

Abstract
Chronic stress causes physiological and hormonal adaptations that lead to neurobiological consequences and behavioral and cognitive impairments. In particular, chronic stress is shown to drive reduced neurogenesis and altered synaptic plasticity in brain regions that regulate mood and motivation. The neurobiological and behavioral effects of stress resemble pathophysiology and symptoms observed in psychiatric disorders, suggesting that there are similar mechanisms.
Accumulating evidence indicates that neuroimmune systems, particularly microglia, have a critical role in regulating the neurobiology of stress. Preclinical models indicate that chronic stress provokes changes in microglia phenotype and increases inflammatory cytokine signaling that affect neuronal function, leading to synaptic plasticity deficits and impaired neurogenesis. More recent work shows that microglia can also phagocytose neuronal elements and contribute to structural remodeling of neurons in response to chronic stress.
In this review we intend to highlight work by the Duman research group (as well as others) that revealed how chronic stress shapes neuroimmune function and, in turn, how inflammatory mediators and microglia contribute to the neurobiological effects of chronic stress. We will also provide considerations to engage neuroimmune systems and improve treatment for psychiatric disorders.
Keywords: Stress, Neuroimmune, Microglia, Cytokines, Synapse, Neurogenesis, Psychiatric Disorders
Clinical and preclinical evidence linking chronic stress, neuroimmune dysregulation, and psychiatric disorders
Psychiatric disorders are a major source of disability and cause considerable economic and health burdens globally (1, 2). Despite significant efforts to define genetic and biological mechanisms that underlie psychiatric disorders, the etiology and pathophysiology of these disorders have not yet been fully characterized (3–5). Clinical studies suggest that symptoms of psychiatric disorders are caused by structural and neurochemical alterations in corticolimbic and mesolimbic circuits, which include the hippocampus (HPC), prefrontal cortex (PFC), nucleus accumbens (NAc), and amygdala (4–7). While there is some genetic risk to developing psychiatric disorders, substantial evidence indicates that severe and/or prolonged exposure to psychosocial or environmental stress increases risk of onset and recurrence of affective disorders (4, 5, 8–11). In particular, stress exposure increases the risk for developing major depressive disorder (MDD) and posttraumatic stress disorder (PTSD), thus these will be referred to as stress-related psychiatric disorders (10, 11).
Stress is experienced when stimuli cause a perceived or actual threat to an organism, and if these ‘stressors’ persist it can overwhelm adaptive capacities (12, 13). Stress exposure is accompanied by activation of the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system, leading to release of glucocorticoids and norepinephrine, respectively. These canonical stress pathways have been shown to drive the neurobiological effects of chronic stress (14). In support of clinical findings, rodent models have demonstrated that chronic stress exposure causes cellular and/or synaptic plasticity deficits (e.g., dendritic or synaptic atrophy, impaired neurogenesis), and neurochemical dysregulation (e.g., neurotransmitter dysregulation, neuroinflammation) in corticolimbic and mesolimbic brain regions (4, 15–18). These preclinical studies indicate that stress-induced neurobiological effects contribute to behavioral and cognitive impairments (e.g., anhedonia, behavioral despair, working memory deficit, social withdrawal, etc.) that are relevant to stress-related psychiatric disorders (7, 19–22). Indeed, several preclinical models of stress, which include chronic social defeat stress, chronic unpredictable mild stress, or stress-enhanced fear learning, reiterate cardinal neurobiological mechanisms that contribute to onset and susceptibility of anxiety- and depressive-like behaviors (15, 22–24)
Importantly, a wealth of clinical studies indicates that markers of low-grade inflammation (i.e., elevated cytokines and other mediators) are also observed in patients with mood (e.g. MDD) and anxiety-related disorders (e.g. PTSD). Meta-analyses of the literature have reported that peripheral interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and C-reactive protein (CRP) are the most reliable biomarkers of inflammation in depression (25, 26) and in PTSD patients (27–29). This is relevant because clinical studies show that levels of inflammatory cytokines, IL-1β and TNF-α, were associated with more severe depressive symptoms (30). Other reports show that inflammatory factors are elevated in the cerebrospinal fluid of patients with MDD (31, 32). Some postmortem data suggest that suicide is associated with microglia activation in the PFC (33) and that increased levels of inflammatory cytokine gene expression in the PFC of MDD subjects (34). Other studies have sought to examine markers of microglia or neuroimmune activation in clinical populations using positron emission tomography (PET) for the translocator protein (TSPO). This research has yielded compelling results as depressed individuals showed broad increases in TSPO levels across corticolimbic brain regions (35). Other studies suggested that the highest levels of TSPO are detected in unmedicated MDD patients (36). Interestingly, a recent report showed that PTSD patients have lower prefrontal-limbic TSPO levels compared to controls, and that TSPO levels were negatively correlated with symptom severity (37). These contrasting findings highlight the heterogeneity of these disorders and suggest that TSPO binding may not be a definitive marker of neuroimmune activation (38).
There are likely several factors that promote immune dysregulation in psychiatric conditions (including genetic polymorphisms), but there is clear evidence that severe and prolonged stress exposure is associated with elevated levels of IL-6, IL-1β, TNF-α, and/or CRP and increased risk for developing depression, anxiety, and other psychiatric conditions (8, 39). Further studies indicate that stressful life experiences promote transcriptional pathways that increase immune reactivity, and these molecular changes like precipitate neuroimmune dysregulation (40–42). Interestingly, there is evidence that antidepressant medication can normalize the elevated levels of IL-1β, IL-6, and TNF-α in depressed patients, and reductions in cytokine levels are associated with improvement in depressive symptoms following antidepressant treatment (31, 43, 44). With that, it is important to note that elevated inflammatory markers are observed in a subset of depressed indviduals (45, 46). In addition, other studies have shown that antidepressant treatments have minimal or opposite effects on inflammatory markers (47). These results suggest that neuroimmune systems may be relevant therapeutic targets in a subset of patients. In this context, it may be useful to examine immunophenotypes across psychiatric conditions to optimize treatment strategies (48). Another critical consideration is sex differences in neuroimmune function and how this may affect risk of stress-related psychiatric disorders. Emerging evidence indicates that depressed males and females have divergent transcriptional changes in the PFC (49, 50). Regarding neuroinflammation, women that have enhanced responses following immune challenge or stress are at higher risk of depressive symptoms as compared to men (51, 52). Unfortunately, these sex differences have been under-studied so additional work is needed to define the sex-specific role of neuroinflammation in psychiatric disease.
Building on these clinical findings, studies using rodent models indicate that dysregulation of microglia in ‘stress-responsive’ brain regions as well as peripheral immune signaling contribute to the neurobiological deficits caused by chronic stress (29, 53). In particular, rodent models demonstrate that chronic stress alters microglia function and increases inflammasome activity, which is a protein complex that can detect diverse danger signals and promotes release of inflammatory cytokines (54–56). This is relevant because inflammatory cytokines or their relevant signaling pathways (e.g., NF-κB) mediate stress-induced behavioral deficits in animal models (57–62). Other studies suggest that stress-induced mobilization and recruitment of peripheral immune cells leads to inflammatory cytokine release in the brain as well (63). More recent work indicates that microglia also contribute to structural remodeling of neurons in response to chronic stress (15, 63). In this review, we will focus on mechanisms that alter neuroimmune systems in response to chronic stress, and subsequently how microglia disrupt synaptic and cellular functions in corticolimbic and mesolimbic brain regions and result in emotional and behavioral consequences.
Soluble factors regulate neuron-microglia interactions
Microglia are tissue-resident macrophages that migrate to the brain early in neurodevelopment (63), and their primary role is to maintain brain homeostasis and support neuronal function (65). In physiological conditions, microglia display a surveying phenotype that enables them to rapidly respond to the changes in neuronal homeostasis or brain injury (64, 65). Microglia processes extend and retract, presenting a constant motility of their ramifications (64, 65). This microglia surveillance is guided by local neuronal activity as well as activity-dependent release of purines (i.e., UDP, uridine triphosphate; ATP, adenosine triphosphate) from neurons (64, 66) (Figure 1A). Notably, microglia processes are attracted to and directly contact hyper-activated neurons, and this interaction reduces neuronal activity (67). Extracellular ATP from neurons is detected by microglia through the puringeric receptor P2RY12, guiding movement of microglial processes (64, 65, 68). Indeed microglia in mice lacking P2RY12 show impaired mobility toward sites of neuronal damage when nucleotides are released (68) and have impaired neuroplasticity in the visual cortex that influences ocular dominance. These data suggest that activity-dependent neuron-microglia interactions are important for shaping neurocircuitry.
Figure 1. Bidirectional microglia–neuron interactions in the healthy or stressed brain.
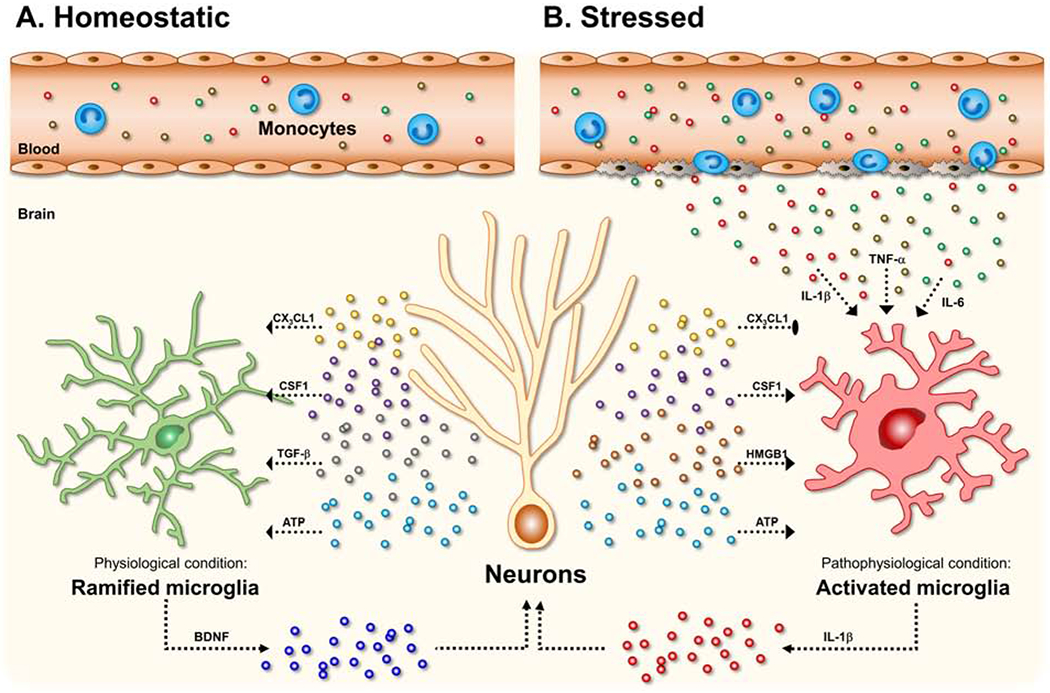
(A) Under physiological or homeostatic conditions, neurons can produce and release soluble factors such as CX3C chemokine ligand 1 (CX3CL1, also named fractalkine), colony-stimulating factor 1 (CSF1), and transforming growth factor-β (TGF-β), which bind to their receptors on microglia to maintain their functional state (e.g., ramified phenotype). In particular, ATP released from neurons guides movement of microglial processes by binding to microglial P2RY12 (purinergic receptor P2Y, G-protein coupled, 12). Further, microglia have been suggested to involve synaptogenesis through microglia-derived BDNF. In these steady-state conditions, there are low levels of circulating monocytes patrolling for infection or tissue damage in the vasculature. (B) Under pathological conditions and chronic stress, microglia alter their morphology with increased cell body size and thickened processes. In this state, CX3CL1 from neurons can regulate microglia activation by binding to its receptor CX3CR1 on microglia. In contrast, elevated levels of CSF1, high-mobility group box 1 (HMGB1) and adenosine triphosphate (ATP) in the pathological conditions such as stress exposure has been implicated in neuronal plasticity and psychiatric disorders. Notably, DAMP-associated pathways (e.g., HMGB1-RAGE, ATP-P2X7R signaling) trigger a critical convergent molecular pathway, termed the NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome. Finally, the activation of NLRP3 inflammasome in microglia results in the production and release of the pro-inflammatory cytokines, including IL-1β. In response to chronic stress, monocytes can be released into circulate and traffic to the brain. The peripheral monocytes in the brain converge with activated microglia to amplify inflammatory signaling. Increased peripheral cytokines (i.e., IL-1β, TNF-α, IL-6) also can penetrate into the brain via passive or active mechanisms and activate microglial stress responses.
Other factors released by neurons, including CX3CL1 (CX3C chemokine ligand 1, also named fractalkine) (69), transforming growth factor (TGF)-β (70), and colony-stimulating factor 1 (CSF1) (71) promote the unique microglia phenotype (Figure 1A). These neuron-derived factors regulate microglia functions through binding to CX3CR1, TGF-β receptor (TGFβR), CSF1 receptor (CSF1R), respectively. As noted, these neuron-derived factors are essential to maintain the molecular profile of microglia (72). More recent studies demonstrate that microglia morphology and surveillance is ‘restricted’ by increased levels of norepinephrine in wakeful states. In particular, microglia show retracted processes and less process motility when mice are in wakeful states, and microglia-specific depletion of the β2-adrenergic receptor prevented this effect (73). In all, these findings indicate that microglia are dynamic macrophages that develop unique phenotypes to respond to and support neuronal function.
Soluble factors, including cytokines, released by microglia have a fundamental role in modulating neuronal function (74). A growing body of studies have elucidated the diverse ways that microglia-derived cytokines and other immune mediators influence neuronal function and neural plasticity. In physiological conditions, basal levels of IL-1β are required for long-term potentiation (75) and TNF-α release is necessary for homeostatic synaptic scaling (76). Other studies have shown that microglia can be a source of neurotrophic factors, such as brain-derived neurotrophic factors (BDNF) that support neuroplasticity and learning (77), and it has been reported that microglia increase BDNF release in response to activating stimuli (78) (Figure 1A). In this context, alterations in microglia function following chronic stress, which may contribute to varied stress-induced BDNF levels in amygdala and NAc compared to PFC and HPC (79, 80). Recent research has also identified a novel pathway by which microglia can ‘inhibit’ neuronal activity. This study showed that elevated neuronal activity triggers microglial release of adenosine, which suppresses neuronal activity via a negative feedback mechanism (81). Altogether this work indicates that microglia-derived cytokines and other soluble factors contribute to homeostatic neuroplasticity in those brain areas (15) (Figure 1A).
Contact-dependent pathways shape neuron-microglia interactions
Recent advances in neuroimmunology have revealed that microglia impact neuronal function through contact-dependent mechanisms. It is now well-established that microglia contribute to synaptic pruning through active phagocytosis during neurodevelopment (82, 83). Further work showed that microglia-mediated synaptic remodeling in neurodevelopment occurred in an activity- and complement-dependent manner (84). Complement factor C3 and C1q bind to synapses with reduced activity and initiate microglia-mediated phagocytosis via complement receptor 3 (CR3, also known as CD11b) (84). Other studies have expanded on these findings, showing that microglia processes can contact neuronal structures in the adult brain. In particular, microglia processes interact frequently with active synapses, and in a model of ischemia extended microglial contacts resulted in synapse loss (85). Several other studies demonstrate that microglial contacts influence the excitability of neural networks (86), promote development of new synapses (87), as well as prevent activity-dependent neurotoxicity (88). Furthermore, recent high resolution microscopy approaches showed that in physiological conditions microglia can partially engulf (referred to as trogocytosis) pre-synaptic elements in HPC slice cultures (89). Although it is yet to be elucidated how microglia target specific neuronal compartments in response to altered neuronal activity, these findings demonstrate that reciprocal neuron-microglia interactions are critical mediators of synaptic plasticity and structural remodeling of neurons.
Microglia activation – it’s all relative
Disruptions in brain homeostasis, such as infection, injury, disease, or stress, lead to changes in microglia phenotype and function. In particular, microglia undergo several cellular adaptions: processes often move rapidly towards sites of injury or infection, the soma is enlarged, and the diameter of primary processes is increased while distal processes are shortened. Altogether these adaptations lead to substantial changes in microglia morphology. In severe pathological conditions, fully activated microglia show complete retraction of all processes and amoeboid-like phenotype. These cellular changes are often accompanied by altered molecular signaling that promotes cytokine and chemokine signaling, increased proliferative activity, and enhanced levels of phagocytic activity (90, 91). It is important to note that microglia activation exists on a continuum, and the magnitude and duration of these microglial responses are determined by the context (i.e., infection, injury, or stress) (92). Indeed cutting-edge molecular approaches, including single-cell RNA sequencing and mass cytometry (CyTOF), have uncovered varied functional profiles in homeostatic (i.e., across development) and disease (i.e., aging or neurodegeneration) states (93). Further studies are likely to provide unique insight into how microglia phenotypes shape neurobiology and behavior.
Microglia activation can be triggered by a variety of peripheral and central molecules. Pathogen-associated molecular patterns (PAMPs; e.g., endotoxin), damage (or danger)-associated molecular patterns (DAMPs; e.g., ATP, HMGB1), inflammatory cytokines, and other mediators can activate microglia in the brain (15, 94). In the absence of blood-brain barrier disruption, these factors can affect the brain function by being transported directly across the blood-brain barrier (BBB) or by binding specific receptors on the BBB and initiating release of secondary messengers (95, 96). In response to these inflammatory challenges, brain-derived PAMPs, DAMPs, cytokines, chemokines, inflammatory mediators, various neurotransmitters, as well as centrally-acting peripheral hormones such as glucocorticoids, can affect microglia function by binding to specific receptors (15, 94) (Figure 1B). Activated microglia can drive neuropathology, in particular, through release of pro-inflammatory cytokines (e.g., IL-1β, TNF-α) that propagate neuroinflammation, recruit peripheral immune cells, and lead to neuronal injury and/or death (97) (Figure 1B).
Canonical stress pathways drive microglia activation
Early studies demonstrated that repeated stress altered the morphology of microglia in ‘stress-responsive’ brain regions, suggesting that stress exposure shifted the functional state of microglia (98, 99) (Figure 1B). Further studies indicated that stress-induced glucocorticoids as well as noradrenergic signaling drive alterations in microglia phenotype. Indeed preclinical studies using adrenalectomy or treatment with the GC receptor antagonist RU486 reduced microglia activation following inescapable stress (100). Other rodent studies showed that blockade of stress-induced noradrenergic signaling reduced pro-inflammatory cytokine levels in the brain (101). Further studies supported this work as treatment with the β-adrenergic receptor antagonist propranolol diminished microglia activation and reduced pro-inflammatory cytokine levels in microglia after repeated social defeat (102). In the repeated social defeat model, microglia activation is accompanied by increased monocyte trafficking to the brain, and these monocytes contribute to elevated inflammatory signaling as well (103–106). While many studies showed that stress caused microglia activation, other work suggested that chronic unpredictable stress caused microglia dystrophy in the dentate gyrus of the HPC (107). These findings indicate that stress-induced neuroendocrine and sympathetic pathways drive brain region-specific alterations in microglia function that may contribute to divergent neurobiological effects observed in these models.
Stress promotes inflammasome activation and inflammatory cytokine release
Several lines of evidence demonstrate that microglial NLRP3 (nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3) inflammasome, a multiprotein complex that is activated by diverse danger signals, plays a critical role in neuroimmune responses to chronic stress (54). In the brain, the NLRP3 inflammasome is activated following pattern recognition receptor (PRR) binding of PAMPs and/or DAMPs (e.g., ATP). These pathways may be initiated by stress-related processes (108) and, in turn, NLRP3 signaling leads to caspase 1 activation, which induces cleavage and eventual release of pro-inflammatory cytokines such as IL-1β (109) (Figure 2A). Animal studies showed that exposure to chronic unpredictable stress leads to activation of microglial NLRP3 inflammasome and increased production of IL-1β in the PFC, inducing depressive-like behavior in rats. The chronic stress-induced activation of the NLRP3 inflammasome and its relevant inflammatory risk factors in PFC and depressive-like behaviors are reversed by chronic fluoxetine treatment (110). Similarly, chronic stress-induced hippocampal IL-1β and its consequential depressive-like behaviors are blocked by pharmacological inhibition of NLRP3 inflammasome in mice (111).
Figure 2. Microglia shape the neurobiology of stress by reducing neurogenesis and remodeling neurons.
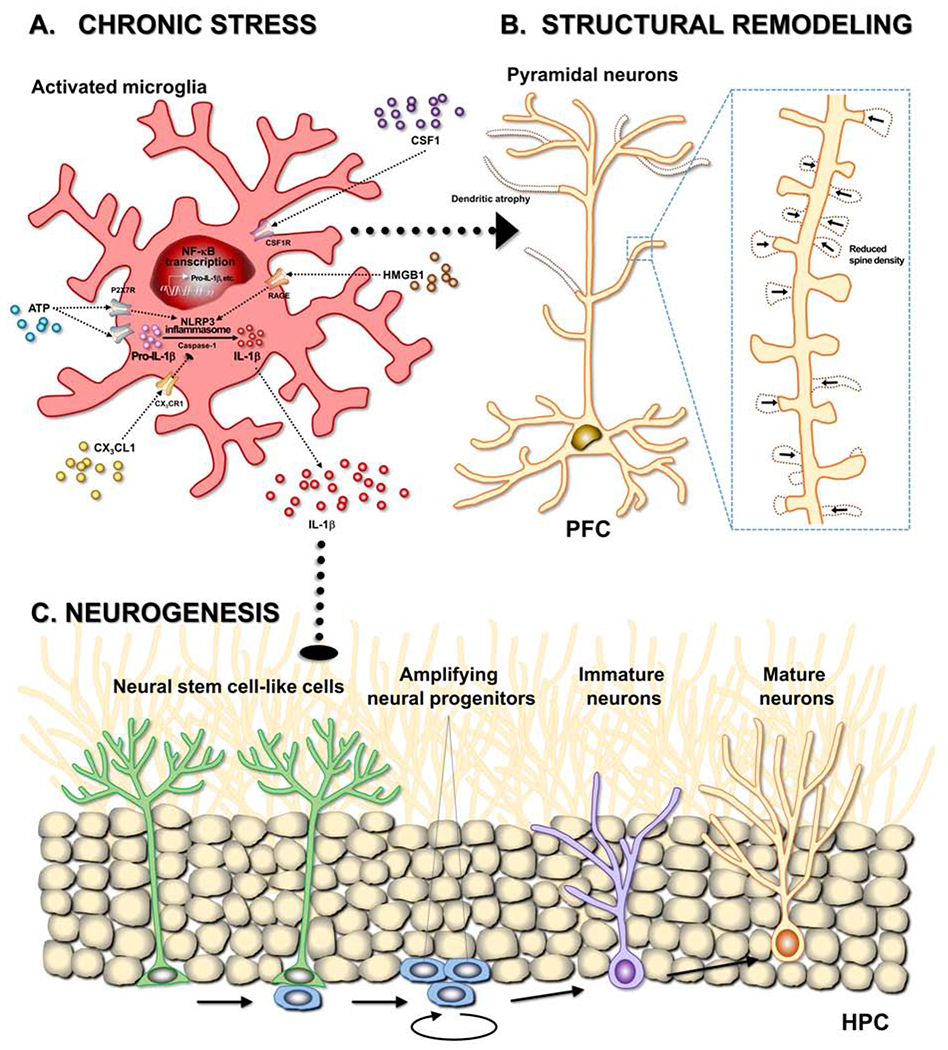
Chronic stress exposure alters microglial morphology and function in various stress-responsive brain areas, including corticolimbic and mesolimbic circuits. In response to chronic stress, ATP and HMGB1 from neurons and binds to the P2X7 receptor (P2X7R) and the receptor for advanced glycation end products (RAGE), respectively. These bindings of the danger associated molecular patterns (DAMPs) to pattern recognition receptors (PRRs) activate NLRP3 and recruit adaptor proteins and pro-caspase-1, which thereby makes a large multiprotein complex termed the inflammasome. The NLRP3 inflammasome cleaves pro-caspase-1 to mature caspase-1, which cleaves pro-IL-1β to mature IL-1β. Released IL-1β is thought to induce tumor necrosis factor alpha (TNF-α) release. Collectively, the stress-induced microglial activation produces and releases pro-inflammatory cytokines, including IL-1β and TNF-α, through activation of NLRP3 inflammasome. The stress-induced pro-inflammatory cytokines elicit neuronal dystrophy such as dendritic atrophy and reduction of spine density in glutamatergic pyramidal neurons in the prefrontal cortex (PFC). Microglia, in response to chronic stress, mediate the structural remodeling by increasing microglia-neuron contact and engulfment of synaptic structures. Neuron-derived factors, CSF1 and CX3CL1, regulate microglial phagocytosis of neuronal elements. In addition, chronic stress impairs proliferation of neural stem-like cells in the hippocampus (HPC) through activation of neuro-immune system (e.g., IL-1β/NF-κB signaling). Of note, microglia actively phagocytize the apoptotic newborn cells in the neuronal niche.
In addition, multiple studies have implicated DAMPs in stress effects on microglia. For example, prolonged stress caused release of HMGB1 (high mobility group box 1), which is a DAMP that binds to PRRs such as receptor for advanced glycation end products (RAGE). This HMGB1 signaling promotes NLRP3 activation and pro-inflammatory cytokine production by microglia in the HPC (112). These effects may be mediated by the NLRP3 inflammasome (Figure 2A), as blockade of the HMGB1-RAGE complex show reduced microglia activation and dampened IL-1β levels after stress exposure (110, 111). These findings are consistent with another study that showed stress increased extracellular ATP levels in the HPC, which is proposed to activate microglia and increase neuroinflammation via the purinergic receptor P2RX7 (55) (Figure 2A). In these studies, stress-induced increased IL-1β and TNF-α levels in the HPC were blunted by a P2RX7 antagonist. This work indicates that stress causes dysregulation of neuron-microglia interactions, which leads to functional alterations in microglia that can increase release of inflammatory mediators that, in turn, can influence neuroplasticity.
Effects of inflammatory cytokines on neurogenesis
There is ample research demonstrating that the stress-induced microglial activation and inflammatory cytokines regulate mood and cognition through direct actions on neurons (15, 20, 21, 25). Relevant to this review, hippocampal neurogenesis has been evaluated as a candidate mechanism for the pathophysiology of depression and as a target for antidepressant action (17, 113). Microglial activation inhibits hippocampal neurogenesis under inflammatory and stressful conditions (114, 115). Consistently, inflammatory challenges such as LPS or irradiation suppress hippocampal neurogenesis (115, 116). In parallel, chronic stress-induced hippocampal IL-1β signaling mediates the anti-neurogenic and depressive-like behavioral effects of chronic stress (58, 59) (Figure 2C). Further studies revealed that the stress-induced impairment of hippocampal neurogenesis and depression-like behaviors are mediated by NF-κB signaling (58, 60). In addition to IL-1β, other inflammatory cytokines, including IL-6, and TNF-α, can reduce adult hippocampal neurogenesis and/or promote depressive-like behaviors (116–118). In contrast, antidepressants increase the number of adult-born neurons in the HPC (119, 120). The adult-born neurons are required for antidepressant effects on some, but not all, depressive-like behaviors (121–123) and for the normal expression of those behaviors following acute stress (124). In parallel, enhanced hippocampal neurogenesis is sufficient for reducing depressive-like behaviors (125). Antidepressants can also normalize markers of neuroinflammation in rodent models (126, 127), and treatment-associated reductions in cytokines are linked to improved mood in MDD patients (31, 43, 44). Despite this, further studies are needed to directly tease apart the effects of antidepressants on neurogenesis and neuroinflammation.
The suppressive effects of activated microglia on neurogenesis are mainly related to the ‘survival’ of newborn neurons rather than their proliferation or differentiation. Microglia actively eliminate excess apoptotic newborn cells in the neuronal niche through phagocytosis (114). Previous studies showed that the number of newborn neurons is inversely proportional to the number of activated microglia under various inflammatory conditions (115, 116). Above all, in vitro experiments demonstrated that conditioned media from LPS-challenged microglia induces death of hippocampal neuroblasts, which is mediated by the secretion of IL-6 (116) or TNF-α (128). On the other hand, inflammatory cytokines regulate neurogenesis by reducing neural stem cell (NSC) proliferation and neuronal differentiation, and by prompting gliogenesis at the expense of neurogenesis (60, 118, 129, 130). For instance, overexpression of IL-6 in mice brains reduces proliferation, survival and neuronal differentiation (118). Similarly, systemic administration of TNF-α decreases NSC proliferation in the dentate gyrus (131). TNF-α administration to cultured hippocampal cells induces cell death (132) as well as an increase in the astrocytic differentiation at the expense of the neuronal differentiation (130). Furthermore, IL-1β treatment over cultured hippocampal NSCs reduces cell proliferation and neurosphere formation (133). Such effects of pro-inflammatory cytokines on neurogenesis may result from altered integration of newly produced neurons in adult brain. For example, an in vitro study found an elevated inhibitory synaptic inputs in newly born neurons matured under a chronic inflammatory environment (134). In parallel, disruption in signaling of CX3CL1, an endogenous neuronal regulator of microglial IL-1β, TNF-α, and IL-6 in the brain (135, 136), have shown reduced neurogenesis (137) (Figure 2C), lipopolysaccharide (LPS)-induced sickness-like behavior (138), and stress-induced depressive-like behavior (139). This work suggests that CX3CL1 regulates microglial function, inducing stress resilience. Thus, adult hippocampal neurogenesis may reciprocally regulate inflammatory signaling and affective behaviors.
In the animal models of PTSD, which is associated with overgeneralized memories of traumatic experiences, hippocampal neurogenesis plays a beneficial role (140). Enhanced adult hippocampal neurogenesis by treatment with memantine (MEM) facilitates forgetting of remote contextual fear memory following prolonged re-exposure to the conditioning context (141, 142) and forgetting of social avoidance memory after repeated exposure to social defeat stress, improving PTSD (anxiety)-like behavior (143). In contrast, blocking hippocampal neurogenesis after conditioning stabilizes or strengthens existing HPC-dependent memories (141, 144). A recent transcriptome and epigenome analyses using a social defeat stress model revealed that activated inflammatory signaling pathways and inhibited pathways involved in neuronal growth factors (e.g., neurogenesis) in brain regions implicated in fear memory and extinction (145). These data support the notion that neuroinflammatory signaling may promote this behavioral phenotype, potentially by inhibiting neurogenesis (145).
Exposure to chronic stress provokes microglia-mediated neuronal remodeling
With their ability to shape neuronal function through phagocytosis of synaptic components, microglia may contribute to stress-induced alterations in synaptic plasticity and associated behavioral consequences (146). Indeed, recent work shows that chronic stress alters neuron-microglia interactions, leading to increased microglial engulfment of synaptic elements. In separate studies, chronic unpredictable stress (CUS) is shown to increase microglia-neuron contact and engulfment of neuronal elements (e.g., pre- and post-synaptic structures) by microglia in the PFC (147) and HPC (108). In particular, microglia in the PFC showed increased engulfment of neuronal elements that was linked to synapse loss on apical dendrites of pyramidal neurons and development of depressive-like behaviors (147). At the molecular level, chronic stress increased neuronal CSF1, and viral-mediated knockdown of neuronal CSF1 in the PFC reduced microglia-mediated neuronal remodeling and associated behavioral consequences. Of note, gene expression analyses in postmortem dorsolateral PFC showed that depressed individuals have increased Csf1 mRNA levels as well (147) (Figure 2B). Follow-up studies have revealed that stress-induced glucocorticoid signaling promotes neuronal CSF1 signaling as well as increased microglia-mediated neuronal remodeling in the PFC (148). More recent studies indicate that elevated neuronal activity in response to stress leads to alterations in microglia function. Indeed pre-treatment with the GABAA receptor modulator diazepam dampened neuronal activity following CUS, which was associated with decreased CSF1 levels and decreased microglia-mediated neuronal remodeling in the PFC (149). Ongoing studies are aimed at determining the factors that direct microglia to engulf specific neuronal elements in the PFC. Beyond direct effects on synaptic structures, recent studies suggest that microglia can also alter neuroplasticity through remodeling of extracellular matrix. In particular, microglia, triggered by the cytokine IL-33, phagocytose extracellular matrix proteins that stabilize synapses and this enables growth or reshaping of specific synapses (150). While these studies were conducted in physiological conditions, it is intriguing to consider how this pathway may contribute to structural remodeling of neurons following chronic stress. Altogether, this work provides strong evidence that chronic stress increases microglial phagocytosis of synaptic and associated structures, and that neuron-microglia interactions have a formative role in the neurobiological and behavioral effects of stress.
Summary – translational implications and future directions
Taken together, there is ample evidence that stress-related psychiatric disorders are associated with excess or dysregulated neuroimmune function. The neurobiological effects of chronic stress, including reduced neurogenesis in the HPC and synapse loss in the PFC, underlie behavioral and cognitive impairments, including anhedonia, behavioral despair, and/or social withdrawal (4, 15, 17, 19, 20, 22, 44). Clinical and preclinical studies examining other stress-related disorders such as PTSD are at the initial stages to understand how immune factors can contribute to their related symptoms (16, 21, 28, 29). While preclinical studies indicate that altered neuroimmune function, particularly changes in microglia, contribute to the neurobiology of stress, it is important to recognize that complex multi-cellular processes underlie the pathophysiology of associated behavioral and cognitive consequences. However, with the limited efficacy of current treatments for psychiatric disorders, it is worth exploring other therapeutic targets such as neuroimmune systems (9, 151). When considering neuroimmune systems as potential therapeutic targets, broad inhibition of cytokine signaling may not be warranted as these pathways support neuroplasticity (97, 152). In addition, a recently published study using mass cytometry (CyTOF) suggests that microglia from depressed individuals do not display an inflammatory phenotype, but instead have increased expression of ‘homeostatic’ markers (153). In this context, it is relevant to consider what mechanisms shift the homeostatic functions of microglia. In particular, novel approaches will be needed to regulate neuron-microglia interactions. Any such interventions will need to be precise – targeting impaired circuits, while leaving other circuits unaffected. To accomplish this clinical and preclinical research will need to refine diagnostic tools and researchers will need to consider the heterogeneous functions of microglia across different brain regions. Further, more comprehensive approaches (i.e., single-cell RNA-Seq) will be needed to fully understand the dynamic neuroimmune processes that contribute to the neurobiology of stress-related psychiatric disorders. With this, we may work towards the goal of more targeted therapeutics for subtypes of patients.
Acknowledgements:
The authors would like to express their sincere gratitude for the professional guidance as well as genuine care that Ron provided to them (and everyone). He is missed every day. His legacy will continue to grow not only through his significant contributions to neuroscience, but also through the kindness he extended to all those that met him. This work was supported by the Korea Brain Research Institute Basic Research Program (Grant No. 20-BR-02-06 [to J.W.K]), National Research Foundation of Korea funded by the Ministry of Science and ICT Brain Research Program and Biomedical Technology Development Program (Grant Nos. 2017M3C7A1048089 and 2018M3C7A1024150 [to J.W.K]), and the National Institutes of Health (Grant Nos. R01MH123545, R21MH120614 [to E.S.W.]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors have declared no conflict of interest exists.
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Konnopka A, Konig H (2020): Economic Burden of Anxiety Disorders: A Systematic Review and Meta-Analysis. Pharmacoeconomics. 38:25–37. [DOI] [PubMed] [Google Scholar]
- 2.Konig H, Konig HH, Konnopka A (2019): The excess costs of depression: a systematic review and meta-analysis. Epidemiol Psychiatr Sci. 29:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charney DS, Manji HK (2004): Life stress, genes, and depression: multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE. 2004:re5. [DOI] [PubMed] [Google Scholar]
- 4.McEwen BS, Nasca C, Gray JD (2016): Stress Effects on Neuronal Structure: Hippocampus, Amygdala, and Prefrontal Cortex. Neuropsychopharmacology. 41:3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hariri AR, Holmes A (2015): Finding translation in stress research. Nat Neurosci. 18:1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherin JE, Nemeroff CB (2011): Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci. 13:263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duman RS, Aghajanian GK, Sanacora G, Krystal JH (2016): Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 22:238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kessler RC, McLaughlin KA, Green JG, Gruber MJ, Sampson NA, Zaslavsky AM, et al. (2010): Childhood adversities and adult psychopathology in the WHO World Mental Health Surveys. Br J Psychiatry. 197:378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otte C, Gold SM, Penninx BW, Pariante CM, Etkin A, Fava M, et al. (2016): Major depressive disorder. Nat Rev Dis Primers. 2:16065. [DOI] [PubMed] [Google Scholar]
- 10.Martin EI, Ressler KJ, Binder E, Nemeroff CB (2009): The neurobiology of anxiety disorders: brain imaging, genetics, and psychoneuroendocrinology. Psychiatr Clin North Am. 32:549–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saveanu RV, Nemeroff CB (2012): Etiology of depression: genetic and environmental factors. Psychiatr Clin North Am. 35:51–71. [DOI] [PubMed] [Google Scholar]
- 12.Selye H (1976): Further thoughts on “stress without distress”. Med Times. 104:124–144. [PubMed] [Google Scholar]
- 13.McEwen BS (1998): Protective and damaging effects of stress mediators. N Engl J Med. 338:171–179. [DOI] [PubMed] [Google Scholar]
- 14.de Kloet ER, Joels M, Holsboer F (2005): Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 6:463–475. [DOI] [PubMed] [Google Scholar]
- 15.Wohleb ES, Franklin T, Iwata M, Duman RS (2016): Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci. 17:497–511. [DOI] [PubMed] [Google Scholar]
- 16.Deslauriers J, Powell S, Risbrough VB (2017): Immune signaling mechanisms of PTSD risk and symptom development: insights from animal models. Curr Opin Behav Sci. 14:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pittenger C, Duman RS (2008): Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 33:88–109. [DOI] [PubMed] [Google Scholar]
- 18.Duman RS, Sanacora G, Krystal JH (2019): Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron. 102:75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Planchez B, Surget A, Belzung C (2019): Animal models of major depression: drawbacks and challenges. J Neural Transm (Vienna). 126:1383–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn AJ, Swiergiel AH, de Beaurepaire R (2005): Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 29:891–909. [DOI] [PubMed] [Google Scholar]
- 21.Aspesi D, Pinna G (2019): Animal models of post-traumatic stress disorder and novel treatment targets. Behav Pharmacol. 30:130–150. [DOI] [PubMed] [Google Scholar]
- 22.Krishnan V, Nestler EJ (2008): The molecular neurobiology of depression. Nature. 455:894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koo JW, Chaudhury D, Han MH, Nestler EJ (2019): Role of Mesolimbic Brain-Derived Neurotrophic Factor in Depression. Biol Psychiatry. 86:738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blouin AM, Sillivan SE, Joseph NF, Miller CA (2016): The potential of epigenetics in stress-enhanced fear learning models of PTSD. Learn Mem. 23:576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller AH, Maletic V, Raison CL (2009): Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 65:732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valkanova V, Ebmeier KP, Allan CL (2013): CRP, IL-6 and depression: a systematic review and meta-analysis of longitudinal studies. J Affect Disord. 150:736–744. [DOI] [PubMed] [Google Scholar]
- 27.Speer K, Upton D, Semple S, McKune A (2018): Systemic low-grade inflammation in post-traumatic stress disorder: a systematic review. J Inflamm Res. 11:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. (2015): Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2:1002–1012. [DOI] [PubMed] [Google Scholar]
- 29.Hori H, Kim Y (2019): Inflammation and post-traumatic stress disorder. Psychiatry Clin Neurosci. 73:143–153. [DOI] [PubMed] [Google Scholar]
- 30.Zou W, Feng R, Yang Y (2018): Changes in the serum levels of inflammatory cytokines in antidepressant drug-naive patients with major depression. PLoS One. 13:e0197267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine J, Barak Y, Chengappa KN, Rapoport A, Rebey M, Barak V (1999): Cerebrospinal cytokine levels in patients with acute depression. Neuropsychobiology. 40:171–176. [DOI] [PubMed] [Google Scholar]
- 32.Lindqvist D, Janelidze S, Hagell P, Erhardt S, Samuelsson M, Minthon L, et al. (2009): Interleukin-6 is elevated in the cerebrospinal fluid of suicide attempters and related to symptom severity. Biol Psychiatry. 66:287–292. [DOI] [PubMed] [Google Scholar]
- 33.Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. (2008): Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 42:151–157. [DOI] [PubMed] [Google Scholar]
- 34.Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, et al. (2011): Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry. 16:751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. (2015): Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 72:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richards EM, Zanotti-Fregonara P, Fujita M, Newman L, Farmer C, Ballard ED, et al. (2018): PET radioligand binding to translocator protein (TSPO) is increased in unmedicated depressed subjects. EJNMMI Res. 8:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhatt S, Hillmer AT, Girgenti MJ, Rusowicz A, Kapinos M, Nabulsi N, et al. (2020): PTSD is associated with neuroimmune suppression: evidence from PET imaging and postmortem transcriptomic studies. Nat Commun. 11:2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Notter T, Coughlin JM, Sawa A, Meyer U (2018): Reconceptualization of translocator protein as a biomarker of neuroinflammation in psychiatry. Mol Psychiatry. 23:36–47. [DOI] [PubMed] [Google Scholar]
- 39.Tursich M, Neufeld RW, Frewen PA, Harricharan S, Kibler JL, Rhind SG, et al. (2014): Association of trauma exposure with proinflammatory activity: a transdiagnostic meta-analysis. Transl Psychiatry. 4:e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powell C, Grant AR, Cornblath E, Goldman D (2013): Analysis of DNA methylation reveals a partial reprogramming of the Muller glia genome during retina regeneration. Proc Natl Acad Sci U S A. 110:19814–19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, et al. (2008): A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 64:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cole SW, Hawkley LC, Arevalo JM, Cacioppo JT (2011): Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci U S A. 108:3080–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hannestad J, DellaGioia N, Bloch M (2011): The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology. 36:2452–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raison CL, Capuron L, Miller AH (2006): Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 27:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Felger JC, Miller AH (2020): Identifying Immunophenotypes of Inflammation in Depression: Dismantling the Monolith. Biol Psychiatry. 88:136–138. [DOI] [PubMed] [Google Scholar]
- 46.Lynall ME, Turner L, Bhatti J, Cavanagh J, de Boer P, Mondelli V, et al. (2020): Peripheral Blood Cell-Stratified Subgroups of Inflamed Depression. Biol Psychiatry. 88:185–196. [DOI] [PubMed] [Google Scholar]
- 47.Kopschina Feltes P, Doorduin J, Klein HC, Juarez-Orozco LE, Dierckx RA, Moriguchi-Jeckel CM, et al. (2017): Anti-inflammatory treatment for major depressive disorder: implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J Psychopharmacol. 31:1149–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. (2013): A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 70:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seney ML, Huo Z, Cahill K, French L, Puralewski R, Zhang J, et al. (2018): Opposite Molecular Signatures of Depression in Men and Women. Biol Psychiatry. 84:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Labonte B, Engmann O, Purushothaman I, Menard C, Wang J, Tan C, et al. (2017): Sex-specific transcriptional signatures in human depression. Nat Med. 23:1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derry HM, Padin AC, Kuo JL, Hughes S, Kiecolt-Glaser JK (2015): Sex Differences in Depression: Does Inflammation Play a Role? Curr Psychiatry Rep. 17:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI (2015): Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology. 40:1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller AH, Raison CL (2016): The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 16:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwata M, Ota KT, Duman RS (2013): The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 31:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, et al. (2016): Psychological Stress Activates the Inflammasome via Release of Adenosine Triphosphate and Stimulation of the Purinergic Type 2X7 Receptor. Biol Psychiatry. 80:12–22. [DOI] [PubMed] [Google Scholar]
- 56.Dong Y, Li S, Lu Y, Li X, Liao Y, Peng Z, et al. (2020): Stress-induced NLRP3 inflammasome activation negatively regulates fear memory in mice. J Neuroinflammation. 17:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jones ME, Lebonville CL, Barrus D, Lysle DT (2015): The role of brain interleukin-1 in stress-enhanced fear learning. Neuropsychopharmacology. 40:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koo JW, Duman RS (2008): IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 105:751–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. (2008): Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 13:717–728. [DOI] [PubMed] [Google Scholar]
- 60.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS (2010): Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 107:2669–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu YN, Peng YL, Liu L, Wu TY, Zhang Y, Lian YJ, et al. (2015): TNFalpha mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw. 26:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, et al. (2017): Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: a possible role of gut-microbiota-brain axis. Transl Psychiatry. 7:e1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wohleb ES (2016): Neuron-Microglia Interactions in Mental Health Disorders: “For Better, and For Worse”. Front Immunol. 7:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. (2005): ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 8:752–758. [DOI] [PubMed] [Google Scholar]
- 65.Nimmerjahn A, Kirchhoff F, Helmchen F (2005): Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308:1314–1318. [DOI] [PubMed] [Google Scholar]
- 66.Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA (2014): Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci. 34:10511–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Du XF, Liu CS, Wen ZL, Du JL (2012): Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. 23:1189–1202. [DOI] [PubMed] [Google Scholar]
- 68.Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB, et al. (2006): The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci. 9:1512–1519. [DOI] [PubMed] [Google Scholar]
- 69.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, et al. (2006): Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 9:917–924. [DOI] [PubMed] [Google Scholar]
- 70.Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. (2014): Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. (2014): Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 82:380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA (2017): Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron. 94:759–773 e758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, Whitelaw BS, et al. (2019): Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci. 22:1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yirmiya R, Goshen I (2011): Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 25:181–213. [DOI] [PubMed] [Google Scholar]
- 75.Schneider H, Pitossi F, Balschun D, Wagner A, del Rey A, Besedovsky HO (1998): A neuromodulatory role of interleukin-1beta in the hippocampus. Proc Natl Acad Sci U S A. 95:7778–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stellwagen D, Malenka RC (2006): Synaptic scaling mediated by glial TNF-alpha. Nature. 440:1054–1059. [DOI] [PubMed] [Google Scholar]
- 77.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. (2013): Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakajima K, Tohyama Y, Kohsaka S, Kurihara T (2002): Ceramide activates microglia to enhance the production/secretion of brain-derived neurotrophic factor (BDNF) without induction of deleterious factors in vitro. J Neurochem. 80:697–705. [DOI] [PubMed] [Google Scholar]
- 79.Yu H, Wang DD, Wang Y, Liu T, Lee FS, Chen ZY (2012): Variant brain-derived neurotrophic factor Val66Met polymorphism alters vulnerability to stress and response to antidepressants. J Neurosci. 32:4092–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. (2006): Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 311:864–868. [DOI] [PubMed] [Google Scholar]
- 81.Badimon A, Strasburger HJ, Ayata P, Chen X, Nair A, Ikegami A, et al. (2020): Negative feedback control of neuronal activity by microglia. Nature. 586:417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. (2007): The classical complement cascade mediates CNS synapse elimination. Cell. 131:1164–1178. [DOI] [PubMed] [Google Scholar]
- 83.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. (2011): Synaptic pruning by microglia is necessary for normal brain development. Science. 333:1456–1458. [DOI] [PubMed] [Google Scholar]
- 84.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. (2012): Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009): Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci. 29:3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akiyoshi R, Wake H, Kato D, Horiuchi H, Ono R, Ikegami A, et al. (2018): Microglia Enhance Synapse Activity to Promote Local Network Synchronization. eNeuro. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, et al. (2016): Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun. 7:12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kato G, Inada H, Wake H, Akiyoshi R, Miyamoto A, Eto K, et al. (2016): Microglial Contact Prevents Excess Depolarization and Rescues Neurons from Excitotoxicity. eNeuro. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. (2018): Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007): Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 10:1538–1543. [DOI] [PubMed] [Google Scholar]
- 91.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011): Physiology of microglia. Physiol Rev. 91:461–553. [DOI] [PubMed] [Google Scholar]
- 92.DiSabato DJ, Quan N, Godbout JP (2016): Neuroinflammation: the devil is in the details. J Neurochem. 139 Suppl 2:136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Masuda T, Sankowski R, Staszewski O, Prinz M (2020): Microglia Heterogeneity in the Single-Cell Era. Cell Rep. 30:1271–1281. [DOI] [PubMed] [Google Scholar]
- 94.Yirmiya R, Rimmerman N, Reshef R (2015): Depression as a microglial disease. Trends Neurosci. 38:637–658. [DOI] [PubMed] [Google Scholar]
- 95.Quan N, Banks WA (2007): Brain-immune communication pathways. Brain Behav Immun. 21:727–735. [DOI] [PubMed] [Google Scholar]
- 96.Maier SF, Watkins LR (1998): Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol Rev. 105:83–107. [DOI] [PubMed] [Google Scholar]
- 97.Harry GJ, Kraft AD (2008): Neuroinflammation and microglia: considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab Toxicol. 4:1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hinwood M, Morandini J, Day TA, Walker FR (2012): Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex. 22:1442–1454. [DOI] [PubMed] [Google Scholar]
- 99.Walker FR, Nilsson M, Jones K (2013): Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr Drug Targets. 14:1262–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Frank MG, Thompson BM, Watkins LR, Maier SF (2012): Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun. 26:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, et al. (2005): Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 135:1295–1307. [DOI] [PubMed] [Google Scholar]
- 102.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. (2011): beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 31:6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. (2018): Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. 23:1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. (2014): Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 75:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013): Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 33:13820–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wohleb ES, McKim DB, Sheridan JF, Godbout JP (2014): Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. 8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kreisel T, Frank MG, Licht T, Reshef R, Ben-Menachem-Zidon O, Baratta MV, et al. (2014): Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry. 19:699–709. [DOI] [PubMed] [Google Scholar]
- 108.Milior G, Lecours C, Samson L, Bisht K, Poggini S, Pagani F, et al. (2016): Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav Immun. 55:114–125. [DOI] [PubMed] [Google Scholar]
- 109.Schroder K, Tschopp J (2010): The inflammasomes. Cell. 140:821–832. [DOI] [PubMed] [Google Scholar]
- 110.Pan Y, Chen XY, Zhang QY, Kong LD (2014): Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun. 41:90–100. [DOI] [PubMed] [Google Scholar]
- 111.Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, et al. (2015): NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int J Neuropsychopharmacol. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015): Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci. 35:316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sahay A, Hen R (2007): Adult hippocampal neurogenesis in depression. Nat Neurosci. 10:1110–1115. [DOI] [PubMed] [Google Scholar]
- 114.Sierra A, Beccari S, Diaz-Aparicio I, Encinas JM, Comeau S, Tremblay ME (2014): Surveillance, phagocytosis, and inflammation: how never-resting microglia influence adult hippocampal neurogenesis. Neural Plast. 2014:610343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ekdahl CT (2012): Microglial activation - tuning and pruning adult neurogenesis. Front Pharmacol. 3:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Monje ML, Toda H, Palmer TD (2003): Inflammatory blockade restores adult hippocampal neurogenesis. Science. 302:1760–1765. [DOI] [PubMed] [Google Scholar]
- 117.Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, et al. (2006): Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 26:9703–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vallieres L, Campbell IL, Gage FH, Sawchenko PE (2002): Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 22:486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, et al. (2009): Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 34:2376–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000): Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 20:9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. (2003): Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science. 301:805–809. [DOI] [PubMed] [Google Scholar]
- 122.David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. (2009): Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 62:479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, et al. (2009): The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry. 14:764–773, 739. [DOI] [PubMed] [Google Scholar]
- 124.Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA (2011): Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 476:458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hill AS, Sahay A, Hen R (2015): Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology. 40:2368–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lu Y, Ho CS, Liu X, Chua AN, Wang W, McIntyre RS, et al. (2017): Chronic administration of fluoxetine and pro-inflammatory cytokine change in a rat model of depression. PLoS One. 12:e0186700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang N, Yu HY, Shen XF, Gao ZQ, Yang C, Yang JJ, et al. (2015): The rapid antidepressant effect of ketamine in rats is associated with down-regulation of pro-inflammatory cytokines in the hippocampus. Ups J Med Sci. 120:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cacci E, Claasen JH, Kokaia Z (2005): Microglia-derived tumor necrosis factor-alpha exaggerates death of newborn hippocampal progenitor cells in vitro. J Neurosci Res. 80:789–797. [DOI] [PubMed] [Google Scholar]
- 129.Carpentier PA, Palmer TD (2009): Immune influence on adult neural stem cell regulation and function. Neuron. 64:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Keohane A, Ryan S, Maloney E, Sullivan AM, Nolan YM (2010): Tumour necrosis factor-alpha impairs neuronal differentiation but not proliferation of hippocampal neural precursor cells: Role of Hes1. Mol Cell Neurosci. 43:127–135. [DOI] [PubMed] [Google Scholar]
- 131.Seguin JA, Brennan J, Mangano E, Hayley S (2009): Proinflammatory cytokines differentially influence adult hippocampal cell proliferation depending upon the route and chronicity of administration. Neuropsychiatr Dis Treat. 5:5–14. [PMC free article] [PubMed] [Google Scholar]
- 132.Hofer S, Grandgirard D, Burri D, Frohlich TK, Leib SL (2011): Bacterial meningitis impairs hippocampal neurogenesis. J Neuropathol Exp Neurol. 70:890–899. [DOI] [PubMed] [Google Scholar]
- 133.Green HF, Treacy E, Keohane AK, Sullivan AM, O’Keeffe GW, Nolan YM (2012): A role for interleukin-1beta in determining the lineage fate of embryonic rat hippocampal neural precursor cells. Mol Cell Neurosci. 49:311–321. [DOI] [PubMed] [Google Scholar]
- 134.Jakubs K, Bonde S, Iosif RE, Ekdahl CT, Kokaia Z, Kokaia M, et al. (2008): Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 28:12477–12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Biber K, Neumann H, Inoue K, Boddeke HW (2007): Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 30:596–602. [DOI] [PubMed] [Google Scholar]
- 136.Jurgens HA, Johnson RW (2012): Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp Neurol. 233:40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bachstetter AD, Morganti JM, Jernberg J, Schlunk A, Mitchell SH, Brewster KW, et al. (2011): Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging. 32:2030–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Corona AW, Huang Y, O’Connor JC, Dantzer R, Kelley KW, Popovich PG, et al. (2010): Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J Neuroinflammation. 7:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hellwig S, Brioschi S, Dieni S, Frings L, Masuch A, Blank T, et al. (2016): Altered microglia morphology and higher resilience to stress-induced depression-like behavior in CX3CR1-deficient mice. Brain Behav Immun. 55:126–137. [DOI] [PubMed] [Google Scholar]
- 140.Besnard A, Sahay A (2016): Adult Hippocampal Neurogenesis, Fear Generalization, and Stress. Neuropsychopharmacology. 41:24–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Akers KG, Martinez-Canabal A, Restivo L, Yiu AP, De Cristofaro A, Hsiang HL, et al. (2014): Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science. 344:598–602. [DOI] [PubMed] [Google Scholar]
- 142.Ishikawa R, Fukushima H, Frankland PW, Kida S (2016): Hippocampal neurogenesis enhancers promote forgetting of remote fear memory after hippocampal reactivation by retrieval. Elife. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ishikawa R, Uchida C, Kitaoka S, Furuyashiki T, Kida S (2019): Improvement of PTSD-like behavior by the forgetting effect of hippocampal neurogenesis enhancer memantine in a social defeat stress paradigm. Mol Brain. 12:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Epp JR, Silva Mera R, Kohler S, Josselyn SA, Frankland PW (2016): Neurogenesis-mediated forgetting minimizes proactive interference. Nat Commun. 7:10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Muhie S, Gautam A, Chakraborty N, Hoke A, Meyerhoff J, Hammamieh R, et al. (2017): Molecular indicators of stress-induced neuroinflammation in a mouse model simulating features of post-traumatic stress disorder. Transl Psychiatry. 7:e1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lenz KM, Nugent BM, Haliyur R, McCarthy MM (2013): Microglia Are Essential to Masculinization of Brain and Behavior. J Neurosci. 33:2761–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wohleb ES, Terwilliger R, Duman CH, Duman RS (2018): Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biol Psychiatry. 83:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Horchar MJ, Wohleb ES (2019): Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav Immun. 81:329–340. [DOI] [PubMed] [Google Scholar]
- 149.Bollinger JL, Horchar MJ, Wohleb ES (2020): Diazepam limits microglia-mediated neuronal remodeling in the prefrontal cortex and associated behavioral consequences following chronic unpredictable stress. Neuropsychopharmacology. 45:1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nguyen PT, Dorman LC, Pan S, Vainchtein ID, Han RT, Nakao-Inoue H, et al. (2020): Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell. 182:388–403 e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Priller J, Prinz M (2019): Targeting microglia in brain disorders. Science. 365:32–33. [DOI] [PubMed] [Google Scholar]
- 152.Alboni S, Maggi L (2015): Editorial: cytokines as players of neuronal plasticity and sensitivity to environment in healthy and pathological brain. Front Cell Neurosci. 9:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bottcher C, Fernandez-Zapata C, Snijders GJL, Schlickeiser S, Sneeboer MAM, Kunkel D, et al. (2020): Single-cell mass cytometry of microglia in major depressive disorder reveals a non-inflammatory phenotype with increased homeostatic marker expression. Transl Psychiatry. 10:310. [DOI] [PMC free article] [PubMed] [Google Scholar]