

Abstract
Translocator Protein 18 kDa (TSPO) is a well-known outer mitochondrial membrane protein and it is widely used as a biomarker of neuroinflammation and brain injury. While it is thought that TSPO plays key roles in a multitude of host cell functions, including steroid biosynthesis, apoptosis, generation of reactive oxygen species, and proliferation, some of these functions have recently been questioned. Here, we report the unexpected finding that circulating immune cells differentially express basal levels of TSPO on their cell surface, with a high percentage of monocytes and neutrophils expressing cell surface TSPO. In vitro stimulation of monocytes with lipopolysaccharide (LPS) significantly increases the frequency of cells with surface TSPO expression in the absence of altered gene expression. Importantly, the LPS-increase in TSPO cell surface expression in monocytes appears to be selective for LPS since two other distinct monocyte activators failed to increase the frequency of cells with surface TSPO. Finally, when we quantified immune cell TSPO surface expression in antiretroviral therapy (ART)-treated HIV+ donors, a chronic inflammatory disease, we found significant increases in the frequency of TSPO surface localization, which could be pharmacologically suppressed with ∆9-tetrahydrocannabinol (THC). These findings suggest that cell surface TSPO in circulating leukocytes could serve as a peripheral blood-based biomarker of inflammation.
Keywords: TSPO, inflammation, HIV, Circulating leukocytes, Δ9-tetrahydrocannabinol
Graphical Abstract
Summary Sentence:
Detection of TSPO on the cell surface of monocytes which was increased by activation with LPS and by HIV infection in circulating leukocytes.
Introduction
Translocator Protein 18 kDa (TSPO) is a nuclear encoded mitochondrial protein primarily localized in the outer mitochondrial membrane (OMM) [1, 2]. Due to its role as a mitochondrial protein, TSPO is ubiquitously expressed to varying degrees by most, if not all host cells [3]. TSPO was initially identified as a binding target of diazepam [4]. However, it was determined that TSPO was a distinctly unique diazepam-binding protein from the neuronally expressed central benzodiazepine receptor (CBR), which localizes to the plasma membrane of neuronal cells [4]. TSPO has been colloquially referred to as an ‘orphan protein searching for a function’, owed in part to the evolving controversies surrounding TSPO expression, function, and it’s implication in disease processes [5].
TSPO was initially described as a cholesterol translocator involved in the mitochondrial import of cholesterol as part of the rate limiting step in steroidogenesis [2]. This was due in part to the fact that TSPO contains a canonical cholesterol binding domain and cells that principally produce and secrete endogenous steroids express the highest levels of TSPO [2, 6]. Further, initial attempts to create TSPO global knockout mice were embryonically lethal, making genetic approaches to studying TSPO function difficult. However, as recently as 2014, Morohaku and colleagues as well as Tu and colleagues succeeded in generating both conditional and global TSPO knockout mice, respectively [7–9]. Studies using isolated cells or whole animals from these model systems unexpectedly revealed that TSPO was not necessary for de novo steroidogenesis, calling into question one of the most studied biological function(s) of TSPO. Follow-up studies have suggested various distinct molecular and cellular functions for TSPO both in healthy and disease states, ranging from putative roles in proliferation, apoptosis, reactive oxygen species (ROS) generation, cell differentiation, immune modulation, and tetrapyrrole biosynthesis [10–15].
Despite the lack of evidence for a clear molecular function of TSPO, it has been widely used as a biomarker of neuroinflammation and brain injury using Positron Emission Tomography (PET) [16]. Studies have correlated increased expression of TSPO in glial cells and infiltrating macrophages in the brains of human patients and animals models of various neuroinflammatory diseases such as HIV-associated neurodegeneration and neurodegenerative disorders such as Parkinson’s disease (See [5] and [16] for review). Consequently, the use of TSPO autoradiography and immunohistochemical methods has allowed robust visualization and correlation of TSPO in glial cells and infiltrating monocytes in the brains of living humans who are experiencing inflammatory-mediated brain diseases [5, 17, 18] making TSPO an extremely attractive biomarker of neuroinflammation and a potential druggable target.
It has been hypothesized that most, if not all, neuroinflammatory processes are initiated in the periphery as either an immune response to an infection or an autoimmune response [19, 20]. Further, monocytes/macrophages have been readily described in several neuroinflammatory diseases as being one of the earliest and most prominent immune cells to infiltrate the brain and initiate local neuroinflammation [21–23]. Indeed, our own studies of HIV associated neurocognitive disorders (HAND) have shown that resting monocytes respond to inflammatory signals in the periphery and rapidly acquire CD16 and a unique effector profile marked by the secretion of proinflammatory IL-1β and IL-6 [24]. These inflammatory cytokines then act on astrocytes to initiate an inflammatory amplification loop which results in increased accumulation of inflammatory monocytes and T cells in the brains of HIV patients experiencing neurocognitive decline [24].
No previous study has directly interrogated the connection between immune cell TSPO surface expression and neuroinflammation. Therefore, we assessed TSPO as an immune-specific biomarker for inflammatory immune cell activation in the periphery. Here, we report for the first time, the novel finding that not only is TSPO detectable on various immune cell types, but it is also readily detectable on the surface of the plasma membrane. Furthermore, monocyte activation with proinflammatory lipopolysaccharide (LPS) increased the frequency of monocytes with detectable cell surface TSPO with no change in intracellular TSPO or TSPO mRNA levels. Finally, we explored the relationship of cell surface TSPO to a relevant, chronic inflammatory disease, HIV. We observed significantly increased basal cell surface TSPO expression on various circulating immune cell types including monocytes and plasmacytoid dendritic cells (pDC) from peripheral blood of HIV infected donors, suggesting potential utility as a peripheral biomarker of chronic inflammation. Furthermore, treatment with anti-inflammatory Δ9-tetrahydrocannabinol (THC) reduced surface TSPO expression on monocytes from HIV− and HIV+ subjects.
Materials and Methods
Human donors and Isolation of mononuclear cells (PBMC):
PBMC were isolated from human leukocyte packs (Gulf Coast Regional Blood Center, Houston, TX) without differentiating between male and female donors. Blood was diluted 1:1 with HBSS (Gibco™). Diluted blood was layered on 15 ml Ficoll Paque Plus (GE Healthcare Life Sciences, Pittsburgh, PA) using SepMate 50 ml conical tubes by StemCell Technologies (Vancouver, BC, Canada) and centrifuged at 1300 × g for 25 min at 4 °C. The PBMC layer was removed and washed with HBSS twice. After the last wash PBMC were resuspended in RPMI1640 (Gibco™) containing 5% Human AB Serum (Valley Biomedical, Winchester, VA), and 1% Penicillin-Streptomycin (Gibco™). PBMC were seeded in 96 well plates at a density of 1 × 106 cells/well in 200 μl of media. Pan monocytes were isolated by negative selection (Human Pan Monocyte Isolation Kit) from Miltenyi Biotec, per manufacturer's direction.
Male whole blood HIV− samples were purchased from the Stanford Blood Center (Palo Alto, CA) in heparin tubes and shipped overnight at room temperature. Male HIV+ donors voluntarily enrolled via the Mid-Michigan HIV Consortium (MMHC) under the Institutional Review Board–approved protocol (Institutional Review Board protocol no. 11-202) and into the MMHC Registry. HIV+ subjects were males that have been receiving ART medication for at least a year and were between the ages of 28 and 66 years, with an average age of 51 years, who had CD4+ counts above 500 counts/ml blood, CD4/CD8 ratios >1, did not report to use cannabinoids, had HIV viral burdens below the detectable limit (<5 HIV mRNA copies/ml blood), and were not coinfected with any strain of hepatitis. HIV+ whole blood was collected in heparin tubes and stored overnight at room temperature. The status of medicinal cannabinoid use was determined by self-reporting and was verified via plasma detection of THC metabolites using the THC ELISA (enzyme-linked immunosorbent assay) Forensic Kit (Neogen Corporation, Lansing, MI). Any subjects that had detectable levels of THC or its metabolites were removed from the study. HIV+ donors received the standard of care and were not asked to change any lifestyle habits to participate. All subject questionnaire and abstracted medical record data of the MMHC are managed using the Research Electronic Data Capture (REDCap) (Vanderbilt University, Nashville, TN), which supports 21 Code of Federal Regulations Part 11 compliance for clinical research and trials data and Health Insurance Portability and Accountability Act of 1996 guidelines. Phenotyping and TSPO surface level detection was performed on 2ml of heparinized whole blood that was lysed with ACK buffer. In brief, 20ml of ACK lysis buffer was added to 2ml of whole blood and incubated for 10 min on ice. After incubation, samples were centrifuge at 300xg for 10 min and the pelleted leukocytes were washed two more times in FACS buffer (HBSS, 1% BSA, 0.1% NaN3). After the final wash, leukocytes were used for phenotyping and TSPO staining as stated above. For the activation studies with whole blood, PBMC were isolated from diluted whole blood using Ficoll Paque Plus as stated above.
Cell culture and activation:
PBMC (5×106 cells/mL) were cultured in complete RPMI media (see above) and were stimulated with LPS from S. typhosa (0.1–100μg/ml, Sigma Aldrich, St. Louis, MO), CpG (10μg/ml, Invivogen, San Diego, CA), Universal Type I Interferon Alpha (10-1000U/ml, PBL Assay Science, Piscataway Township, NJ) or anti-human CD3 and CD28 (5 μg/ml, BD Biosciences, San Jose, CA) and incubated for the desired time at 37°C and 5% CO2.
Flow cytometry:
Before surface phenotyping of PBMC, red blood cells were removed using an ACK lysis buffer protocol as stated above. FACS buffer was used to wash cells in between staining and fixing steps. First, cells were incubated with FACS containing 20% human AB serum to block Fc receptors. For surface staining, cells were first incubated with antibodies and LIVE/DEAD™ Fixable Dead Cell Stain (Thermo Fisher Scientific, Waltham, MA) followed by fixation with BD Cytofix™ (BD Biosciences). For intracellular TSPO staining, cells were first surfaced stained with the antibodies listed below to identify specific immune cells and fixed. After fixation, cells were permeabilized with PERM wash (BD Biosciences) and stained with the TSPO antibody. After intracellular staining, cells were washed in PERM wash three times and resuspend in FACS buffer for analysis. Cells were analyzed on a FACS BD Canto II™ (BD Biosciences). It is noteworthy that the photomultiplier tube voltage setting for TSPO surface stained samples was approximately 2 fold higher than the TSPO intracellular settings due to high intracellular TSPO levels. Antibodies included anti-TSPO (Abcam, Cambridge, MA), anti-CD14 to identify monocytes, anti-CD16 to identify subtypes of monocytes and neutrophils, anti-CD3 to identify T cells, anti-CD19 to identify B cells, anti-CD56 and anti-CD57 to identify NK cells (BioLegend, San Diego, CA) and CD303 (Miltenyi Biotec, Bergisch Gladbach, Germany) to identify pDCs. After acquisition, data analysis was performed using FLOWJO v10 (Tree Star Inc., Ashland, OR) software.
Image Flow Cytometry and Confocal Microscopy:
Both surface and intracellular staining for TSPO was performed as stated above. DAPI was added 10 min prior to analysis on the Mark 4 Amnis Image flow cytometer to visualize the nuclei. After acquisition, data analysis was preformed using IDEAS software. Plasma membrane versus internal expression of TSPO was quantified using the IDEAS internalization feature. This feature works by creating a mask to identify the cytoplasmic region of each cell and calculates the median pixel value of these cellular compartments and creates a ratio of cytoplasmic to membrane fluorescence. The ratio is log transformed to generate a Gaussian distribution such that when the fluorescence is equally on the surface and in the cytoplasm, the internalization value is zero. Surface expression is denoted by negative values.
For confocal microscopy analysis, PBMC were isolated as stated above and activated with 10μg/ml LPS for 24 hours in a 8 well Lab-Tek chamber slide™ (Nunc, Thermo Fisher Scientific, Rochester, NY) treated with poly-D-Lysine. Surface and intracellular staining for TSPO was performed as stated above except the surface TSPO was stained with an anti-TSPO antibody conjugated to AF488 and the anti-TSPO antibody used for the intercellular stain was conjugated to PE. Selected wells on the slide were stained with Cell Mask™ Deep Red plasma membrane stain (Invitrogen, Thermo Fisher Scientific) and all wells were stained with DAPI for identification of nuclei. Slides were mounted with Prolong™ Glass Antifade Mountant (Invitrogen). Confocal images were acquired on the Nikon A1 Rsi Confocal Laser Scanning Microscope, configured on a Nikon Elipse Ti inverted microscope with a 100x Apo TIRF objective (NA 1.49).
Statistical analysis:
Statistical analysis was performed using Prism 6 (GraphPad, San Diego, CA). The experimental data was graphed as the mean +/− SEM. The statistical tests performed for each experiment are indicated in the figure legends.
Results
TSPO is readily detectable on the cell surface of human circulating immune cells.
In order to begin to investigate the utility of using TSPO expression as a potential peripheral marker of inflammation, we began by quantifying ex vivo the relative protein expression of TSPO on various immune cells found in circulation. Briefly, peripheral blood cells were obtained from commercially available human whole blood in heparin coated tubes, permeabilized, and stained for intracellular TSPO (Fig 1A). Cells were also stained for TSPO in the absence of permeabilization as a negative control (Fig.1B). As shown in representative flow plots in Figure 1A, virtually every immune cell expressed intracellular TSPO. As macrophage expression of TSPO has been reported in the literature to be detectable in brain inflammation using PET [25, 26], we found that cells expressing CD14, a marker of monocytes/macrophage, were one of the highest intracellular TSPO expressing cell types (Fig. 1A).
Figure 1. Comparison of intra- and surface TSPO expression in human monocytes by flow cytometry.
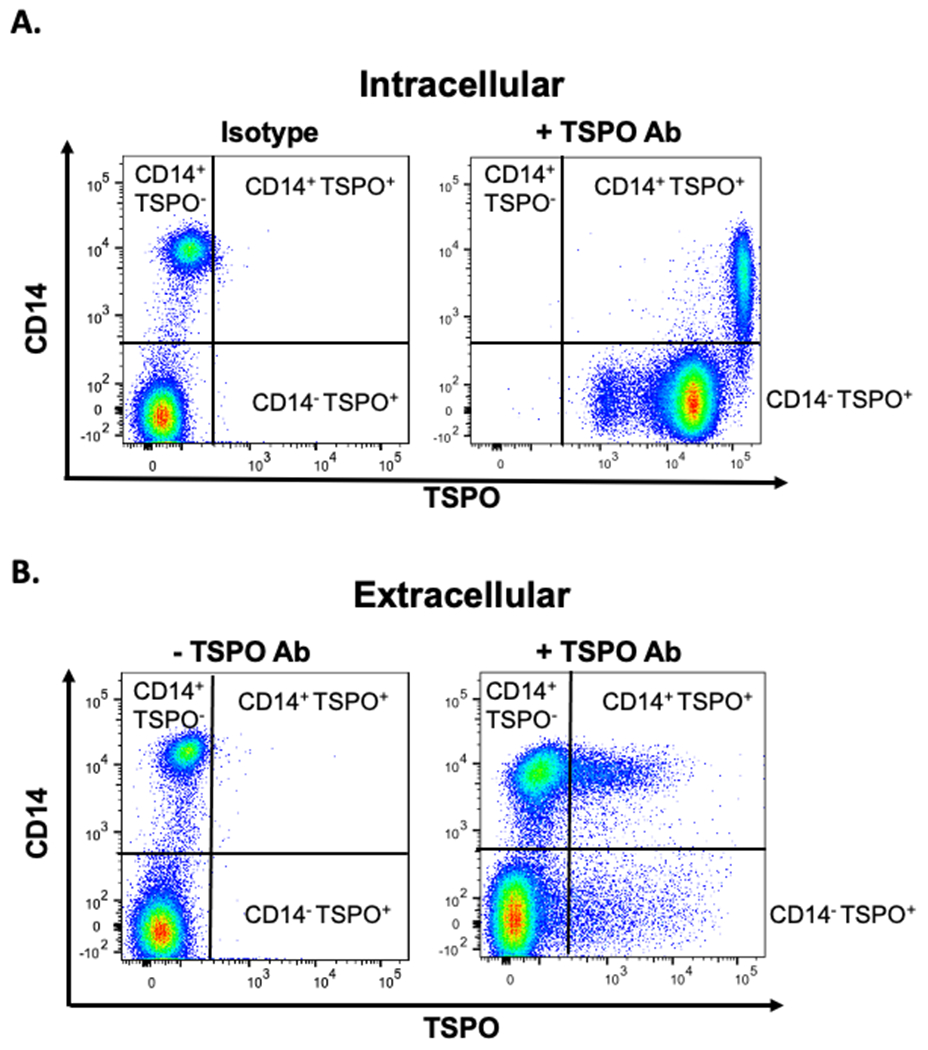
Human PBMC were isolated from commercially available leukocyte packs by ficoll-density gradient centrifugation and stained for CD14 and TSPO expression either (A) intracellular or (B) surface. Representative flow plots are shown in figure 1. Positive TSPO populations are indicated.
We observed that in the absence of cell permeabilization, TSPO was detectable on the cell surface of approximately 7% of all immune cells (Fig. 1B & Fig. 2). This finding was novel since a limited number of cell types have been reported as expressing cell surface TSPO [27, 28]. It is generally accepted that TSPO is abundantly expressed in the mitochondrial OMM and is rarely present on the cell surface [28]. In fact, when we subdivided cells into general immune cell populations, we found that all immune cells expressed cell surface TSPO to varying degrees, with neutrophil and monocytes exhibiting the highest frequencies ex vivo with cell surface TSPO (Fig. 2). Together these data suggest that a significant number of circulating immune cells readily express TSPO on their cell surface, with myeloid derived granulocytes, such as neutrophils and monocytes, having the highest ex vivo expression.
Figure 2. TSPO is expressed on the cell surface on all major circulating immune cells, with neutrophils and monocytes having the highest basal expression.
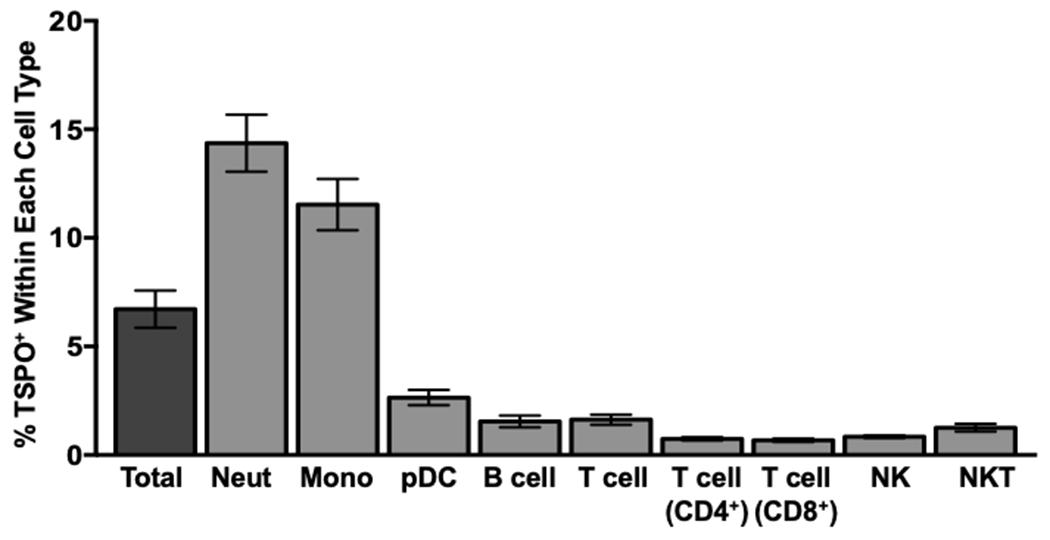
Human whole blood was collected and red blood cells were lysed with an erythrocyte lysing agent (ACK). Immune cells were stained for cell surface TSPO and the following immune cell markers: CD14 to identify monocytes, CD16 to identify neutrophils, CD3, CD4, and CD8 to identify T cells, CD19 to identify B cells, and CD56 and CD57 to identify NK cells. The percentage of surface TSPO positivity was calculated within each individual immune cell population, the average of at least 2 independent experiments with 16 individual human donors is shown. Values are mean ± SEM.
Monocytes increase TSPO surface expression in a concentration- and time-dependent manner after LPS stimulation
As PBMC represent a heterogenous immune cell population comprising innate and adaptive immune cells, as well as memory, effector, and naïve immune cells, it was important to identify the cellular profile underpinning the diverse expression of cell surface TSPO observed in Figure 2. Given the finding of cell surface TSPO expression on immune cells, it was important to determine the kinetics of cell surface TSPO expression with respect to immune cell activation. We focused on CD14 expressing monocytes in circulation as monocytes are known to be involved in inflammation [29, 30]. Furthermore, monocytes were the second highest population of cells expressing TSPO on the cell surface (Fig. 2).
First, we determined whether monocytes increased cell surface TSPO expression in a concentration dependent manner in response to a proinflammatory stimulus, such as exposure to LPS stimulation. PBMC isolated as described previously were cultured with increasing concentrations of LPS for 24 hours ex vivo and subsequently stained for surface TSPO. As shown in figure 3A, we detected minimal surface expression of TSPO (5-6%) on freshly isolated monocytes and monocytes receiving no LPS stimulation (Fig. 3A). Moreover, monocyte TSPO surface expression increased in a LPS concentration-dependent manner following stimulation for 24 hours, with 1μg/mL being sufficient to significantly increase the frequency of monocytes expressing surface TSPO (Fig. 3A). The 10 μg/mL concentration elicited the greatest increase in the percentage of monocytes with surface TSPO (Fig. 3A).
Figure 3. Human monocytes increase surface TSPO expression in a concentration- and time-dependent manner in response to LPS stimulation.
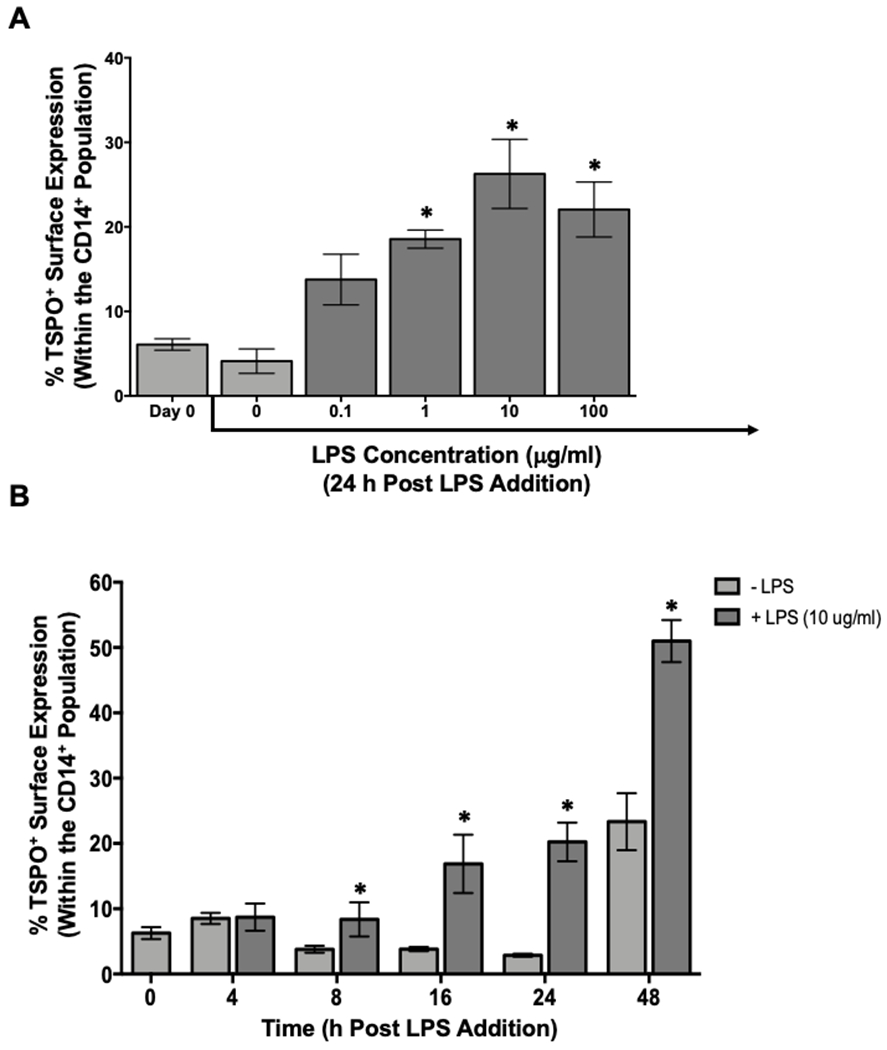
Human PBMC were isolated from leukocyte packs and stimulated with increasing concentrations of LPS (0.1–100μg/ml) for 24 hours, collected and stained for surface TSPO and CD14 expression. (A) Percentage of surface CD14+ and TSPO+ cells. (B) PBMC stimulated with 10μg/mL of LPS for different times. Average surface CD14 and TSPO at indicated time points of incubation. Results shown are the average of 2 independent experiments assessing a total of 6 human donors. * indicate significant differences at the p <0.05 level when compared to (A) Day 0 determined by a one-way ANOVA with a Dunnett’s multiple comparisons post-test and (B) No LPS at each time point as determined by an unpaired t-test. Values are mean ± SEM.
Next, we established the temporal relationship between LPS stimulation and monocyte-specific TSPO surface expression. As shown in Figure 3B, we found that the percentage of monocytes expressing surface TSPO doubled following LPS stimulation in as little as 8 hours after exposure. Likewise, the percentage of monocytes with cell surface TSPO expression was significantly increased at every time point after 8 hours (Fig. 3B). It is noteworthy that by 48 hours post LPS stimulation, the frequency of monocytes expressing cell surface TSPO continued to increase; however, even monocytes receiving no LPS began to express TSPO on their cell surface (Fig. 3B). This increase was not due to non-specific cell death during the culture period. Rather, this is likely due to the non-specific activation of monocytes in protracted culture, which has been reported previously [31]. These data demonstrate that the frequency of monocytes expressing TSPO on their cell surface increased in a LPS concentration- and time-dependent manner (Fig. 3B). The aforementioned finding would suggest that immune cell surface expression of TSPO is an indicator of immune cell activation, confirming the potential utility of TSPO as a peripheral marker of inflammation.
Increased frequency of monocyte-specific TSPO surface expression is not due to increased levels of surface TSPO on a per cell basis.
The previous results indicate that activation with LPS increased the frequency of monocytes expressing cell surface TSPO. We next sought to determine if the relative levels of surface TSPO increased over time. Isolated PBMC were treated with 10 μg/mL of LPS for 24 and 48 hours and surface stained for TSPO. As shown in representative flow plots in Figure 4A, both the frequency and mean fluorescence intensity (MFI) of TSPO surface expression increased following treatment with LPS (Fig. 4B and C). We also noted a decrease in CD14 expression following 24 hours of LPS activation, an effect that has been described previously in the literature [32]. This is also true in averaged data across multiple donors where both the frequency and MFI of surface TSPO doubles after 24h LPS stimulation (Fig. 4B and C). However, despite the continued increase in the frequency of monocyte-specific TSPO surface expression 48h post LPS treatment, we did not detect further increases of surface TSPO on a per cell basis. This suggests that once a cell has become positive for TSPO on the cell surface, TSPO protein levels remain the same and do not further increase as evidenced by MFI (Fig. 4C).
Figure 4. The frequency and density of monocyte TSPO surface expression increases with LPS stimulation and is not due to cell death.
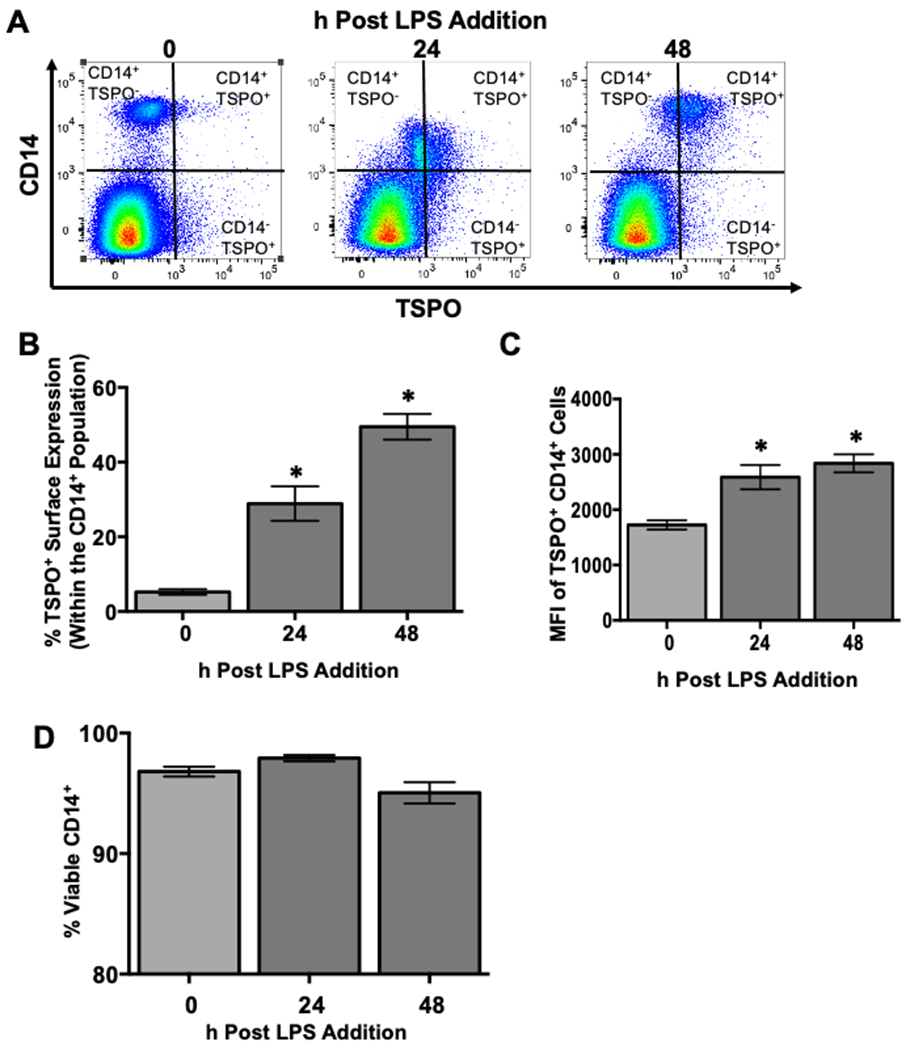
To determine if monocyte TSPO surface expression increased over the course of the 48 hour activation period on a per cell basis and whether this increase corresponded with increased cell death, human PBMC were isolated and treated with 10μg/mL LPS. At the indicated time points, cells were collected and stained extracellularly for CD14 and TSPO followed by incubation with a cell viability dye. (A) Representative flow plots for CD14 and TSPO surface expression. (B) Average change in the percentage of monocytes with TSPO surface expression following LPS activation. (C) Average change in surface TSPO (mean fluorescence intensity). (D) Monocyte cell viability was calculated by subtracting the frequency of live cells from the total to yield a viability percentage. Results are from 2 independent experiments assessing a total of 6 human donors. * indicate significant differences at the p <0.05 level when compared to time 0 as determined by a one-way ANOVA using a Dunnett’s multiple comparisons post-test. Values are mean ± SEM.
Increased frequency of monocyte-specific TSPO surface expression is not a result of increased cell death.
It has been suggested in the literature that one of the roles of mitochondrial TSPO is to facilitate the opening of the mitochondrial permeability transition pore to facilitate cell death in response to cell injury as well as various cell stress pathways [33, 34]. Therefore, one potential explanation for the increase in monocyte-specific cell surface TSPO expression is that these cells are undergoing cell death in response to LPS-mediated activation over time. Furthermore, as immune cells become activated, a percentage of cells go on to die as a result of activation-induced cell death (AICD), which could lead to a compromised cell plasma membrane that could potentially allow for the intercalation of TSPO-specific antibody in the absence of cell permeabilization. To test these possibilities, PBMC were isolated and activated with LPS, and cells were stained for CD14, TSPO, and a cell viability dye at time 0 and 24 and 48 hours post activation with LPS. As shown in Figure 4D, we did not observe any significant loss of cell viability over the course of the 48 hour activation. Given that we previously found significantly more monocytes expressing surface TSPO in as little as 8 hours after LPS treatment, these data would suggest that monocyte-specific surface localization of TSPO is not due, in part, to cell death.
The LPS-induced increase in frequency of monocytes with TSPO surface expression is not due to changes in TSPO mRNA levels.
While it is clear that TSPO is readily detectable on the surface of immune cells and monocytes specifically following stimulation with LPS. However, the underlying mechanism of this change is not known. A potential explanation is that nuclearly encoded TSPO is translated and alternatively trafficked to the plasma membrane rather than to the mitochondria of monocytes following LPS stimulation. To test this possibility, PBMC were isolated and treated with LPS for 24 hours as previously described. After LPS activation, cells were stained to assess cell surface and intracellular TSPO and an aliquot of cells were lysed for RNA extraction and subsequent qRT-PCR for TSPO mRNA expression. As shown in representative flow plots in Figure 5A, there was no measurable increase in intracellular TSPO expression at any given time point, even when TSPO MFI was quantified across multiple donors and time points (Fig. 5B).
Figure 5. The increase in the percentage of monocytes with TSPO surface expression is not due to increased expression of intracellular TSPO or increased gene expression.
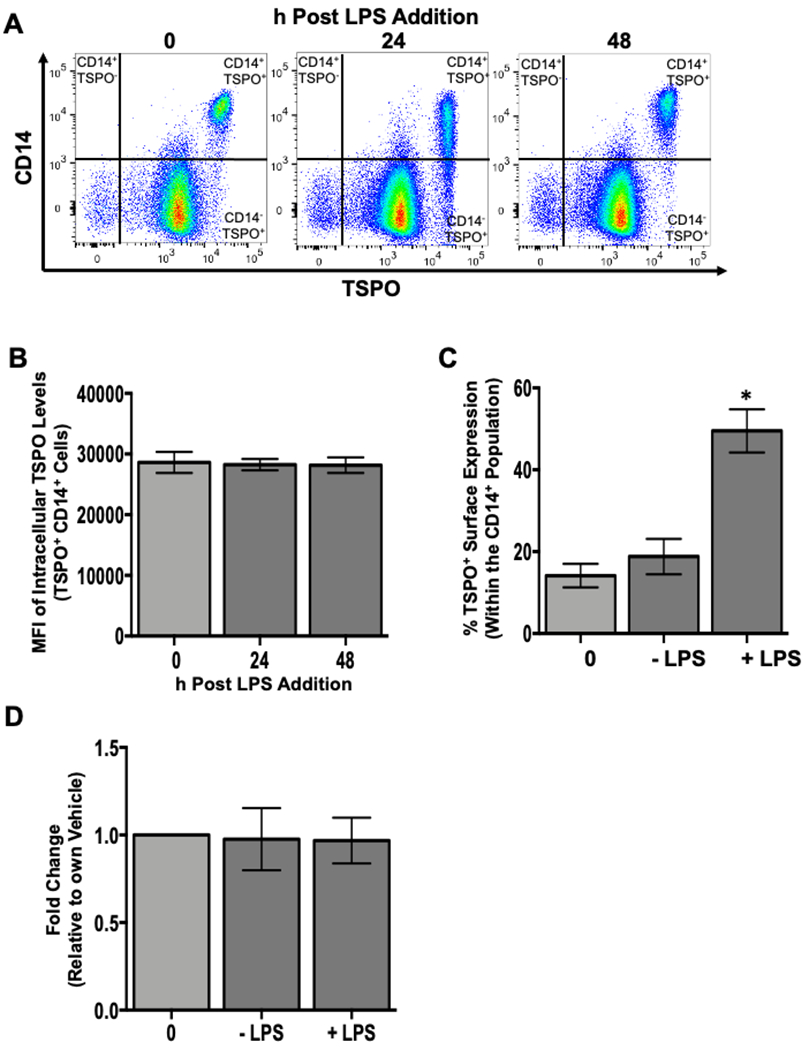
To assess whether the increased frequency of TSPO surface expression on monocytes following LPS treatment was due, in part, to increased intracellular expression, human PBMC were isolated and activated with LPS as described previously. At the indicated time points, cells were collected and stained for surface CD14, permeabilized, and stained intracellularly for TSPO. Purified monocytes were isolated and activated with LPS for 24 hours and stained for surface CD14 and TSPO to verify the increased frequency of TSPO surface expression following LPS treatment. Corresponding samples were also lysed at 24 hours post LPS treatment, mRNA was extracted from these isolated monocytes and qRT-PCR was used to determine TSPO gene expression. (A) Representative flow plots of intracellular TSPO staining at indicated time points. (B) Average mean fluoresence intensity (MFI) for intracellular TSPO. (C) Average increase in TSPO surface expression in purified monocytes. (D) Average fold change in TSPO mRNA expression in purified monocytes at 24 hours with and without LPS treatment. All data are from 2 independent experiments assessing a total of 6 human donors. * indicate significant differences at the p <0.05 level when compared to the time 0 control as determined by a one-way ANOVA with a Dunnett’s multiple comparisons post-test. Values are mean ± SEM.
Despite this finding, it is possible that intracellular TSPO expression is so high that we lack the sensitivity to quantify changes in intracellular TSPO accurately on a per cell basis. To this end, we employed qRT-PCR to determine if TSPO gene expression changed with LPS treatment. To confirm our previous finding, samples from the same donors used for gene expression were also surface stained for TSPO. As shown in Figure 5C, the increase in the frequency of monocytes expressing surface TSPO following LPS stimulation was confirmed. This increase did not correlate with increased TSPO mRNA levels in monocytes at 24 hours post LPS treatment (Fig. 5D) and is likely due to the high basal expression level in resting monocytes. These data would argue against the hypothesis that the TSPO found on the cell surface of monocytes following LPS stimulation is newly synthesized as a response to activation. One potential explanation is that TSPO is being transported from the mitochondrial outer membrane to the cellular plasma membrane through a yet to be determined mechanism.
Immunofluorescence imaging confirms plasma membrane TSPO localization and shows a punctate pattern of TSPO cell surface expression.
We have shown that TSPO is expressed on the cell surface of monocytes at basal levels and that LPS activation increases the frequency of monocytes with TSPO cell surface expression. However, the pattern of TSPO surface localization is less evident. To this end, cells were isolated and activated as previously described and surface stained for CD14 and TSPO expression (i.e. in the absence of permeabilization) as well as counterstained with DAPI to identify cell nuclei. As a control, cells were also stained for intracellular TSPO to visualize the normal intracellular distribution. As seen in representative immunofluoresence images shown in Figure 6A, intracellular TSPO expression was diffuse throughout the cell and intense, corroborating intracellular MFI measurements using flow cytometry [see Figure 5A, B]. A similar pattern of diffuse expression was observed with CD14 surface expression where CD14 appeared to be ubiquitously expressed on the cell surface of monocytes as shown in representative images in Figure 6A. It is noteworthy that TSPO surface expression, with or without LPS treatment, was punctate suggesting labeling of specific cell surface microdomains (Fig. 6A). This profile of cell surface expression is reminiscent of cell surface receptor localization or vesicles. Moreover TSPO surface expression did not show a clear pattern of colocalization with CD14, a coreceptor for LPS.
Figure 6. TSPO surface expression is punctate on monocytes following LPS activation and the use of flow imaging software confirms surface localization.
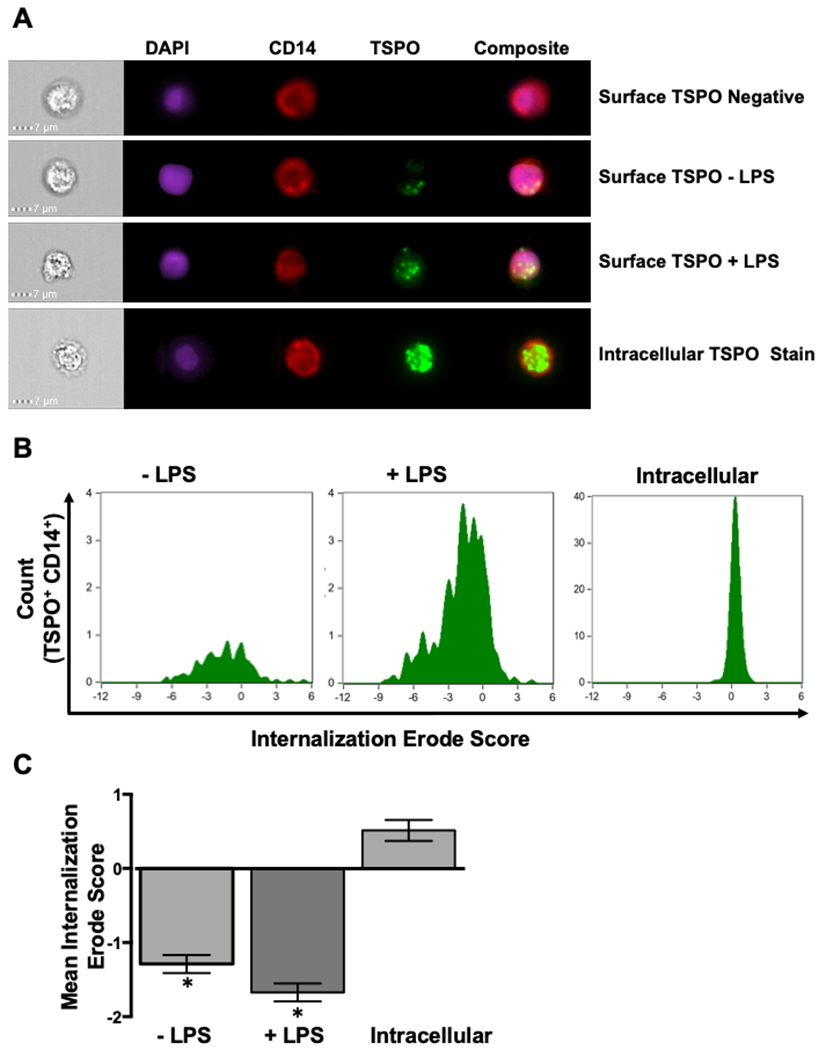
Given that this is one of the first reports of TSPO surface expression on immune cells, we wanted to determine the pattern of TSPO surface expression and verify in an unbiased manner that TSPO was being expressed on the cell surface. To accomplish this, human PBMC were isolated and treated with LPS as described previously. Following 24 hours of activation, cells were collected and stained for surface CD14, surface TSPO, intracellular TSPO and counter stained with DAPI to identify cell nuclei. Cells were then run on a Mark 4 AMNIS flow cytometer to generate single cell IFA images and analyzed using the IDEAS software to calculate TSPO surface expression from a series of cell images. (A) Representative IFA images. (B) Representative histograms of Internalization/Erode score for a given donor. (C) Averaged values of the Internalization/Erode score. * indicate significant differences at the p <0.05 level compared to the intracellular stain as determined by a one-way ANOVA with a Tukey’s multiple comparisons post-test. Values are mean ± SEM. Images were collected from N=3 different individual donors.
Next, single cell immunofluoresence images were used to quantify in an unbiased, high-throughput manner the increased frequency of TSPO surface expression following LPS stimulation. To accomplish this, an algorithm within the image analysis software IDEAS was used in which it creates a digital mask denoting the intracellular space of each image. Pixel density was used to assign an Internalization/Erode score where more positive values denoted intracellular localization and negative values corresponded to surface expression. When values across all cells in each treatment group where quantified, significant differences were observed in the Internalization/Erode scores for TSPO surface expression compared to intracellular demonstrating that TSPO is associated with the cell surface as compared to the inside of the cell (Figs. 6B and 6C). While visual inspection of immunofluorescence images further suggest TSPO surface expression, it is important to emphasize these measurements provide an unbiased, high-throughput approach to confirm this phenomenon quantitatively from thousands of images.
While imaging flow cytometry of single cells provides a unique opportunity to quantify TSPO surface localization from thousands of images in an unbiased manner, it lacks the capabilities of more traditional confocal microscopy to include other types of cell stains as well as allow sectioning of images along the z-axis [35]. To this end, we utilized traditional multi-color confocal microscopy of LPS treated PBMC to: a) further confirm the finding of surface TSPO using another visualization method; b) to localize surface TSPO with a membrane stain; and c) to capitalize on using anti-TSPO antibodies conjugated to differing fluorophores to further distinguish between surface and intracellular TSPO. Briefly, isolated PBMC were seeded into glass chamber slides and treated with LPS for 24 hours. Following the treatment period, cells were stained for AF488-conjugated surface TSPO, plasma membrane dye, and counterstained with DAPI to identify nuclei. In indicated wells, cells were also permeabilized and subsequently stained intracellularly with PE-conjugated TSPO antibodies.
When we examined the overlap between surface TSPO (green) and the plasma membrane (magenta) in the absence of cellular permeabilization, we observed considerable overlap as indicated by red arrows (Fig. 7A). Interestingly, the majority of surface TSPO expression appeared distal to the nucleus with little overlap between the two as evidenced by the overlayed images (Fig. 7A). Next we compared the staining pattern of surface (green) and intracellular TSPO (red), with the key difference being cells were permeabilized following surface TSPO staining. As shown in Figure 7B, we again saw minimal overlap between surface TSPO and cell nuclei. Conversely, intracellular TSPO appeared to more closely overlap with cell nuclei as indicated in the representative cell in the red box (Fig. 7B&C). Indeed, the lack of overlap is suggested by the absence of yellow fluorescence detection, which would be apparent if both surface and intracellular TSPO overlapped (Fig. 7B&C). Taken together, these data as well as the single cell flow cytometry imaging data strongly suggest that indeed TSPO is found on the surface of immune cells following treatment with LPS.
Figure 7. Confocal imaging shows TSPO surface expression is differentially localized relative to intracellular TSPO.
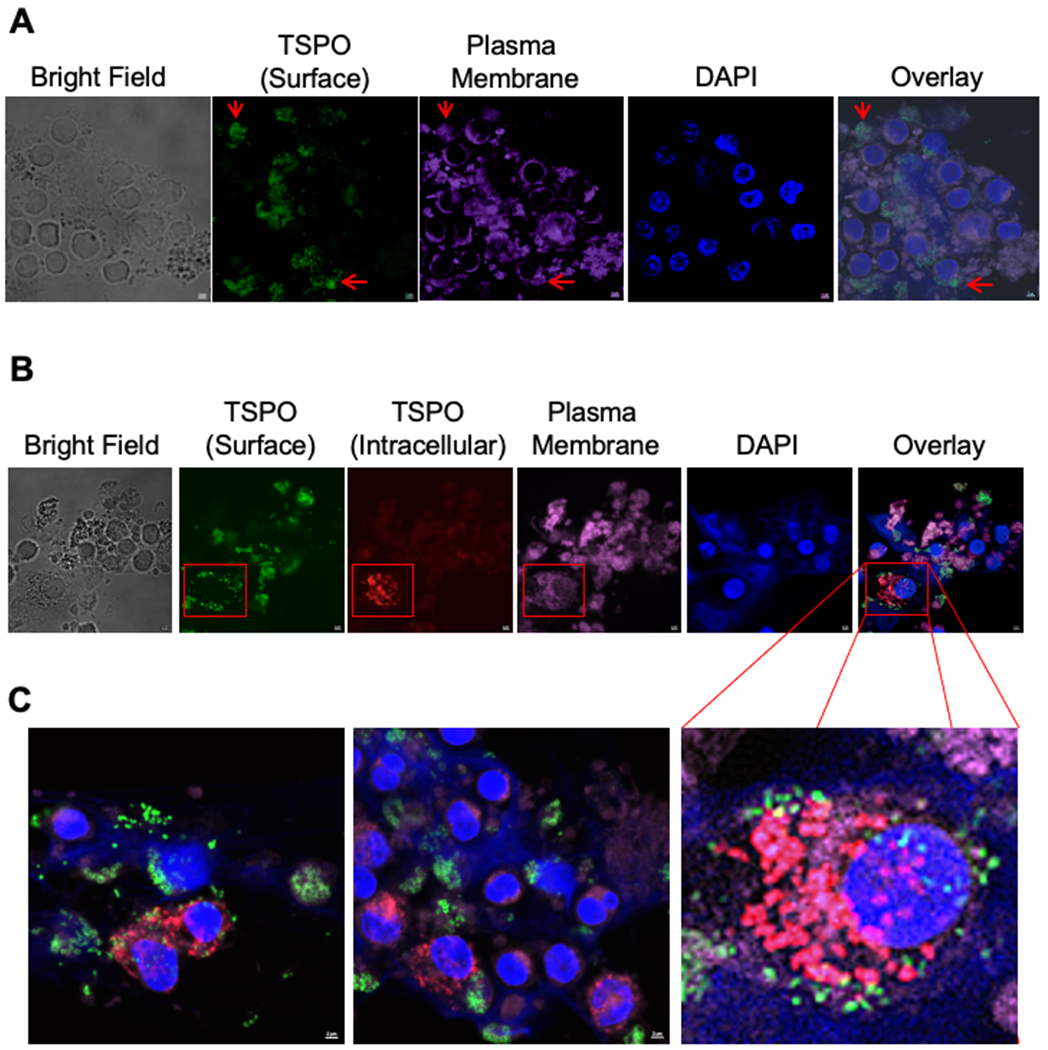
To further confirm that TSPO was localized on the cell surface relative to intracellular TSPO, we used confocal imaging using the same TSPO antibody but conjugated to two different fluorophores. To accomplish this, isolated human PBMC were placed in an 8 well glass slide treated with poly-D-Lysine and treated with LPS (10μg/ml). After 24 hours of activation, cells were directly stained for surface TSPO (AF488) and the plasma membrane (deep red), fixed and then stained for intracellular TSPO (PE) and counter stained with DAPI to identify cell nuclei.. Confocal images were acquired on the Nikon A1 Rsi Confocal Laser Scanning Microscope. (A) Representative image of surface TSPO (green) with the plasma membrane stain. (B) Representative image of surface TSPO (green), intercellular TSPO (red) with the plasma membrane stain. (C) Magnified representative images from other slides showing surface TSPO (green) along with intercellular TSPO (red) staining. Images were collected from N=3 different individual donors.
Detection of surface TSPO expression is not due solely to immune cell activation.
The finding of TSPO expression on the cell surface of immune cells in circulation and the increase in the frequency of cell surface TSPO mediated by LPS treatment suggests that TSPO translocation to the cell surface is a consequence of immune cell activation. To test this premise, we used two different stimuli, interferon α and the TLR9 agonist, CpG, to determine if monocyte activation through these two different pathways also increased the frequency of cell surface TSPO. Our results show that monocytes treated with increasing concentrations of IFNα or CpG for 48 hours showed no increase in surface TSPO expression (Fig. 8A and B). These data suggest that in monocytes, TSPO surface expression is not a global phenomenon in which any monocyte-specific activator can induce TSPO cell surface translocation, demonstrating some degree of specificity.
Figure 8. Increased percentage of TSPO surface expression on immune cells is not due solely to immune cell activation.
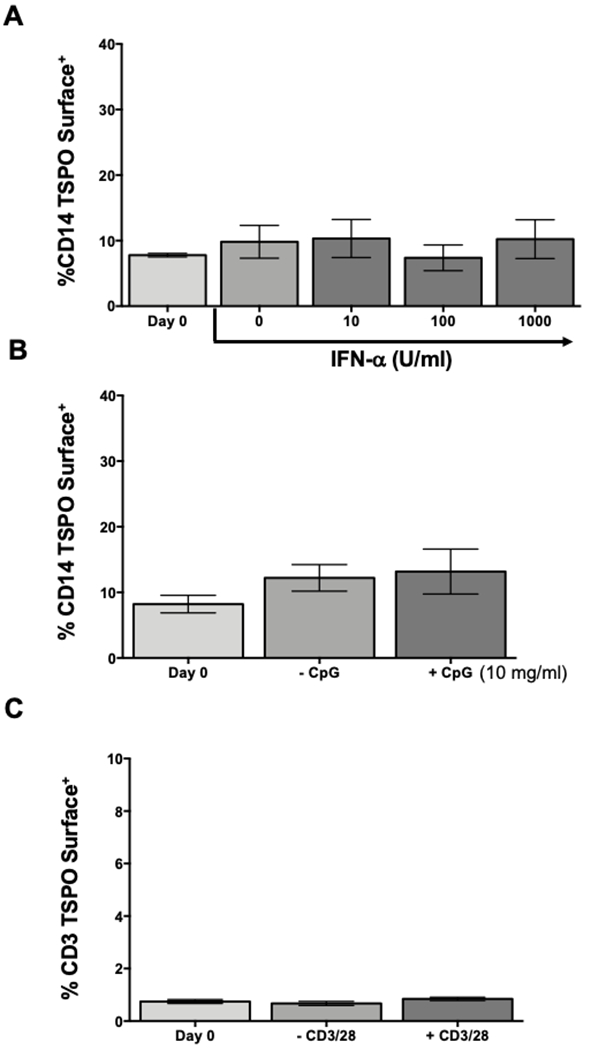
The data presented so far suggest that TSPO surface expression increases following monocyte activation with LPS. Next, we wanted to determine if this was a general hallmark of immune cell activation or if it was selective. To test this possibility, human PBMC were isolated as previously described and activated with either increasing concentrations of interferon α, 10μg/mL of CpG, or 5μg/mL of anti-CD3/CD28, a selective activator of T cells. Following 24 hours of activation, cells were collected and stained for surface CD14, CD3, or TSPO. (A) Frequency of TSPO surface expression on CD14 monocytes treated with increasing concentration of IFNα. (B) Frequency of monocyte TSPO surface expression after CpG activation. (C) Average increase of surface TSPO on CD3+ T cells after CD3/CD28 activation. Results are from 2 independent experiments assessing a total of 6 human donors. * indicate significant differences at the p <0.05 level compared to time 0 control as determined by a one-way ANOVA with a Dunnett’s multiple comparisons post-test. Values are mean ± SEM.
Since myeloid lineage cells exhibited the highest cell surface TSPO expression, additional experiments were conducted to determine if immune cell activation was sufficient to increase TSPO surface expression on all immunocompetent cells. We chose T cells, a lymphoid lineage immune cell with unique requirements for immune cell activation. To this end, we activated T cells with anti-CD3/CD28 stimulation, a T cell specific activator, to determine if T cell surface TSPO expression changed with activation. As shown in figure 8C, no change in the frequency of TSPO surface expression was observed on T cells above background levels. Taken together, these findings confirm that increased TSPO expression in the cell surface is not solely a consequence of immune cell activation but appears to be specific for LPS.
The frequency of circulating immune cells expressing surface TSPO is significantly elevated in HIV-infected donors.
The results presented above clearly show that immune cells express basal surface TSPO and that the number of cells with TSPO surface expression increased with inflammatory LPS stimulation. Thus, we sought to investigate the potential utility of surface TSPO expression in peripheral blood as a biomarker of inflammation in chronic inflammatory diseases like HIV. HIV is accompanied by protracted, chronic inflammation in the periphery as a consequence of the host immune response to the virus as well as other negative events associated with disease progression such as the intercalation of bacterial components into circulation through the loosening of the gut barrier epithelium [36]. Further, previous studies have suggested that the chronic inflammation in the periphery alters monocyte differentiation and promotes their trafficking across the blood brain barrier where they initiate local inflammation in the central nervous system, a condition termed HAND [37]. Given that monocytes were the second cell type with the highest TSPO surface expression, we wanted to quantify and compare monocytic TSPO surface expression in HIV+ donors and HIV− negative controls to evaluate the utility of using cell surface TSPO as a biomarker of inflammation.
Human donors’ positive for HIV were recruited and selected based on inclusion criteria [24] and matched with HIV− controls. Immune cells were obtained from whole blood and then stained for surface TSPO as well as individual markers for specific immune cell subsets, i.e. CD14. When we compared TSPO surface expression on total immune cells from HIV+ and HIV− individuals, we observed significantly more TSPO surface expression in peripheral immune cells from HIV+ subjects, with an approximately two-fold increase in the background frequency of TSPO surface expression (Fig. 9). When TSPO surface expression was quantified by immune cell type, we observed the general trend that nearly all immune cells from HIV+ donors displayed higher frequencies of TSPO surface expression compared to HIV− controls (Fig. 9). Moreover, we found significant increases in the frequency of TSPO surface expression on monocytes and plasmacytoid dendritic cells (pDC) culminating in a 2- and 20-fold increase, respectively (Fig. 9). Furthermore, surface TSPO expression was also significantly elevated in HIV+ donors on every immune cell type tested, albeit with much more modest increases, with the sole exception being T cells (Fig. 9). These findings were striking as both monocytes and pDC have been identified as critical early initiators of HIV-associated inflammation and have been reported to be the earliest responding immune cells to HIV-dependent increases in peripheral inflammation associated with chronic HIV infection [38]. Taken together, these findings support that immune cell TSPO surface expression may be upregulated during disease states in which chronic peripheral inflammation is present.
Figure 9. Circulating non-T immune cells from HIV+ donors have significantly elevated surface TSPO expression compared to HIV− donors.
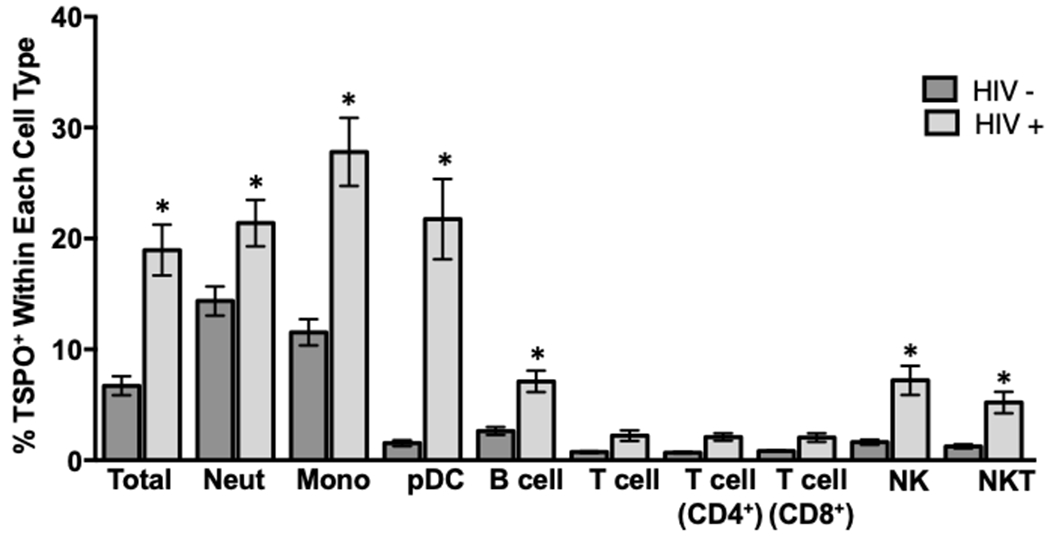
As we have demonstrated that surface TSPO expression increases under certain stimuli on immune cells, we wanted to extend these results to examine immune cell TSPO surface expression in the context of a relevant, inflammatory human disease. To accomplish this, immune cells were collected from lysed, whole blood from HIV+ and HIV− human donors. Cells were then stained extracellularly for TSPO and the following immune cell markers: CD14 to identify monocytes, CD16 to identify neutrophils, CD3 to identify T cells, CD19 to identify B cells, and CD56 and CD57 to identify NK cells. The frequency of TSPO surface positivity was calculated within each individual immune cell population, the average of 2 independent experiments with 16 individual human donors is shown. * indicate significant differences at the p <0.05 level as determined using an unpaired t-test comparing between HIV− and HIV+ within each type of immune cell. Values are mean ± SEM.
Monocytes from HIV+ donors showed an increased frequency of TSPO surface expression in response to LPS activation, which was attenuated by pretreatment with increasing concentrations of Δ9-tetrahydrocannabinol (Δ9-THC).
The finding that a greater frequency of immune cells isolated from HIV+ donors expressed surface TSPO compared to HIV− donors was striking and supports the premise that TSPO surface localization may be associated with chronic inflammation. However, it remains unclear if immune cells from HIV+ donors can further increase TSPO surface frequency in response to further inflammatory stimuli, such as LPS. Moreover, our laboratory has demonstrated that treatment with increasing concentrations of Δ9-THC can suppress many of the inflammatory effector functions of monocytes; in particular monocytes obtained from HIV+ donors, by suppressing monocyte activation [24]. Therefore, we examined the effect of Δ9-THC, an anti-inflammatory compound, on TSPO surface expression in monocytes from HIV+ and HIV− donors. Monocytes from HIV− donors were stimulated with LPS for 48 hours and confirmed an approximately 2 to 3-fold increase in the frequency of TSPO surface expression (Fig. 10). Furthermore, when monocytes from both HIV+ and HIV− donors were treated with increasing concentration of Δ9-THC prior to LPS stimulation, Δ9-THC significantly decreased TSPO surface expression in a concentration-dependent manner (Fig. 10). This result suggests that the frequency of monocytes with TSPO surface expression may be correlated with the state of immune cell activation. It is notable that surface TSPO levels while decreased, with Δ9-THC treatment, did not revert to resting levels, indicating that the relationship between immune cell activation and TSPO surface localization may not be exclusively binary.
Figure 10. The frequency of TSPO surface expression on monocytes from HIV+ and HIV− donors can be reduced by treatment with Δ9-tetrahydrocannabinol (Δ9-THC).
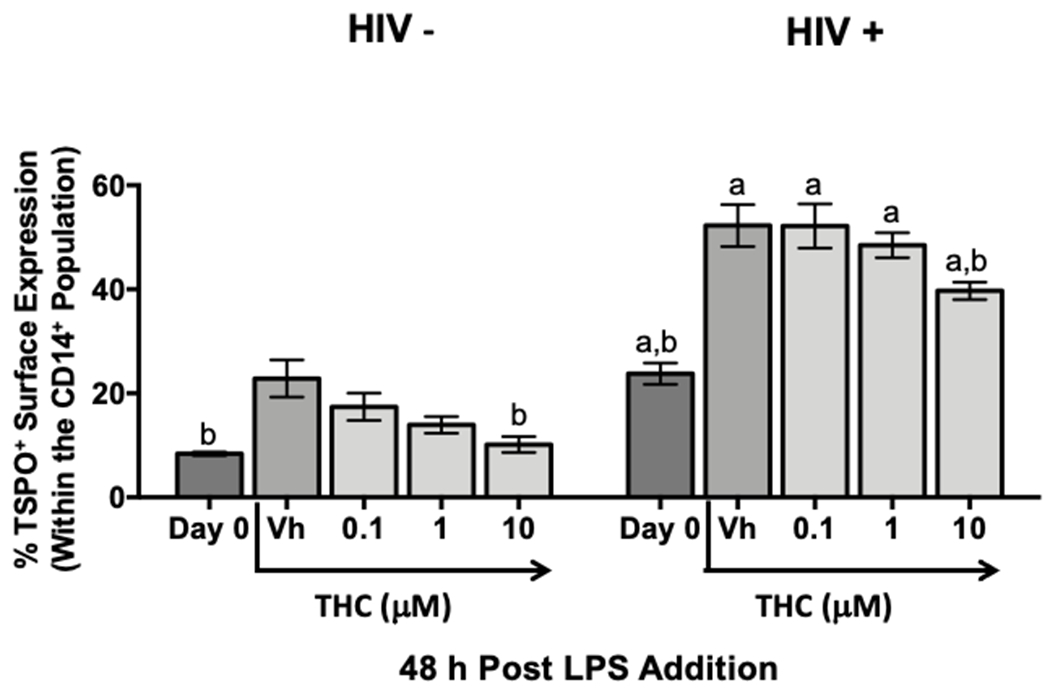
In order to determine if the increase in TSPO surface expression on monocytes following activation with LPS was transient, we used a well characterized, potent suppressor of monocyte immune cell activation, Δ9-THC. Briefly, immune cells from lysed, whole blood from HIV+ and HIV− donors were treated with increasing concentrations of Δ9-THC and activated for 48 hours with LPS. Following the activation period, cells were collected and surface stained for CD14 and TSPO. The average TSPO surface expression on monocytes from HIV+ and HIV− donors with indicated concentrations of THC are shown. Data are from 2 independent experiment assessing 6 human donors. (a) indicates significant differences between HIV+ and - donors within respective treatment groups and (b) indicates significant differences within HIV+ and - to their own vehicle control at the p <0.05 level as determined by a two-way ANOVA with an uncorrected Fisher’s LSD. Values are mean ± SEM.
Discussion
In this report, we present for the first time the finding of TSPO expression on the cell surface of immune cells, preferentially those of the myeloid lineage. We also show that the frequency of monocytes expressing surface TSPO increases after exposure to LPS and in immune cells from HIV+ donors. The increase in TSPO surface localization can be significantly suppressed by treatment with Δ9-THC, a known inhibitor of inflammatory immune cell activation [39].
TSPO was originally identified in 1977 as the Peripheral Benzodiazepine receptor (PBR) and was named for its high level of expression in cells from peripheral tissues such as heart, kidney, testis, skin, etc [4] compared to a closely related receptor called the Central Benzodiazepine receptor (CBR), which is primarily expressed in neuronal tissue [4]. Cell fractionation studies determined that TSPO was mostly associated with the outer mitochondrial membrane [2]. However, TSPO cell surface expression has been reported in the literature in a limited number of cell types [27, 28]. For example, Oke and colleagues described positive immunohistochemical staining for TSPO in the plasma membrane of cells isolated from adrenal gland tissues sections taken from mouse [27].
Oke and coworkers were likely the first to report the instance of TSPO being expressed as a cell surface protein. Since this discovery, TSPO has been reported on the surfaces of a few other cell types such as erythrocytes [40] as well as some prokaryotic bacterial species [28]. It should be noted that these studies were conducted in 1988, at a time in which flow cytometry and TSPO-specific antibodies were not available. Olson and colleagues made this observation using tritium labeled PK11195 in binding studies, a high affinity ligand for TSPO. However, PK11195 is a small molecule that is lipophilic and easily diffuses across cellular membranes. Since TSPO had been reported previously to be primarily associated with mitochondria [2], these data offered little reason to assume that [3H]-PK11195 binding to TSPO could also include a plasma membrane component. Indeed, it is tempting to speculate that the initial studies that identified TSPO localization to the OMM may have been biased by the extreme TSPO content found in the OMM. Even in our present studies using flow cytometry, it was difficult to compare intracellular TSPO expression to surface expression as it was necessary to utilize differential voltage settings (~2 fold) to accommodate the high concentration of intracellular TSPO. As many studies subsequently used radiolabeled, small molecule TSPO ligands to identify TSPO expressing cells, they failed to detect cell surface localization of TSPO on immune cells. Further, it is widely established that large molecules such as antibodies, do not readily pass through the cellular membrane in the absence of either active import or permeabilization of the cell membrane [41, 42]. Given our findings of TSPO surface detection in the absence of permeabilization and the distinct staining patterns between surface and intracellular TSPO, we conclude that TSPO is found on the surface of immune cells and is not an artifact of antibody intercalation inside the cell. This novel finding in cells of myeloid lineage has recently been confirmed in primary microglia as shown by Loth and coworkers using flow cytometry [43]. Furthermore, another study in BV2 cells, a microglia cell line, has also shown that BV2 cells express plasma membrane TSPO that can be increased by LPS activation ([44]).
An important aspect of the study by Shimoyama and colleagues was that they performed isolations of subcellular fractions using differential centrifugation and Western blotting of subcellular fractions [44]. These two studies confirm our current findings in peripheral immune cells that TSPO surface expression can be modulated by LPS, particularly in cells of the myeloid lineage.
While we conclusively report that TSPO is found on the surface of virtually all immune cells and that, in monocytes, LPS activation increases this frequency, the potential biological implications of this observation are not currently known. One of the first reported biological roles for TSPO was that of a cholesterol transporter involved in steroidogenesis [1]. This was based on two key observations: (a) that TSPO contained a cholesterol binding domain; and (b) that TSPO density was high in steroid synthesizing cells such as Leydig cells [45, 46]. The hypothesis that immune cell surface expression of TSPO is involved in steroidogenesis in these cells would be highly unlikely as immune cells produce little to no steroid proteins [47]. The potential role of TSPO in cholesterol transport, however, could have biological implications for immune cells with TSPO on their plasma membrane.
Many immune cells utilize cholesterol embedded into their surface plasma membranes as a means of modulating the fluidity of the cellular membrane [48, 49]. For example, T lymphocytes utilize cholesterol-rich lipid rafts to shuttle the disparate components of the T cell receptor complex together to form the immunological synapse with antigen-major histocompatibility complexes to facilitate antigen receptor signaling and T cell activation [50]. This provides T lymphocytes with a physical level of control over aberrant T cell activation by maintaining the major components of the antigen receptor separate from one another [50]. As a result of such a control mechanism, the ability to rapidly shuttle these components around the plasma membrane is required [50]. Indeed, similar events occur in monocytes in which the CD14 coreceptor for LPS moves about the plasma membrane in order to signal with TLR4 upon LPS engagement [51]. Cholesterol content in the plasma membranes of immune cells also allows for changes in cell shape, morphology, and trafficking [52–54]. Given the crucial role of cholesterol in immune cell function, it’s tempting to speculate if the role of TSPO in the plasma membrane is a part of this process. However, since the initial proposal that TSPO was involved in cellular cholesterol transport, this hypothesis has come into question as studies utilizing TSPO knockout mice and cell-specific conditional TSPO knockdown have shown that cholesterol transport as well as cholesterol-dependent steroid production are largely unaffected, strongly arguing against TSPO being necessary for those functions [7, 8]. In the absence of more direct evidence, the hypothesis that surface TSPO on immune cells is regulating cholesterol transport or steroid synthesis is unlikely.
More recently, studies have begun to suggest a biological function of TSPO involved with the generation of ROS [14, 15, 55]. This is primarily due to direct evidence of subcellular localization of TSPO preferentially to the OMM, a region of the mitochondria known to be involved in the generation of cellular ROS [2, 14]. Moreover, studies have also identified subcellular localization of TSPO with other components involved in ROS production, such as NAPDH oxidases, in extra-mitochondrial spaces such as vesicles and peroxisomes [56–58]. The hypothesis that TSPO could be facilitating ROS generation in monocytes, particularly following stimulation with LPS is highly attractive and supported by the recent finding of a TSPO-NOX2 interaction in primary microglia [43]. Indeed, one of the first events in monocytes and macrophage that have been stimulated with bacterial-derived components such as LPS is the upregulation of protein components involved in ROS generation [59]. This is due to the fact that monocytes and neutrophil utilize ROS generation to kill phagocytosed pathogens. Considering that it has been reported that both monocytes and neutrophil express some of the highest levels of TSPO, this hypothesis is likely. Indeed, our own findings that TSPO surface expression in monocytes appears to be unique to TLR4-mediated monocyte activation confirm this possibility. The TLR4-specific ligand, LPS, is a component of gram-negative bacterial cell wall and is one of the first bacterially derived components that monocytes encounter [60]. Further, LPS orchestrates a very specific differentiation program in human monocytes. For example, LPS treatment of monocytes initiates’ rapid upregulation of ROS generating machinery, induces monocyte differentiation toward macrophage, and prepares the cells to phagocytose and kill extracellular pathogens [60, 61]. In contrast, stimulation of monocytes with activators that mimics intracellular pathogens such as viruses (CpG), drive monocytes down an alternative pathway which results in proliferation as well as secretion of pro-inflammatory cytokines such as IL-1β and IL-6 [62]. However, monocytes that undergo this activation pathway do not produce as much ROS, differentiate into macrophage, or upregulate the cellular machinery necessary to carry out phagocytosis [62]. Given that activation of human monocytes with either IFNα or CpG did not induce any increases in the frequency of TSPO surface detection compared to LPS, strongly suggests that TSPO surface expression may be changing in order to facilitate ROS generation in these cells. Along these lines, our results with THC treatment also support this premise as THC has been shown to suppress monocyte activation as well as its ability to phagocytose and produce ROS [63]. Other studies also support this premise as treatment with TSPO ligands was shown to enhance ROS generation, suggesting a role for TSPO [64]. Furthermore, it has recently been shown that oxidation and ROS generation is important for various immune cells, for example the finding that ROS can regulate T cell activation and effector functions [65]. As such, TSPO surface localization on immune cells may be, in part, a means of regulating internal and external ROS to ensure optimal immune responses. However, we did not detect increased frequency of TSPO surface translocation on T cells following activation, therefore, the involvement of TSPO surface expression as a general indicator of immune cell activation is unlikely. Given the potential role of TSPO and ROS generation, more studies to directly test this hypothesis are warranted.
Another likely, albeit less specific, hypothesis for TSPO translocation to the plasma membrane of immune cells could be due to mitochondrial communication with the host cell. Mitochondria are unique organelles in that they encode their own genome and some of their own proteins, which is likely due to the theory that mitochondria arose as a separate organism which formed an endosymbiotic relationship with the earliest eukaryotic progenitor [66]. As such, mitochondria regularly communicate with the host cell through the import and export of mitochondrial proteins [67–69]. Proteins exported from the mitochondria can occur via the export of mitochondrial RNA, which is translated by host machinery in the cytosol, or via the export of fully formed mitochondrial proteins via vesicles (Reviewed in [67] and [70]). Since this process can be accelerated with activation of immune cells, it is possible that TSPO surface expression may be the result of mitochondrial proteins being transported inter- and intracellularly. Mitochondria contain their own plasma membrane and release protein containing vesicles in a similar manner as the host cell. As a result, any proteins decorating the budding plasma membrane will be incorporated into the subsequently formed vesicle [67, 71]. If that vesicle fuses with the host cell plasma membrane, releasing its cargo, any proteins imbedded in the mitochondrial vesicle membrane will become embedded in the host cell plasma membrane as the vesicle fuses [67, 71]. Given that monocytes and neutrophil had the highest frequency of surface TSPO, this explanation makes sense if the hypothesis that TSPO co-localized to vesicles with NADPH oxidases as a part of ROS generation and release were trafficking to the cell surface [72]. This would also explain the identification of surface TSPO in the absence of specific immune activation, as a part of the normal turnover of this process [72].
Finally, we characterized TSPO expression by circulating immune cells to test the potential that TSPO may serve as a peripheral biomarker of inflammation given the interest to TSPO in brain injury and neuroinflammation (Reviewed in [5]). Most contemporary studies of TSPO have focused on its usage in conjunction with neuroimaging techniques, such as PET, to identify areas of the brain undergoing inflammation [73–75]. The utility of such an approach has been demonstrated in various models of brain injury as well as models of neuroinflammatory diseases unassociated with traumatic brain injury [76–78]. Further, injection of human subjects with subclinical doses of LPS resulted in increased TSPO detection in the brain using PET [79] suggesting that peripheral inflammatory insults can and do impact the inflammatory state of the CNS.
Our own findings lend support to the notion that TSPO may be a biomarker of ongoing peripheral inflammation. The use of peripheral immune cells from ART-treated HIV+ donors, which are known to have increased, chronic, peripheral inflammation compared to HIV− donors, supports this hypothesis. HIV-associated neuroinflammation is one of the most common complications of HIV infection in the era of antiretroviral medications, with up to 50% of chronically infected patients experiencing some level of decline [80]. It is thought that the transference of bacterially-derived molecules, such as LPS, leaking from the gut as a result of the loosening of the tight-junctions of the gut epithelium in response to antiretroviral medications [81]. Our observation that TLR ligands such as LPS, which are derived from bacteria, induced TSPO surface expression supports this possibility. As we observed significant upregulation of TSPO surface expression on monocytes and pDC, two critical mediators of HAND, it is possible that TSPO surface expression on immune cells may serve as a peripheral marker of chronic inflammation in response to loss of lumenal integrity in the gastrointestinal tract resulting in bacterial component translocation (i.e., leaky gut), adding to a growing list of biomarkers such as sCD14 and sCD163. Based on the results reported here, it is tempting to speculate on the utility of detection of surface TSPO on circulating immune cells. While we reported the finding that TSPO surface expression was enhanced on peripheral immune cells from HIV+ donors, this could have implications far reaching beyond HIV patient care. As mentioned prior, there is extensive interest in the use of TSPO as a biomarker of brain injury and neuroinflammation. However, even with TSPO-PET imaging, the procedures are costly, time consuming, and impractical to use on a routine basis. Our results suggest that identification of cell surface TSPO expression on peripheral immune cells may be a practical, low cost, rapid means of assessing systemic inflammation and could potentially serve as a surrogate biomarker for neuroinflammation. Indeed, it has been speculated that the overall levels of systemic inflammation can be predictive of poor patient outcomes and more severe disease [82–85].
In conclusion, we have shown that TSPO is ubiquitously expressed on the cell surface of various immune cell types, with monocytes and neutrophils having the highest frequency of TSPO surface expression. Further, activation of monocytes with proinflammatory LPS increases the frequency of TSPO surface expression in the absence of altered TSPO gene expression. Also, TSPO surface expression is selectively induced by LPS compared to other immune cell activators and increased frequency of TSPO surface localization is significantly enhanced in a chronic inflammatory disease such as chronic HIV infection. Lastly, TSPO surface localization may be intrinsically tied to immune cell activation or function as treatment with immune suppressive THC reduces monocyte surface TSPO expression providing novel insights into the biology and function of TSPO on immune cells.
Acknowledgements:
Supported by grants NIH R01 DA047180 to NEK and R01 ES007062-20 to TRG
Abbreviations:
- ACK
Ammonium Chloride Potassium
- AICD
Activation Induced Cell Death
- ART
Anti-retroviral Therapy
- CBR
Central Benzodiazepine Receptor
- CD
Cluster of Differentiation
- CNS
Central Nervous System
- DAPI
4, 6-diamidino-2-phenylindole
- ELISA
Enzyme--Linked Immunosorbent Assay
- FACS
Fluoresence Activated Cell Sorting
- HAND
HIV-associated Neurocognitive Disorders
- HBSS
Hanks Buffered Saline Solution
- HIV
Human Immunodeficiency Virus
- IL
Interleukin
- LPS
lipopolysaccharide
- MFI
Mean Fluoresence Intensity
- mRNA
messenger Ribo Nucleic Acid
- NADPH
Nicotinamide Adenine Dinucleotide Phosphate
- OMM
Outer Mitochondrial Membrane
- PBMC
Peripheral Blood Mononuclear Cells
- PBR
Peripheral Benzodiazepine Receptor
- pDC
Plasmacytoid Dendritic Cell
- PET
Positron Emission Tomography
- qRT-PCR
quantitative Real Time- Polymerase Chain Reaction
- ROS
Reactive Oxygen Species
- THC
Δ9-tetrahydrocannabinol
- TLR
Toll-Like Receptor
- TSPO
Translocator Protein 18 kDa
Bibliography
- 1.Mukhin AG, et al. , Mitochondrial benzodiazepine receptors regulate steroid biosynthesis. Proc Natl Acad Sci U S A, 1989. 86(24): p. 9813–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krueger KE and Papadopoulos V, Peripheral-type benzodiazepine receptors mediate translocation of cholesterol from outer to inner mitochondrial membranes in adrenocortical cells. J Biol Chem, 1990. 265(25): p. 15015–22. [PubMed] [Google Scholar]
- 3.Bonsack F and Sukumari-Ramesh S, TSPO: An Evolutionarily Conserved Protein with Elusive Functions. Int J Mol Sci, 2018. 19(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braestrup C and Squires RF, Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc Natl Acad Sci U S A, 1977. 74(9): p. 3805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guilarte TR, TSPO in diverse CNS pathologies and psychiatric disease: A critical review and a way forward. Pharmacol Ther, 2019. 194: p. 44–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, et al. , Cholesterol binding at the cholesterol recognition/ interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc Natl Acad Sci U S A, 2001. 98(3): p. 1267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tu LN, et al. , Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem, 2014. 289(40): p. 27444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morohaku K, et al. , Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology, 2014. 155(1): p. 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu LN, et al. , PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology, 2015. 156(3): p. 1033–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papadopoulos V, Structure and function of the peripheral-type benzodiazepine receptor in steroidogenic cells. Proc Soc Exp Biol Med, 1998. 217(2): p. 130–42. [DOI] [PubMed] [Google Scholar]
- 11.Hirsch JD, et al. , Mitochondrial benzodiazepine receptors mediate inhibition of mitochondrial respiratory control. Mol Pharmacol, 1989. 35(1): p. 157–63. [PubMed] [Google Scholar]
- 12.Hirsch T, et al. , PK11195, a ligand of the mitochondrial benzodiazepine receptor, facilitates the induction of apoptosis and reverses Bcl-2-mediated cytoprotection. Exp Cell Res, 1998. 241(2): p. 426–34. [DOI] [PubMed] [Google Scholar]
- 13.Lee DH, et al. , Effects of peripheral benzodiazepine receptor ligands on proliferation and differentiation of human mesenchymal stem cells. J Cell Physiol, 2004. 198(1): p. 91–9. [DOI] [PubMed] [Google Scholar]
- 14.Veenman L, Papadopoulos V, and Gavish M, Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des, 2007. 13(23): p. 2385–405. [DOI] [PubMed] [Google Scholar]
- 15.Choi J, et al. , Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia, 2011. 59(2): p. 219–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen MK and Guilarte TR, Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther, 2008. 118(1): p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werry EL, et al. , Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int J Mol Sci, 2019. 20(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persson A, et al. , Imaging of [11C]-labelled Ro 15-1788 binding to benzodiazepine receptors in the human brain by positron emission tomography. J Psychiatr Res, 1985. 19(4): p. 609–22. [DOI] [PubMed] [Google Scholar]
- 19.Huang C, et al. , Evidence of the impact of systemic inflammation on neuroinflammation from a non-bacterial endotoxin animal model. J Neuroinflammation, 2018. 15(1): p. 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabrera-Pastor A, et al. , Peripheral inflammation induces neuroinflammation that alters neurotransmission and cognitive and motor function in hepatic encephalopathy: Underlying mechanisms and therapeutic implications. Acta Physiol (Oxf), 2019. 226(2): p. e13270. [DOI] [PubMed] [Google Scholar]
- 21.Klein RS, et al. , Neuroinflammation During RNA Viral Infections. Annu Rev Immunol, 2019. 37: p. 73–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wohleb ES and Delpech JC, Dynamic cross-talk between microglia and peripheral monocytes underlies stress-induced neuroinflammation and behavioral consequences. Prog Neuropsychopharmacol Biol Psychiatry, 2017. 79(Pt A): p. 40–48. [DOI] [PubMed] [Google Scholar]
- 23.Minogue AM, Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: Effects on cognition, learning and affective behaviour. Prog Neuropsychopharmacol Biol Psychiatry, 2017. 79(Pt A): p. 15–18. [DOI] [PubMed] [Google Scholar]
- 24.Rizzo MD, et al. , HIV-infected cannabis users have lower circulating CD16+ monocytes and IFN-gamma-inducible protein 10 levels compared with nonusing HIV patients. AIDS, 2018. 32(4): p. 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abourbeh G, et al. , Imaging microglial/macrophage activation in spinal cords of experimental autoimmune encephalomyelitis rats by positron emission tomography using the mitochondrial 18 kDa translocator protein radioligand [(1)(8)F]DPA-714. J Neurosci, 2012. 32(17): p. 5728–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venneti S, Lopresti BJ, and Wiley CA, Molecular imaging of microglia/macrophages in the brain. Glia, 2013. 61(1): p. 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oke BO, et al. , Cell surface localization of the peripheral-type benzodiazepine receptor (PBR) in adrenal cortex. Mol Cell Endocrinol, 1992. 87(1-3): p. R1–6. [DOI] [PubMed] [Google Scholar]
- 28.Batoko H, Veljanovski V, and Jurkiewicz P, Enigmatic Translocator protein (TSPO) and cellular stress regulation. Trends Biochem Sci, 2015. 40(9): p. 497–503. [DOI] [PubMed] [Google Scholar]
- 29.Xie X, et al. , Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J Neurochem, 2017. 141(2): p. 222–235. [DOI] [PubMed] [Google Scholar]
- 30.Kratofil RM, Kubes P, and Deniset JF, Monocyte Conversion During Inflammation and Injury. Arterioscler Thromb Vasc Biol, 2017. 37(1): p. 35–42. [DOI] [PubMed] [Google Scholar]
- 31.Kelley JL, et al. , Activation of human blood monocytes by adherence to tissue culture plastic surfaces. Exp Mol Pathol, 1987. 46(3): p. 266–78. [DOI] [PubMed] [Google Scholar]
- 32.Landmann R, et al. , Human monocyte CD14 is upregulated by lipopolysaccharide. Infect Immun, 1996. 64(5): p. 1762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vercesi AE, et al. , Mitochondrial calcium transport and the redox nature of the calcium-induced membrane permeability transition. Free Radic Biol Med, 2018. 129: p. 1–24. [DOI] [PubMed] [Google Scholar]
- 34.Chelli B, et al. , Peripheral-type benzodiazepine receptor ligands: mitochondrial permeability transition induction in rat cardiac tissue. Biochem Pharmacol, 2001. 61(6): p. 695–705. [DOI] [PubMed] [Google Scholar]
- 35.St Croix CM, Shand SH, and Watkins SC, Confocal microscopy: comparisons, applications, and problems. Biotechniques, 2005. 39(6 Suppl): p. S2–5. [DOI] [PubMed] [Google Scholar]
- 36.Simeonova D, et al. , Recognizing the Leaky Gut as a Trans-diagnostic Target for Neuroimmune Disorders Using Clinical Chemistry and Molecular Immunology Assays. Curr Top Med Chem, 2018. 18(19): p. 1641–1655. [DOI] [PubMed] [Google Scholar]
- 37.Veenstra M, et al. , Mechanisms of CNS Viral Seeding by HIV(+) CD14(+) CD16(+) Monocytes: Establishment and Reseeding of Viral Reservoirs Contributing to HIV-Associated Neurocognitive Disorders. MBio, 2017. 8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coleman CM and Wu L, HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology, 2009. 6: p. 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagarkatti P, et al. , Cannabinoids as novel anti-inflammatory drugs. Future Med Chem, 2009. 1(7): p. 1333–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson JM, et al. , Presence of peripheral-type benzodiazepine binding sites on human erythrocyte membranes. Eur J Pharmacol, 1988. 152(1-2): p. 47–53. [DOI] [PubMed] [Google Scholar]
- 41.Joshi S and Yu D, Immunofluorescence, in Basic Science Methods for Clinical Researchers. 2017, Elsevier. p. 135–150. [Google Scholar]
- 42.Jamur MC and Oliver C, Permeabilization of cell membranes. Methods Mol Biol, 2010. 588: p. 63–6. [DOI] [PubMed] [Google Scholar]
- 43.Loth MK, et al. , A Novel Interaction of Translocator Protein 18 kDa (TSPO) with NADPH Oxidase in Microglia. Mol Neurobiol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimoyama S, et al. , Lipopolysaccharide induces mouse translocator protein (18 kDa) expression via the AP-1 complex in the microglial cell line, BV-2. PLoS One, 2019. 14(9): p. e0222861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H and Papadopoulos V, Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology, 1998. 139(12): p. 4991–7. [DOI] [PubMed] [Google Scholar]
- 46.Papadopoulos V, et al. , The peripheral-type benzodiazepine receptor is functionally linked to Leydig cell steroidogenesis. J Biol Chem, 1990. 265(7): p. 3772–9. [PubMed] [Google Scholar]
- 47.Bohm M, et al. , KU812 basophils express urocortin, CRH-R, MC1R and steroidogenic enzymes and secrete progesterone. Exp Dermatol, 2012. 21(7): p. 541–3. [DOI] [PubMed] [Google Scholar]
- 48.Spector AA and Yorek MA, Membrane lipid composition and cellular function. J Lipid Res, 1985. 26(9): p. 1015–35. [PubMed] [Google Scholar]
- 49.Rivnay B, Globerson A, and Shinitzky M, Peturbation of lymphocyte response to concanavalin A by exogenous cholesterol and lecithin. Eur J Immunol, 1978. 8(3): p. 185–9. [DOI] [PubMed] [Google Scholar]
- 50.Janes PW, et al. , The role of lipid rafts in T cell antigen receptor (TCR) signalling. Semin Immunol, 2000. 12(1): p. 23–34. [DOI] [PubMed] [Google Scholar]
- 51.Triantafilou M, et al. , Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci, 2002. 115(Pt 12): p. 2603–11. [DOI] [PubMed] [Google Scholar]
- 52.Simons K and Toomre D, Lipid rafts and signal transduction. Nat Rev Mol Cell Biol, 2000. 1(1): p. 31–9. [DOI] [PubMed] [Google Scholar]
- 53.Simons K and Vaz WL, Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct, 2004. 33: p. 269–95. [DOI] [PubMed] [Google Scholar]
- 54.Kwik J, et al. , Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc Natl Acad Sci U S A, 2003. 100(24): p. 13964–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lenaz G, Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta, 1998. 1366(1-2): p. 53–67. [DOI] [PubMed] [Google Scholar]
- 56.Guilarte TR, Loth MK, and Guariglia SR, TSPO Finds NOX2 in Microglia for Redox Homeostasis. Trends Pharmacol Sci, 2016. 37(5): p. 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boujrad N, et al. , The peroxisome proliferator perfluorodecanoic acid inhibits the peripheral-type benzodiazepine receptor (PBR) expression and hormone-stimulated mitochondrial cholesterol transport and steroid formation in Leydig cells. Endocrinology, 2000. 141(9): p. 3137–48. [DOI] [PubMed] [Google Scholar]
- 58.Gatliff J, et al. , A role for TSPO in mitochondrial Ca(2+) homeostasis and redox stress signaling. Cell Death Dis, 2017. 8(6): p. e2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu HY and Wen MH, Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. J Biol Chem, 2002. 277(25): p. 22131–9. [DOI] [PubMed] [Google Scholar]
- 60.Dahl TA, Midden WR, and Hartman PE, Comparison of killing of gram-negative and gram-positive bacteria by pure singlet oxygen. J Bacteriol, 1989. 171(4): p. 2188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guha M and Mackman N, LPS induction of gene expression in human monocytes. Cell Signal, 2001. 13(2): p. 85–94. [DOI] [PubMed] [Google Scholar]
- 62.Klinman DM, et al. , CpG DNA: recognition by and activation of monocytes. Microbes Infect, 2002. 4(9): p. 897–901. [DOI] [PubMed] [Google Scholar]
- 63.Han KH, et al. , CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc Res, 2009. 84(3): p. 378–86. [DOI] [PubMed] [Google Scholar]
- 64.Larcher JC, et al. , Effects of peripheral benzodiazepines upon the O2 consumption of neuroblastoma cells. Eur J Pharmacol, 1989. 161(2-3): p. 197–202. [DOI] [PubMed] [Google Scholar]
- 65.Belikov AV, Schraven B, and Simeoni L, T cells and reactive oxygen species. J Biomed Sci, 2015. 22: p. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Archibald JM, Endosymbiosis and Eukaryotic Cell Evolution. Curr Biol, 2015. 25(19): p. R911–21. [DOI] [PubMed] [Google Scholar]
- 67.Scanlon DP and Salter MW, Strangers in strange lands: mitochondrial proteins found at extra-mitochondrial locations. Biochem J, 2019. 476(1): p. 25–37. [DOI] [PubMed] [Google Scholar]
- 68.Murcha MW, et al. , Adaptations required for mitochondrial import following mitochondrial to nucleus gene transfer of ribosomal protein S10. Plant Physiol, 2005. 138(4): p. 2134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duarte FV, Palmeira CM, and Rolo AP, The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. Adv Exp Med Biol, 2015. 888: p. 123–54. [DOI] [PubMed] [Google Scholar]
- 70.Kotiadis VN, Duchen MR, and Osellame LD, Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim Biophys Acta, 2014. 1840(4): p. 1254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ardehali H, et al. , Targeting of the mitochondrial membrane proteins to the cell surface for functional studies. Biochem Biophys Res Commun, 2005. 338(2): p. 1143–51. [DOI] [PubMed] [Google Scholar]
- 72.Casbon AJ, et al. , Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J Immunol, 2009. 182(4): p. 2325–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuhlmann AC and Guilarte TR, Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem, 2000. 74(4): p. 1694–704. [DOI] [PubMed] [Google Scholar]
- 74.Kuhlmann AC and Guilarte TR, Regional and temporal expression of the peripheral benzodiazepine receptor in MPTP neurotoxicity. Toxicol Sci, 1999. 48(1): p. 107–16. [DOI] [PubMed] [Google Scholar]
- 75.Chen MK and Guilarte TR, Imaging the peripheral benzodiazepine receptor response in central nervous system demyelination and remyelination. Toxicol Sci, 2006. 91(2): p. 532–9. [DOI] [PubMed] [Google Scholar]
- 76.Banati RB, et al. , The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain, 2000. 123 ( Pt 11): p. 2321–37. [DOI] [PubMed] [Google Scholar]
- 77.Cagnin A, et al. , In vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain, 2001. 124(Pt 10): p. 2014–27. [DOI] [PubMed] [Google Scholar]
- 78.Guilarte TR, et al. , Methamphetamine-induced deficits of brain monoaminergic neuronal markers: distal axotomy or neuronal plasticity. Neuroscience, 2003. 122(2): p. 499–513. [DOI] [PubMed] [Google Scholar]
- 79.Sandiego CM, et al. , Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci U S A, 2015. 112(40): p. 12468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heaton RK, et al. , HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol, 2011. 17(1): p. 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chung CY, et al. , Progressive proximal-to-distal reduction in expression of the tight junction complex in colonic epithelium of virally-suppressed HIV+ individuals. PLoS Pathog, 2014. 10(6): p. e1004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dolan RD, et al. , The role of the systemic inflammatory response in predicting outcomes in patients with operable cancer: Systematic review and meta-analysis. Sci Rep, 2017. 7(1): p. 16717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dolan RD, et al. , The role of the systemic inflammatory response in predicting outcomes in patients with advanced inoperable cancer: Systematic review and meta-analysis. Crit Rev Oncol Hematol, 2017. 116: p. 134–146. [DOI] [PubMed] [Google Scholar]
- 84.Chao A, et al. , The admission systemic inflammatory response syndrome predicts outcome in patients undergoing emergency surgery. Asian J Surg, 2013. 36(3): p. 99–103. [DOI] [PubMed] [Google Scholar]
- 85.Dhar R and Diringer MN, The burden of the systemic inflammatory response predicts vasospasm and outcome after subarachnoid hemorrhage. Neurocrit Care, 2008. 8(3): p. 404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]