) confers resistance to Verticillium wilt in cotton. Plant Biotechnology Journal, 16, 1629–1643. 10.1111/pbi.12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Z. , Liu, H. , Li, Z. , Ke, X. , Dou, D. , Gao, X. et al. (2015) Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytologist, 208, 1202–1216. 10.1111/nph.13544 [DOI] [PubMed] [Google Scholar]
- Yu, L. , Xiong, D. , Han, Z. , Liang, Y. & Tian, C. (2019) The mitogen‐activated protein kinase gene CcPmk1 is required for fungal growth, cell wall integrity and pathogenicity in Cytospora chrysosperma . Fungal Genetics and Biology, 128, 1–13. 10.1016/j.fgb.2019.03.005 [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Jin, Y. , Zhao, J.‐H. , Gao, F. , Zhou, B.‐J. , Fang, Y.‐Y. et al. (2016) Host‐induced gene silencing of the target gene in fungal cells confers effective resistance to the cotton wilt disease pathogen Verticillium dahliae . Molecular Plant, 9, 939–942. 10.1016/j.molp.2016.02.008 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Choi, Y.E. , Zou, X. & Xu, J.R. (2011) The FvMK1 mitogen‐activated protein kinase gene regulates conidiation, pathogenesis, and fumonisin production in Fusarium verticillioides . Fungal Genetics and Biology, 48, 71–79. 10.1016/j.fgb.2010.09.004 [DOI] [PubMed] [Google Scholar]
- Zheng, L. , Campbell, M. , Murphy, J. , Lam, S. & Xu, J.R. (2000) The BMP1 gene is essential for pathogenicity in the gray mold fungus Botrytis cinerea . Molecular Plant‐Microbe Interactions, 13, 724–732. 10.1094/MPMI.2000.13.7.724 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Phylogenetic analysis of Fus3/Kss1/Pmk1 from different species. The detailed information of the Fus3/Kss1/Pmk1 orthologs is listed in Tables 1 and S1
FIGURE S2 Principal component analysis of read counts of the wild‐type Cytospora chrysosperma and CcPmk1 deletion mutant revealed the clear separation of the two tested conditions and the proximity of biological replicates
FIGURE S3 Global view of gene expression level in CcPmk1 deletion mutant and wild type
FIGURE S4 GO annotation of overlapped down‐regulated genes in both CcPmk1 deletion mutant and MoPmk1 inhibition mutant in Magnaporthe oryzae. The yellow dotted box represents the significant enrichment GO terms. BP, biological process; CC, cellular component; MF, molecular function
FIGURE S5 Fluorescence microscopy of the hyphae treated with the artificial small interfering RNA, indicating that the small interfering RNA was successfully taken up by the hyphae
FIGURE S6 Sequence alignment of BcPmk1 and CcPmk1 fragments used for virus‐induced gene silencing. The same residues are shadowed in black
TABLE S1 The percentage of sequence identity between CcPmk1 and Pmk1 homologues in nonpathogenic fungi
TABLE S2 The secondary metabolism gene clusters in Cytospora chrysosperma predicted by antiSMASH
TABLE S3 The list of transcription factors regulated by CcPmk1
TABLE S4 The list of putative effector genes regulated by CcPmk1
TABLE S5 Primers used in this study
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.