

Abstract
The development of new chemical tools with improved properties is essential to chemical and cell biology. Of particular interest is the development of mimics of small molecules with important cellular function that allow the direct observation of their trafficking in a cell. To this end, a novel 15-azasterol has been designed and synthesized as a luminescent cholesterol mimic for the monitoring of cholesterol trafficking. The brightness of this probe, which is ~32-times greater than the widely used dehydroergosterol probe, is combined with resistance to photobleaching in solution and in human fibroblasts and an exceptionally large Stokes-like shift of ~150–200 nm. The photophysical properties of the probe have been studied experimentally and computationally, suggesting an intersystem crossing to the triplet excited state with subsequent phosphorescent decay. Molecular dynamics simulations show a similar binding mode of cholesterol and the azasterol probe to NPC proteins, demonstrating the structural similarity of the probe to cholesterol.
Graphical Abstract

INTRODUCTION
Cholesterol plays a vital role in the cell processes of eukaryotic organisms. It is important in cell signaling, regulating membrane fluidity, and biosynthesis of steroid hormones.1 Furthermore, the disruption of cholesterol trafficking results in its accumulation, which can lead to diseases such as Niemann-Pick type C (NPC) disease and other lipid-associated disorders.2 The understanding of cholesterol processing and transport in cells and organisms is therefore very important from both the basic chemical biology standpoint and the study of numerous disease states. The need for a method of monitoring cholesterol trafficking on a cellular level has resulted in the development of luminescent molecules that can be used as biological tools for the tracking of cholesterol. Recent reports of new probes confirm that this is an area of intense interest,3 with dozens of studies published every year. A major problem in the field is that strongly luminescent probes incorporate large fluorophores and thus fail to preserve key structural features of cholesterol, which can result in dissimilar interactions with proteins or in lipid bilayers.4 On the other hand, the photophysical properties of commonly used structural analogs of sterols leave much to be desired, especially under the conditions of long time-scale microscopy on living cells,4 during which the brightness of a probe fades significantly due to photodecomposition and photobleaching.
To serve as an effective cholesterol mimic, a chemical probe must be strongly luminescent at excitation wavelengths >300 nm and have high structural similarity to cholesterol. Dehydrosterols are a widely used type of probe for cholesterol tracking. Compounds such as dehydroergosterol (DHE) and cholestatrienol (CTL) (Figure 1) are structurally similar to cholesterol and intrinsically fluorescent, but their photophysical properties are not optimal.5–8 The low extinction coefficient (10500 M−1 cm−1 in methanol) and low fluorescence quantum yield (0.024 in methanol) of DHE combine to give low molecular brightness,9 which is even further diminished by rapid photobleaching. DHE is known to rapidly decompose upon exposure to ultraviolet (UV) light, even in deaerated solutions.10 These pitfalls can lead to poor imaging or require the use of large quantities of the probes. Consequently, there is an urgent need for a better analog that incorporates a strong fluorophore while avoiding photobleaching pathways.
Figure 1.

Structures of common dehydrosterols.
A variety of azasteroids have been reported to have biological activities, suggesting that they can interact with cholesterol binding sites. Common azasteroid scaffolds include functionalization of the aliphatic side chain as well as incorporation of amide,11–13 amine,11–14 imine,14–17 or pyrazole18 moieties in the carbocyclic steroid core (Figure 2a). These azasteroids have mainly been studied for their antibacterial and antifungal activities. Although some analogs incorporate fluorophores, their luminescent properties have to the best of our knowledge not been examined or exploited. A common type of luminescent azasteroid is based on an 8-azasteroid scaffold (Figure 2b), the fluorescent and phosphorescent properties of which have been extensively studied.19–25 However, their photophysical properties have not been optimized for use as chemical probes. In addition, their structures are not well-suited as probes in the study of cholesterol trafficking due to the lack of an aliphatic side chain at C17. This side chain plays a notable role in binding of cholesterol to protein targets26,27 and in membrane interactions.28 We thus designed a new luminescent azasterol probe that would possess the key structural features of cholesterol as well as superior photophysical properties. Desirable photophysical properties include strong emission upon irradiation with wavelengths above 300 nm to prevent interfering absorption by other cell components and UV damage to the cells under study, and stability for extended periods of time under irradiation to prevent photobleaching or undesired side reactions.
Figure 2.

(a) Representative examples of reported azasteroids and (b) luminescent 8-azasteroid scaffold.
RESULTS AND DISCUSSION
Our probe design is based on the replacement of the A and B rings of cholesterol by a naphthalene conjugated π-system as a means of introducing a fluorophore. We envisioned an improvement of the photophysical properties through this incorporation of an aromatic system with extended conjugation. A second design element to enhance the luminescent properties was to introduce a push–pull effect29,30 by replacing C15 with an imine nitrogen. These structural changes in analog 1 are highlighted in color in Figure 3. As discussed in the computational studies, we predicted that a conjugated imine system would undergo rapid intersystem crossing (ISC) to the triplet excited state, resulting in a lower energy emission that would be desirable to separate the excitation and detection wavelengths, thus increasing the signal/noise ratio in fluorescence microscopy. Rapid ISC is well-known for aromatic aldehydes and ketones, such as benzophenone,31 and has been more recently reported for an aromatic donor–acceptor imine.32
Figure 3.

Structure of cholesterol and proposed azasterol (1).
This design prompted the development of a convergent synthesis for 1 that is based on a Michael addition of an enolate to a nitroalkene to give nitroketone 7 (Scheme 1).33 Nitroalkene 5 could be obtained in 67% yield over four steps on a gram scale without intermediate chromatographic purification starting from readily available 1-bromo-3-methyl-butane and tert-butyl acrylate. Copper-catalyzed conjugate addition of isopentylmagnesium bromide to tert-butyl acrylate yielded ester 2, which was subjected to reduction with DIBAL to give aldehyde 3 in very good yield. Henry reaction of 3 with nitromethane followed by dehydration of nitrol 4 afforded nitroalkene 5 in excellent yield. The other building block, ketone 6, was obtained by a modified literature procedure from 6-methoxy-1-tetralone and methyl 4-bromocrotonate.16,34 The conjugate addition of the potassium enolate of 6 to 5 gave nitroketone 7 in 93% yield as a 4:1 mixture of diastereomers. Following Raney nickel-catalyzed hydrogenation of the nitro group and spontaneous cyclization to form the conjugated imine, the diastereomers could be separated via chromatography or crystallization. The relative stereochemistry of the major diastereomer was confirmed via X-ray crystallography (Figure S7). Deprotection of the methyl ether moiety of 8 with boron tribromide yielded 1 as a crystalline racemate in 22% overall yield over seven linear steps. The final three steps could be conducted on millimole scales without significant reduction in yield, allowing access to hundreds of milligrams of (±)-1, which is readily available for biological applications. While this racemate is useful for certain applications, the enantiomers may be separated by chiral HPLC (see Supporting Information) for cases in which the properties are dependent on stereochemistry. Absolute configuration assignments were made by X-ray crystallography (Figure S8) and circular dichroism (CD) spectroscopy (Figure S9).
Scheme 1.

Synthesis of Azasterol 1
Studies were performed to confirm that (±)-1 possessed desired luminescent properties. To give insight into the photophysical behavior of 1, we compared the luminescent properties in methanol and water as examples for hydrophilic environments and 1-octanol as a model for lipophilic environments such as membranes. The absorbance of 1 was similar in each solvent (Figure 4), and the absorbance maximum (λabs) was above 300 nm, which is important to reduce interfering absorption, autofluorescence, and UV damage in cell studies. While the longest wavelength absorption is similar to the known probes DHE9 and CTL,35 the molar extinction coefficient (ε) is 40–50% higher (Table 1). The long wavelength absorption in aryl imines is generally thought to derive from a n → π* excitation,36 which is confirmed by computational studies (Figure S11).
Figure 4.

Absorbance (top) and emission (bottom, λex = 310 nm) spectra of 10 μM solutions of 1 in water (blue), methanol (red), and octanol (green), measured at 22 °C.
Table 1.
Photophysical Properties of 1 and Other Luminescent Sterols
| probe | λabs (nm) | ε (M−1 cm−1) | λem (nm) | Φ |
|---|---|---|---|---|
| 1a | 321 | 15 000 | 504 | 0.03 |
| 1b | 308 | 15 000 | 514 | 0.54 |
| 1c | 316 | 15 100 | 526 | 0.10 |
| DHEa,9 | 324.4 | 10 500 | 372.5 | 0.024 |
| CTLd,35 | 324 | 11 250 | 374 | - |
Water as solvent.
Methanol as solvent.
Octanol as solvent.
Ethanol as solvent.
Significant differences in emission were observed under continuous irradiation in the different solvents (Figures 4 and 5). While emission was brightest in methanol, we were pleased to find that it was readily detectable in both water and octanol. Furthermore, the luminescence quantum yield (Φ) of 1 was 0.54 in methanol, giving a brightness that is 32-times that of DHE. The quantum yield of 1 in octanol remains higher than that of DHE in various solvents,9 suggesting that the improved brightness holds in lipophilic environments. Resistance to photobleaching was demonstrated by the ability of 1 to luminesce after 30 min of UV irradiation (Figure 5). Although the probe was designed to have a large shift in emission relative to excitation, the observed shift of ~200 nm was much larger than anticipated, exceeding the criteria of mega-Stokes shifts (>100 nm) desired for bioimaging.37,38 Mega-Stokes shifts of this magnitude are unusual for organic fluorophores, prompting further investigation of the photophysics of 1, which suggested that the observed shift in emission was not a true Stokes shift due to participation of a triplet excited state.
Figure 5.

Irradiation with hand-held UV lamp (254 nm) of 10 μM solutions of (left to right) 1 in water, 1 in methanol, 1 in octanol, DHE in methanol, and 1 in methanol after 30 min of UV irradiation.
Given the remarkable properties of 1, flash photolysis experiments were conducted to give more detailed information about the photophysical behavior. A deaerated solution of (±)-1 in methanol was irradiated at 308 nm in a stationary cell to produce the transient absorption spectrum shown in Figure 6. A trace was recorded at λob = 360 nm to show the decay of this signal with second order kinetics (Figure S3). The decay of the emission spectrum at λob = 360 nm approximately follows second-order kinetics, with small deviations due to some contribution of decay with first-order kinetics (Figure S4). The second-order kinetics observed for the decay of the transient absorption and emission spectra can be attributed to a triplet–triplet annihilation reaction. The accessibility of the triplet state for aryl imines is well-known in the literature36 and confirms our original design of a system undergoing rapid ISC. It should also be noted that prior to deaeration, the luminescence was slightly quenched by triplet oxygen, giving further evidence for the presence of a triplet excited state. It is important to note that the stability of the baseline absorbance in the stationary cell confirmed that 1 was resistant to photobleaching. This is observed even in the presence of oxygen (Figure 5) and is another important quality for the use of 1 as a biological probe and a significant improvement over the DHE and CTL probes.
Figure 6.

Transient absorption of (±)-1 in methanol upon irradiation at 308 nm.
In an effort to gain a better understanding of the observed photophysical properties of 1, specifically the structural elements responsible for the large Stokes-like shift in emission and the unusual stability of the triplet state even in the presence of oxygen, a computational study was completed using Gaussian 16.39 After benchmarking a number of functionals and basis sets (see Tables S1 and S2 and Figure S5 in the Supporting Information) against the experimentally observed spectra, the structure of 1 in the S0 state was optimized using B3LYP-D3/6-311++G** and solvent effects of n-octanol were included using the polarizable continuum model (PCM). Vertical excitation energies and oscillator strengths were calculated for the 15 lowest singlet and triplet excited states using time-dependent density functional theory (TDDFT) (Table S3). These results were used to simulate absorption spectra using Gaussian functions with a full width at half-maximum (fwhm) of 0.3 eV to convolute the peaks. The calculated absorption spectrum shows good agreement with experiment (Figure 7). As expected, the first excited state corresponds to the n → π* transition (Figure S6).
Figure 7.

Experimental (green) and calculated (black) absorption spectra of 1 in 1-octanol. Gaussian functions with a fwhm of 0.3 eV were used to convolute the calculated stick spectrum.
The structures of the S1 and T1 states in 1-octanol were optimized using TDDFT/PCM/B3LYP-D3/6-311++G** and ∆SCF/PCM/B3LYP-D3/6-311++G**, respectively. Vertical emission energies were calculated and compared to experiment (Table 2). On the basis of these results, the higher energy emission at λ = 365 nm in octanol is assigned to the emission from the S1 state. This emission is not observed experimentally in methanol or water, both of which show only emission from the T1 state with wavelength longer than 500 nm. In analogy to the photochemistry of benzophenone,40 the T1 (n → π*) state is generated through rapid ISC from the S1 (n → π*) and then undergoes radiative decay (phosphorescence) to the S0 state. This observation that an emission from the S1 state is observed in octanol but not in water or methanol may be explained by a narrowing of the singlet–triplet energy gap in more polar solvents, which is known to increase the rate of intersystem crossing.41
Table 2.
Comparison of Experimental and Calculated Vertical Emission Wavelengths in 1-Octanol
| transition | λexp (nm) | λcalc (nm) |
|---|---|---|
| S0 ← S1 | 365 | 374 |
| S0 ← T1 | 527 | 581 |
We considered two hypotheses to explain the large Stokes-like shift in emission wavelength observed for the decay from T1 to S0: (i) significant changes in the electron distribution could lead to a solvent reorganization that further lowers the energy of the T1 state relative to the S1 state and (ii) changes in the geometry of the molecule could lead to a vibronic relaxation upon ISC. Comparisons of Hirshfeld charges and geometries of S0, S1, and T1 showed no notable changes in charges. In contrast, significant differences in the geometries were calculated for the three states, especially in the length of the C8–C9 bond, as shown in Figure 8 for the S0 and T1 states. This bond is elongated upon excitation from the S0 to the S1 state and is longer yet in the optimized geometry of the T1 state. Meanwhile, the C8–C14 bond shortens and the C14–N15 bond lengthens. These changes can be represented by the resonance structure shown in Figure 8, which is also supported by our calculation that the greatest spin density is on C9 and N15. The aromaticity in the ring is decreased, but conjugation is extended through participation of the imine moiety, which has adopted a more enamine-like geometry. These results are in line with the low energy of the T1 state leading to the large Stokes shift as well as its relative stability in the presence of oxygen.
Figure 8.

Highest occupied molecular orbitals of 1 in the optimized geometries of the S0 and T1 states. The most notable change is in the length of the C8–C9 bond (denoted by arrow). We rationalize this change in geometry with contribution from the resonance structure shown.
Live cell imaging and bleaching analysis studies were performed to demonstrate the improved photophysical properties of 1 in a cellular environment compared to CTL. In human fibroblasts, 1 showed weak emission in the UV but very strong emission over the whole spectral range when excited between 320 and 340 nm. Furthermore, 1 was shown to bleach very slowly compared to CTL (Figure 9). Pixel-wise bleaching analysis using a stretched exponential fitting function showed that CTL bleaches slightly faster than exponential and fades after a few seconds, whereas bleaching of 1 has a strongly compressed exponential with a much longer time constant (Figure S10). Because of a combination of the strong signal in the absence of a UV emission filter and the long delay before significant photobleaching takes place, we conclude that 1 can be imaged for a much longer time than probes such as CTL.
Figure 9.

Photobleaching kinetics of azasterol and cholestatrienol (CTL) in cells acquired in the UV channel are shown in A. A corresponding bleach profile for a bleach stack of azasterol acquired without emission filter is shown in B.
Having demonstrated the superior photophysical properties of 1 over DHE and CTL, we sought to investigate whether our probe is sufficiently similar to cholesterol for use in protein binding studies. To study the similarity of 1 to cholesterol and their interaction to biomedically relevant cholesterol binding proteins, we compared the binding of 1 and cholesterol to NPC1 and NPC2, two key lysosomal proteins essential for maintenance of proper cholesterol homeostasis.42 The crystal structures of cholesterol bound to the N-terminal domain of NPC1 (PDB ID: 3GKI, 1.80 Å)43 and to NPC2 (PDB ID: 2HKA, 1.81 Å)42 have been determined. It is known from these structures that the hydroxyl group and the aliphatic side chain of cholesterol are key in binding to NPC1 and NPC2, respectively, suggesting that these moieties need to be preserved in a successful cholesterol mimic. Docking followed by 100 ns molecular dynamics (MD) simulations of 1 and cholesterol to NPC1 and NPC2 proteins showed that 1 overlays well with cholesterol in the binding sites (Figure 10 and Figures S11–S15 in the Supporting Information). Like the H-bonding interactions observed for cholesterol with ASN 41 of NPC1, the hydroxyl group of 1 also indicated H-bonding with the side chain amide of the ASN 41 residue (2.06 Å). Moreover, one of the aromatic rings of 1 also indicated π–π interactions with the PHE 203 residue of NPC1 (Figure S11). The free energies of binding for 1 and cholesterol in NPC proteins were estimated using MM-GBSA rescoring of snapshots from a 100 ns MD simulation44 and were found to be similar (Table S5), further supporting our hypothesis that 1 is an effective mimic of cholesterol. These results strongly suggest that modification of the polycyclic core and removal of the C21 methyl group do not significantly affect binding.
Figure 10.
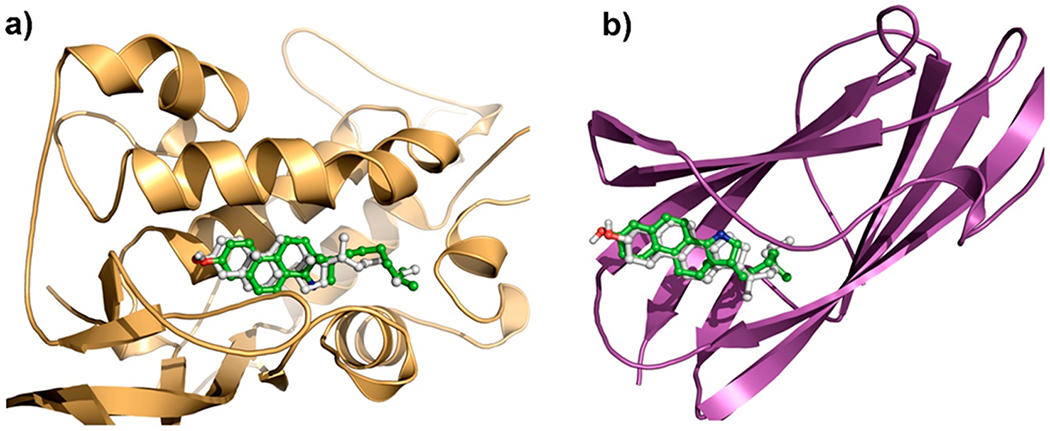
Overlay of cholesterol (gray) and azasterol (1, green) docked in (a) NPC1 and (b) NPC2.
CONCLUSIONS
A novel 15-azasterol 1 was designed as a biochemical tool for the study of cholesterol trafficking and synthesized in 22% overall yield in seven linear steps in high purity and on a scale of 100s of mg. Our probe incorporates a strong fluorophore as well as key structural properties to serve as an effective luminescent cholesterol mimic without issues of photobleaching. The probe was shown to have desirable photophysical properties including absorption at ~310–320 nm, a large Stokes-like shift of ~150–200 nm due to ISC and vibronic relaxation, emission in the visible region at ~450–525 nm with a quantum yield of 0.54 (methanol), and resistance to photobleaching upon UV irradiation in a stationary cell and in human fibroblasts. This new probe has the potential to be used in a variety of applications related to lysosomal storage disorders and other cholesterol-related diseases, including time-lapse microscopy, for which use of the DHE probe is very limited.4,45 Our probe is applicable to the study of intracellular cholesterol trafficking but may also prove useful in other realms in which luminescent sterols are used such as the study of atherosclerosis and Alzheimer’s disease.46 Efforts toward an asymmetric synthesis of 1 are currently underway to increase synthetic efficiency for applications where only the native sterol stereochemistry is desired. Future work will also include experimental investigation of the binding of 1 in NPC proteins. Further studies in biological systems will be published in the future.
EXPERIMENTAL SECTION
General Methods.
All reactions were conducted in oven-dried glassware under an atmosphere of argon unless otherwise specified. Tetrahydrofuran, toluene, and dichloromethane were purified by passage through an Inert PureSolv Micro solvent purification system. Other solvents were purified by literature procedures or used as received. All other reagents were used as received from commercial sources unless otherwise stated. MPLC was performed to obtain analytically pure material using a Biotage Isolera Prime (Version 1.5.2) system with Silicycle, Inc. SiliaSep cartridges or Biotage SNAP KP-SIL cartridges. Corrected melting points were obtained using a Mel-Temp II device. 1H and 13C spectra were obtained using either a Bruker AVANCE III HD 400 instrument operating at 400 and 101 MHz, respectively, or a Bruker AVANCE II 800 instrument operating at 800 and 201 MHz, respectively. All 1H and 13C spectral data are reported in ppm (δ) relative to the residual solvent peak for CDCl3 at 7.26 and 77.23 ppm, respectively. Coupling constants (J) are reported in Hz. High-resolution mass spectrometry (HRMS) was performed on a Bruker micrOTOF-Q II instrument. Infrared spectra were recorded on a Thermo Nicolet IR 200 spectrometer. X-ray diffraction collections were obtained on a Bruker APEX-II diffractometer or a Bruker PHOTON-II diffractometer. Absorbance spectra were collected using an Evolution 201 UV–vis spectrometer with ThermoInsight software. Luminescence spectra were collected using a Horiba Fluoromax-4 fluorometer with FluorEssence software. Chiral HPLC was performed using a Waters 1525 Binary HPLC pump and Waters 2487 dual λ absorbance detector with Breeze software and a Chiralcel OZ-H column purchased from Chiral Technologies, Inc. Circular dichroism (CD) spectra were collected using a Jasco J-815 spectrometer. 7-Methoxy-3,4-dihydrophenanthren-1(2H)-one was prepared from 6-methoxy-1-tetralone and methyl 4-bromocrotonate according to a literature procedure.34
tert-Butyl 6-methylheptanoate (2).
A 100 mL round-bottom flask with a stir bar was charged with freshly ground turnings of magnesium (365 mg, 15 mmol) and a crystal of iodine. The flask was capped with a septum, evacuated, and backfilled with argon (3×). THF (15 mL) was added, and 1-bromo-3-methylbutane (1.8 mL, 15 mmol) was added dropwise via syringe with stirring. The yellow solution turned colorless and then dark and cloudy with heat generation upon initiation. The reaction was stirred for 2 h at 22 °C. Then the septum was removed, and DMAP (2.443 g, 20 mmol) and copper(I) bromide-dimethyl sulfide complex (206 mg, 1 mmol) were quickly added. The reaction turned dark blue immediately. The flask was fitted with an addition funnel, which was capped with a septum and placed under an argon atmosphere. The flask was cooled to −78 °C, and the addition funnel was charged with tert-butylacrylate (1.47 mL, 10 mmol), TMSCl (2.54 mL, 20 mmol), and THF (2 mL). This solution was added dropwise with vigorous stirring, and then the addition funnel was rinsed with additional THF (2 mL). The reaction mixture was allowed to warm to 22 °C and stirred for 14 h. The reaction was quenched with 1 M HCl (70 mL) and extracted with diethyl ether (3 × 50 mL). The combined organic layers were dried over anhyd Na2SO4 and concentrated on a rotary evaporator to give a slightly yellow oil. The crude material was passed through a silica plug, eluting with 20% diethyl ether in hexanes, and then concentrated to give 1.67 g (83%) of the title compound as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 2.20 (t, J = 7.5 Hz, 2H), 1.59–1.49 (m, 3H), 1.44 (s, 9H), 1.33–1.25 (m, 2H), 1.20–1.14 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (201 MHz, CDCl3) δ 173.5, 80.0, 38.8, 35.9, 28.3, 28.0, 27.0, 25.6, 22.8; HRMS (ESI) calcd for C12H24NaO2 [M + Na] 223.1669, found 223.1661; IR (neat) 2955, 2933, 2870, 1731, 1366, 1151 cm−1.
6-Methylheptanal (3).
A 100 mL round-bottom flask with a stir bar was charged with ester 2 (2.145g, 10.7 mmol). The flask was capped with a septum, evacuated, and backfilled with argon (3×). DCM (27 mL) was added and the solution cooled to −78 °C. DIBALH (10.7 mL of 1 M solution in DCM, 10.7 mmol) was added dropwise via syringe. The reaction was stirred at −78 °C for 15 min, then warmed to 22 °C and stirred for an additional 5 min. Next, 1 M HCl was added carefully until all aluminum salts were dissolved. The layers were separated, and the aq layer was extracted with DCM (3 × 25 mL). The combined organic layers were dried over anhyd Na2SO4 and concentrated to give a clear fragrant liquid, which was partially purified by passing through a silica plug with DCM and carefully concentrated to give the title compound (1.213 g, 88%) as a clear, colorless liquid. Because of volatility issues, this compound was used directly in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 9.76 (t, J =1.9, 1H), 2.42 (td, J = 7.4, 1.9 Hz, 2H), 1.61 (quint, J = 7.5 Hz, 2H), 1.53 (nonet, J = 6.6 Hz, 1H), 1.36–1.28 (m, 2H), 1.21–1.15 (m, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (201 MHz, CDCl3) δ 203.1, 44.2, 38.9, 28.0, 27.2, 22.8, 22.5; IR (neat) 2954, 2931, 2869, 2716, 1726, 1467, 1366, 1155 cm−1. (lit.47 1H NMR, 13C NMR)
(E)-7-Methyl-1-nitrooct-1-ene (5).
A 100 mL round-bottom flask with a stir bar was charged with aldehyde 3 (1.201 g, 9.4 mmol) and methanol (3.7 mL) and cooled to 0 °C in an ice bath. Nitromethane (502 μL, 9.4 mmol) was added dropwise with stirring, and then a solution of sodium hydroxide (588 mg, 14.7 mmol) in water (7.3 mL) was added dropwise. The reaction was stirred at 0 °C for 1 h and then diluted with water (9.2 mL). Conc hydrochloric acid (2.1 mL) was added slowly, and the mixture was extracted with diethyl ether. The combined organic layers were washed with brine, dried over anhyd Na2SO4, and concentrated to give a pale yellow oil (1.725 g, 97%), which was used directly in the next step without further purification. An analytically pure sample was obtained by MPLC (7–10% ethyl acetate in hexane) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.43 (dd, J = 12.7, 2.7 Hz, 1H), 4.37 (dd, J = 12.7, 8.3 Hz, 1H), 4.33–4.27 (m, 1H), 2.65 (s, 1H), 1.58–1.41 (m, 4H), 1.38–1.24 (m, 3H), 1.17 (q, J = 7.1 Hz, 2H), 0.86 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 80.9, 68.9, 38.9, 34.0, 28.1, 27.3, 25.6, 22.8, 22.7; IR (neat) 3411, 2952, 2930, 2867, 1551, 1384 cm−1.
A 100 mL round-bottom flask with a stir bar was charged with nitrol 4 (1.725 g, 9.1 mmol) and DCM (5.4 mL). Trifluoroacetic anhydride (1.27 mL, 9.1 mmol) was added dropwise with stirring. Then the reaction was cooled to −10 °C in an ice/brine bath and stirred for 2 min. Triethylamine (2.54 mL, 18.2 mmol) was added slowly dropwise over several min. The reaction was stirred at −10 °C for an additional 30 min and then was diluted with DCM (6.1 mL) and washed with satd aq ammonium chloride. The organic layer was dried over anhyd Na2SO4 and concentrated to give a crude yellow–orange oil. The crude material was purified by MPLC (2–5% DCM in hexane) to give the title compound (1.482 g, 95%) as a colorless oil. The yield over three steps was 81% based on the starting ester. 1H NMR (400 MHz, CDCl3) δ 7.28 (dt, J = 13.7, 7.2 Hz, 1H), 6.98 (d, J = 13.4 Hz, 1H), 2.27 (qd, J = 7.2, 1.3 Hz, 2H), 1.58–1.46 (m, 3H), 1.37–1.30 (m, 2H), 1.21–1.15 (m, 2H), 0.87 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 143.0, 139.8, 38.7, 28.7, 28.2, 28.0, 27.1, 22.7; HRMS (ESI) calcd for C9H17NO2 [M + H] 172.1332, found 172.1337; IR (neat) 3105, 2954, 2930, 2867, 1650, 1524, 1467, 1350 cm−1.
7-Methoxy-2-methyl-3,4-dihydrophenanthren-1(2H)-one(6)
A 10 mL crimp cap vial with a stir bar was capped, evacuated, and backfilled with argon (3×). LiHMDS (0.42 mL of 1 M solution in THF, 0.42 mmol) and THF (2.5 mL) were added, and the solution was cooled to 0 °C. A solution of 7-methoxy-3,4-dihydrophenanthren-1(2H)-one34 (91 mg, 0.40 mmol in 1.5 mL THF) was added dropwise. The solution was stirred at 0 °C for 15 min, and then iodomethane (27.4 μL, 0.44 mmol) was added dropwise to the enolate solution. The reaction mixture was warmed to 22 °C, stirred for 14 h, quenched with satd aq NH4Cl, and extracted 3 times with DCM. The combined organic layers were washed with brine, dried over anhyd Na2SO4, and concentrated to a give an orange oil that solidified upon standing. The crude material was purified by MPLC (7–15% ethyl acetate in hexane) to give 80 mg (83%) of the title compound as a white crystalline solid, which was recrystallized from hexane to give colorless needles. Mp = 108–109 °C; 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.20 (dd, J = 9.2, 2.2 Hz, 1H), 7.13 (d, J = 2.3 Hz, 1H), 3.93 (s, 3H), 3.47 (dt, J = 17.2, 4.1 Hz, 1H), 3.20 (ddd, J = 17.3, 10.9, 4.8 Hz, 1H), 2.65 (m, 1H), 2.34 (dq, J = 13.1, 4.4 Hz, 1H), 1.93 (qd, J = 20.2, 4.5 Hz, 1H), 1.31 (d, J = 6.7 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 200.9, 159.6, 142.6, 137.6, 128.2, 126.5, 126.0, 124.1, 119.1, 107.1, 55.6, 41.7, 30.9, 25.2, 15.6. (lit.16 1H NMR)
7-Methoxy-2-methyl-2-(7-methyl-1-nitrooctan-2-yl)-3,4-di-hydrophenanthren-1(2H)-one (7).
A 25 mL round-bottom flask with a stir bar was charged with ketone 6 (240 mg, 1 mmol). The flask was capped with a septum, evacuated, and backfilled with argon (3×). THF (13.5 mL) was added, and the solution was cooled to −78 °C. KHMDS (2.2 mL of 0.5 M solution in toluene, 1.1 mmol) was added dropwise via syringe, and the solution turned dark red. The solution was stirred at −78 °C for 30 min, and then a solution of nitroalkene 5 (188 mg, 1.1 mmol in 1 mL THF) was added dropwise to the enolate solution. The solution turned from dark red to yellow and was allowed to stir at −78 °C for an additional 30 min. The reaction mixture was warmed to 22 °C, quenched with satd aq NH4Cl, and extracted 3 times with DCM. The combined organic layers were washed with brine, dried over anhyd Na2SO4, and concentrated to a give a pale yellow oil. The crude material was purified by MPLC (2–7% ethyl acetate in hexane) to give 381 mg (93%) of the title compound in 4:1 d.r. as a thick colorless oil. The diastereomers were characterized as a mixture. Major diastereomer: 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 9.2 Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.23 (dd, J = 9.2, 2.4 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 4.55 (dd, J = 13.3, 4.8 Hz, 1H), 4.30 (dd, J = 13.3, 6.5 Hz, 1H), 3.95 (s, 3H), 3.42–3.25 (m, 1H), 2.94–2.89 (m, 1H), 2.33 (ddd, J = 13.9, 8.2, 5.6 Hz, 1H), 2.05–1.99 (m, 1H), 1.56–1.43 (m, 2H), 1.42–1.26 (m, 4H), 1.20 (s, 3H), 1.16–1.11 (m, 3H), 0.85 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 201.1, 159.9, 141.2, 137.8, 126.52, 126.47, 126.3, 124.6, 119.4, 107.2, 77.3, 55.6, 47.1, 41.0, 38.9, 30.6, 30.4, 28.5, 28.1, 27.8, 22.8, 21.9, 19.5; Minor diastereomer: 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.7 Hz, 1H), 8.02 (d, J = 9.2 Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.23 (dd, J = 9.2, 2.4 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 4.68 (dd, J = 13.0, 4.1 Hz, 1H), 4.43 (dd, J = 13.0, 7.4 Hz, 1H), 3.95 (s, 3H), 3.52–3.44 (m, 1H), 2.70 (sept, J = 3.9 Hz, 1H), 2.26 (ddd, J = 13.9, 8.2, 5.6 Hz, 1H), 2.12–2.07 (m, 1H), 1.56–1.43 (m, 2H), 1.42–1.26 (m, 4H), 1.24 (s, 3H), 1.07–1.05 (m, 3H), 0.79 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (101 MHz, CDCl3) δ 201.4, 141.1, 137.7, 137.3, 127.4, 127.1, 126.6, 119.5, 78.5, 42.3, 38.8, 31.8, 29.6, 28.6, 28.0, 27.6, 22.73, 22.67, 22.2, 19.7, other resonances may be indistinguishable from those corresponding to the major diastereomer; HRMS (ESI) calcd for C25H34NO4 [M + H] 412.2482, found 412.2491; IR (neat) 2932, 2866, 1666, 1619, 1550, 1223, 1033, 857, 821, 780 cm−1.
7-Methoxy-11a-methyl-1-(5-methylhexyl)-2,10,11,11a-tetrahydro-1H-naphtho[1,2-g]indole (8).
A 250 mL round-bottom flask with a stir bar was charged with nitroketone 7 (380 mg, 0.92 mmol) and ethanol (10 mL), evacuated, and backfilled with argon. Raney nickel (880 mg of ~50% slurry in water) was washed with water (3 × 2 mL) and then ethanol (3 × 2 mL). The washed Raney nickel was suspended in ethanol (4 mL) and transferred via pipet into the reaction flask with rapid stirring. The flask was evacuated and backfilled with a balloon of hydrogen gas (3×). The reaction was allowed to stir under H2 (1 atm) at 22 °C for 7 h. The flask was evacuated and backfilled with argon, and the reaction mixture was diluted with DCM, filtered through Celite, and concentrated to give a pale yellow oil. The crude material was purified by MPLC (15% ethyl acetate in hexane) to give 310 mg (92%) combined yield of the separated diastereomers of the title compound as a white solid. The major diastereomer was recrystallized from hexane to yield colorless crystals for characterization. Single crystals suitable for X-ray diffraction analysis were obtained as colorless plates via cooling of a concentrated solution in diethyl ether. mp = 97–98 °C; 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.63 (d, J = 8.6 Hz, 1H), 7.19 (dd, J = 9.2, 2.1 Hz, 1H), 7.15 (d, J = 2.1 Hz, 1H), 4.17 (dd, J = 15.2, 7.4 Hz, 1H), 3.93 (s, 3H), 3.47 (dd, J = 15.1, 10.8 Hz, 1H), 3.39 (dd, J = 17.8, 5.7 Hz, 1H), 3.24 (ddd, J = 17.7, 12.5, 5.3 Hz, 1H), 2.25 (dd, J = 12.8, 4.8 Hz, 1H), 2.05 (quint, J =8.1 Hz, 1H), 1.84 (td, J = 12.7, 5.8 Hz, 1H), 1.56 (sept, J = 6.6Hz, 1H), 1.49–1.43 (m, 1H), 1.40–1.32 (m, 4H), 1.24–1.19 (m, 2H), 0.97 (s, 3H), 0.89 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (201 MHz, CDCl3) δ 179.3, 158.6, 136.6, 136.3, 127.2, 126.1, 126.0, 125.2, 124.1, 118.8, 107.3, 63.9, 55.6, 50.7, 47.9, 39.2, 35.3, 29.3, 28.2, 28.11, 28.07, 23.6, 22.89, 22.85, 14.2; HRMS (ESI) calcd for C25H34NO [M + H] 364.2635, found 364.2646; IR (solid) 2924, 2843, 1615, 1248, 1033, 855 cm−1.
11a-Methyl-1-(5-methylhexyl)-2,10,11,11a-tetrahydro-1H-naphtho[1,2-g]indol-7-ol (1).
A 25 mL round-bottom flask with a stir bar was charged with methyl ether 8 (271 mg, 0.75 mmol). The flask was capped, evacuated, and backfilled with argon (3×). DCM (7.5 mL) was added, and the solution was stirred at 0 °C in an ice bath. Boron tribromide (1.5 mL of 1 M solution in DCM, 0.198 mmol) was added dropwise over 5 min via syringe, the yellow solution was allowed to warm to 22 °C, and then it was heated to 30 °C in a sand bath for 6 h. The reaction mixture was quenched with water and extracted 3 times with DCM. The organic layers were combined, dried over anhyd Na2SO4, and concentrated on a rotary evaporator to give a crude yellow solid. The crude material was purified by MPLC (3–10% methanol in DCM) to give the title compound as a powdery solid (252 mg, 96%), which was recrystallized from methanol/DCM to give colorless crystals of a single diastereomer. Mp = 206–207 °C (decomp.); 1H NMR (800 MHz, CDCl3) δ 8.03 (d, J = 8.7 Hz, 1H), 7.95 (d, J = 9.2 Hz, 1H), 7.41 (d, J = 8.7 Hz, 1H), 7.16 (dd, J = 9.1, 2.6 Hz, 1H), 7.09 (d, J = 2.6 Hz, 1H), 4.19 (dd, J = 14.8, 7.4 Hz, 1H), 3.50 (dd, J = 14.4, 10.8 Hz, 1H), 3.40 (dd, J = 17.6, 4.8 Hz, 1h), 3.25 (ddd, J = 17.8, 12.4, 5.5 Hz, 1H), 2.27 (ddd, J = 12.9, 5.6, 1.5 Hz, 1H), 2.08 (dddd, J = 10.5, 9.2, 7.4, 5.9 Hz, 1H), 1.87 (td, J = 12.9, 5.8 Hz, 1H), 1.58–1.51 (m, 2H), 1.47–1.43 (m, 1H), 1.39–1.31 (m, 4H), 1.22–1.18 (m, 2H), 0.99 (s, 3H), 0.88 (d, J = 6.6 Hz, 6H); 13C{1H} NMR (201 MHz, CDCl3) δ 179.8, 155.4, 137.0, 136.5, 126.9, 126.4, 125.9, 124.6, 124.1, 118.4, 111.0, 63.5, 51.1, 50.6, 48.1, 39.2, 35.2, 29.3, 28.2, 28.1, 23.6, 22.89, 22.86, 14.2; HRMS (ESI) calcd for C24H32NO [M + H] 350.2478, found 350.2481; IR (solid) 3100, 2955, 2927, 2857, 1613, 1391, 1231, 1028, 857 cm−1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. A. Oliver for crystallographic assistance, Dr. B. Smith for access to a fluorometer, J. Burton for assistance with the synthesis, and the Biophysics Instrumentation Core Facility for use of the CD spectrometer. E.M.W. thanks the Arthur J. Schmitt Foundation for fellowship support. D.W. acknowledges technical assistance from Maria Szomek and thanks Dr. Douglas Covey (Washington University, St. Louis) for kindly providing CTL. We gratefully acknowledge financial support from the National Institutes of Health (NS092653), the Ara Parseghian Medical Research Foundation, and the Danish Council for Independent Research (Grant No. DFF-7014-00054).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c02460.
Analytical data and NMR spectra for all new compounds; photophysical studies; live cell imaging and bleaching analysis; computational details (PDF)
Accession Codes
CCDC 2016041 and 2022200 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
DEDICATION
We dedicate this paper to Professor Robert M. Carlson on the occasion of his 80th birthday in 2020.
Contributor Information
Emily M. Work, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States
Guillermo Ferraudi, Radiation Research Laboratory, University of Notre Dame, Notre Dame, Indiana 46556, United States.
Luke Kiefer, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
Gang Liu, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
Michael Grigalunas, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
Atul Bhardwaj, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
Rasmin Kaur, Department of Biochemistry and Molecular Biology, University of Southern Denmark, DK-5230 Odense M, Denmark.
Janel M. Dempsey, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States
Daniel Wüstner, Department of Biochemistry and Molecular Biology, University of Southern Denmark, DK-5230 Odense M, Denmark.
Paul Helquist, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
Olaf Wiest, Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46566, United States.
REFERENCES
- (1).Hu J; Zhang Z; Shen WJ; Azhar S Cellular Cholesterol Delivery, Intracellular Processing and Utilization for Biosynthesis of Steroid Hormones. Nutr. Metab 2010, 7, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Maxfield FR; Tabas I Role of Cholesterol and Lipid Organization in Disease. Nature 2005, 438, 612–621. [DOI] [PubMed] [Google Scholar]
- (3).Gupta A; Rivera-Molina F; Xi Z; Toomre D; Schepartz A Endosome Motility Defects Revealed at Super-Resolution in Live Cells Using HIDE Probes. Nat. Chem. Biol 2020, 16, 408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Maxfield FR; Wüstner D Analysis of Cholesterol Trafficking with Fluorescent Probes. Methods Cell Biol. 2012, 108, 367–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mukherjee S; Zha X; Tabas I; Maxfield FR Cholesterol Distribution in Living Cells: Fluorescence Imaging Using Dehydroergosterol as a Fluorescent Cholesterol Analog. Biophys. J 1998, 75, 1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Scheidt HA; Müller P; Herrmann A; Huster D The Potential of Fluorescent and Spin-Labeled Steroid Analogs to Mimic Natural Cholesterol. J. Biol. Chem 2003, 278, 45563–45569. [DOI] [PubMed] [Google Scholar]
- (7).Solanko KA; Modzel M; Solanko LM; Wüstner D Fluorescent Sterols and Cholesteryl Esters as Probes for Intracellular Cholesterol Transport. Lipid Insights 2015, 2015, 95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sezgin E; Can FB; Schneider F; Clausen MP; Galiani S; Stanly TA; Waithe D; Colaco A; Honigmann A; Wüstner D; Platt F; Eggeling C A Comparative Study on Fluorescent Cholesterol Analogs as Versatile Cellular Reporters. J. Lipid Res 2016, 57, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Smutzer G; Crawford BF; Yeagle PL Physical Properties of the Fluorescent Sterol Probe Dehydroergosterol. Biochim. Biophys. Acta, Biomembr 1986, 862, 361–371. [DOI] [PubMed] [Google Scholar]
- (10).Morton RA; Heilbron IM; Spring FS Absorption Spectra in Relation to Vitamin A. Biochem. J 1930, 24, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Martins-Duarte ÉS; Lemgruber L; Lorente SO; Gros L; Magaraci F; Gilbert IH; De Souza W; Vommaro RC Evaluation of Three Novel Azasterols against Toxoplasma Gondii. Vet. Parasitol 2011, 177, 157–161. [DOI] [PubMed] [Google Scholar]
- (12).Gigante F; Kaiser M; Brun R; Gilbert IH SAR Studies on Azasterols as Potential Anti-Trypanosomal and Anti-Leishmanial Agents. Bioorg. Med. Chem 2009, 17, 5950–5961. [DOI] [PubMed] [Google Scholar]
- (13).Gros L; Lorente SO; Jimenez CJ; Yardley V; Rattray L; Wharton H; Little S; Croft SL; Ruiz-Perez LM; Gonzalez-Pacanowska D; Gilbert IH Evaluation of Azasterols as Anti-Parasitics. J. Med. Chem 2006, 49, 6094–6103.17004723 [Google Scholar]
- (14).Alonso F; Cirigliano AM; Cabrera GM; Ramírez JA Synthesis and Preliminary Biological Screening of Sterol Analogues as New Antifungal Agents against Plant Pathogens. Steroids 2010, 75, 659–664. [DOI] [PubMed] [Google Scholar]
- (15).Woloshuk CP; Sisler HD; Dutky SR Mode of Action of the Azasteroid Antibiotic 15-Aza-24-Methylene-D-Homocholesta-8,14-Dien-3B-Ol in Ustilago Maydis. Antimicrob. Agents Chemother 1979, 16, 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Berlin KD; Morgan JG; Durham NN; Chestnut RW Synthesis of 1,10,11,11a-Tetrahydro-11a-Methyl-2H-Naphth[1,2-g]-Indol-7-Ol, an Equilenin-Like 15-Aza Steroid. J. Org. Chem 1971, 36, 1599–1602. [DOI] [PubMed] [Google Scholar]
- (17).Chesnut RW; Durham NN; Brown RA; Mawdsley EA; Berlin KD Antibacterial Activity of 15-Azasteroids Alone and in Combination with Antibiotics. Steroids 1976, 27, 525–541. [DOI] [PubMed] [Google Scholar]
- (18).Mawdsley EA; Berlin KD; Chesnut RW; Durham NN Synthesis and Structure-Activity Relations of Heterocyclic Compounds Containing a Trimethoxyarene Function. J. Med. Chem 1976, 19, 239–243. [DOI] [PubMed] [Google Scholar]
- (19).Borisevich NA; Raichyonok TF; Sukhodola AA; Tolstorozhev GB; Shashilov VA Absorption and Fluorescence of 8-Azasteroids in the Gas Phase. J. Appl Spectrosc 2005, 72, 49–58. [Google Scholar]
- (20).Dubovskii VL; Gulyakevich OV; Mikhal’chuk AL; Raichenok TF; Tikhomirov SA; Tolstorozhev GB Spectral and Kinetic Properties of an Immunoactive 8-Azasteroid and a Product of Its Photochemical Transformation in Aqueous Solutions. Opt. Spectrosc 2008, 104, 875–885. [Google Scholar]
- (21).Borisevich NA; Raichyonok TF; Sukhodola AA; Tolstorozhev GB Delayed Fluorescence and Phosphorescence of 8-Aza-D-Homogonane in the Gas and Condensed Phases. J. Fluoresc 2006, 16, 649–653. [DOI] [PubMed] [Google Scholar]
- (22).Dubouski VL; Gulyakevich OV; Mikhal’chuk AL; Raichenok TF; Tikhomirov SA; Tolstorozhev GB Spectral and Luminescent Properties of 2,3-Dimethoxy-8-Aza-D-Homogona-1,3,5(10),13-Tetraene-12,17a-Dione and Its Delta9,11 Derivative. Opt. Spectrosc 2006, 101, 882–888. [Google Scholar]
- (23).Bazyl OK; Artyukhov VY; Mayer GV A Quantum-Chemical Study of the Spectral and Luminescent Properties of Photoproducts of Some 8-Azasteroids. Opt. Spectrosc 2008, 104, 172–179. [Google Scholar]
- (24).Basyl OK; Artykhov VY; Mayer GV Proton-Acceptor Properties of 8-Azasteroids. Russ. Phys. J 2005, 48, 196–199. [Google Scholar]
- (25).Bagnich SA; Khropik NN; Knyukshto VN; Mikhalchuk AL; Rubinov DB Phosphorescence of 8-Azasteroids and Related Compounds. Spectrochim. Acta, Part A 2005, 61, 2161–2167. [DOI] [PubMed] [Google Scholar]
- (26).Fantini J; Barrantes FJ How Cholesterol Interacts with Membrane Proteins: An Exploration of Cholesterol-Binding Sites Including CRAC, CARC, and Tilted Domains. Front. Physiol 2013, 4, 1 – 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bukiya AN; Dopico AM Common Structural Features of Cholesterol Binding Sites in Crystallized Soluble Proteins. J. Lipid Res 2017, 58, 1044–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Scheidt HA; Meyer T; Nikolaus J; Baek DJ; Haralampiev I; Thomas L; Bittman R; Müller P; Herrmann A; Huster D Cholesterol’s Aliphatic Side Chain Modulates Membrane Properties. Angew. Chem. Int. Ed 2013, 52, 12848–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Abbotto A; Beverina L; Bozio R; Bradamante S; Ferrante C; Pagani GA; Signorini R Push-Pull Organic Chromophores for Frequency-Upconverted Lasing. Adv. Mater 2000, 12, 1963–1967. [Google Scholar]
- (30).Bureš F Fundamental Aspects of Property Tuning in Push-Pull Molecules. RSC Adv. 2014, 4, 58826–58851. [Google Scholar]
- (31).Dormán G; Nakamura H; Pulsipher A; Prestwich GD The Life of Pi Star: Exploring the Exciting and Forbidden Worlds of the Benzophenone Photophore. Chem. Rev 2016, 116, 15284–15398. [DOI] [PubMed] [Google Scholar]
- (32).Uraguchi D; Tsuchiya Y; Ohtani T; Enomoto T; Masaoka S; Yokogawa D; Ooi T Unveiling Latent Photoreactivity of Imines. Angew. Chem., Int. Ed 2020, 59, 3665–3670. [DOI] [PubMed] [Google Scholar]
- (33).Seebach D; Goliński J Synthesis of Open-Chain 2,3-Disubstituted 4-nitroketones by Diastereoselective Michael-addition of (E)-Enamines to (E)-Nitroolefins. A Topological Rule for C, C-bond Forming Processes between Prochiral Centres. Preliminary Communication. Helv. Chim. Acta 1981, 64, 1413–1423. [Google Scholar]
- (34).Morgan JG; Berlin KD; Durham NN; Chestnut RW Syntheses of Some Indazoles Structurally Related to Equilenin. J. Heterocycl Chem 1971, 8, 61–63. [Google Scholar]
- (35).Fischer RT; Stephenson FA; Shafiee A; Schroeder F ∆5,7,9(10)-Cholestatrien-3β-Ol: A Fluorescent Cholesterol Analogue. Chem. Phys. Lipids 1984, 36, 1–14. [DOI] [PubMed] [Google Scholar]
- (36).Padwa A Photochemistry of the Carbon-Nitrogen Double Bond. Chem. Rev 1977, 77, 37–68. [Google Scholar]
- (37).Martin A; Long C; Forster RJ; Keyes TE Near IR Emitting BODIPY Fluorophores with Mega-Stokes Shifts. Chem. Commun 2012, 48, 5617–5619. [DOI] [PubMed] [Google Scholar]
- (38).Más-Montoya M; Montenegro MF; Espinosa Ferao A; Tárraga A; Rodríguez-López JN; Curiel D Rigid π-Extended Boron Difluoride Complex with Mega-Stokes Shift for Bioimaging. Org. Lett 2020, 22 (9), 3356–3360. [DOI] [PubMed] [Google Scholar]
- (39).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 16, revision B.01; Gaussian, Inc.: Wallingford, CT, 2016. [Google Scholar]
- (40).Wilkinson F Luminescence in Chemistry; Bowen, E. J, Ed.; Van Nostrand: London, 1968; pp 155–182. [Google Scholar]
- (41).Yguerabide J Nanosecond Fluorescence Spectroscopy of Biological Macromolecules and Membranes. In Fluorescence Techniques in Cell Biology; Thaer AA, Sernetz M, Eds.; Springer: Berlin, Heidelberg, 1973; pp 311–331. [Google Scholar]
- (42).Xu S; Benoff B; Liou H; Lobel P; Stock AM Structural Basis of Sterol Binding by NPC2, a Lysosomal Protein Deficient in Niemann-Pick Type C2 Disease. J. Biol. Chem 2007, 282, 23525–23531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kwon HJ; Abi-Mosleh L; Wang ML; Deisenhofer J; Goldstein JL; Brown MS; Infante RE Structure of N-Terminal Domain of NPC1 Reveals Distinct Subdomains for Binding and Transfer of Cholesterol. Cell 2009, 137, 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shen Y; Liu J; Estiu G; Isin B; Lee D-S; Barabási A-L; Kapatral V; Wiest O; Oltvai ZN; et al. A Blueprint for Antimicrobial Hit Discovery Targeting Metabolic Networks. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 1082–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Berzina Z; Solanko LM; Mehadi AS; Jensen MLV; Lund FW; Modzel M; Szomek M; Solanko KA; Dupont A; Nielsen GK; Heegaard CW; Ejsing CS; Wüstner D Niemann-Pick C2 Protein Regulates Sterol Transport between Plasma Membrane and Late Endosomes in Human Fibroblasts. Chem. Phys. Lipids 2018, 213, 48–61. [DOI] [PubMed] [Google Scholar]
- (46).Chaudhuri A; Anand D Cholesterol: Revisiting Its Fluorescent Journey on 200th Anniversary of Chevruel’s “Cholesterine. Biomed. Spectrosc. Imaging 2017, 6, 1–24. [Google Scholar]
- (47).Steinhaus M Characterization of the Major Odor-Active Compounds in the Leaves of the Curry Tree Bergera Koenigii L. by Aroma Extract Dilution Analysis. J. Agric. Food Chem 2015, 63, 4060–4067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.