

Abstract
Post-traumatic stress disorder (PTSD), an anxiety-related syndrome, is associated with increased risk for cardiovascular diseases. The present study investigated whether predator scent (PS) stress, a model of PTSD, induces sensitization of hypertension and anxiety-like behaviors and underlying mechanisms related to renin–angiotensin systems (RAS) and inflammation. Coyote urine, as a PS stressor, was used to model PTSD. After PS exposures, separate cohorts of rats were studied for hypertensive response sensitization (HTRS), anxiety-like behaviors, and changes in plasma levels and mRNA expression of several components of the RAS and proinflammatory cytokines (PICs) in the lamina terminalis (LT), paraventricular nucleus (PVN), and amygdala (AMY). Rats exposed to PS as compared to control animals exhibited (1) a significantly greater hypertensive response (i.e., HTRS) when challenged with a slow-pressor dose of angiotensin (ANG) II, (2) significant decrease in locomotor activity and increase in time spent in the closed arms of a plus maze as well as general immobility (i.e., behavioral signs of increased anxiety), (3) upregulated plasma levels of ANG II and interleukin-6, and (4) increased expression of message for components of the RAS and PICs in key brain nuclei. All the PS-induced adverse effects were blocked by pretreatment with either an angiotensin-converting enzyme antagonist or a tumor necrosis factor-α inhibitor. The results suggest that PS, used as an experimental model of PTSD, sensitizes ANG II-induced hypertension and produces behavioral signs of anxiety, probably through upregulation of RAS components and inflammatory markers in plasma and brain areas associated with anxiety and blood pressure control.
Keywords: Predator scent stress, Behavior, Blood pressure, Renin–angiotensin system, Inflammation
Introduction
Post-traumatic stress disorder (PTSD) is an anxiety-related syndrome triggered by stressful events (Nemeroff et al. 2006). There is significant comorbidity between PTSD and cardiovascular diseases. Most notably, there is a twofold increase in the prevalence of hypertension in PTSD patients in contrast to those without the psychological disorder (Cohen et al. 2015; Burg and Soufer 2016; Edmondson and von Kanel 2017).
PTSD and hypertension are associated with one another through activation of common mediators. Both PTSD and hypertension activate the renin–angiotensin system (RAS) (Bali and Jaggi 2013; Duchemin et al. 2013; Young and Davisson 2015; Nakagawa et al. 2020) and the immune system, increasing peripheral and central angiotensin (ANG ) II and proinflammatory cytokines [PICs, e.g., tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β and IL-6] (Saavedra et al. 2011; Patki et al. 2013; Winklewski et al. 2015; Lee et al. 2016; Marina et al. 2016; Michopoulos et al. 2017; Weber et al. 2017). Wilson and colleagues showed that exposure of rats to a cat upregulated mRNA and protein expression of PICs in the hippocampus, pre-frontal cortex, and amygdala (AMY) (Wilson et al. 2013). Many types of stress also increase brain ANG II formation and upregulate brain ANG II type 1 receptor (AT1-R) expression in the hypothalamic paraventricular nucleus (PVN) and the subfornical organ (SFO) (Saavedra et al. 2011). These brain areas have been shown to be associated with experimentally induced PTSD and contain nuclei that are involved in integrating information that affects cardiovascular and behavioral responses to stressful stimuli (Lucassen et al. 2014; Dampney 2016; Michopoulos et al. 2017). Reduction of RAS activity and inflammation by anti-hypertensive and anti-inflammatory drugs has been shown to improve mood and reduce blood pressure (BP) in various pathological conditions (Saavedra et al. 2011; Khoury et al. 2012; Levkovitz et al. 2015; Fontes et al. 2016).
Many physiological stressors have been shown to induce hypertensive response sensitization (HTRS). Physiological stressors also induce upregulation of message for several prohypertensive components of the brain RAS and of central innate immune system (Xue et al. 2012a; Xue et al. 2012b; Clayton et al. 2014; Johnson et al. 2015; Xue et al. 2016a; Xue et al. 2016b; Xue et al. 2019; Johnson and Xue 2018; Xue et al. 2020; Hurley et al. 2020). Such changes in the central nervous system (CNS) have been found in key components of the neural network such as the lamina terminalis (LT) and PVN that controls sympathetic nervous system tone affecting BP. Furthermore, a previous study from our laboratory using a resident-intruder psychosocial stress model demonstrated that a psychosocial stressor induces HTRS accompanied by upregulation of RAS and PIC expression of mRNA in the LT and PVN. The resident-intruder model involves introducing one male rat, the intruder, into the cage of a second male rat. In this stress model, administration of an angiotensin-converting enzyme (ACE) inhibitor, captopril (Cap), or the TNF-α synthesis inhibitor, pentoxifylline (PTX), reduced the physiological and molecular effects in the psychosocially stressed intruder (Johnson and Xue 2018; Xue et al. 2019).
The predator scent (PS) paradigm is regarded as a valid model of PTSD (Whitaker and Gilpin 2015; Albrechet-Souza and Gilpin 2019), which involves no physical contact between the stressor and the subject. We tested whether PS induces HTRS and this PS-induced HTRS is associated with augmented RAS and immune activity in the LT, PVN, and AMY that might contribute to the hypertensive and the behavioral responses. To further assess the role of the RAS and PICs in HTRS and PTSD-like anxiety, we determined whether systemic treatment with Cap or PTX would block sensitization and have anxiolytic effects.
Materials and Methods
Animals
Sprague Dawley rats (9 weeks old, ENVIGO) were housed in standard plastic microisolator cages and maintained in a temperature (23 ± 2 °C) and light (12-h light/dark cycle)-controlled animal facility, with access to standard rat chow and water ad libitum. A total of one hundred seven male rats were used for the experiments. Rats were divided into 4 groups including (1) control, (2) predator (coyote) urine (The Pee Mart, Bucksport, ME) exposure, (3) pretreatment of ACE inhibitor (Cap, 40 mg/kg/day, Sigma) plus urine exposure, or (4) pretreatment of TNF-α synthesis inhibitor (PTX, 100 mg/kg/day, Sigma) plus urine exposure. The dosages of Cap and PTX used above were chosen based on published studies (Moe et al. 1984; Thunhorst et al. 1989; Banfi et al. 2004; Plotnikov et al. 2017), and they were dissolved in the animals’ drinking water. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Iowa Institutional Animal Care and Use Committee (protocol #: 8081509).
PS Stressor Exposure
Rats were given normal drinking water or were pretreated with either an ACE inhibitor or a TNF-α synthesis inhibitor in the drinking water for two weeks. Beginning the second week, following a modified predator scent (PS) stressor paradigm (Albrechet-Souza and Gilpin 2019), rats in the three PS conditions were individually placed in a wire mesh metal cage. Coyote urine was sprayed onto a gauze sponge and placed under the wire mesh. The exposure period lasted 20 min and three sessions were given with each treatment separated by two days
Test for HTRS and Locomotor Activity After PS
The Induction-Delay-Expression (I-D-E) paradigm was used to test the HTRS as previously described (Xue et al. 2012a). Prior to beginning control, Cap or PTX pretreatments, all rats were chronically instrumented with telemetry probes (HD-S10; DSI) through the femoral artery allowing for continuous monitoring of BP, HR, and locomotor activity. After recovery from surgery, drug pretreatment was paralleled by baseline BP monitoring of all rats for one week. The PS induction procedure was performed during the second week of BP monitoring (induction period). After a one-week delay following the last predator scent exposure (delay period), control and PS-treated rats received a subcutaneous infusion of ANG II (120 ng/kg/min, Sigma) through implantation of an osmotic pump (2002 model, Alzet) in the back of animals for two weeks (expression period).
Behavioral Testing
Three days after the last PS exposure, all rats underwent an elevated plus maze (EPM) test to assess anxiety-like behavior. The EPM test is a well-established, thoroughly validated paradigm used in rodents (Pellow et al. 1985; Rodgers and Dalvi 1997). The test relies on placing an animal in a situation of conflict between an innate fear of high open areas versus their motivation to explore new environments.
In the present experiment, rats were placed in the center of an EPM facing an open arm and allowed to explore freely for 10 min. Locomotor behavior was recorded by an overhead camera. The number of open and closed arm entries and the time spent in the center, open arms, and closed arms were measured. The hind paws crossing the border between the center section of the EPM and an arm of the maze was scored as an entry into that arm. The time spent in the center was defined as the amount of time spent at the start of the trial before making the choice of entering an arm. Between each trial, 95% ethanol was used to clean the maze.
Blood Plasma Analysis
After plus maze testing, trunk blood was collected in sodium heparin tube (BD vacutainer) and centrifuged. The plasma was used for biochemical assays. Plasma levels of ANG II (Cat, CEA005Ra, Cloud Clone, Wuhan, China) and IL-6 (Cat, R6000B, R&D Systems, Minneapolis, MN) were measured with commercial ELISA kits according to the manufacturers’ instructions.
Real-Time PCR Analysis
Additional studies were performed to assess the effect of the PS on RAS and PIC components and microglial activity in the LT, PVN, and AMY. The brains of control rats, PS stressed rats, and PS stressed rats pretreated with Cap or PTX rats were collected on day 7 after the last PS exposure, corresponding to the time at which ANG II infusion was initiated in HTRS Experiment. All rats were decapitated, and the brains were quickly removed and put in ice saline for 1 min. Then, the brains were cut into coronal sections of approximately 300 μm thickness, and the target tissues, including the LT and both sides of the PVN or AMY, were punched with a 15-gauge needle stub (inner diameter: 1.5 mm). Some immediately surrounding tissue was usually included in the punch biopsies. The structures lying along the LT include the SFO, the median preoptic nucleus (MnPO), and the organum vasculosum (OVLT). Because each of the structures lying along the LT is very small and they are located at same level of a brain section in the coronal plane, we collected these structures together and analyzed their mRNA expression as a whole. Total RNA was isolated from the LT, PVN, or AMY using the Trizol method (Invitrogen) and treated with DNase I (Invitrogen, Carlsbad, CA, USA) to remove any genomic DNA contamination. RNA integrity was checked by gel electrophoresis. Total RNA was reverse transcribed following the manufacturer’s instructions (Applied Biosystems, Foster City, CA, USA). Real-time PCR was conducted using 200–300 ng of cDNA and 500 nM of each primer in a 20-μl reaction with iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Amplification cycles were conducted at 95 °C for 3 min, followed by 40 cycles of 95 °C for 15 s and annealing/extension at 60 °C for 30 s. Reactions were performed in duplicate and analyzed using a C1000 thermocycler system (Bio-Rad). Messenger RNA levels for RAS components (ACE, AT1-R), PICs (TNF-α, IL-1β, and IL-6), microglial marker (CD11b), and GAPDH were analyzed with SYBR Green real-time RT-PCR. The values were corrected by GAPDH, and the final concentration of mRNA was calculated using the formula x = 2(−ΔΔCt), where x = fold difference relative to control. Primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). The sequences of the primers are shown in Table 1.
Table 1.
Primer sequences for real-time PCR
| Gene | Forward primer | Reverse primer | Product size (bp) |
|---|---|---|---|
| GAPDH | TGACTCTACCCACGGCAAGTTCAA | ACGACATACTCAGCACCAGCATCA | 141 |
| ACE | GTGTTGTGGAACGAATACGC | CCTTCTTTATGATCCGCTTGA | 187 |
| AT1-R | CTCAAGCCTGTCTACGAAAATGAG | GTGAATGGTCCTTTGGTCGT | 188 |
| TNF-α | GCCGATTTGCCACTTCATAC | AAGTAGACCTGCCCGGACTC | 209 |
| IL-6 | GCCTATTGAAAATCTGCTCTGG | GGAAGTTGGGGTAGGAAGGA | 160 |
| IL-1β | AGCAACGACAAAATCCCT GT | GAAGACAAACCGCTTTTCCA | 209 |
| CD11b | TTACCGGACTGTGTGGACAA | AGTCTCCCACCACCAAAGTG | 239 |
ACE angiotensin-converting enzyme 1, AT1-R angiotensin II type 1 receptor, TNF-α tumor necrosis factor-α, IL-1β interleukin-1β, IL-6 interleukin-6, CD11b cluster of differentiation molecule 11b
Statistical Analysis
Mean arterial pressure (MAP), HR, and locomotor activity, obtained from the telemetry recordings, are presented as mean daily values. Differences for MAP and HR were calculated for each animal based on the mean of a 7-day baseline subtracted from the mean of the final 5 days of ANG II treatment. Mean locomotor activities were averaged from consecutive days for baseline, induction, delay, and expression, respectively, and are presented as mean values. One-way ANOVAs for the experimental groups were then conducted on the means of calculated differences for MAP and HR and on averaged locomotor activity. After establishing a significant ANOVA, post hoc analyses were performed with Tukey multiple comparison tests between pairs of mean changes (Graph-pad Prism 8.0). One-way ANOVAs and post hoc Tukey analyses were also used to test for behavioral measures, the differences in plasma levels and mRNA expression of the RAS and PIC components and microglial marker in the blood, LT, PVN, and AMY, respectively. All data are expressed as means ± SE. Statistical significance was set at p < 0.05.
Results
PS-Induced Sensitization of the Hypertensive Response to Systemic Ang II Infusion and the Effects of Pretreatment with Cap or PTX
PS exposure had no effects on basal mean arterial pressure (MAP) and heart rate (HR). During infusion with the slow-pressor dose of ANG II, the PS-exposed rats showed a significantly enhanced hypertensive response (Δ45.7 ± 5.5 mmHg) compared with non-stressed control rats (Δ21.1 ± 3.7 mmHg, p < 0.05, Fig. 1a and b). Pretreatment with either the ACE inhibitor Cap or the TNF-α synthesis inhibitor PTX significantly reduced the ANG II-elicited pressor response in the stressed rats (Cap, Δ18.7 ± 5.2 mmHg; PTX, Δ21.3 ± 5.1 mmHg, p < 0.05, Fig. 1a and b).
Fig. 1.

Pressor effects (Fig. 1a and b) and heart rate (HR) (Fig. 1c and d) changes induced by angiotensin (ANG) II in control, coyote urine scent-exposed, and coyote urine scent-exposed plus pretreatment plus either captopril (Cap) or pentoxifylline (PTX) rats. The enhanced pressor effect in coyote urine scent-exposed rats was attenuated by either Cap or PTX pretreatment. Baseline recordings are denoted by B’s. (n = 7–10/group; *p < 0.05 vs baseline; #p < 0.05 vs control or coyote urine scent-exposed rats plus Cap or PTX pretreatment; ǂp < 0.05 vs coyote urine scent-exposed or coyote urine scent-exposed rats with PTX pretreatment; §p < 0.05 vs control, coyote urine scent-exposed or coyote urine scent-exposed rats with PTX pretreatment)
The slow-pressor dose of ANG II produced significant decreases in HR in the unstressed control rats (− 25.7 ± 3.6 beats/min, p < 0.05) and the stressed rats pretreated with Cap (− 62.6 ± 4.8 beats/min, p < 0.05), but not in the untreated stressed rats (− 12.9 ± 3.9 beats/min) or in the PS-exposed rats pretreated with PTX (− 8.9 ± 3.7 beats/min) (Fig. 1c and d).
Cap and PTX both had significant effects on baseline variables. Basal MAP (103.8 ± 1.3 mmHg, p < 0.05) was lower in the groups pretreated with Cap, compared to groups without pretreatment (control, 113.5 ± 1.0 mmHg; urine alone, 114.4 ± 1.2 mmHg) or pretreated with PTX (114.6 ± 1.7 mmHg) (Fig. 1a). Basal HR was higher in the groups pretreated with Cap (387.5 ± 7.3 beats/min, p < 0.05) and lower in the group pretreated with PTX (341.3 ± 2.7 beats/min, p < 0.05) when compared to groups without pretreatment (control, 360.5 ± 5.2 beats/min; urine alone, 354.4 ± 4.2 beats/min) (Fig. 1c).
PS-Induced Locomotor Activity Changes and the Effects of Pretreatment with Cap or PTX
There were no differences in baseline locomotor activity in the rats with normal drinking water or pretreated with PTX (p > 0.05). However, the rats pretreated with Cap exhibited a significant decrease in baseline locomotor activity (p < 0.05). PS treatment induced a significant reduction in locomotor activity through induction, delay, and expression periods (p < 0.05), and pretreatment with either Cap or PTX and ANG II infusion had no effects on this PS-induced locomotor activity (p > 0.05, Fig. 2a and b).
Fig. 2.

Locomotor activity changes (Fig. 2a and b) induced by coyote urine scent exposure in rats with normal drinking water or pretreated with either captopril (Cap) or pentoxifylline (PTX). Coyote urine scent exposure significantly reduced rat locomotor activity in all groups. Baseline activity is denoted by B’s. (n = 7–10/group; *p < 0.05 vs baseline; #p < 0.05 vs normal drinking water or pretreatment with PTX)
PS Produced Anxiety-Like Behavior and the Effects of Pretreatment with Cap or PTX
The results of the elevated plus maze (EPM) test showed that rats with PS exposure spent more time in the closed arms (607.7 ± 8.2 s, p < 0.05, Fig. 3a) and less time in the open arms (9.0 ± 4.5 s, p < 0.05, Fig. 3b) and exhibited less open arm entries (0.6 ± 0.3 times, p < 0.05, Fig. 3c) than control rats (closed arms, 527.3 ± 18.7 s; open arms, 85.8 ± 16.4 s; open arm entries, 3.9 ± 0.7 times). There were no differences in entries into the closed arms in all groups (Fig. 3c). Pretreatment with either Cap or PTX significantly reversed the PS-induced increase in the time spent in the closed arms (Cap, 570.1 ± 11.1 s; PTX, 554.8 ± 16.9 s, p < 0.05, Fig. 3a) and decreased the time spent in the open arms (Cap, 37.7 ± 10.4 s; PTX, 55.3 ± 14.5 s, p < 0.05, Fig. 3b), while only pretreatment with PTX increased open arm entries in PS-exposed rats (2.6 ± 0.6 times, p < 0.05, Fig. 3c).
Fig. 3.
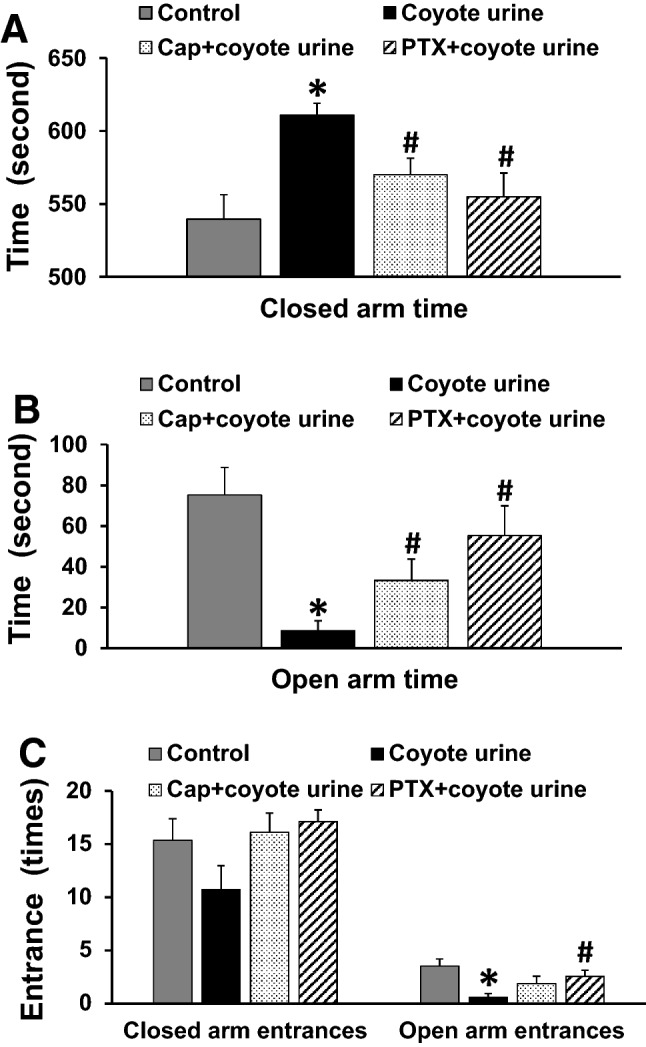
Assessment of anxiety-like behavior in control or coyote urine scent-exposed rats given either normal drinking water or an ACE inhibitor (captopril, Cap) or a TNF-α inhibitor (pentoxifylline, PTX) in the drinking water, including time in the closed arms (Fig. 3a), time in the open arms (Fig. 3b), and entries in the closed or open arms (Fig. 3c). (n = 9–16/group; *p < 0.05 vs. control rats; #p < 0.05 vs. coyote urine scent-exposed rats)
PS-Induced Changes in Brain RAS, PIC, and Microglial Activity and Effect of Cap or PTX
In LT tissues collected from rats on day 3 after the last PS exposure, stressed rats exhibited increased mRNA expression of RAS component (ACE) and the PICs (TNF-α, IL-6) when compared with non-stressed rats (p < 0.05, Fig. 4a). Pretreatment with either ACE inhibitor Cap or TNF-α synthesis inhibitor PTX blocked PS-induced increases in gene expression of ACE, TNF-α, and IL-6. Pretreatment with the ACE inhibitor upregulated the mRNA expression of AT1-R while pretreatment with the TNF-α synthesis inhibitor attenuated the expression of the microglial marker CD11b.
Fig. 4.

Quantitative comparison of the mRNA expression of renin–angiotensin system components, proinflammatory cytokines, and microglial marker in the lamina terminalis (LT, Fig. 4a), paraventricular nucleus (PVN, Fig. 4b), and amygdala (AMY, Fig. 4c) in control, coyote urine scent-exposed, coyote urine scent-exposed, and pretreatment with either captopril (Cap) or pentoxifylline (PTX) rats before angiotensin II infusion. (n = 5–7/group; *p < 0.05 vs control rats; #p < 0.05 vs. coyote urine scent-exposed rats without pretreatment).
In the PVN, only PS-treated rats showed upregulation of mRNA expression of TNF-α, IL-6, and CD11b. Both Cap and PTX pretreatments reversed the increased expression of TNF-α and CD11b, while only PTX pretreatment blocked the increased expression of IL-6. Like the effects on LT tissues, pretreatment with Cap also upregulated the expression of AT1-R in the PVN (p < 0.05, Fig. 4b).
In AMY tissues, PS exposure resulted in a significant increase in mRNA expression of ACE, TNF-α, IL-6, IL-1β, and CD11b when compared with the control group (p < 0.05, Fig. 3c). Pretreatment with either the ACE inhibitor or the TNF-α synthesis inhibitor blocked these PS-induced increases in gene expression (p < 0.05, Fig. 4c).
PS-Induced Changes in Plasma Levels of ANG II and IL-6 and the Effects of Cap or PTX
Plasma levels of ANG II (Fig. 5a) and IL-6 (Fig. 5b) were markedly higher in PS-exposed rats than in the control rats (p < 0.05). Pretreatment with either the ACE inhibitor or the TNF-α synthesis inhibitor reversed the PS-induced increases in plasma levels of these humoral factors (p < 0.05, Fig. 5a and b).
Fig. 5.

Plasma levels of renin–angiotensin system component (angiotensin II, ANG II, Fig. 5a), proinflammatory cytokine (interleukin-6, IL-6, Fig. 5b) in control, coyote urine scent-exposed, coyote urine scent-exposed plus pretreatment with either captopril (Cap) or pentoxifylline (PTX) rats before ANG II administration. The elevated plasma levels of ANG II or IL-6 in coyote urine scent-exposed rats were attenuated by either Cap or PTX pretreatment. (n = 8–12/group; *p < 0.05 vs. control rats; #p < 0.05 vs. coyote urine scent exposed without pretreatment)
Discussion
The current studies investigated the capacity of experimental anxiety to induce HTRS. The stressor employed was PS, which has been used previously to study experimentally induced PTSD (Albrechet-Souza and Gilpin 2019). Under the same PS conditions used to induce HTRS, anxiety-related behavioral changes were characterized in the elevated plus maze. Circulating levels of ANG II and cytokines and putative CNS molecular and cellular mediators in brain structures implicated in controlling sympathetic activity and mediating anxiety were also measured after PS. Finally, to assess the role of increased activity of the systemic and central RASs and inflammatory mechanisms, the capacity of RAS antagonism and TNF-α inhibition by Cap or PTX to prevent the PS-induced changes in functional and biochemical end-points was studied. The major findings of the present study were that PS (1) induced HTRS, (2) reduced locomotor activity, increased the preference for the closed arms in the plus maze, and increased the time of immobility, which are considered to be behavioral signs of anxiety, (3) increased plasma levels of ANG II and IL-6, and (4) increased mRNA expression of PICs and several prohypertensive components of the RAS in the LT, PVN, and AMY. Most of the PS-induced effects were blocked by pretreatment with either ACE or TNF-α inhibition. These results indicate that PS-induced HTRS and heightened anxiety-like behaviors are likely to be mediated by increased RAS and inflammatory activity in the periphery and in the brain.
Stressful challenges are classified as physiological or psychosocial stressors. Physiological stressors (AKA homeostatic or interoceptive stressors) are challenges that result from a clear disruption of homeostasis, such as hypoxia, hypovolemia, hypertonicity, hypoglycemia hypertension, or hypotension (Herman and Cullinan 1997; Sawchenko et al. 2000; Pacak and Palkovits 2001). In comparison, psychosocial stressors (AKA psychological, processive, neurogenic, mental, or exteroceptive stressors) have no immediate effects on homeostasis but are stimuli perceived as potential threats to one’s physical or psychological integrity (Herman and Cullinan 1997; Sawchenko et al. 2000; Pacak and Palkovits 2001). Previous studies from our laboratory have demonstrated that a wide range of physiological stressors induce HTRS (Johnson et al. 2015; Johnson and Xue 2018). Recently, we began to investigate the capacity of psychosocial stressors to induce HTRS (Xue et al. 2019).
Our previous studies demonstrated that HTRS can be induced in an intruder rat by repeated introduction into the cage of a resident animal (i.e., the resident-intruder psychosocial stress paradigm) (Xue et al. 2019). When confronted by a resident in the resident-intruder model, the intruder displays a pattern of cardiovascular and behavioral responses along with accompanying behavioral signs of defeat that can be best characterized as the classic fight-flight or defense response (Viken et al. 1991). One limitation of the resident-intruder model to investigate the effects of a psychosocial stressor is the possibility that even minor injury resulting from the confrontation has the potential to activate the immune system and the RAS in the intruder. Also, in our previous study, anxiety-related behaviors were not studied and shown to be induced by the resident-intruder procedures we used as a psychosocial stressor. The PS paradigm obviates any concern about physical contact or injury-inducing changes in candidate blood-borne or brain molecular mediators of HTRS or anxiety-like behaviors. The primary purpose of the present studies was to test the generality of the capacity of psychosocial stressors to induce HTRS and behavior changes in a model of PTSD that eliminates the possibility of any physical insult.
In the present studies, we found that ANG II and IL-6 levels in the plasma were increased after PS exposure. Information indicative of increased activity of the systemic RAS and of the immune system is communicated to the CNS by both humoral and neural afferent signaling (Ericsson et al. 1994; Banks et al. 1995; Goehler et al. 2000; Dantzer 2018). Blood-borne ANG II or PICs acting on the SFO have been shown to activate the RAS and increase inflammation in the PVN that receives input via SFO efferents. This peripheral-central, humoral-neural coupling increases sympathetic drive and in turn, increased BP (Wei et al. 2009, 2015; Wei et al. 2018). Therefore, prior stressor exposure may elevate the levels of the RAS and PICs in systems that enhance CNS activity to provide a physiological foundation for the sensitization of hypertension. In PS animals, the induction of HTRS was blocked by systemic administration of either ACE or TNF-α inhibitors. This is consistent with other findings from our laboratory indicating that mutual activity of the brain RAS and PICs is necessary for the induction of HTRS by many different types of stressors (Johnson et al. 2015; Johnson and Xue 2018; Xue et al. 2020).
The brain contains both a tissue RAS and an innate immune system involving microglia with associated PICs (de Git and Adan 2015; Nakagawa et al. 2020). The activation of the brain RAS or immune system can independently or synergistically lead to hypertension and behavioral disorders (Saavedra et al. 2011; Duchemin et al. 2013; Michopoulos et al. 2017). Shi and colleagues demonstrated that activity of microglia increased during ANG II-induced hypertension. Rats treated with a microglia inhibitor, minocycline, exhibited lower PIC levels that resulted in reduced ANG II-induced hypertension (Shi et al. 2010).
Psychological disorders such as anxiety and depression have high levels of comorbidity with hypertension and other cardiovascular diseases (Grippo and Johnson 2002, 2009; Kibler et al. 2009). PTSD is not only a psychological stress-related disorder, but it is also associated with an increased risk of cardiovascular diseases including hypertension (Cohen et al. 2015; Burg and Soufer 2016; Edmondson and von Kanel 2017). PS induces heightened anxiety, exaggerated startle, and impaired cognition, all of which are common symptoms seen in humans with PTSD (Cohen et al. 2012; Maria-Rios and Morrow 2020). PS is associated with activation of the RAS and elevated inflammation in both the periphery and the CNS (Bali and Jaggi 2013; Wilson et al. 2013). Administration of angiotensin II receptor blockers (ARBs) and ACE-I inhibitors or anti-inflammatory drugs (minocycline, ibuprofen) significantly attenuates stress responses in humans and animal models (Khoury et al. 2012; Levkovitz et al. 2015; Lee et al. 2016). Following the exact PS protocol that produced HTRS, we confirmed that behavioral changes associated with anxiety are produced by PS. Rats exposed to the scent of coyote urine exhibited a significant reduction in locomotor activity throughout induction–delay–expression paradigm. Furthermore, these rats spent significantly more time in the closed arms and less time in the open arms as well as less open arm entries than controls in the elevated plus maze. Treatment with either ACE or TNF-α inhibitors reversed most of these anxiety-like behaviors. Treatment with PTX appears to be more effective than CAP, since it also restored the number of open arm entries. These results are consistent with previous studies showing that both the RAS and inflammation are associated with PS-induced anxiety-like behaviors (Whitaker and Gilpin 2015; Albrechet-Souza and Gilpin 2019). This finding receives further support from our results from the measurement of increased plasma levels and brain expression of PICs and RAS components.
The LT, PVN, and AMY play roles in the regulation of sympathetic activity and BP by integrating information derived from many different types of physiological and psychosocial stressors (Lucassen et al. 2014; Dampney 2016). RAS activation, inflammation, and microglia are involved in these structures in processing and the storage of information (i.e., memories of earlier events). In the present study, PS significantly activated the RAS and induced inflammation through an increase of ACE mRNA expression and the upregulation of PICs, including TNF-α, IL-6, and IL-1β in the LT, PVN, and AMY. The microglial marker CD11b was also upregulated in the PVN and AMY, which is consistent with the idea that increased microglial activity will increase PICs. Either Cap or PTX reduced the upregulation of both ACE and PICs. Taken together, these findings are consistent with other results from our laboratory (Johnson et al. 2015; Johnson and Xue 2018) indicating that there is a requirement for mutual activity of the brain RAS and PICs for the induction of HTRS by a wide range of stressors.
Interestingly, Cap treatment upregulated AT1-R expression in the LT and PVN, whereas PS exposure did not. It is possible that the AT1-R upregulation represents a counter-regulatory response to the inhibition of ACE production and presumed decrease in ambient ANG II. However, the compensatory increase in AT1-R expression might not be sufficient to counteract the effects of downregulation of PICs and CD11b since Cap treatment still attenuated the HTRS induced by PS. We also found that Cap treatment resulted in decreased basal BP and locomotor activity, an increase in basal HR, and a significant decrease in HR during ANG II infusion when compared with other groups. These results are consistent with a previous study (Maliszewska-Scislo et al. 2003), but whether the locomotor activity and hemodynamic changes in the Cap-treated group are related to upregulation of AT1-R in the brain or to other central mechanisms need further study.
In summary, PS as a psychosocial stress model of PTSD induces HTRS as well as increases in anxiety-like behaviors probably through upregulation of RAS and PICs components in key brain areas associated with BP control and anxiety (Fig. 6). Reducing activity of the RAS and inflammation is effective in blocking the induction of HTRS and as an anxiolytic intervention. This study provides insights into mechanisms which increase the predisposition for the development of hypertension under stressful conditions and suggests therapeutic strategies for the prevention and treatment of hypertension.
Fig. 6.
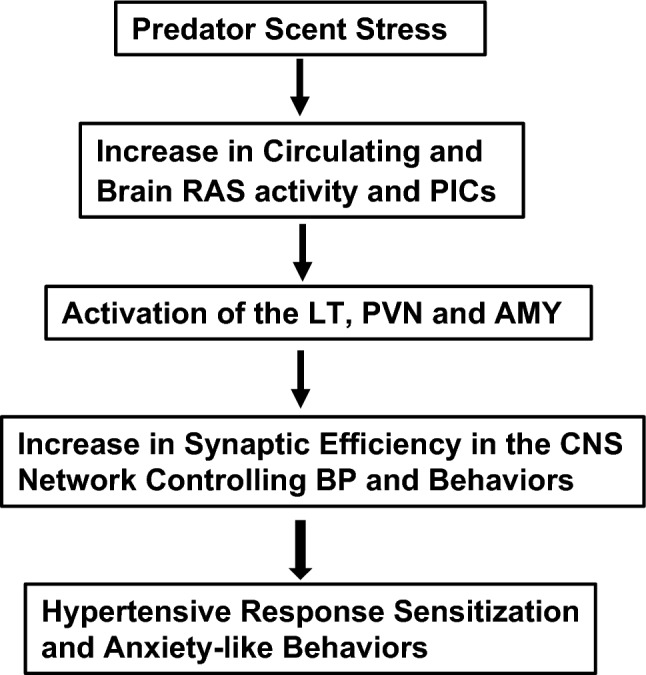
Schematic representation of predator scent (PS) stress-elicited hypertensive response sensitization (HTRS) and anxiety-like behaviors through upregulation of renin–angiotensin system (RAS) and inflammatory cytokines (PICs) in peripheral and central nervous system (CNS) network controlling blood pressure (BP) and behaviors including the lamina terminalis (LT), paraventricular nucleus of hypothalamus (PVN), and amygdala (AMY)
Author Contributions
BX and AKJ designed the experiments; BX, JX, YY, and TB performed the experiments and analyzed the data; BX and JX wrote the manuscript; BX, JX, YY, SGW, RF, and AKJ revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the NIH grants HL-139575 (AKJ & BX), HL-139521 (SGW), and HL-073986 (RBF).
Compliance with Ethical Standards
Conflict of interest
The authors declared that they have no conflicts of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reference
- Albrechet-Souza L, Gilpin NW (2019) The predator odor avoidance model of post-traumatic stress disorder in rats. Behav Pharmacol 30 (2 and 3-Spec Issue):105-114. https://doi.org/10.1097/FBP.0000000000000460 [DOI] [PMC free article] [PubMed]
- Bali A, Jaggi AS (2013) Angiotensin as stress mediator: role of its receptor and interrelationships among other stress mediators and receptors. Pharmacol Res 76:49–57. 10.1016/j.phrs.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Banfi C, Sironi L, De Simoni G, Gelosa P, Barcella S, Perego C, Gianazza E, Guerrini U, Tremoli E, Mussoni L (2004) Pentoxifylline prevents spontaneous brain ischemia in stroke-prone rats. J Pharmacol Exp Ther 310(3):890–895. 10.1124/jpet.104.067090 [DOI] [PubMed] [Google Scholar]
- Banks WA, Kastin AJ, Broadwell RD (1995) Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 2(4):241–248. 10.1159/000097202 [DOI] [PubMed] [Google Scholar]
- Burg MM, Soufer R (2016) Post-traumatic stress disorder and cardiovascular disease. Curr Cardiol Rep 18(10):94. 10.1007/s11886-016-0770-5 [DOI] [PubMed] [Google Scholar]
- Clayton SC, Zhang Z, Beltz T, Xue B, Johnson AK (2014) CNS neuroplasticity and salt-sensitive hypertension induced by prior treatment with subpressor doses of ANG II or aldosterone. Am J Physiol Regul Integr Comp Physiol 306(12):R908-917. 10.1152/ajpregu.00010.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen BE, Edmondson D, Kronish IM (2015) State of the art review: depression, stress, anxiety, and cardiovascular disease. Am J Hypertens 28(11):1295–1302. 10.1093/ajh/hpv047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Kozlovsky N, Alona C, Matar MA, Joseph Z (2012) Animal model for PTSD: from clinical concept to translational research. Neuropharmacology 62(2):715–724. 10.1016/j.neuropharm.2011.04.023 [DOI] [PubMed] [Google Scholar]
- Dampney RA (2016) Central neural control of the cardiovascular system: current perspectives. Adv Physiol Educ 40(3):283–296. 10.1152/advan.00027.2016 [DOI] [PubMed] [Google Scholar]
- Dantzer R (2018) Neuroimmune interactions: from the brain to the immune system and vice versa. Physiol Rev 98(1):477–504. 10.1152/physrev.00039.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Git KC, Adan RA (2015) Leptin resistance in diet-induced obesity: the role of hypothalamic inflammation. Obes Rev 16(3):207–224. 10.1111/obr.12243 [DOI] [PubMed] [Google Scholar]
- Duchemin S, Belanger E, Wu R, Ferland G, Girouard H (2013) Chronic perfusion of angiotensin II causes cognitive dysfunctions and anxiety in mice. Physiol Behav 109:63–68. 10.1016/j.physbeh.2012.10.005 [DOI] [PubMed] [Google Scholar]
- Edmondson D, von Kanel R (2017) Post-traumatic stress disorder and cardiovascular disease. Lancet Psychiatr 4(4):320–329. 10.1016/S2215-0366(16)30377-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson A, Kovacs KJ, Sawchenko PE (1994) A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J Neurosci 14(2):897–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes MA, Martins Lima A, Santos RA (2016) Brain angiotensin-(1–7)/Mas axis: a new target to reduce the cardiovascular risk to emotional stress. Neuropeptides 56:9–17. 10.1016/j.npep.2015.10.003 [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, Watkins LR (2000) Vagal immune-to-brain communication: a visceral chemosensory pathway. Auton Neurosci 85(1–3):49–59. 10.1016/S1566-0702(00)00219-8 [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Johnson AK (2002) Biological mechanisms in the relationship between depression and heart disease. Neurosci Biobehav Rev 26(8):941–962. 10.1016/s0149-7634(03)00003-4 [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Johnson AK (2009) Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress 12(1):1–21. 10.1080/10253890802046281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Cullinan WE (1997) Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci 20(2):78–84 [DOI] [PubMed] [Google Scholar]
- Hurley SW, Beltz TG, Guo F, Xue B, Johnson AK (2020) Amphetamine-induced sensitization of hypertension and lamina terminalis neuroinflammation. Am J Physiol Regul Integr Comp Physiol 318(3):R649–R656. 10.1152/ajpregu.00233.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AK, Xue B (2018) Central nervous system neuroplasticity and the sensitization of hypertension. Nat Rev Nephrol 14(12):750–766. 10.1038/s41581-018-0068-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AK, Zhang Z, Clayton SC, Beltz TG, Hurley SW, Thunhorst RL, Xue B (2015) The roles of sensitization and neuroplasticity in the long-term regulation of blood pressure and hypertension. Am J Physiol Regul Integr Comp Physiol 309(11):R1309-1325. 10.1152/ajpregu.00037.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury NM, Marvar PJ, Gillespie CF, Wingo A, Schwartz A, Bradley B, Kramer M, Ressler KJ (2012) The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatr 73(6):849–855. 10.4088/JCP.11m07316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibler JL, Joshi K, Ma M (2009) Hypertension in relation to posttraumatic stress disorder and depression in the US National Comorbidity Survey. Behav Med 34(4):125–132. 10.3200/BMED.34.4.125-132 [DOI] [PubMed] [Google Scholar]
- Lee B, Sur B, Yeom M, Shim I, Lee H, Hahm DH (2016) Effects of systemic administration of ibuprofen on stress response in a rat model of post-traumatic stress disorder. Korean J Physiol Pharmacol 20(4):357–366. 10.4196/kjpp.2016.20.4.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkovitz Y, Fenchel D, Kaplan Z, Zohar J, Cohen H (2015) Early post-stressor intervention with minocycline, a second-generation tetracycline, attenuates post-traumatic stress response in an animal model of PTSD. Eur Neuropsychopharmacol 25(1):124–132. 10.1016/j.euroneuro.2014.11.012 [DOI] [PubMed] [Google Scholar]
- Lucassen PJ, Pruessner J, Sousa N, Almeida OF, Van Dam AM, Rajkowska G, Swaab DF, Czeh B (2014) Neuropathology of stress. Acta Neuropathol 127(1):109–135. 10.1007/s00401-013-1223-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliszewska-Scislo M, Scislo TJ, Rossi NF (2003) Effect of blockade of endogenous angiotensin II on baroreflex function in conscious diabetic rats. Am J Physiol Heart Circ Physiol 284(5):H1601-1611. 10.1152/ajpheart.00578.2002 [DOI] [PubMed] [Google Scholar]
- Maria-Rios CE, Morrow JD (2020) Mechanisms of shared vulnerability to post-traumatic stress disorder and substance use disorders. Front Behav Neurosci 14:6. 10.3389/fnbeh.2020.00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Teschemacher AG, Kasparov S, Gourine AV (2016) Glia, sympathetic activity and cardiovascular disease. Exp Physiol 101(5):565–576. 10.1113/EP085713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michopoulos V, Powers A, Gillespie CF, Ressler KJ, Jovanovic T (2017) Inflammation in fear- and anxiety-based Disorders: PTSD, GAD, and Beyond. Neuropsychopharmacology 42(1):254–270. 10.1038/npp.2016.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe KE, Weiss ML, Epstein AN (1984) Sodium appetite during captopril blockade of endogenous angiotensin II formation. Am J Physiol 247(2 Pt 2):R356-365. 10.1152/ajpregu.1984.247.2.R356 [DOI] [PubMed] [Google Scholar]
- Nakagawa P, Gomez J, Grobe JL, Sigmund CD (2020) The renin-angiotensin system in the central nervous system and its role in blood pressure regulation. Curr Hypertens Rep 22(1):7. 10.1007/s11906-019-1011-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB, Bremner JD, Foa EB, Mayberg HS, North CS, Stein MB (2006) Posttraumatic stress disorder: a state-of-the-science review. J Psychiatr Res 40(1):1–21. 10.1016/j.jpsychires.2005.07.005 [DOI] [PubMed] [Google Scholar]
- Pacak K, Palkovits M (2001) Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr Rev 22(4):502–548. 10.1210/edrv.22.4.0436 [DOI] [PubMed] [Google Scholar]
- Patki G, Solanki N, Atrooz F, Allam F, Salim S (2013) Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res 1539:73–86. 10.1016/j.brainres.2013.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin P, File SE, Briley M (1985) Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods 14(3):149–167. 10.1016/0165-0270(85)90031-7 [DOI] [PubMed] [Google Scholar]
- Plotnikov MB, Aliev OI, Shamanaev AY, Sidekhmenova AV, Anfinogenova Y, Anishchenko AM, Fomina TI, Arkhipov AM (2017) Effects of pentoxifylline on hemodynamic, hemorheological, and microcirculatory parameters in young SHRs during arterial hypertension development. Clin Exp Hypertens 39(6):570–578. 10.1080/10641963.2017.1291662 [DOI] [PubMed] [Google Scholar]
- Rodgers RJ, Dalvi A (1997) Anxiety, defence and the elevated plus-maze. Neurosci Biobehav Rev 21(6):801–810. 10.1016/s0149-7634(96)00058-9 [DOI] [PubMed] [Google Scholar]
- Saavedra JM, Sanchez-Lemus E, Benicky J (2011) Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation and ischemia: therapeutic implications. Psychoneuroendocrinology 36(1):1–18. 10.1016/j.psyneuen.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE, Li HY, Ericsson A (2000) Circuits and mechanisms governing hypothalamic responses to stress: a tale of two paradigms. Prog Brain Res 122:61–78 [DOI] [PubMed] [Google Scholar]
- Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK (2010) Brain microglial cytokines in neurogenic hypertension. Hypertension 56(2):297–303. 10.1161/HYPERTENSIONAHA.110.150409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thunhorst RL, Fitts DA, Simpson JB (1989) Angiotensin-converting enzyme in subfornical organ mediates captopril-induced drinking. Behav Neurosci 103(6):1302–1310 [DOI] [PubMed] [Google Scholar]
- Viken RJ, Johnson AK, Knutson JF (1991) Blood pressure, heart rate, and regional resistance in behavioral defense. Physiol Behav 50(6):1097–1101. 10.1016/0031-9384(91)90567-8 [DOI] [PubMed] [Google Scholar]
- Weber MD, Godbout JP, Sheridan JF (2017) Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 42(1):46–61. 10.1038/npp.2016.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Felder RB (2018) Blood-borne interleukin-1beta acts on the subfornical organ to upregulate the sympathoexcitatory milieu of the hypothalamic paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 314(3):R447–R458. 10.1152/ajpregu.00211.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB (2009) Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol 296(5):H1425-1433. 10.1152/ajpheart.00942.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei SG, Yu Y, Zhang ZH, Felder RB (2015) Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension 65(5):1126–1133. 10.1161/HYPERTENSIONAHA.114.05112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker AM, Gilpin NW (2015) Blunted hypothalamo-pituitary adrenal axis response to predator odor predicts high stress reactivity. Physiol Behav 147:16–22. 10.1016/j.physbeh.2015.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, McLaughlin LD, Nair A, Ebenezer PJ, Dange R, Francis J (2013) Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post-traumatic stress disorder in a predator exposure animal model. PLoS One 8(10):e76146. 10.1371/journal.pone.0076146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklewski PJ, Radkowski M, Wszedybyl-Winklewska M, Demkow U (2015) Brain inflammation and hypertension: the chicken or the egg? J Neuroinflammation 12:85. 10.1186/s12974-015-0306-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Thunhorst RL, Yu Y, Guo F, Beltz TG, Felder RB, Johnson AK (2016) Central Renin-Angiotensin System Activation and Inflammation Induced by High-Fat Diet Sensitize Angiotensin II-Elicited Hypertension. Hypertension 67(1):163–170. 10.1161/HYPERTENSIONAHA.115.06263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Yu Y, Wei SG, Beltz TG, Guo F, Felder RB, Johnson AK (2019) Stress-induced sensitization of angiotensin II hypertension is reversed by blockade of angiotensin-converting enzyme or tumor necrosis factor-alpha. Am J Hypertens. 10.1093/ajh/hpz075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Yu Y, Zhang Z, Guo F, Beltz TG, Thunhorst RL, Felder RB, Johnson AK (2016) Leptin mediates high-fat diet sensitization of angiotensin II-Elicited Hypertension By Upregulating The Brain Renin-Angiotensin System And Inflammation. Hypertension 67(5):970–976. 10.1161/HYPERTENSIONAHA.115.06736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Zhang Y, Johnson AK (2020) Interactions of the brain renin-angiotensin-system (RAS) and inflammation in the sensitization of hypertension. Front Neurosci 14:650. 10.3389/fnins.2020.00650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Zhang Z, Johnson RF, Johnson AK (2012) Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension 59(2):459–466. 10.1161/HYPERTENSIONAHA.111.185116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue B, Zhang Z, Roncari CF, Guo F, Johnson AK (2012) Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension 60(4):1023–1030. 10.1161/HYPERTENSIONAHA.112.196576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN, Davisson RL (2015) Angiotensin-II, the Brain, and Hypertension: An Update. Hypertension 66(5):920–926. 10.1161/HYPERTENSIONAHA.115.03624 [DOI] [PMC free article] [PubMed] [Google Scholar]