

Abstract
The bicyclic molecule indole has been in the limelight because of its numerous pharmacological potencies. It is used as an excellent scaffold in drug discovery of medicinal drugs such as antimicrobials, anticancer agents, antihypertensives, anti-proliferative agents and anti-inflammatory agents. In spite of its diverse therapeutic activity, it is used as a key pharmacophore in synthesizing the most potent biological agents. Besides, viral infections are ubiquitous and their prevention and cure have become a great challenge. In this regard, the design of indole-containing antiviral drugs is accomplished to combat viral infections. A lot of research is being carried out towards antiviral drug discovery by many researchers round the clock. Herein, the antiviral activity of recently discovered indole scaffolds is compiled and critically evaluated to give a meaningful summary. In addition, the structure–activity relationship of remarkable antiviral agents is discussed. Also, the structural motifs attributed to noteworthy antiviral properties are highlighted to guide future antiviral research.
The antiviral properties of indole derivatives discovered recently are described considering their inhibitory values, cytotoxicity and SAR studies. The study helps researchers to carry out further investigation and to design efficient antiviral agents.
1.1. Introduction
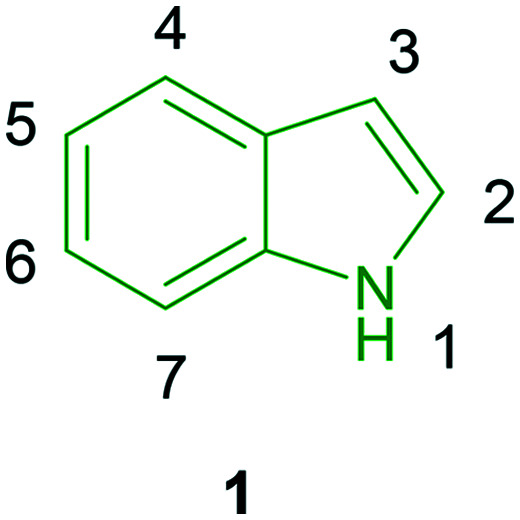
Although a plethora of heterocyclic compounds is available for the design of pharmaceutical drugs, indole finds itself in an apt place in drug discovery. In the field of drug design of antiviral agents, heterocyclic molecules such as benzimidazoles,1,2 coumarins,3–5 carbazoles,6,7 oxadiazoles,8,9 thiazoles,10,11etc. have been used to construct efficient drug molecules. In particular, indole forms a group of attractive pharmacological agents because of which researchers are working determinedly to improve the antiviral potencies of novel indole derivatives. Indole scaffolds have been reported to possess antibacterial activity,12 α-amylase and monooxime inhibitory activity,13 antimalarial properties14 and anti-tumor activity.15 Structurally, indole is a bicyclic molecule wherein pyrrole is fused to benzene at the 2,3-positions (1, Fig. 1).
Fig. 1. Illustration of the structure of indole.
Indole may be substituted with a wide range of substituents at the N-1, C-2 to C-6, or C-7 positions to avail of diverse indole scaffolds. This fact is utilized in obtaining versatile indole derivatives. In this review, recent antiviral research results are critically assimilated to obtain an idea of the current progress of the discovery of antivirals since the viruses are a bit difficult to understand. Further, antiviral drugs inhibit virulence by retarding virus-cell proliferation, while another class of drugs, viricides, deactivate or destroy the virus. Having considered only antiviral drugs because of their harmlessness to the host, these are extensively investigated. Parallelly, vaccine discovery is in progress, however despite the success of these vaccines, their use is limited because of allergic reactions, inadvertent attacks on a host, high cost, herd immunity, etc. Therefore, the design of antiviral drugs is given higher priority. Antiviral drugs may be designed to function by various mechanisms, such as (i) entry inhibition, (ii) uncoating inhibition, (iii) viral synthesis inhibition and (iv) viral enzyme inhibition.
Antiviral drug design and discovery has become a great challenge because of acquired antiviral resistance and this issue has posed a major obstacle to antiviral therapy. Hence, antiviral drug discovery is a continuous process wherein old drugs become obsolete and new drugs must replace them.
1.1.1. Hepatitis C virus and its inhibitors
Hepatitis C virus (HCV) poses a major health threat and is associated with a high risk of liver diseases. HCV is a single-stranded RNA virus that causes chronic inflammatory liver diseases including liver cirrhosis and hepatocellular carcinoma. Approximately 350 000–500 000 individuals die due to HCV-related liver infection, as reported by the WHO. HCV infection is responsible for approximately 40–45% of all liver transplantations performed. There are many HCV genotypes; amongst those, eleven HCV genotypes have been identified.16 Amongst the HCV gt 1–6 genotypes, HCV gt 1 and HCV gt 4 are hard to treat as they result in an aggressive form of the disease. After revealing the HCV life cycle, efforts were made to discover anti-HCV drugs targeting HCV proteins. Further, NS3 protease, NS5B polymerase, and NS5A proteins are also targeted.17 In addition, the main drawback of recently approved drugs is high cost. Also, the high mutation rate of HCV and the rapid emergence of drug resistance have presented major obstacles to anti-HCV drug discovery.18,19 The unavailability of an efficient vaccine for HCV is also a major problem.20,21 Further, the current standard therapy involving a combination of pegylated interferon-α and ribavirin22 is unaffordable because of the high price tags and such treatment is also associated with undesirable side effects23,24 such as flu-like symptoms, anemia, and depression in patients.
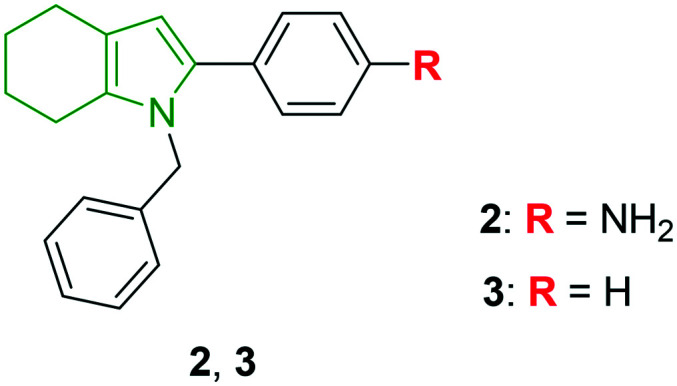
An urgent need for efficient antiviral drugs towards HCV with the least side effects and low cost has arisen. In view of this, I. A. Andreev et al. have synthesized 2-phenyl-4,5,6,7-tetrahydro-1H-indole derivatives targeting HCV.25 In total, 44 derivatives have been prepared and investigated for HCV gt 1b and gt 2a genotypes. A few indole derivatives showed a decent anti-HCV profile wherein tetrahydroindole involving benzene amine and benzyl moieties at the 1,2-positions, respectively (2, Fig. 2), has good antiviral properties towards gt 1b and gt 2a with EC50 values of 12.4 μM and 8.7 μM, respectively. With low cytotoxicity (CC50 = 109.9 μM), compound 2 is found to be a hit molecule. Further, phenyl- and benzyl-substituted tetrahydroindole 3 has exhibited the most elegant anti-HCV properties (gt 1b: EC50 = 7.9 μM, gt 2a: EC50 = 2.6 μM). Amongst the synthesized tetrahydroindoles, N-benzyl-substituted scaffolds have rendered higher anti-HCV properties. Replacement of the N-benzyl moiety with non-aromatic groups has led to deteriorated activity towards gt 1b. Concerning C-2 substitution in the N-benzyl derivatives, the presence of a simple phenyl ring (compound 3) or 4-substituted phenyl rings is responsible for the remarkable activity, inferring that the simple ring is well tolerated. On the other hand, the presence of meta-substitutions and ortho-OH substitutions has contributed to increased cytotoxicity. Compound 3 was evaluated for enzyme inhibitory properties and it was found to be inactive towards NS5B and could not inhibit HCV NS3 to unwind DNA even at a concentration as high as 500 μM. The results reveal that compound 3 could not inhibit HCV by targeting the protein. Overall, the benzyl tetrahydroindole derivatives might be interesting scaffolds in designing good anti-HCV derivatives.
Fig. 2. Depiction of N-benzylated tetrahydroindoles [reproduced from ref. 25 with permission from Elsevier, copyright 2020].
To identify the inhibitors of NS5A resistance-associated polymorphisms, tetracyclic indole-based HCV NS5A inhibitors are developed utilizing the HCV NS5A inhibitor, MK-8742.26 Amongst the open-chain alkyl-substituted derivatives, methyl-substituted tetracyclic indole 4 (Fig. 3) has exhibited a potent anti-HCV profile, while poor activity is observed towards genotypes 4a, 1a Y93H, and 1a L31V (Table 1). Also, ethyl analog 5 is found to possess an almost similar anti-HCV profile compared to compound 4. Further, an increase in the alkyl chain length has resulted in comparable results with that of compound 4. Although the introduction of a fluoro atom has no improvement, however, appending either a cyano or an ester group to the alkyl chain diminished the potency. In addition, HCV inhibitory properties are investigated for the cycloalkane-substituted tetracyclic indoles. The cyclopropyl analog 6 is depicted as an efficient anti-HCV molecule. Reduced anti-HCV potencies are noticed for most of the derivatives with increased ring sizes, indicating the unfavorable effects of increased number of methylene units in the case of cycloalkane ring-substituted tetracyclic indoles. Apart from these, the isobutyl analog 7 has exhibited extraordinary anti-HCV activity towards some genotypes. In addition, the cycloalkyl derivatives in the form of spiro-scaffolds showed almost similar activity. Finally, the introduction of polar functionalities such as acid, amide, CN, etc. have yielded weak activity.
Fig. 3. Structures of aminal analogs of MK-8742 [reproduced from ref. 26 with permission from Elsevier, copyright 2020].

Anti-HCV properties of tetracyclic indole derivatives.
| Compd | Anti-HCV activity (EC50, nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotypes | ||||||||
| 1a | 1b | 2a | 2b | 3a | 4a | 1a Y93H | 1a L31V | |
| 4 | 0.003 | 0.003 | 0.007 | 23 | 0.09 | 0.03 | 8 | 7 |
| 5 | 0.002 | 0.002 | 0.001 | 2 | 0.3 | 0.04 | 4 | 1 |
| 6 | 0.003 | 0.004 | 0.002 | 1 | 0.02 | 0.03 | 5 | 1 |
| 7 | 0.0008 | 0.0007 | 0.008 | 10 | 0.4 | 0.02 | 11 | 0.8 |
The research on the anti-HCV activity of MK-8742 is continued wherein the tetracyclic indole core is modified in addition to an aminal position.27 Initially, a series of derivatives comprising fluoro/chloro groups on the tetracyclic indole core is synthesized. Indole derivative 8 (Fig. 4) connected with the fluoro group at the C-1 position is reported to render a potent anti-HCV profile (Table 2) and derivatives with fluoro substitutions at other positions have not yielded anti-HCV activity up to the mark. Meanwhile, tetracyclic indole scaffold 9 with a 12-F group has emerged as a noteworthy inhibitor of all the HCV genotypes. Having checked the necessity of the fluoro group at the 1 and 12 positions, the effect of substitution of alkyl moieties at the C-1 and C-12 positions is investigated. The anti-HCV activity reveals the elite activity of indole-cyclopropyl analog 10 (Fig. 4) towards all the genotypes. A cyclopropyl ring at the C-1 position has failed to exhibit the finest activity. Also, fluoroproline-containing KM-8742 derivatives have produced worthy anti-HCV results to be mentioned. A combination of fluoroproline with an alkoxy phenyl ring on the tetracyclic indole moiety showed a synergistic effect towards the anti-HCV activity. Furthermore, one more series of KM-8742 derivatives comprising alkoxy phenyl motifs at the 6-position rendered decent inhibitory activity wherein 3,5-dimethoxy phenyl analog 11 (Table 2) exhibited the most potent anti-HCV activity.
Fig. 4. Structures of halogenated MK-8742 derivatives [reproduced from ref. 27 with permission from Elsevier, copyright 2020].

Anti-HCV potency of MK-8742 derivatives towards various genotypes.
| Compd | Anti-HCV activity (EC50, nM) | |||||||
|---|---|---|---|---|---|---|---|---|
| Genotypes | ||||||||
| 1a | 1b | 2a | 2b | 3a | 4a | 1a Y93H | 1a L31V | |
| 8 | 0.005 | 0.003 | 0.15 | 18 | 0.4 | 0.04 | 34 | 0.4 |
| 9 | 0.007 | 0.007 | 0.004 | 15 | 0.3 | 0.06 | 25 | 0.7 |
| 10 | 0.03 | 0.004 | 0.0005 | 122 | 10 | 0.3 | 100 | 8 |
| 11 | 0.004 | — | 0.06 | — | 0.06 | — | 3 | 0.1 |
In the category of thumb site-I inhibitors, a new generation HCV NS5B thumb site inhibitor 12 (Fig. 5) has been in clinical trials with remarkable HCV inhibitory activity (gt 1a: EC50 = 0.043 μM, gt 1b: EC50 = 0.017 μM).28 Except for some mild symptoms, good tolerability is observed in 41 out of 46 subjects. The non-zwitterionic indole derivative 13 is reported as a nanomolar cellular inhibitor (EC50 = 77 nM) with low cytotoxicity (CC50 > 20 μM)28 and further showed high affinity and persistent binding to NS5B polymerase that favors in vivo properties in preclinical species. With enhanced HCV NS3 protease inhibitory effects (gt 1a: EC50 = 0.003 μM, gt 1b: EC50 = 0.006 μM), an alkyl-bridged piperazine carboxamide analog 14 is reported as a noteworthy anti-HCV molecule.28
Fig. 5. Structures of noteworthy indole-containing HCV inhibitors [reproduced from ref. 28 with permission from Elsevier, copyright 2020].

Using chemical genetics, three structurally different groups of indole derivatives are synthesized and evaluated for HCV inhibitory potency.29 In this study, firstly, unsubstituted indole acryl amides attached to different amines are evaluated for anti-HCV activity. t-Butylaniline-appended acrylamide derivative 15 (Fig. 6) has exhibited the highest ratio of cell viability (60.1%) to HCV replication (2.1%). The corresponding EC50 value for compound 11 is reported to be 1.9 μM. Because of the potent anti-HCV activity of compound 15, further derivatization is considered wherein N-(4-t-butylphenyl) amide and 2-methylacryl moieties are retained while substituting the indole ring. Amongst the indole 2-methacrylate analogs designed, 4-t-butylaniline acrylamide 16 connected with a –CN group at the indole 5-position exhibited the strongest anti-HCV activity (EC50 = 1.1 μM) in addition to lower cytotoxicity (CC50 = 61.6 μM) and the selectivity index is found to be 56.9. The observed activity is eightfold higher compared with that of clemizole. Further, the electron-donating group-substituted derivatives have yielded lower activity; interestingly, 6-isopropyl indole showed a better activity profile (EC50 = 1.1 μM, CC50 = 24.8 μM), suggesting that the lipophilic group is crucial for the anti-HCV activity. However, the selectivity index is less than that of compound 16. In addition, compound 16 is tested on the reporter virus-free gt 2a infectious clone system using J6/JFH1 transfected Huh7.5 cells. The results indicate a similar EC50 value of 1.9 μM to that of the luciferase-linked reporter virus. To check the cross-genotype anti-HCV activity, compound 16 is allowed to inhibit gt 2b wherein 3.2-fold greater potency (EC50 = 0.6 μM) is observed compared to that of gt 2a; however, compound 16 needed a 1.8-fold longer period to reduce HCV RNA levels. Thereby, compound 16 is said to possess cross-genotype potency towards both gt 1b and gt 2a. The previously reported potent HCV active compound 16 has been demonstrated to induce transcriptional activation of several pro-inflammatory as well as antiviral cytokine genes including CXCL-8, IL-α, TNF-α, IL-3, IRAK-1, DDX58, and TLR-7.30 Also, compound 16 stimulated the secretion of soluble factors that play a prominent role in the suppression of HCV replication in HCV-positive neighboring cells. The data suggests that compound 16 could inhibit HCV replication through the induction of several pro-inflammatory genes. Additionally, no additive or synergistic down-regulation of HCV replication is noticed with the co-treatment of compound 16 with ribavirin. Apart from these, treatment of compound 16 (5 μM mL−1) increased the extracellular concentration of CXCL-8 protein up to 25 ng ml−1, indicating that CXCL-r protein alone may not be able to suppress the HCV replication. Rather, it may need to work together with pro-inflammatory cytokines such as IL-1α, TNF-α, IL-3, IRAK-1, and DDX58 to accomplish complete suppression. Hence, such an indole derivative may form potent anti-HCV therapeutics with a novel mode of action.
Fig. 6. Structures of t-butylaniline acrylamide analogs of indole.30.

With a view to develop improved HCV replication inhibitory activity and enhanced water solubility, indole acrylamide scaffolds with N-substitution are synthesized and evaluated for anti-HCV activity.31 Herein, sulfonyl analog 17 (Fig. 7) with an unsubstituted indole ring rendered noteworthy anti-HCV activity (EC50 = 1.56 μM) with lower cytotoxicity of EC50 = 54.1 μM and corresponding selectivity index of 34.7. The selectivity index (SI = 59.5) is enhanced for the N-benzoyl analog 18 (EC50 = 1.16 μM, CC50 = 69.0 μM). Although, higher anti-HCV activity (EC50 = 0.98 μM) is observed for N-acetyl analog 19, its cytotoxicity is higher (CC50 = 40.74 μM), inferring a lower selectivity index (41.6). Further, the solubility of compounds 17–19 is evaluated and compound 19 is reported to exhibit higher solubility at pH 2 and pH 7.4 with a solubility of 103.0 μM and 101.3 μM, respectively. The low solubility of compound 18 might be due to the benzene carbonyl group appended to the indole N-atom. Hence, compound 19 is considered to be the most potent anti-HCV molecule.
Fig. 7. Illustration of structures of N-substituted acrylamide indole derivatives [reproduced from ref. 31 with permission from Wiley, copyright 2020].

Based on the hit compounds, N-substituted indole derivatives, indole N-substituted pyrazolone derivatives are designed.32 One of the series involving N-methylene indole pyrazolones failed to exhibit potent selectivity indices. Although a few derivatives possessed good EC50 values, their cytotoxic values are higher. The results indicate that the glycidyl group may disfavor the anti-HCV activity due to its high chemical reactivity to nucleophiles. Hence, oxirane ring-opened derivatives are designed and evaluated for the activity. Amongst the different secondary amines used to ring open in addition to the phenyl ring on pyrazolone moiety, the morpholine analog showed good activity. Therefore, retaining the alkylmorpholine moiety, substituted phenyl rings are attached to pyrazolone rings and the resulting derivatives have shown interesting potencies. Accounting for the cytotoxicity, most of the morpholine analogs are not efficient anti-HCV molecules. However, 4-fluorophenyl ring-appended indole derivative 20 (Fig. 8) is found to be a decent anti-HCV drug (EC50 = 1.02 μM, CC50 = 46.47 μM and SI = 45.56). Despite the strongest anti-HCV activity (EC50 = 0.92 μM) of 3-fluorophenyl analog 21, its higher cytotoxicity (CC50 = 21.46 μM) led to a lower selectivity index (23.33). Since the chiral compounds 20 and 21 exist as R- and S-isomers, the isomers are individually investigated for their antiviral potency. The R-isomers have exhibited exclusively stronger anti-HCV activity compared to S-isomers (Table 3).
Fig. 8. Chemical structures of indole-pyrazolone derivatives [reproduced from ref. 32 with permission from Elsevier, copyright 2020].

Anti-HCV properties of isomers of compound 20 and 21.
| Compd | Anti-HCV activity | CC50, μM | SI |
|---|---|---|---|
| EC50, μM | |||
| (R) 20 | 0.72 | >50 | >69.44 |
| (S) 20 | 7.12 | 34.57 | 4.86 |
| (R) 21 | 0.74 | 31.06 | 41.97 |
| (S) 21 | 5.87 | 23.31 | 3.97 |
A set of sugar-modified nucleosides derived from 4-substituted 6-chloropyrimido [4,5-b]indole ribonucleosides is synthesized to explore their anti-HCV activity.33 Moderate to poor anti-HCV results towards genotype 1b are noted for the designed derivatives comprising three series of pyrimidoindole nucleosides. A comparable EC50 value is observed for the 4-thiomethylpyrimidoindole connected with fluoro nucleoside 22 (Fig. 9 and Table 4) with that of standard mericitabine. However, it cannot be considered as a potent HCV 1b inhibitor due to its higher cytotoxicity and lower selectivity index. In this series, an almost similar activity profile has been exhibited by the methoxy derivative 23. Further, the 2-thienyl analog 24 is reported to have enhanced activity compared to compound 23. Again, 2-furyl scaffold 25 is found to render decent anti-HCV activity which is comparable with that of compound 23.
Fig. 9. Illustration of potent inhibitors of HCV 1b genotype.33.

Anti-HCV properties of pyrimidoindole-ribonucleoside derivatives.
| Compd | Anti-HCV activity (gt 1b) | Cytotoxicity | SI |
|---|---|---|---|
| IC50, μM | CC50, μM | ||
| 22 | 1.6 | 22.9 | 14.3 |
| 23 | 3.1 | >44.4 | >14.3 |
| 24 | 2.5 | >44.4 | >17.8 |
| 25 | 3.0 | >44.4 | >14.8 |
| 26 | 2.3 | >44.4 | >19.3 |
| Mericitabine | 1.2 | >44.4 | >37 |
Meanwhile, the combination of 2-furyl and a fluorinated sugar unit (compound 26) resulted in the strongest anti-HCV gt activity. In the case of inhibitory activity towards HCV gt 2a, poor activity is observed compared to mericitabine (SI >44.4) wherein the highest selectivity index of evaluated molecules is >2.8, revealing that the pyrimidoindole-ribonucleoside analogs are not tolerable to HCV gt 2a.
Tetracyclic indole derivatives (MK-8742) are once again modified to discover efficient anti-HCV drugs.34 Broad changes are brought about concerning amino acid end caps. Initially, the amino acid side chain is substituted with alkyl/cycloalkyl moieties. When these derivatives were evaluated against HCV genotypes 1a, 2b, 1a Y93H, and 1a L31V, a promising gt 1a inhibitory activity was observed (IC50 = 0.006–0.10 nM), while poor activity is exhibited by most of the designed molecules against other genotypes. However, introduction of a tetrahydropyran ring in compound 27 (Fig. 10) showed some significance (2b: EC50 = 1.2 μM, 1a Y93H: EC50 = 23 μM, and 1a L31V: EC50 < 0.2 μM). Removal of the amino acid side chain, while retaining pyrrolidine and subsequently substituting at its N-position, led to a series of tetracyclic indole derivatives with much-diminished activity. However, mixed alkyl/cycloalkyl motifs substituted on the carbamate chains improved inhibitory results across the genotypes 2b, 1a Y93H, and 1a L31V compared to MK-8742. In particular, the combination of tetrahydropyran and isopropyl moieties (compounds 28 and 29, Fig. 10) has elicited striking activity (Table 5). Conjugation of a methylene unit to the tetrahydropyran in compound 30 reduced its activity compared to compounds 28 and 29, while a lot of enhancement of activity is observed against gt 1a.
Fig. 10. Structures of MK-8742 derivatives with mixed caps [reproduced from ref. 34 with permission from Elsevier, copyright 2020].

HCV inhibitory properties of mixed capped tetracyclic indole derivatives.
| Compd | Anti-HCV activity (EC50, μM), genotype | |||
|---|---|---|---|---|
| 1a | 2b | 1a Y93H | 1a L31V | |
| 27 | 0.060 | 9 | 11 | 0.6 |
| 28 | 0.061 | 4 | 11 | 0.4 |
| 29 | 0.014 | 8.9 | 24 | 0.33 |
Tricyclic indole carboxylic acid scaffolds are prepared to anticipate greater anti-HCV activity.35 The C-7 fluoro analog 31 (Fig. 11) has demonstrated a decent activity (IC50 = 17 nM); however, the cellular activity is found to be diminished. Further, the C-6 fluoro derivative 32 showed improvement in the activity (IC50 = 2 nM, EC50 = 90 nM). Despite the potent activity of fluoro derivatives, compound 32 is developed further. Since the introduction of 2-fluoro-5-carbamoyl benzyl at the N-1 position has exhibited enhanced cellular activity, the particular pharmacophore is incorporated in compound 32, resulting in derivative 33 that rendered improved cellular potency (IC50 = 4 nM, EC50 = 15 nM). Presuming that the presence of primary amide is responsible for the reduced oral absorption of compound 33, the carboxamide functionality is replaced with bicyclic heterocyclic isosteres containing hydrogen bond donors. In this process, most of the resulting derivatives have shown decent inhibitory activity. Amongst the various modifications, incorporation of the 3-amino-6-fluoro-1H-indazolylmethyl group led to a remarkable derivative 34 (gt 1b: IC50 = 4 nM, EC50 = 3 nM) (Fig. 5). Also, similar anti-HCV activity (gt 1b: IC50 = 3 nM, EC50 = 4 nM) is noted when the 6-fluoro-1H-indazole motif is incorporated (compound 35). 6-Methyl analog 36 has emerged as the strongest gt 1b inhibitor (IC50 = 2 nM). These facts infer that the indazole moiety has tolerated well against HCV activity.
Fig. 11. Depiction of structures of tricyclic fluoroindole derivatives [reproduced from ref. 35 with permission from Elsevier, copyright 2020].

1.1.2. HIV inhibitors
HIV belongs to a Lentivirus species, a subgroup of a retrovirus, and results in acquired immune deficiency syndrome over time in humans.36 This condition would further lead to progressive failure of the immune system that allows life-threatening opportunistic infections to thrive.37 HIV virions particularly target vital cells in the immune system such as helper T cells (specifically CD4+ T cells), macrophages, and dendritic cells.38 Thus, HIV infection results in low levels of CD4+ T cells through various mechanisms. Regarding its genome, the single-stranded RNA codes for nine genes that are enclosed in protein capsid p24, and the single-stranded RNA is tightly bound to nucleoplasmid proteins, p7, and enzymes required for the development of the virion such as reverse transcriptase, proteases, ribonuclease, and integrase. In addition, the treatment of HIV/AIDS normally includes multiple antiretroviral drugs. However, HIV latency and the consequent viral reservoir in CD4+ T cells, dendritic cells, and macrophages have become a major obstacle for HIV eradication.39 Antiretroviral therapy comprising a combination of drugs of NNRTI, nucleoside reverse transcriptase (NRTI), protease, and integrase inhibitor classes has enhanced the quality of life of AIDS patients. Despite antiretroviral therapy, the side effects and rapid emergence of drug resistance40 are limitations that have hindered the continuous development of new anti-HIV compounds. To combat the drug-resistant HIV strains, antiviral agents that function through innovative mechanisms are required. Amongst the wide variety of heterocycles available for HIV drug discovery, indole derivatives have attracted a great deal of attention.
As umifenovir analogs have been reported to possess a broad-spectrum antiviral activity, the conformationally restricted structural analogs of umifenovir are studied and their anti-HIV activity is evaluated using umifenovir and rac-MC-1501 as reference compounds.41 Cyclopropyl phenyl analogs have demonstrated stronger anti-HIV activity compared to umifenovir but the activity is inferior compared to rac-MC-1501. The presence of the cyclopropyl phenyl motif at the indole 2-position in compound 37 (Fig. 12) is responsible for the good anti-HIV-1 activity (EC50 = 3.58 μM). The switching of methyl and cyclopropyl phenyl rings between the N-1 and the C-2 positions of indole (compound 38) led to diminished potency (EC50 = 5.91 μM). In addition, hydroxy and acetyl groups have further reduced the activity. Compounds 37 and 38 have a higher cytotoxicity of >298 μM and >250 μM, respectively, and the corresponding selectivity indices are found to be >83.3 and >42.3, respectively. In the case of HIV-2 activity, compared to both reference compounds, compound 37 has rendered greater activity (EC50 = 5.4 μM); however, the HIV-2 activity is lost for compound 38, revealing the importance of the positions of methyl and cyclopropyl phenyl moieties. Furthermore, appending hydroxy and acetoxy groups (compound 39) has contributed to considerable activity (EC50 = 11.091 μM) but with higher cytotoxicity (CC50 = 50 μM). The selectivity index (>55.2) of compound 39 is superior to that of rac-MC-1501 (>3.6). The data indicates the importance of the position of substituents methyl and cyclopropyl motifs on the indole bicycle.
Fig. 12. Illustration of structural analogs of umifenovir [reproduced from ref. 41 with permission from Elsevier, copyright 2020].

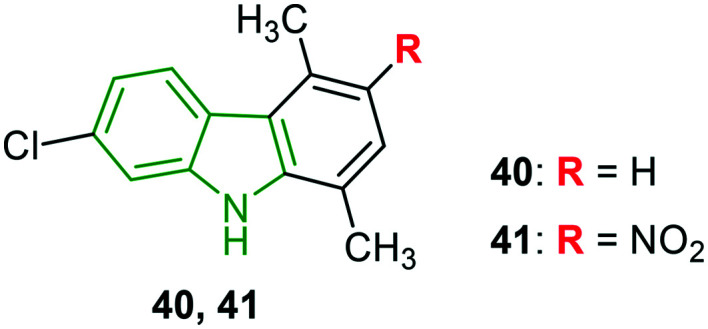
Halogenated carbazole scaffolds composed of dimethyl phenyl rings are prepared to investigate anti-HIV activity towards NL4.3X4 and Bal R5 variants.42 Weak anti-HIV properties have been rendered by 2,5-dimethylcarbazole analogs connected with 7-chloro and 8-chloro groups, while 8-chlorocarbazole 40 (Fig. 13) without a –NO2/NH2 group elicited moderate NL4.3X4 inhibitory activity (IC50 = 3.5 μM) and stronger Bal R5 inhibitory potency (IC50 = 3.8 μM) compared to maraviroc (IC50 = 4.2 μM). Further, the introduction of the nitro group (compound 41) resulted in the most potent activity (IC50 = 1.4 μM) towards the NL4.3X4 variant. In addition, it could also exhibit comparable Bal R5 activity (IC50 = 5.3 μM) to maraviroc (IC50 = 4.2 μM). With cytotoxicity of 22.7 μM, compound 41 showed a good selectivity index (16.4).
Fig. 13. Structures of remarkable NL4.3X4 and Bal R5 inhibitory agents.42.
By incorporating a broad range of structural variants into compound 42 (Fig. 14), a series of indole-based HIV-1 attachment inhibitors is prepared.43 The antiviral activity reveals that indole-3yl analog 43 involving (R)-Me at the piperazine 2-position is an elite inhibitor (EC50 = 4.0 nM) with favorable cytotoxicity (CC50 = 200 μM). Removal of (R)-Me has led to much-diminished activity. Comparatively reduced antiviral activity is observed for indole-2-yl analogs. Pyridine-fused indole derivative 44 has elicited promising activity (EC50 = 17 nM) but with higher cytotoxicity (CC50 = 45 μM). In addition, glyoxamide derivatives with an indole moiety connected at farther positions have resulted in lower activity. Apart from these, the effect of the introduction of the methoxy motif in various positions of indole is investigated wherein the 6-methoxyindole analog expressed almost comparable activity (EC50 = 4.8 nM) to compound 43, while the 5-methoxy derivative and 4,6-dimethoxy analogs have failed to render the expected results, inferring that 6-methoxy indole has a more favorable HIV-1 inhibitory potency.
Fig. 14. Structure of the indole glyoxamide derivatives [reproduced from ref. 43 with permission from Elsevier, copyright 2020].

Some selected compounds from an in-house DHICA library have been tested for the ability to inhibit HIV-1 integrase and RNase H activities.44 Poor activity is noted for most of the derivatives. The simple molecule 5,6-dihydroxyindole-2-carboxylic acid showed significant anti-HIV-1 activity (IC50 = 7.0 μM). Amongst the few potent inhibitors, 5,6-dihydroxyindole carboxamide derivative 45 (Fig. 15) has exerted the strongest anti-HIV-1 integrase LEDGF/p75-dependent activity (IC50 = 1.4 μM), while bis(dihydroxyindole carboxamide) 46 has rendered a slightly diminished activity compared to compound 45 (IC50 = 2.1 μM). The compound resulting from the introduction of an ethylene unit into bis-indole derivative 46 has lost its activity. However, the incorporation of a piperazine ring has resulted in significant activity (IC50 = 7.0 μM). The potent derivatives are utilized for further studies wherein dihydroxyindole-2-carboxylic acid has demonstrated the strongest inhibition of HIV-1 (strain IIIB) replication (EC50 = 12 μM) in MT-4 cells, while compound 45 and 46 have exhibited diminished activity. However, compound 45 is found to be an excellent inhibitor of HIV-1 mutant integrases wt IN and A128T IN with IC50 values of 1.4 μM and 2.8 μM, respectively. These values are comparable with that of ALLINI-2. Further, compound 45 exhibited decent HIV-1-associated RNase H inhibitory activity (IC50 = 17 μM), and slightly abated activity (IC50 = 24 μM) has been shown by the dimer 46. In the inhibition of the RT-associated RNase H activity of mutated HIV-1 RTs, surprisingly greater activities (wt RT: IC50 = 17.5 μM, L503F: IC50 = 65 μM) are evident for compound 45 compared to the standard RDS1759 (wt RT: IC50 = 24.1 μM, L503F: IC50 > 100 μM). These results indicate that the compound 45 is a potential anti-HIV drug candidate for future development.
Fig. 15. Depiction of structures of dihydroxyindole carboxylic acid derivatives [reproduced from ref. 44 with permission from Elsevier, copyright 2020].

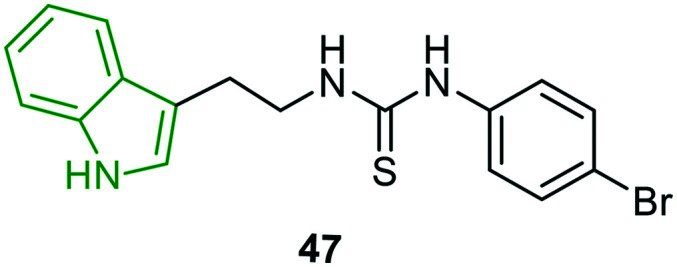
Considering that an electron acceptor group such as a halogen in a strategic part of a molecule significantly increases lipophilicity and in turn enhances biological activity,45,46 indole derivatives incorporated with a 1,3-disubstituted thiourea moiety are synthesized and evaluated for anti-HIV activity.47 Out of the evaluated molecules, only 4-bromoaniline analog 47 (Fig. 16) is demonstrated as a remarkable HIV-1 inhibitor (EC50 = 8.7 μM) with moderate cytotoxicity (CC50 = 45 μM). However, the activity is inferior compared to that of efavirenz (EC50 = 0.002 μM). The other derivatives have turned out to be poor HIV-1 inhibitors.
Fig. 16. Structure of a remarkable anti-HIV-1 inhibitor.47.
N-substituted indoles with substitution on the 3-position are designed to investigate HIV integrase inhibitory properties.48 The HIV-1 integrase strand transfer activity reveals that the derivatives with halogens at C-5 (Br, Cl) possessed good inhibitory properties, suggesting that they possessed apt conformation to fit into the enzyme pocket. In particular, fluoro N-benzyl-substituted indole 48 (Fig. 17) is reported as a noteworthy integrase inhibitor (IC50 = 0.2 μM) towards the WT strain. In the case of mutant variants G140S and Y143R, again compound 48 is an elite inhibitor with IC50 values of 0.2 μM and 0.37 μM, respectively. In addition, lower cytotoxicity (CC50 >200 μM) has benefited the anti-integrase activity, while the 2,4-difluorobenzyl analog has rendered comparatively reduced activity; it might be due to the absence of a H-bond with E-152 because of steric hindrance which is not observed in the case of compound 48. Further, despite the presence of the 2,4-difluorophenyl moiety, 5-fluoroindole scaffold 49 has exerted promising WT HIV integrase inhibitory potency (IC50 = 0.5 μM). However, it showed abated activity against mutant variants (G140S: IC50 = 3.4 μM, Y143R: IC50 = 5.7 μM). The dioxolan derivatives expressed moderate to poor activity; however, N-tosyl-5-bromoindole derivative 50 exhibited remarkable activity (WT: IC50 = 5.7 μM, G140S: IC50 = 4.7 μM, and Y143R: IC50 = 5.0 μM). Furthermore, the potent antiviral agents 48 and 49 are evaluated for activity against RAL resistant mutant protein. In this activity, the compounds 48 and 49 showed excellent inhibitory activity with EC50 values of 0.13 μM and 0.15 μM, respectively. Overall, compound 48 comprising a Br atom at C-5, β-hydroxy, and a δ-oxocarbonyl moiety has emerged as a potent anti-HIV integrase inhibitor.
Fig. 17. Structures of halo-indole derivatives with HIV integrase inhibitory activity [reproduced from ref. 48 with permission from Wiley, copyright 2020].

Indolylarylsulfones (IASs) are being developed to address the drug-resistance issue. The 2-carboxamide moiety of IAS analogs is modified, making use of a wide variety of substituents which subsequently enhanced the HIV inhibitory potency.49 Motivated by such decorated IAS scaffolds, the focus is shifted towards the synthesis of indole analogs with benzyl/phenylethyl linked carboxamide at the indole 2-position.50 All the designed molecules exhibited superior anti-HIV-1 NL4-3 inhibitory activity compared to nevirapine. Among those, some compounds (4-methoxyphenyl 51, 4-aminobenzyl 52, and isobutylphenyl 53 analogs) (Fig. 18) have rendered extraordinary activity (EC50 = 0.21 nM) with 7.6-fold higher activity compared to AZT (EC50 = 3.7 nM). All these derivatives have possessed highly electron-donating groups. While the 3-fluorobenzyl 54 and 4-fluorobenzyl 55 analogs have exerted slightly lower activity (EC50 = 0.68 nM), they exhibited a higher selectivity index than that of compounds 51–53. In the case of HIV-1 IIIB strain, compounds 54 and 55 have been reported to be the strongest inhibitors with EC50 values of 3.6 nM and 4.3 nM, respectively. A diminished HIV-2 inhibitory profile is noted for the evaluated molecules. Furthermore, the anti-HIV activity of the screened molecules revealed noteworthy potencies towards mutant variants. In particular, compounds 52 (K103N: EC50 = 4.3 nM, Y181C: EC50 = 11 nM) and 54 (K103N: EC50 = 4.4 nM, Y181C: EC50 = 4.3 nM) have been demonstrated as exclusive inhibitors of mutant variants K103N and Y181C. Both the derivatives have superior activity compared to AZT. Compound 54 is a decent inhibitor (EC50 = 5.0 nM) of variant L1001. Towards the mutant variant Y181C, moderate inhibitory potencies are observed. Since isopropyl phenyl analog 56 is in racemic form, it is separated into the corresponding isomers (R and S forms) and evaluated individually wherein the (R)-isomer has exhibited higher activity against all the mutant variants, while the (S)-isomer has rendered poor activity compared to the racemic mixture.
Fig. 18. Structures of the most potent anti-HIV agents [reproduced from ref. 50 with permission from Elsevier, copyright 2020].

A series of novel IAS scaffolds with a heterocyclic tail is designed and investigated for anti-HIV properties.51 An excellent anti-HIV profile has been showcased by the screened molecules. Amongst them, the derivatives 57–60 (Fig. 19) have exhibited extraordinary activity (Table 6) against HIV-1 NL4-3 and HIV-1 IIIB strains. Presumably, nitrogen-containing 6-membered heterocycles have exhibited stronger activity. Unfortunately, no single molecule could exhibit remarkable anti-HIV-2 ROD activity. Towards mutant HIV-1 strains, 2-methyl aryl analog 63 (Fig. 7) is responsible for incredible inhibitory properties (Table 7). In addition, 4-pyridyl (61) and 3-methylpyrimidyl (62) analogs are also reported to be mighty anti-HIV inhibitors. 4-Ethylpyridyl- (EC50 = 4.7 nM) and 2-ethylthiophenyl-containing (EC50 = 5.4 nM) scaffolds have emerged as promising inhibitors of HIV-1 L100I. Other derivatives are reported to possess good to moderate potencies.
Fig. 19. Structures of indolylarylsulfones with an aromatic/heterocyclic tail [reproduced from ref. 51 with permission from American Chemical Society, copyright 2020].

Anti-HIV-1 NL4-3 and anti-HIV-1 IIIB inhibitory properties of IASs.
| Compd | HIV-1 NL4-3 | HIV-1 IIIB | ||
|---|---|---|---|---|
| CC50 (nM) | EC50 (nM) | SI | EC50 (nM) | |
| 57 | 23 769 | 0.22 | 89 643 | 17 |
| 58 | 30 216 | 0.2 | 151 078 | 4.1 |
| 59 | 54 589 | 0.6 | 90 981 | 3.2 |
| 60 | 25 350 | 0.62 | 40 887 | 4.1 |
Inhibitory properties of mutant HIV-1 strains.
| Compd | EC50 (nM) | ||
|---|---|---|---|
| K103N | Y181C | Y188L | |
| 61 | 0.23 | 16 | ns |
| 62 | 0.22 | 2.20 | 50.6 |
| 63 | 0.2 | 0.8 | 45 |
| AZT | 16 | 6.0 | 33 |
Furthermore, the anti-HIV-1 activity against WT RT and mutant RTs affirms that compound 62 has the most potent Y181I RT inhibitory activity (IC50 = 4.2 nM) with 40-fold higher activity compared to ETV (IC50 = 164 nM) and moderately active towards WT RT and K103N RT. Meanwhile, compound 63 has striking inhibitory properties against WT RT (IC50 = 3 nM) and K103N (IC50 = 9 nM) strains; the activities displayed are threefold and twofold stronger respectively compared to ETV. The anti-HIV-1 properties discussed so far suggest that these molecules could be developed into potential anti-HIV drug molecules.
The past research involving cyclopropyl-indole derivatives as NNRTIs is further optimized against resistant strains of the virus. The focus is emphasized on the ester functionality located at the indole 2-position.52 The presence of the carbethoxy group at the C-2 position and phenyl and hydroxy moieties at the C-3 position has resulted in reduced HIV-1 activity (IC50 = 1.15 μM) compared to the parent compound. Ethylation of the hydroxy group (compound 64) (Fig. 20) has exhibited enhanced activity (IC50 = 0.03 μM). Further, the trifluorosulfonyl analog showed diminished potency. In addition, 3,5-dimethyl phenyl-containing indole 65 rendered similar activity compared to compound 64; interestingly, lower cytotoxicity is observed (CC50 = 36.3 μM), making compound 65 a decent anti-HIV-1 agent. In addition, indole derivatives unsubstituted at the C-2 position have reported lower activity; however, 3,5-dimethoxy phenyl analog 66 rendered some significant potency (IC50 = 0.3 μM), revealing the importance of the carbethoxy functionality.
Fig. 20. Depiction of structures of 5-fluoroindole derivatives as anti-HIV agents [reproduced from ref. 52 with permission from Wiley, copyright 2020].

1.1.3. Anti-dengue activity
Dengue is a mosquito-borne neglected tropical disease spread by female mosquitos of Aedes type, principally A. aegypti,53 and caused by the dengue virus. The symptoms may include a high fever, headache, vomiting, muscle and joint pain, and a characteristic skin rash. In addition, in some cases, it develops into severe dengue. Dengue has become a global threat since the Second World War. Dengue virus is an RNA virus containing about 11 000 nucleotide bases which code for the three different types of protein molecules such as C, prM, and E that are responsible for the formation of a virus particle. Additionally, seven other non-structural protein molecules (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) are observed in infected host cells only which are required for virus replication.54 Based on the antigenicity,55 four serotypes of dengue viruses are reported, namely DENV-1, DENV-2, DENV-3, and DENV-4.56,57 With regard to inhibition of dengue, several plausible targets include inhibition of viral RNA-dependent RNA polymerase and inhibition of viral protease, entry inhibitors, etc. The use of the first developed dengue vaccine CYD-TDV by Sanofi Pasteur is limited due to low efficacy and safety issues.58,59 Hence, the design and discovery of efficient drugs for dengue treatment is of prime concern.
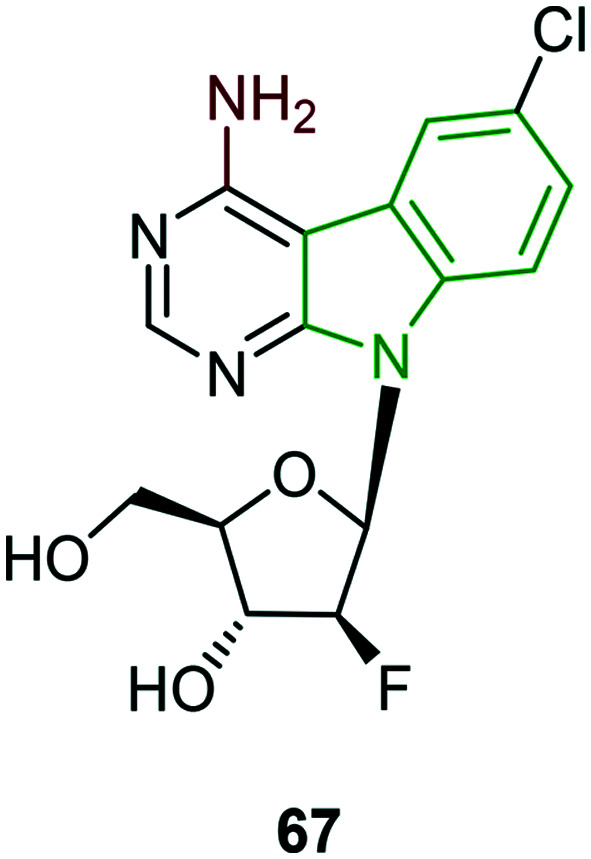
Sugar-modified nucleoside derivatives which have exhibited remarkable anti-HCV activity are also tested for dengue type 2 inhibitory activity.33 In this activity, only a few compounds have rendered activity and other scaffolds are completely inactive. Fluoronucleoside-appended amine-containing scaffold 67 (Fig. 21) has exhibited moderate activity (EC50 = 10.8 μM) and it is the strongest compared to other potent molecules, while almost similar activity (EC50 = 10.5 μM) but with lower cytotoxicity (CC50 = 39.0 μM) is noted for compound 26. In addition, two other series involving nucleoside analogs lacking a fluorine atom and nucleoside derivatives with fluorine below the plane have ended with poor activity.
Fig. 21. Structure of nucleoside analog with moderate anti-DENV activity.33.
Spiro-triazolopyrimidine-indolone derivatives are designed and evaluated for inhibition of various dengue stereotypes (DENV-1–DENV-4).60 Most of the derivatives have moderate activity towards DENV-1 and DENV-4. No single derivative has displayed the strongest inhibitory activity against all the DENV stereotypes. However, to mention a single compound with potential against all the four dengue virus types, N-isopentylindolinone analog 68 (Fig. 22) is one, and its inhibitory activity (IC50 values) against DENV-1, DENV-2, DENV-3, and DENV-4 is found to be 0.78 μM, 0.16 μM, 0.035 μM, >5.0 μM, and >10 μM, respectively. In the case of individual inhibitory properties, compound 68 is the strongest inhibitor of DENV-1, whereas N-methyl pyrrolidinone derivative 69 is most potent against DENV-2 (EC50 = 0.019 μM). Unfortunately, compound 69 is a weak inhibitor of DENV-1 and DENV-4. Further, N-(N,N-dimethylpropylamine)indolinone scaffold 70 and isopropyl analog 71 have demonstrated the most potent DENV-3 (EC50 = 0.001 μM) and DENV-4 (EC50 = 0.78 μM) inhibitory properties, respectively. Overall observation reveals that indolone N-substitution is beneficial towards DENV-2 and DENV-3 inhibitory activity. Also, the various functional groups exhibited almost similar potencies. Alkyl groups of a particular chain length are conducive to DENV-1 inhibitory property, while DENV-4 inhibitory activity is benefitted by long and bulky alkyl groups. In some derivatives, substitution on the indolinone N-position or pyridinone N-position has resulted in diminished DENV-2 and DENV-3 inhibitory activity.
Fig. 22. Structures of spiro-indolinone scaffolds [reproduced from ref. 60 with permission from American Chemical Society, copyright 2020].

Considering the recently discovered anti-DENV hit molecule, its structural analogs are designed to study the structure–activity relationship in the case of DENV-2 activity.61 In the first series of aniline-modified derivatives, 3,5-dimethoxyaniline analog 72 (Fig. 23) has elicited tenfold higher activity (EC50 = 0.007 μM) compared to the hit molecule. Investigation of dimethoxyaniline derivatives indicates that at least one methoxy group in the meta-position of aniline is necessary for DENV-2 nanomolar activity. The modification of the phenyl part led to interesting facts; in particular, 4-fluorophenyl derivative 73 elicited an attractive DENV-2 property (EC50 = 0.004 μM), higher than that of compound 72. The result infers that the 4-fluoro atom has a greater impact on DENV-2 activity. Furthermore, the indole core is decorated in combination with 3-methoxyaniline/3,5-dimethoxyaniline wherein fluoro and chloroindole analogs have expressed decent activity. A synergistic effect (EC50 = 0.007 μM) is observed for compound 74 comprising 3,5-dimethylaniline and 5-fluoroindole pharmacophores. The individual isomers (72a and 72b) are isolated from the racemic mixture 72 and subsequently evaluated for DENV-2 inhibitory activity. Surprising results are noticed wherein the (+)-enantiomer 72a has exhibited extraordinary activity (EC50 = 0.001 μM), while inferior activity has been noticed for the (−)-enantiomer 72b relative to 72a.
Fig. 23. Illustration of structures of indoles connecting diaryl rings [reproduced from ref. 61 with permission from the American Chemical Society, copyright 2020].

Apart from these, the remarkable inhibitors 72 and 74 are investigated for broad-spectrum DENV inhibitory activities (Table 8); again, compound 72a is found to possess a noteworthy anti-DENV profile. Additionally, oxypropanol analog 75 [(+)-enantiomer] showed the finest DENV-2 inhibitory activity.
Broad-spectrum inhibitory properties of compounds 72a and 74.
| Compd | Anti-DENV activity (μM) | |||
|---|---|---|---|---|
| DENV-1 | DENV-2 | DENV-3 | DENV-4 | |
| 72a | 0.047 | 0.001 | 0.092 | 1.9 |
| 74 | 0.247 | 0.007 | 0.680 | 4.8 |
The compounds 72 and 75 have demonstrated good pharmacokinetic properties. Solubility (48 μM) and cytotoxicity (CC50 = 105 μM) of compound 75 are good compared to compound 73 (solubility = 9 μM, CC50 = 16 μM). These potentials suggest that the structural features of compound 76 are most beneficial for efficient anti-dengue drug discovery.
Based on the enzyme activities of NS5 RdRp polymerase and NS3 protease,62,63 potent DENV inhibitors are identified.64 Amongst the prepared molecules, two indole analogs have been investigated for enzyme inhibitory activities. 4-Hydroxy-3,5-dimethyl-appended 5-chloroindole analog 76 (Fig. 24) displayed the best enzyme inhibitory activity (Table 9) and it is superior to compound 77 which is structurally devoid of a 4-hydroxy group on the phenyl ring and a carbonyl moiety flanking the indole and aryl rings. The results in Table 9 reveal that the compounds 76 and 77 suppress DENV replication by directly inhibiting DENV protease activity. With beneficial (low) cytotoxicity, compound 77 could be an efficient anti-DENV molecule.
Fig. 24. Structures of 5-chloroindole derivatives with decent anti-DENV-2 activity.64.

RNA and enzyme inhibitory activities of compounds 76 and 77.
| Compd | DENV-2 inhibitory activity | ||||
|---|---|---|---|---|---|
| RNA | NS3 | ||||
| CC50 (μM) | EC50 (μM) | SI | Cell-based EC50 (μM) | Enzyme based EC50 (μM) | |
| 76 | 181 | 4.6 | 39.3 | 6.7 | 4.7 |
| 77 | >200 | 7.3 | 27.4 | 7.9 | 6.9 |
The remarkable inhibitor 76 is evaluated for its ability to inhibit DENV in a DENV-infected ICR sucking mouse model. Treatment with compound 76 reduced DENV-induced pathology, including ruffled fur, anorexia, severe paralysis, and lethargy in DENV-infected mice within 4–6 days post-infection. After exhibiting all these potencies, compound 76 could inhibit DENV replication and represent prototypical DAA molecules.
Natural products from marine microorganisms have been isolated and found to possess anti-DENV activity.65 In particular, the isolates (scequinadoline 78, quinadoline A 79, and quinadoline E 80, Fig. 25) from Dichotomomyces cejpii F31-1 culture are explored for their ability to inhibit DENV-2.66 In the initial cytotoxicity studies, no significant cytotoxicity is observed in any of the isolates. Compounds 79 and 80 have not shown an effect on DENV virus production, while treatment with compound 78 revealed a significant reduction in virus output (approximately 40%). However, compounds 78–80 have not resulted in the reduction of infection level which is the same as the control.
Fig. 25. Chemical structures of dihydroindole-containing natural products.66.

Natural and synthetic β-carboline analogs are explored for their anti-DENV properties.67 Most of the evaluated molecules have expressed higher cytotoxicity and are unfavorable. Despite these, harmol 81 (Fig. 26) and 9-methylharmine 82 have bestowed good anti-DENV-2 activity (Table 10); particularly harmol 81 has demonstrated a greater selectivity index. Good DENV-2 inhibitory properties of compounds 81 and 82 inspired to check the ability to inhibit other DENV serotypes wherein again compound 81 is found to be a better anti-dengue molecule towards DENV-1 and DENV-3 and moderate activity towards DENV-4.
Fig. 26. Depiction of structures of harmol and 9-methylharmine [reproduced from ref. 67 with permission from Elsevier, copyright 2020].

DENV-2 inhibitory properties of harmol and 9-methylharmine.
| Compd | DENV-2 inhibitory activity | DENV-1 | DENV-3 | DENV-4 | ||
|---|---|---|---|---|---|---|
| CC50 (μM) | EC50 (μM) | SI | EC50 (μM) | |||
| 81 | 203.3 | 3.3 | 61.3 | 10.1 | 7.7 | 21.1 |
| 82 | 178.6 | 3.2 | 56.2 | 49.5 | 33.7 | 16.3 |
Additionally, further research on the antiviral properties of compound 82 indicates that it lacks a direct inactivating effect on DENV-2 particles and no viricidal activity is observed. Compound 82 has no significant effect on viral RNA synthesis compared to the reference ribavirin. In addition, 9N-methylharmine-treated cultures indicate that the ratio between intracellular and extracellular viral genomes is fourfold higher compared to control cells, while the ratio between viral genomes and infectious viral particles in the extracellular medium is 3.6-fold higher than that from the control supernatants. The results infer that the treatment with compound 82 does not affect macromolecular synthesis but impairs the maturation and release of infectious virus into the extracellular space.
1.1.4. Anti-HSV activity
HSV stands for herpes simplex virus comprising HSV-1 (human alphaherpesvirus 1) and HSV-2 (human alphaherpesvirus 2) members belonging to the human Herpesviridae family. They are responsible for most of the cold sore (HSV-1) and genital herpes (HSV-2)68,69 infections which are contagious. About 67% of the world population under the age of fifty has been affected by HSV-1. In general, HSV-2 is one of the most general sexually transmitted infections.70 HSV-2 may also lead to a higher risk of acquiring HIV.71 Since HSV-1 is reported to possess a nervous system damaging ability, it may be linked to a greater chance of developing Alzheimer's disease. HSV-1 and HSV-2 particles comprise at least 74 genes in their genomes72 which encode for a variety of proteins involved in forming the capsid, tegument, and envelope of the virus in addition to controlling its replication and infectivity.
HSV causes lifelong infections as it cannot be eradicated from the body. The treatment includes antiviral drugs that interfere with viral replication. However, extensive use of antiherpetic drugs would lead to the development of drug resistance and hence new drugs have to be investigated.
Given the pharmacological importance of the medicinal plant Peganum harmala, and a wide range of pharmacological effects of alkaloids,73–75 the organic extracts of P. harmala are evaluated for anti-HSV-2 activity.76 Methanol extracts exhibited lower cytotoxicity, while 100% viral inhibition is observed in the case of anti-HSV-2 activity. However, the corresponding IC50 value is reported to be a weak activity (161 μg mL−1). The active molecule responsible for the potent activity is identified as compound 83 (Fig. 27). Other extracts are completely inactive, while results of the investigation of the mode of action of methanol seed extract suggest that the extract has inhibited virus replication completely by direct contact (viricidal effect) and also during and after penetration. In addition, the combined effect of compound 83 with the standard antiviral agent acyclovir is explored in HSV-2 replication. The result suggested a synergistic effect towards the virus with a CI value of 0.5 and the effect is interesting as the two compounds have two dissimilar modes of action. The combined anti-HSV-2 activity is higher than that of acyclovir alone.
Fig. 27. Illustration of the active molecule of Peganum harmala83 and dihydropyridoindole derivative (harmaline) 84 [reproduced from ref. 76 and 77 with permission from Elsevier, copyright 2020].

Potent anti-HSV-2 alkaloid harmaline is extracted from O. nicobarica and its anti-HSV properties are evaluated.76,77 The isolated alkaloid harmaline 84 (Fig. 27) is tested on HSV-1 strains wherein potent anti-HSV activity (EC50 = 1.1 μM, CC50 = 30 μM) is rendered and the activity is comparatively better than that of acyclovir (EC50 = 2.1 μM). The time-of-addition and removal assay is performed to identify the possible stage of inhibition of the viral life cycle and results suggest that harmaline has exerted an inhibitory effect during 0–4 h post-infection. The attachment and penetration assays indicate that harmaline could not block the virus entry and it might interfere with the viral early replication process. HCF1-dependent recruitment of LSD1 plays a crucial role in the commencement of HSV infection. Harmaline-treated cells have demonstrated an appreciable reduction of the association between HCF1 and LSD1, confirming its interference with the recruitment of LSD1 by HCF1.
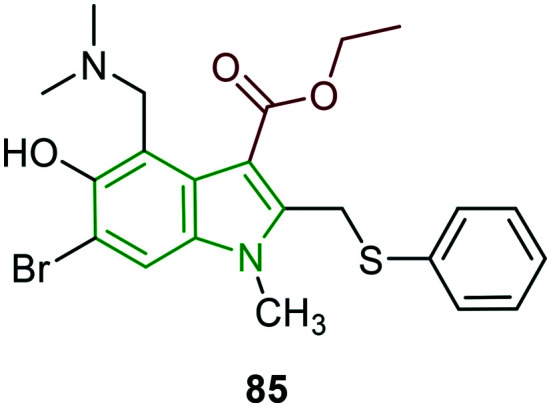
The anti-HSV-1 effects of arbidol 85 (Fig. 28), an indole-based hydrophobic molecule, are investigated.78 In initial cytotoxicity studies, arbidol showed a CC50 value of 30.09 μM. The inhibitory activity of arbidol on HSV-1 replication is reported to be remarkable (EC50 = 10.49 μM). Further, when infected cells are allowed for arbidol treatment, the progeny virus is reduced by 10 000-fold compared to the virus control group with an EC50 value of 4.40 μM. Even at a low arbidol concentration (0.625 μM), plaque numbers are reduced by about 20%, while no reduction of progeny virus is observed, indicating that arbidol at the low concentration could inhibit the formation of virus plaques. The time-of-addition and time-of-removal assays suggest the inhibitory effects when arbidol is added before 12 h p.i, while inactivation assay, attachment assay, and penetration assay infer that arbidol failed to directly kill and inactivate HSV-1 and block HSV-1 entry into Hep-2 cells. In addition, tests performed to check the inhibition of expression of HSV-1 immediate-early, early and late genes reveal that arbidol could strongly affect the expression of IE, E, and L genes during HSV-1 infection. Evaluation of the anti-HSV-1 activity of arbidol in cutaneous HSV-1 infected guinea pig models demonstrates its effectiveness in reducing the severity and duration of lesions in the guinea pig model. In this activity, the thickness of the prickle layer of viral infected skin (130.1 μm) is reduced to 42.9 μm with high-dose arbidol treatment compared to that of acyclovir treatment (31.6 μm).
Fig. 28. Chemical structure of arbidol [reproduced from ref. 78 with permission from Elsevier, copyright 2020].
Based on preliminary results of arbidol derivatives as potent anti-HCV agents, the structural derivatives of arbidol are evaluated for inhibition of HaCat cell growth at different stages of HSV-1 replication.79 Cytotoxic analysis has not revealed any toxic effects on the keratinocyte viability. Plaque assays for arbidol show that arbidol inhibited HSV-1 with an IC50 value of 12 μM when added during HSV-1 infection, while dose-dependent inhibition is observed with an IC50 value of 3 μM when HaCat cells are pre-incubated with arbidol. At a drug concentration of 3 μM, only compounds 86–89 (Fig. 29) could reduce the virus replication compared to HSV-1-infected control. A significant reduction of plaque numbers is observed in the presence of compounds 88 (35%) and 89 (36%) after only 24 h of infection. However, arbidol and compound 87 maintained the inhibitory effect up to 48 h. The significant improvement in the inhibitory activity of compounds 87 and 89 is attributed to the pyrrolidinyl substituent. Arbidol and its derivatives are tested for VP16 and 1CP27 viral proteins in cell lysates collected from HaCat cells at 3 and 6 h post-infection wherein a significant reduction in levels of VP16 protein is noticed after 3 h of infection, while the reduction is more marked after 6 h of treatment. Additionally, arbidol analogs have demonstrated a more intense effect compared to arbidol alone. In the case of cytokine expression inhibition effects, arbidol and its derivatives strongly enhanced expression of the IL-6 gene evaluated in HaCat cells 6 h post-infection. HaCat cells after 6 h of treatment with arbidol or its derivatives showed a marked enhancement of TNF-α and TNF-β.
Fig. 29. Depiction of structures of arbidol derivatives [reproduced from ref. 79 with permission from Microbial Society, copyright 2020].

To study the HSV-2 inhibitory potency of arbidol in vitro and in vivo, the HCE cell model is utilized.80 With an EC50 of 3.386 μM and SI of 9.92, arbidol is reported to be a suitable antiviral agent. The virus yield reduction assay indicates a significant reduction of the progeny virus yield. Also, IFA and western blotting tests demonstrate dramatically lowered HSV-2 gD content in arbidol-treated cells which suggests down-regulation of HSV-2 RNA level and protein, while the binding assay infers that arbidol blocks the entry of the virus but has no effect on virus binding. The results of the activity assay performed to identify the inhibition of phases in the HSV-2 life cycle show that arbidol has remarkable effects in the earlier time of the HSV-2 cycle in viral DNA synthesis; however, it failed to kill the virus directly. Further, the influence of drugs in the viral replication cycle is explored wherein a significant reduction of ICP8 in arbidol-treated cells is observed and the production of E genes of HSV-2 is inhibited. The performance of arbidol in blocking the TLR/NF-kB pathways during the infection demonstrates the down-regulation of TLRs. Additionally, the protein level of NF-kB is alleviated, revealing that arbidol inhibited HSV-2-induced NF-kB activation. Further, significant down-regulation of TNF-α and IL-6 has been exhibited by arbidol treatment compared to control.
Derivatives of fused pyridoindole appended with thiazolones are designed to investigate the anti-HSV-1 inhibitory activity.81 In this activity, thiazolone-tethered fused-pyridoindoles excelled. In particular, 4-N-dimethyl phenyl analog 91 (Fig. 30) elicited excellent activity (EC50 = 0.80 μM). With lower cytotoxicity of 467 μM and SI of 592.5, compound 91 is a promising anti-HSV-1 agent. Slightly diminished inhibitory potencies have been noticed for the phenyl and 3-nitrophenyl scaffolds 90 (EC50 = 2.15 μM) and 92 (EC50 = 2.02 μM), respectively. An almost similar selectivity index is exhibited by compound 92, while the SI of compound 90 is reduced and its antiviral activity is also low. Compounds 91 and 92, possessing good selectivity indices, could be developed into potential anti-HSV-1 molecules.
Fig. 30. Structures of thiazolidinone-appending pyridoindole derivatives [reproduced from ref. 81 with permission from Elsevier, copyright 2020].

Pyridoindole scaffolds have again come in the limelight upon exhibiting decent anti-HSV-1 inhibitory properties.82 Out of the 21 derivatives prepared, 11 molecules have rendered good antiviral activity. Because of higher cytotoxicity, most of the derivatives have not emerged as the finest anti-HSV agents. Nonetheless, compounds 93–95 (Fig. 31) expressed striking anti-HSV-1 activity avoiding cytopathic effects (Table 11). The N-methyl analog 93 exhibited the strongest anti-HSV activity with favorable cytotoxic effect and in turn good selectivity index. The activity is consistent with percentage inhibition. Methylation of fused-pyridine rings resulted in higher cytotoxicity, although the EC50 value is almost similar, suggesting lower antiviral potency. Also, in the case of compound 95 with a 6-methoxy moiety, the EC50 value is not in agreement with percentage inhibition. The test performed to investigate the mode of action indicates that the selected compounds are not viricidal and could not interfere with attachment or penetration of HSV-1. Western blot studies reveal that early and late protein expression is markedly suppressed. Additionally, compounds 93–95 restricted HSV-1 ICP0 localization to the nucleus during later stages of infection, probably affecting its functionality at the cytoplasm. This might inhibit antiviral signaling and subsequently inhibit viral replication.
Fig. 31. Structures of substituted pyridoindole analogs [reproduced from ref. 82 with permission from Elsevier, copyright 2020].

HSV inhibitory properties of pyridoindole analogs.
| Compd | Anti-HSV activity | % inhibition | ||
|---|---|---|---|---|
| CC50 (μM) | EC50 (μM) | SI | ||
| 93 | 435 | 4.9 | 88.8 | 83 |
| 94 | 237 | 5.9 | 40.2 | 69 |
| 95 | 137 | 19.5 | 7.0 | 84 |
1.1.5. Anti-influenza activity
An infectious disease, influenza, or flu, is caused by the influenza virus and symptoms may be mild to severe. Severe conditions of flu would result in viral pneumonia, secondary bacterial pneumonia, sinus infections,83etc. The influenza virus spreads usually through the air from coughs or sneezes. Based on antibody responses,84 to date, four types of influenza viruses have been reported, namely, type A, type B, type C, and type D; amongst these, type D has not been reported to infect humans. Devastating outbreaks of type A have occurred and given rise to a human influenza pandemic. Globally, influenza is responsible for 3–5 million severe complications and approximately 250 000 to 500 000 deaths occur every year. The serotypes of type A confirmed are H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9N2, H7N2, H7N3, H10N7, H7N9, and H6N1. Out of these serotypes, H7N9 is rated as having the highest pandemic potential among the type A subtypes. Type B is the only influenza type that infects exclusively humans;84 however, it is less common than type A. The overall structure of the various influenza viruses is almost similar85 and it applies to their composition also. They are made up of a viral envelope comprising the glycoproteins hemagglutinin and neuraminidase wrapped around a central core. The central core involves the viral RNA genome which unusually for a virus is a set of seven or eight pieces. In the case of influenza A, the genes present on the viral genome code for the 11 proteins HA, NA, NP, M1, M2, NS1, NS2, PA, PB1, PB1-F2, and PB2.86 Despite the high mutation rate of the virus, the influenza vaccine confers protection no more than a few years. Antibiotics are ineffective towards influenza infection, while drugs may be effective if given early. Two classes of antivirals are prevalent, namely, neuraminidase inhibitors and M2 inhibitors.87,88
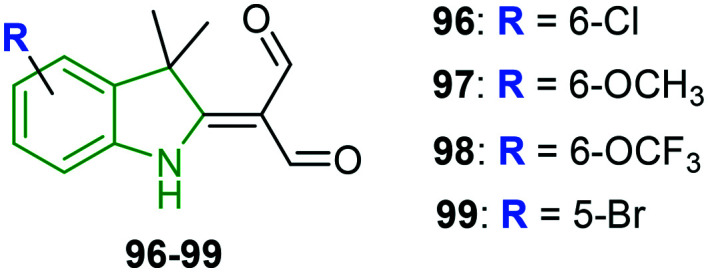
A set of novel indole derivatives are prepared and evaluated for anti-influenza properties.89 The four new synthetic indole derivatives 96–99 (Fig. 32) are checked for cytotoxicity wherein the derivatives are found to be non-toxic (IC50 = 266–442 μM). Pretreatment of A549-PB1 cells with the indole analogs 98 and 99 at a concentration of 50 μM for 4 h has not shown any reduction of eGFP-positive cells. In the case of compound 96 a slight reduction of eGFP-positive cells is noticed, while moderate reduction has been exhibited by the 6-methoxy analog 97. Since the antiviral activity of compound 97 is not due to its cytotoxicity as the cell proliferation is unaffected at 50 μM, it is chosen for further testing. In addition, the antiviral activity of compounds 97–99 is tested on vesicular stomatitis virus wherein all compounds exhibited different levels of reduction of eGFP-positive cells, with compound 96 resulting in the strongest reduction. The effect of compound 97 on replication of IAV is investigated through IAV polymerase activity. In this activity, no significant difference in the vRNA levels including NP, NS1, eGFP, and M1 is detected, suggesting that compound 97 is unable to inhibit the entry of IAV. However, it is demonstrated to inhibit the replication of IAV as the expression of vRNAs is reduced at 6 h p.i. The absence of a significant difference in the expression level of the housekeeping genes (SDHA, β-actin, and 18S) indicates that compound 97 does not inhibit the host cell generation machinery or RNA processing. A great reduction of viral-induced expression of IFNβ, IFIT2, HIL6, and IP10 in compound 97-treated A549-PB1-cells infers that it is incapable of protecting the host cells from IAV infection by enhancing the antiviral response. Further, in the presence of compound 97, IAV-induced phosphorylation of STAT1 is reduced, revealing that compound 97 could inhibit IAV-induced activation of IRF3 and STAT1.
Fig. 32. Depiction of chemical structures of 3,3-dimethylindole derivatives.89.
A series of novel 5-hydroxy-2-aminomethyl-1H-indole-3-carboxylic acids and their analogs are synthesized and the corresponding antiviral effects are screened.90 The cytotoxicity of the prepared molecules (chosen for antiviral activity) is much lower compared to that of arbidol. The 6-fluoroindole derivative connected to N,N-dimethyl amine 102 (Fig. 33) exerted threefold greater activity compared to arbidol and a better therapeutic index (sevenfold higher compared to arbidol) (Table 12). The replacement of the 6-fluoro group with a 3-pyridyl ring resulted in a diminished activity which is evident in compounds 100 and 101. In particular, the C-2 pyrrolidine scaffold 101 exhibited moderate activity with a lower therapeutic index, whereas the N,N-dimethyl amine analog 100 possesses an almost similar activity but a twofold higher therapeutic index than that of arbidol. Furthermore, for compound 102 at a dose of 25 mg kg−1 per day given i.v. and p.o., the mean survival times in mice are 11.4 and 8.3 days, respectively, the activity is comparable with arbidol (i.v. = 5.5 and p.o. = 7.3 days). However, at a higher dose, the survival time is reduced, indicating a higher degree of toxicity. The results show that compound 102 is highly effective in the prophylaxis and treatment of mice with influenza.
Fig. 33. Structures of indole-3-carboxylic acid analogs [reproduced from ref. 90 with permission from springer, copyright 2020].

Anti-influenza effects of indole-3-carboxylic acid analogs.
| Compd | Anti-influenza activity (μM) | ||
|---|---|---|---|
| EC50 | CC50 | TI50 | |
| 100 | 12.7 | 188 | 14.8 |
| 101 | 18.6 | 110 | 5.9 |
| 102 | 4.3 | 151 | 35.1 |
| Arbidol | 12.9 | 97 | 7.5 |
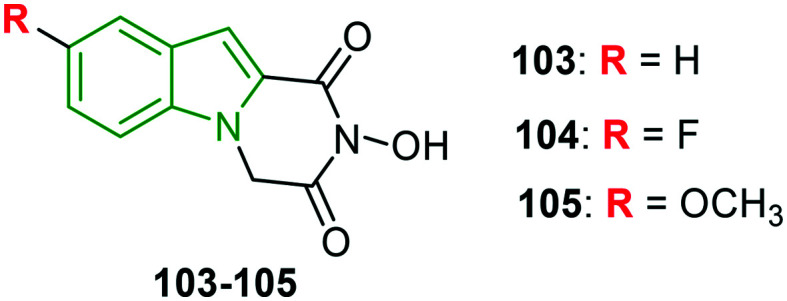
Indole–flutimide heterocycles are synthesized and evaluated for influenza PAN inhibitory effects.91 Except for a few synthesized molecules, all the other derivatives are reported to be significant inhibitors of influenza PAN in an enzymatic assay. Amongst these, compound 103 (Fig. 34) has emerged as the most active PAN inhibitor with an IC50 value of 12.7 μM, while 5-fluoroindole analog 104 is also a good inhibitor of PA endonuclease (IC50 = 17.3 μM) amongst the substituted indole derivatives. This indicates that fluorine is more preferable when compared to bulkier halogens, which is because of optimal van der Waals interactions with the secondary N-terminal cavity of the PA active site. Apart from this, methoxy analog 105 has yielded the best activity (EC50 = 48 μM) in the influenza virus vRNP assay.
Fig. 34. Illustration of structures of indole–flutimide derivatives.91.
Indole-substituted spirothiazolidinones are designed as superior inhibitors of HA-mediated fusion.92 Influenza A inhibitory activity is evaluated in terms of cytopathic effect (CPT) and MTS-based cell viability assay. Almost similar inhibitory potencies are observed in both assays. The spirothiazolidinone analogs lacking a methyl motif on the thiazolidinone ring have failed to render good antiviral activity, while a few methyl-containing spiro derivatives have exhibited the finest activity. Here, compound 106 (Fig. 35) possessing a plane cyclohexane ring showed decent activity (Table 13). Approximately threefold stronger antiviral activity is noticed for compound 107 compared to 106 wherein the spiro-cyclohexane ring is substituted with a methyl group at the 2-position. Further, the shift of the methyl position from the 2- to the 3-position has led to twofold abated activity. However, the replacement of ethyl with the methyl group at the 3-position has resulted in extraordinary activity, suggesting that increased alkyl chain length has a positive impact on anti-influenza A activity. Having said that, still, larger groups have demonstrated deteriorated activity. The same trend in inhibitory potencies is observed in the case of MTS-based cell viability. In the case of the inhibitory effect on HA-mediated membrane fusion in HA-WT, compounds 106–109 are reported as beneficial molecules with EC50 values of 0.88–2.1 μM which are comparable with that of the reference compound. Again, compound 109 has incarnated as the most potent molecule (EC50 = 0.88 μM) compared to a reference (EC50 = 1.1 μM), in agreement with influenza A activity. In the case of HA-E572K and HA-D1122N strains, weak inhibitory values are put up.
Fig. 35. Chemical structures of carboxamide-tethered indole-spirothiazolidinones [reproduced from ref. 92 with permission from Elsevier, copyright 2020].

Anti-influenza A inhibitory effects of indole-substituted spirothiazolidinones.
| Compd | Influenza A inhibitory activity (EC50, μM) | |
|---|---|---|
| CPE | MTS | |
| 106 | 0.085 | 0.063 |
| 107 | 0.031 | 0.023 |
| 108 | 0.063 | 0.045 |
| 109 | 0.0012 | 0.0007 |
| Amantadine | 3.8 | 1.2 |
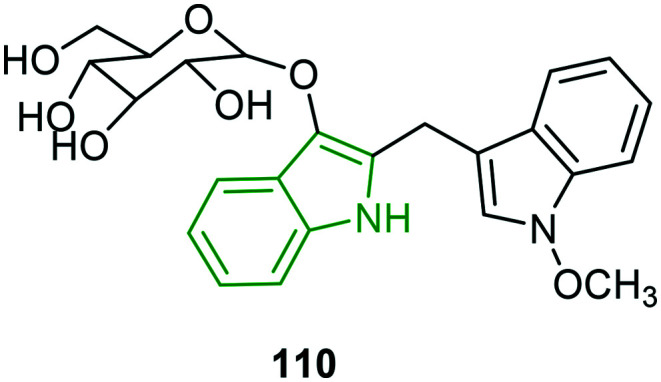
In continuation of the previous studies of the extraction of new alkaloids,93,94 seven new bisindole glucosides or isatindigobisindolosides are isolated and subsequently explored for antiviral properties.95 In the evaluation of anti-influenza activity, moderate to poor potencies are observed compared to ribavarin wherein the N-methoxy analog 110 (Fig. 36) is reported to be a moderate inhibitor (IC50 = 12.6 μM). The antiviral potencies of these molecules suggest that they can be developed into efficient biomimetics.
Fig. 36. Illustration of the structure of a bisindole derivative [reproduced from ref. 95 with permission from Tailor & Francis, copyright 2020].
1.2. Discussion
Having demonstrated the potential pharmacological properties of indole derivatives, the in vitro and in vivo antiviral potencies of indole scaffolds reported recently are discussed briefly. The whole review work is categorized based on the type of virus being inhibited. In the anti-HCV activity, MK-8742 derivatives have been demonstrated as the elite inhibitors; further derivatization on the indole or phenyl ring of the tetracyclic part has not yielded fruitful results. In continuation, the amino acid side chain is decorated with cyclic/acyclic alkyl moieties to enhance the anti-HCV effects. Additionally, a non-zwitterionic indole derivative appended with an alkyl bridged piperazine carboxamide analog 14 has exhibited promising activity. In addition, it is worth mentioning that furan-fused tricyclic indole analogs have rendered nanomolar inhibitory activity. Overall the MK-8742 derivatives have the immense potentiality to develop into future anti-HCV molecules. Anti-HIV investigation reveals that indolyl-glyoxamide derivatives are very good at the inhibitory activity. Astonishingly, incredible results are elicited by the indolearylsulfone (IAS) analogs. These derivatives have also proved to be functional towards the mutant strains. The legacy of IASs towards anti-HIV is continued for other scaffolds wherein the amide side chain is decorated with heteroaryl rings, suggesting that the IASs are the most promising pharmacophores and could be potential anti-HIV candidates, whereas only indole-3-carbonyl scaffolds connected with substituted anilines exhibited the finest anti-dengue activity towards all serotypes, and other derivatives are found to be moderate inhibitors. When it comes to herpes simplex viruses, moderate to decent inhibitory potencies are noticed. Also, the anti-HSV molecules are explored for related protease inhibitory and virus replication inhibitory effects. Most of the compounds have been demonstrated to inhibit HSV through various mechanisms. Some of the indole analogs showed good anti-influenza activity along with influenza-related enzyme inhibitory properties.
Conclusion
In this review, the in vitro and in vivo antiviral properties of various indole derivatives reported recently are discussed. The classification of antiviral agents is made based on the type of virus being inhibited. In addition to the MIC/IC50 values of potent compounds, the structure–activity relationship is established for evaluated derivatives, which helps future researchers to design efficient therapeutic agents. In addition, the pharmacokinetic and pharmacodynamic properties are also considered to check the clinical efficacy of the antiviral drug. The review work indicates that indole is the most promising heterocycle available for antiviral drug discovery and development against drug-resistant viral strains. Overall, the review infers that the MK-8742 derivatives and indole aryl sulfonic acids have attracted much attention by their significant in vitro and in vivo antiviral properties. Considering these derivatives as lead molecules, further derivatization has to be investigated towards the discovery of efficient antiviral molecules.
Conflicts of interest
The author declares there is no potential conflict of interest.
Biography
Atukuri Dorababu.

Dr. Atukuri Dorababu obtained his MSc (2010) and PhD (2017) in chemistry from the Karnatak University, Dharwad. He worked as a research associate in Syngene, Bengaluru, for a short period. Later, he worked as a lecturer for five years (2013–2017) in Govt. Pre-University College, Belgaum. Meanwhile, he has been appointed as an assistant professor in SRMPP GFGC, Huvinahadagali, in 2017 and is presently working there in the same position. He has around 19 publications in various reputed international journals. His research focused on drug discovery, anti-microbial activity, anticancer activity and natural product extraction and their pharmacological activity.
References
- Kanwal A. Ahmad M. Aslam S. Naqvi S. A. R. Saif M. J. Pharm. Chem. J. 2019;53:179–187. doi: 10.1007/s11094-019-01976-3. [DOI] [Google Scholar]
- Dittmer A. Woskobojnik I. Adfeldt R. Drach J. C. Townsend L. B. Voigt S. Bogner E. Antiviral Res. 2017;137:102–107. doi: 10.1016/j.antiviral.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Osman H. Yusufzai S. K. Khan M. S. Abd Razik B. M. Sulaiman O. Mohamad S. Gansau J. A. Ezzat M. O. Parumasivam T. Hassan M. Z. J. Mol. Struct. 2018;1166:147–154. doi: 10.1016/j.molstruc.2018.04.031. [DOI] [Google Scholar]
- Shen Y. F. Liu L. Feng C. Z. Hu Y. Chen C. Wang G. X. Zhu B. Fish Shellfish Immunol. 2018;81:57–66. doi: 10.1016/j.fsi.2018.07.005. [DOI] [PubMed] [Google Scholar]
- Detsi A. Kontogiorgis C. Hadjipavlou-Litina D. Expert Opin. Ther. Pat. 2017;27:1201–1226. doi: 10.1080/13543776.2017.1360284. [DOI] [PubMed] [Google Scholar]
- Caruso A. Ceramella J. Iacopetta D. Saturnino C. Mauro M. V. Bruno R. Aquaro S. Sinicropi M. S. Molecules. 2019;24:1912. doi: 10.3390/molecules24101912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Wu Y. Sun L. Gu Y. Hu L. Eur. J. Med. Chem. 2020;191:112181. doi: 10.1016/j.ejmech.2020.112181. [DOI] [PubMed] [Google Scholar]
- Wang P. Y. Shao W. B. Xue H. T. Fang H. S. Zhou J. Wu Z. B. Song B. A. Yang S. Res. Chem. Intermed. 2017;43:6115–6130. doi: 10.1007/s11164-017-2980-x. [DOI] [Google Scholar]
- Gan X. Hu D. Chen Z. Wang Y. Song B. Bioorg. Med. Chem. Lett. 2017;27:4298–4301. doi: 10.1016/j.bmcl.2017.08.038. [DOI] [PubMed] [Google Scholar]
- Gürsoy E. Dincel E. D. Naesens L. Güzeldemirci N. U. Bioorg. Chem. 2020;95:103496. doi: 10.1016/j.bioorg.2019.103496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamut A. Gjorgjieva M. Naesens L. Liekens S. Lillsunde K. E. Tammela P. Kikelj D. Tomašič T. Bioorg. Chem. 2020;98:103733. doi: 10.1016/j.bioorg.2020.103733. [DOI] [PubMed] [Google Scholar]
- Wang M. Rakesh K. P. Leng J. Fang W. Y. Ravindar L. Channe Gowda D. Qin H. L. Bioorg. Chem. 2018;76:113–129. doi: 10.1016/j.bioorg.2017.11.007. [DOI] [PubMed] [Google Scholar]
- Fang W. Y. Ravindar L. Rakesh K. P. Manukumar H. M. Shantharam C. S. Alharbi N. S. Qin H. L. Eur. J. Med. Chem. 2019;173:117–153. doi: 10.1016/j.ejmech.2019.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C. Rakesh K. P. Ravidar L. Fang W. Y. Qin H. L. Eur. J. Med. Chem. 2019;162:679–734. doi: 10.1016/j.ejmech.2018.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moku B. Ravindar L. Rakesh K. P. Qin H.-L. Bioorg. Chem. 2019;86:513–537. doi: 10.1016/j.bioorg.2019.02.030. [DOI] [PubMed] [Google Scholar]
- Ju W. Yang S. Feng S. Wang Q. Liu S. Xing H. Xie W. Zhu L. Cheng J. Virol. J. 2015;12:109. doi: 10.1186/s12985-015-0341-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes J. Thompson A. Hepatic Med. 2015;2015:51–70. doi: 10.2147/HMER.S55864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal G. Mondal D. Warshel A. J. Phys. Chem. B. 2017;121:6831–6840. doi: 10.1021/acs.jpcb.7b04562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbo M. C. Cento V. di Maio V. C. Howe A. Y. M. Garcia F. Perno C. F. Ceccherini-Silberstein F. Drug Resist. Updates. 2018;37:17–39. doi: 10.1016/j.drup.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Fauvelle C. Lepiller Q. Felmlee D. J. Fofana I. Habersetzer F. Stoll-Keller F. Baumert T. F. Fafi-Kremer S. Microb. Pathog. 2013;58:66–72. doi: 10.1016/j.micpath.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Law L. M. J. Landi A. Magee W. C. Lorne Tyrrell D. Houghton M. Emerging Microbes Infect. 2013;2:79–84. doi: 10.1038/emi.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilby K. J. Partovi N. Ford J. A. E. Greanya E. D. Yoshida E. M. Can. J. Gastroenterol. Hepatol. 2012;26:205–210. doi: 10.1155/2012/751057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T. J. Ghany M. G. N. Engl. J. Med. 2013;368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong A. D. ACS Med. Chem. Lett. 2014;5:214–220. doi: 10.1021/ml500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreev I. A. Manvar D. Barreca M. L. Belov D. S. Basu A. Sweeney N. L. Ratmanova N. K. Lukyanenko E. R. Manfroni G. Cecchetti V. Frick D. N. Altieri A. Kaushik-Basu N. Kurkin A. V. Eur. J. Med. Chem. 2015;96:250–258. doi: 10.1016/j.ejmech.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W. Coburn C. A. Nair A. G. Wong M. Tong L. Dwyer M. P. Hu B. Zhong B. Hao J. Yang D. Y. Selyutin O. Jiang Y. Rosenblum S. B. Kim S. H. Lavey B. J. Zhou G. Rizvi R. Shankar B. B. Zeng Q. Chen L. Agrawal S. Carr D. Rokosz L. Liu R. Curry S. McMonagle P. Ingravallo P. Lahser F. Asante-Appiah E. Nomeir A. Kozlowski J. A. Bioorg. Med. Chem. Lett. 2016;26:3800–3805. doi: 10.1016/j.bmcl.2016.05.041. [DOI] [PubMed] [Google Scholar]
- Yu W. Zhou G. Coburn C. A. Zeng Q. Tong L. Dwyer M. P. Hu B. Zhong B. Hao J. Ji T. Zan S. Chen L. Mazzola R. Kim J. H. Sha D. Selyutin O. Rosenblum S. B. Lavey B. Nair A. G. Heon Kim S. Keertikar K. M. Rokosz L. Agrawal S. Liu R. Xia E. Zhai Y. Curry S. McMonagle P. Ingravallo P. Asante-Appiah E. Chen S. Kozlowski J. A. Bioorg. Med. Chem. Lett. 2016;26:4851–4856. doi: 10.1016/j.bmcl.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Zhao F. Liu N. Zhan P. Jiang X. Liu X. Eur. J. Med. Chem. 2015;94:218–228. doi: 10.1016/j.ejmech.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Jin G. Lee S. Choi M. Son S. Kim G. W. Oh J. W. Lee C. Lee K. Eur. J. Med. Chem. 2014;75:413–425. doi: 10.1016/j.ejmech.2014.01.062. [DOI] [PubMed] [Google Scholar]
- Lee S. Jin G. Kim D. Son S. Lee K. Lee C. Acta Virol. 2015;59:64–77. doi: 10.4149/av_2015_01_64. [DOI] [PubMed] [Google Scholar]
- Son S. Kim D. Lee S. Jin G. Park J. A. Han H. K. Lee K. Lee C. Bull. Korean Chem. Soc. 2015;36:88–98. doi: 10.1002/bkcs.10021. [DOI] [Google Scholar]
- Han Z. Liang X. Wang Y. Qing J. Cao L. Shang L. Yin Z. Eur. J. Med. Chem. 2016;116:147–155. doi: 10.1016/j.ejmech.2016.03.062. [DOI] [PubMed] [Google Scholar]
- Konč J. Tichý M. Pohl R. Hodek J. Džubák P. Hajdúch M. Hocek M. MedChemComm. 2017;8:1856–1862. doi: 10.1039/C7MD00319F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer M. P. Keertikar K. M. Chen L. Tong L. Selyutin O. Nair A. G. Yu W. Zhou G. Lavey B. J. Yang D. Y. Wong M. Kim S. H. Coburn C. A. Rosenblum S. B. Zeng Q. Jiang Y. Shankar B. B. Rizvi R. Nomeir A. A. Liu R. Agrawal S. Xia E. Kong R. Zhai Y. Ingravallo P. Asante-Appiah E. Kozlowski J. A. Bioorg. Med. Chem. Lett. 2016;26:4106–4111. doi: 10.1016/j.bmcl.2016.06.063. [DOI] [PubMed] [Google Scholar]
- Venkatraman S. Velazquez F. Gavalas S. Wu W. Chen K. X. Nair A. G. Bennett F. Huang Y. Pinto P. Jiang Y. Selyutin O. Vibulbhan B. Zeng Q. Lesburg C. Duca J. Heimark L. Huang H. C. Agrawal S. Jiang C. K. Ferrari E. Li C. Kozlowski J. Rosenblum S. Shih N. Y. George Njoroge F. Bioorg. Med. Chem. 2014;22:447–458. doi: 10.1016/j.bmc.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Douek D. C. Roederer M. Koup R. A. Annu. Rev. Med. 2009;60:471–484. doi: 10.1146/annurev.med.60.041807.123549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell M. K. Benková K. Selinger P. Dogo'i M. Luňáčková I. K. Koutníková H. La'tíková J. Roubíčková A. Špùrková Z. Laclová L. Eis V. Šach J. Heneberg P. PLoS One. 2016;11:e0162704. doi: 10.1371/journal.pone.0162704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham A. L. Donaghy H. Harman A. N. Kim M. Turville S. G. Curr. Opin. Microbiol. 2010;13:524–529. doi: 10.1016/j.mib.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Siliciano R. F. Greene W. C. Cold Spring Harbor Perspect. Med. 2011;1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman D. D. Antiviral Res. 2006;71:117–121. doi: 10.1016/j.antiviral.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Schols D. Ruchko E. A. Lavrenov S. N. Kachala V. V. Nawrozkij M. B. Babushkin A. S. Chem. Heterocycl. Compd. 2015;51:978–983. doi: 10.1007/s10593-016-1807-9. [DOI] [Google Scholar]
- Saturnino C. Grande F. Aquaro S. Caruso A. Iacopetta D. Bonomo M. G. Longo P. Schols D. Sinicropi M. S. Molecules. 2018;23:286. doi: 10.3390/molecules23020286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. Wallace O. B. Zhang Z. Fang H. Yang Z. Robinson B. A. Spicer T. P. Gong Y. F. Blair W. S. Shi P. Y. Lin P. F. Deshpande M. Meanwell N. A. Kadow J. F. Bioorg. Med. Chem. Lett. 2019;29:1423–1429. doi: 10.1016/j.bmcl.2019.03.018. [DOI] [PubMed] [Google Scholar]
- Esposito F. Sechi M. Pala N. Sanna A. Koneru P. C. Kvaratskhelia M. Naesens L. Corona A. Grandi N. di Santo R. D'Amore V. M. di Leva F. S. Novellino E. Cosconati S. Tramontano E. Antiviral Res. 2020;174:104671. doi: 10.1016/j.antiviral.2019.104671. [DOI] [PubMed] [Google Scholar]
- Müller K. Faeh C. Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Hagmann W. K. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Sanna G. Madeddu S. Giliberti G. Piras S. Struga M. Wrzosek M. Kubiak-Tomaszewska G. Koziol A. E. Savchenko O. Lis T. Stefanska J. Tomaszewski P. Skrzycki M. Szulczyk D. Molecules. 2018;23:2554. doi: 10.3390/molecules23102554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. Yadav P. Urvashi Tandon V. ChemistrySelect. 2016;1:5471–5478. doi: 10.1002/slct.201601024. [DOI] [Google Scholar]
- Piscitelli F. Coluccia A. Brancale A. la Regina G. Sansone A. Giordano C. Balzarini J. Maga G. Zanoli S. Samuele A. Cirilli R. la Torre F. Lavecchia A. Novellino E. Silvestri R. J. Med. Chem. 2009;52:1922–1934. doi: 10.1021/jm801470b. [DOI] [PubMed] [Google Scholar]
- Famiglini V. la Regina G. Coluccia A. Pelliccia S. Brancale A. Maga G. Crespan E. Badia R. Clotet B. Esté J. A. Cirilli R. Novellino E. Silvestri R. Eur. J. Med. Chem. 2014;80:101–111. doi: 10.1016/j.ejmech.2014.04.027. [DOI] [PubMed] [Google Scholar]
- Famiglini V. la Regina G. Coluccia A. Pelliccia S. Brancale A. Maga G. Crespan E. Badia R. Riveira-Muñoz E. Esté J. A. Ferretti R. Cirilli R. Zamperini C. Botta M. Schols D. Limongelli V. Agostino B. Novellino E. Silvestri R. J. Med. Chem. 2014;57:9945–9957. doi: 10.1021/jm5011622. [DOI] [PubMed] [Google Scholar]
- Müller R. Mulani I. Basson A. E. Pribut N. Hassam M. Morris L. van Otterlo W. A. L. Pelly S. C. Bioorg. Med. Chem. Lett. 2014;24:4376–4380. doi: 10.1016/j.bmcl.2014.08.020. [DOI] [PubMed] [Google Scholar]
- Kularatne S. A. M. BMJ. 2015;351:h4661. doi: 10.1136/bmj.h4661. [DOI] [PubMed] [Google Scholar]
- Rodenhuis-Zybert I. A. Wilschut J. Smit J. M. Cell. Mol. Life Sci. 2010;67:2773–2786. doi: 10.1007/s00018-010-0357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomonides T. Stud. Health Technol. Inform. 2010;159:235. [PubMed] [Google Scholar]
- Messina A. J. P. Brady O. J. Scott T. W. Zou C. Pigott D. M. Duda K. A. Bhatt S. Katzelnick L. Howes R. E. Battle K. E. Simmons C. P. Hay S. I. Trends Microbiol. 2014;22:138–146. doi: 10.1016/j.tim.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M. G. Harris E. Lancet. 2015;385:453–465. doi: 10.1016/S0140-6736(14)60572-9. [DOI] [PubMed] [Google Scholar]
- Vannice K. S. Wilder-Smith A. Barrett A. D. T. Carrijo K. Cavaleri M. de Silva A. Durbin A. P. Endy T. Harris E. Innis B. L. Katzelnick L. C. Smith P. G. Sun W. Thomas S. J. Hombach J. Vaccine. 2018;36:3411–3417. doi: 10.1016/j.vaccine.2018.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekly Epidemiological Record, WHO, 2018, vol. 93, pp. 388–396 [Google Scholar]
- Xu J. Xie X. Ye N. Zou J. Chen H. White M. A. Shi P. Y. Zhou J. J. Med. Chem. 2019;62:7941–7960. doi: 10.1021/acs.jmedchem.9b00698. [DOI] [PubMed] [Google Scholar]
- Bardiot D. Koukni M. Smets W. Carlens G. McNaughton M. Kaptein S. Dallmeier K. Chaltin P. Neyts J. Marchand A. J. Med. Chem. 2018;61:8390–8401. doi: 10.1021/acs.jmedchem.8b00913. [DOI] [PubMed] [Google Scholar]
- Noble C. G. Chen Y. L. Dong H. Gu F. Lim S. P. Schul W. Wang Q. Y. Shi P. Y. Antiviral Res. 2010;85:5450–5462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Lim S. P. Noble C. G. Shi P. Y. Antiviral Res. 2015;119:57–67. doi: 10.1016/j.antiviral.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Pelliccia S. Wu Y. H. Coluccia A. la Regina G. Tseng C. K. Famiglini V. Masci D. Hiscott J. Lee J. C. Silvestri R. J. Enzyme Inhib. Med. Chem. 2017;32:1091–1101. doi: 10.1080/14756366.2017.1355791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira R. R. Pereira W. L. da Silveira Oliveira A. F. C. da Silva A. M. de Oliveira A. S. da Silva M. L. da Silva C. C. de Paula S. O. Molecules. 2014;19:8151–8181. doi: 10.3390/molecules19068151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D. L. Li H. J. Smith D. R. Jaratsittisin J. Xia-Ke-Er X. F. K. T. Ma W. Z. Guo Y. W. Dong J. Shen J. Yang D. P. Lan W. J. Mar. Drugs. 2018;16:229. doi: 10.3390/md16070229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana V. M. Piccini L. E. Panozzo Zénere J. D. Damonte E. B. Ponce M. A. Castilla V. Antiviral Res. 2016;134:26–33. doi: 10.1016/j.antiviral.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Chayavichitsilp P. Buckwalter J. V. Krakowski A. C. Friedlander S. F. Pediatr. Rev. 2009;30:119–129. doi: 10.1542/pir.30-4-119. [DOI] [PubMed] [Google Scholar]
- Sherris Medical Microbiology, ed. K. J. Ryan and C. G. Ray, McGraw Hill, 4th edn, 2004 [Google Scholar]
- Straface G. Selmin A. Zanardo V. de Santis M. Ercoli A. Scambia G. Infect. Dis. Obstet. Gynecol. 2012;2012:385697. doi: 10.1155/2012/385697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looker K. J. Elmes J. A. R. Gottlieb S. L. Schiffer J. T. Vickerman P. Turner K. M. E. Boily M. C. Lancet Infect. Dis. 2017;17:1303–1316. doi: 10.1016/S1473-3099(17)30405-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch D. J. Rixon F. J. Davison A. J. Virus Res. 2006;117:90–104. doi: 10.1016/j.virusres.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Singh V. K. Mishra V. Tiwari S. Khaliq T. Barthwal M. K. Pandey H. P. Palit G. Narender T. Phytomedicine. 2013;20:1180–1185. doi: 10.1016/j.phymed.2013.06.017. [DOI] [PubMed] [Google Scholar]
- Nenaah G. Fitoterapia. 2010;81:779–782. doi: 10.1016/j.fitote.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Tanweer A. J. Chand N. Saddique U. Bailey C. A. Khan R. U. Parasitol. Res. 2014;113:2951–2960. doi: 10.1007/s00436-014-3957-y. [DOI] [PubMed] [Google Scholar]
- Benzekri R. Bouslama L. Papetti A. Hammami M. Smaoui A. Limam F. Microb. Pathog. 2018;114:291–298. doi: 10.1016/j.micpath.2017.12.017. [DOI] [PubMed] [Google Scholar]
- Bag P. Ojha D. Mukherjee H. Halder U. C. Mondal S. Biswas A. Sharon A. van Kaer L. Chakrabarty S. Das G. Mitra D. Chattopadhyay D. Antiviral Res. 2014;105:126–134. doi: 10.1016/j.antiviral.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Li M. K. Liu Y. Y. Wei F. Shen M. X. Zhong Y. Li S. Chen L. J. Ma N. Liu B. Y. Mao Y. D. Li N. Hou W. Xiong H. R. Yang Z. Q. Int. J. Antimicrob. Agents. 2018;51:98–106. doi: 10.1016/j.ijantimicag.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Perfetto B. Filosa R. de Gregorio V. Peduto A. la Gatta A. de Caprariis P. Tufano M. A. Donnarumma G. J. Med. Microbiol. 2014;63:1474–1483. doi: 10.1099/jmm.0.076612-0. [DOI] [PubMed] [Google Scholar]
- Ma N. Shen M. Chen T. Liu Y. Mao Y. Chen L. Xiong H. Hou W. Liu D. Yang Z. Biomed. Pharmacother. 2019;118:109359. doi: 10.1016/j.biopha.2019.109359. [DOI] [PubMed] [Google Scholar]
- Barbosa V. A. Baréa P. Mazia R. S. Ueda-Nakamura T. da Costa W. F. Foglio M. A. Ruiz A. L. T. G. de Carvalho J. E. Vendramini-Costa D. B. Nakamura C. V. Sarragiotto M. H. Eur. J. Med. Chem. 2016;124:1093–1104. doi: 10.1016/j.ejmech.2016.10.018. [DOI] [PubMed] [Google Scholar]
- Gonzalez M. M. Cabrerizo F. M. Baiker A. Erra-Balsells R. Osterman A. Nitschko H. Vizoso-Pinto M. G. Int. J. Antimicrob. Agents. 2018;52:459–468. doi: 10.1016/j.ijantimicag.2018.06.019. [DOI] [PubMed] [Google Scholar]
- Longo D. L., Influenza, in Harrison's principles of internal medicine, McGraw-Hill, New York, 18th edn, 2012, ch. 187 [Google Scholar]
- Hay A. J. Gregory V. Douglas A. R. Yi P. L. Philos. Trans. R. Soc., A. 2001;356:1861–1870. doi: 10.1098/rstb.2001.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsu S. Murakami S. Shindo K. Horimoto T. Sagara H. Noda T. Kawaoka Y. J. Virol. 2018;92:e02084. doi: 10.1128/JVI.02084-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghedin E. Sengamalay N. A. Shumway M. Zaborsky J. Feldblyum T. Subbu V. Spiro D. J. Sitz J. Koo H. Bolotov P. Dernovoy D. Tatusova T. Bao Y. St. George K. Taylor J. Lipman D. J. Fraser C. M. Taubenberger J. K. Salzberg S. L. Nature. 2005;437:1162–1166. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- Gubareva L. V. Besselaar T. G. Daniels R. S. Fry A. Gregory V. Huang W. Hurt A. C. Jorquera P. A. Lackenby A. Leang S. K. Lo J. Pereyaslov D. Rebelo-de-Andrade H. Siqueira M. M. Takashita E. Odagiri T. Wang D. Zhang W. Meijer A. Antiviral Res. 2017;146:12–20. doi: 10.1016/j.antiviral.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawatari M. Saito R. Hibino A. Kondo H. Yagami R. Odagiri T. Tanabe I. Shobugawa Y. PLoS One. 2019;14:e0224683. doi: 10.1371/journal.pone.0224683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. C. Wong W. Y. Ng W. L. Yeo K. S. Begam T. Mohidin M. Lim Y. Y. Lafta F. Ali H. M. Ea C. K. PLoS One. 2017;12:e0170352. doi: 10.1371/journal.pone.0170352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivachtchenko A. V. Yamanushkin P. M. Mitkin O. D. Kisil V. M. Korzinov O. M. Vedenskii V. Y. Leneva I. A. Bulanova E. A. Bichko V. V. Okuń I. M. Ivashchenko A. A. Ivanenkov Y. A. Pharm. Chem. J. 2015;49:151–162. doi: 10.1007/s11094-015-1244-6. [DOI] [Google Scholar]
- Zoidis G. Giannakopoulou E. Stevaert A. Frakolaki E. Myrianthopoulos V. Fytas G. Mavromara P. Mikros E. Bartenschlager R. Vassilaki N. Naesens L. MedChemComm. 2016;7:447–456. doi: 10.1039/C5MD00439J. [DOI] [Google Scholar]
- Cihan-Üstündağ G. Zopun M. Vanderlinden E. Ozkirimli E. Persoons L. Çapan G. Naesens L. Bioorg. Med. Chem. 2020;28:115130. doi: 10.1016/j.bmc.2019.115130. [DOI] [PubMed] [Google Scholar]
- Chen M. Lin S. Li L. Zhu C. Wang X. Wang Y. Jiang B. Wang S. Li Y. Jiang J. Shi J. Org. Lett. 2012;14:5668. doi: 10.1021/ol302660t. [DOI] [PubMed] [Google Scholar]
- Wang X. L. Chen M. H. Wang F. Bu P. B. Lin S. Zhu C. G. Li Y. H. Jiang J. D. Shi J. G. Zhongguo Zhongyao Zazhi. 2013;38:1172. [PubMed] [Google Scholar]
- Liu Y. F. Chen M. H. Guo Q. L. Lin S. Xu C. B. Jiang Y. P. Li Y. H. Jiang J. D. Shi J. G. J. Asian Nat. Prod. Res. 2015;17:689–704. doi: 10.1080/10286020.2015.1055729. [DOI] [PubMed] [Google Scholar]