

Abstract
The early secretory pathway and autophagy are two essential and evolutionarily conserved endomembrane processes that are finely interlinked. Although growing evidence suggests that intracellular trafficking is important for autophagosome biogenesis, the molecular regulatory network involved is still not fully defined. In this study, we demonstrate a crucial effect of the COPII vesicle‐related protein TFG (Trk‐fused gene) on ULK1 puncta number and localization during autophagy induction. This, in turn, affects formation of the isolation membrane, as well as the correct dynamics of association between LC3B and early ATG proteins, leading to the proper formation of both omegasomes and autophagosomes. Consistently, fibroblasts derived from a hereditary spastic paraparesis (HSP) patient carrying mutated TFG (R106C) show defects in both autophagy and ULK1 puncta accumulation. In addition, we demonstrate that TFG activity in autophagy depends on its interaction with the ATG8 protein LC3C through a canonical LIR motif, thereby favouring LC3C‐ULK1 binding. Altogether, our results uncover a link between TFG and autophagy and identify TFG as a molecular scaffold linking the early secretion pathway to autophagy.
Keywords: autophagy, ERGIC, LC3C, TFG
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Trk‐fused gene (TFG) acts as a molecular scaffold linking the early secretory pathway and autophagy in mammalian cells.
Introduction
Macroautophagy (hereafter autophagy) is an evolutionarily conserved catabolic process, by which double‐membrane vesicles deliver intracellular macromolecules and organelles to lysosomes for degradation. Upon induction of autophagy by different stimuli, autophagosomes form de novo and initially appear as small membrane structures referred to as isolation membrane (IM) or phagophores. IM expands, gradually engulfing portions of the cytoplasm, and gives rise to autophagosomes. The formation of an autophagosome requires the action of autophagy‐related (ATG) proteins that are organized into functional complexes (Mizushima et al, 2011), which localize to the IM at some stage of autophagosome formation. The Unc‐51‐like kinase 1 (ULK1) complex and the [(phosphatidylinositol (3)] kinase complex III (PIK3C3), together with the activation of localized phosphatidylinositol (PI) synthase (Nishimura et al, 2017), lead to the production of PI3P, which acts as a platform for the recruitment of effector proteins, including the double FYVE domain‐containing factor DFCP1 (Axe et al, 2008) and WIPI proteins (Funderburk et al, 2010). This phase is followed by the recruitment of the remaining autophagy core machinery involved in phagophore expansion, including ATG9, and two ubiquitin‐like conjugation systems, ATG12 ~ ATG5 and ATG16L1 (Mizushima et al, 2003). These complexes process small ATG8 ubiquitin‐like proteins [microtubule‐associated protein 1A/1B‐light chain 3 (LC3) and GABARAP family members)] (Slobodkin & Elazar, 2013), to membrane‐associated PE. LC3 incorporation represents the maturation step of autophagosomes, which in the end fuse with lysosomes, where their content is digested by lysosomal hydrolases and released into the cytosol for recycling.
Over the years, a strict connection between autophagy and vesicular transport processes is emerging. In particular, the coat protein complex II (COPII), the first component involved in the transport pathway that leads out of the cell, has been linked to autophagosome formation. COPII transport carriers are composed of two distinct layers: an inner coat characterized by SEC23/SEC24 and an outer coat formed by SEC13/SEC31. They originate at the ER exit sites (ERES) and are essential for cellular cargo transport to the ER‐Golgi intermediate compartment (ERGIC). ERES, ERGIC and COPII vesicles have been shown to co‐localize with sites of autophagosome biogenesis (Karanasios et al, 2016; Stadel et al, 2015; Ge et al, 2013; Ge et al, 2014; Jeong et al, 2018), whereas a number of proteins involved in this early secretory pathway are found to directly contribute to autophagy regulation (Moyer et al, 2001; Zoppino et al, 2010). Indeed, in both yeast (sec12, sec16, sec23, sec24) (Ishihara et al, 2001; Davis et al, 2016) and mammals (SEC23A) (Ge et al, 2014), deficiency of SEC proteins was found to impair autophagy (Ishihara et al, 2001). Furthermore, COPII components physically interact with core proteins required for autophagy (Lord et al, 2011; Wang et al, 2014; Davis et al, 2016; Gan et al, 2017). However, how the regulators of COPII vesicle transport are controlled in response to nutrient deprivation, and to allow their impact to autophagosome formation, is largely unknown.
TFG (TRK‐fused gene) is a SEC16‐interacting protein ubiquitously expressed in human cells (Witte et al, 2011). TFG appears to coordinate the distribution of COPII transport carriers, promoting their uncoating < and retaining these vesicles at the region between the ER and the ERGIC, in order to facilitate their fusion with the latter compartment (Witte et al, 2011; Johnson et al, 2015; Hanna et al, 2017). In humans, TFG mutations cause sensory axon degeneration, hereditary spastic paraplegia and Charcot–Marie–Tooth disease type 2 (Takashima et al, 1997; Beetz et al, 2013; Tsai et al, 2014). In these neuronal disorders, the defects appear related to structural changes in the ER‐associated processes, such as ER architecture maintenance and ER export. Besides these, autophagy impairment is also emerging as a central process in the pathogenesis of such complicated forms of neuropathy (Colecchia et al, 2018).
Although very recently, and in a specific immune system model, a link between TFG and autophagy has been found (Steinmetz et al, 2020), no details are known about the molecular mechanisms of this interplay and its control. In the present study, we identify the mechanisms of TFG–autophagy interplay. By using a combination of biochemical, morphological and functional analyses, we clearly demonstrate the interaction between TFG and the autophagy kinase ULK1 and, in particular, the crucial role of TFG in ULK1 proper localization during nutrient deprivation. Moreover, TFG‐deficient cells exhibit decreased LC3B‐II levels and a delay in the association of LC3B to the isolation membrane components, defining a primary role for TFG in autophagosome formation. Consistently, fibroblasts derived from a HSP patient carrying mutated TFG (R106C) show defects in both autophagy and ULK1 puncta accumulation. Last, our report reveals that, via a canonical LC3‐interacting region (LIR) motif, TFG interacts with LC3C; hence, this binding is required for regulating the proper distribution of ULK1 and the formation of autophagosomes. This implies the need for a LIR‐dependent TFG‐LC3C interaction as a positive regulatory step in autophagosome formation.
Results
TFG interacts with ULK1 and regulates its stability and localization, independently of the COPII pathway
By an unbiased proteomics‐based approach to identify ULK1‐interacting proteins, performed in a previous study (Nazio et al, 2016), we identified TFG as a putative ULK1‐interacting protein. The recent discovery of a link between COPII and autophagy (Ge et al, 2014; Jeong et al, 2018) and the fact that TFG is known to be involved in facilitating COPII vesicle export (Johnson et al, 2015) and to prime autophagy in specific experimental immune system models (Steinmetz, et al, 2020) prompted us to investigate the mechanisms of TFG‐ULK1 interaction in autophagy regulation. First, to characterize the interaction at endogenous levels, we isolated endogenous TFG by co‐immunoprecipitation and analysed ULK1‐complex members (ULK1, ATG13 and FIP200). Each member analysed shows interaction with TFG (Fig 1A), as also confirmed by immunofluorescence (IF): further, we detected a tight association between TFG, ULK1 and ATG13 during autophagy induction by starvation (Figs 1B and EV1A). Second, to investigate the functional significance of the TFG‐ULK1 interaction, we examined the effects of TFG depletion on both ULK1 protein levels and activity. Since long periods of TFG depletion result in an inhibition of cellular proliferation and in an increase in cell death (Johnson et al, 2015), we chose the 48–60 h timepoint of siRNA treatment for all subsequent experiments. In Fig 1C‐E, we observed, in TFG‐silenced cells, an increase in the steady‐state levels of ULK1, but no changes in the phosphorylation levels of ULK1 substrates ATG13 and ATG14 (Fig EV1B). Of note, such an effect on protein stability is specific for ULK1, since TFG depletion does not impact at all ATG13 and FIP200 levels (Fig EV1C).
Figure 1. TFG interacts with ULK1 and regulates its stability and localization.
-
AHeLa cells were cultured in complete growth or starvation (STV) media for 30’. Cell lysates were immunoprecipitated (IP) with anti‐TFG antibody or IgG as a negative control. Immunoprecipitated complexes were analysed by WB to detect TFG, ULK1, ATG13 and FIP200 as indicated.
-
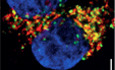
BHeLa cells were cultured in complete growth (FED) or starvation (STV) media for 1 h. Cells were fixed, permeabilized and labelled with anti‐TFG (red) and anti‐ULK1 (green) antibodies, respectively. Hoechst was used to stain nuclei. Co‐localization analysis was performed by ImageJ plugin Jacop and reported in the graph (right). Values of Mander’s coefficient for ULK1 are expressed as mean ± SEM. Significance was assigned by unpaired Student’s t‐test. **P < 0.01 (n = 3 independent experiments, n = 12 fields analysed). Scale bar 5 μm.
-
CHeLa cells were transfected with specific RNAi oligonucleotides (siTFG) or unrelated oligonucleotides as a negative control (siCTR) for 48 h. Protein extracts were analysed by WB to detect ULK1, TFG and ACTIN as indicated. Densitometry analysis of ULK1 over ACTIN is shown (right). All values are mean ± SEM. Statistical analyses were performed by two‐tailed Student’s t‐test. ***P < 0.001 (n = 3 independent experiments).
-
DHeLa cells were co‐transfected with control siRNA and an empty vector (EV), or 3’UTR TFG siRNA together with an EV or HA‐TFG plasmids. Protein extracts were analysed by WB to detect ULK1, TFG and ACTIN as indicated. Densitometry analysis of ULK1 protein levels normalized over ACTIN is also reported (right). All values are expressed as the mean ± SEM. Statistical analyses were performed by one‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 and **P < 0.01 (n = 3 independent experiments).
-
EHeLa cells were transfected as in C), grown in the presence or not of cycloheximide (CHX) for indicated time periods. Protein extracts were analysed by WB to detect ULK1, TFG and ACTIN as indicated. Densitometry analysis of ULK1 normalized over ACTIN is also shown (right). Values are mean ± SEM Statistical analyses were performed by multiple t‐test. *P < 0.05 (n = 3 independent experiments).
-
F, GHeLa cells were treated as in B) and then fixed and stained with ULK1 (green) (F) or ATG13 (red) antibodies (G), respectively. Hoechst was used to stain nuclei. Graphs (right) show the average of both ULK1 (F) (n = 3 independent experiments, n = 11 fields analysed) and ATG13 (n = 3 independent experiments, n ≥ 16 fields analysed) (G) puncta number per cell and both ULK1 (n = 3 independent experiments) (F) and ATG13 (n = 3 independent experiments) (G) fold of increase of the average size, respectively. White arrows indicate most representative puncta of analysed conditions. All data are reported as the mean value ± SEM. Statistical analyses were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 and ****P < 0.0001 or by unpaired Student’s t‐test when two groups were compared. **P < 0.01 and ****P < 0.0001. Scale bar 5 μm.
Source data are available online for this figure.
Figure EV1. TFG regulates ULK1 independently of its function on COPII trafficking.
-
AHeLa cells were grown for 1 h in starvation (STV) medium, then fixed, permeabilized and stained with TFG (green) and ATG13 (red). Hoechst was used to stain nuclei. Representative micrograph is reported. Scale bar 2 μm.
-
BHeLa cells were transfected with the indicated siRNAs and grown in nutrient complete (FED) or starvation (STV) media for 1 h. Protein extracts were analysed by WB to detect p‐ATG13, ATG13, GADPH, p‐ATG14, ATG14, ACTIN and TFG protein levels. Densitometry analysis representing p‐ATG13/ATG13 normalized over GADPH and p‐ATG14/ATG14 normalized over ACTIN of starved samples of both the conditions analysed was also reported. All data are expressed as mean ± SEM. Statistical analyses were obtained by unpaired Student’s t‐test. ns, not significant (n = 3 independent experiments).
-
CHeLa cells were transfected with the indicated siRNAs and grown in the presence or not of cycloheximide (CHX) for indicated time periods. Protein extracts were analysed by WB to detect FIP200, ATG13, TFG and ACTIN. Densitometry analysis of FIP200 over ACTIN (bottom left) and ATG13 over ACTIN (bottom right) is also displayed. Values are mean ± SEM. Statistical analyses were performed by multiple t‐test. ns, not significant (n = 4 independent experiments).
-
DQuantification of ULK1 puncta in HeLa cells transfected with the indicated siRNAs and cultured in complete growth or starvation (STV) media for 1 h is shown. TFG downregulation and ULK1 protein levels were analysed by WB as reported. All data are expressed as mean ± SEM. Statistical analysis was obtained by two‐way ANOVA followed by Tukey’s multiple comparison test ***P < 0.001 and ****P < 0.0001 (n = 3 independent experiments).
-
EHeLa cells were grown for 1 h in starvation (STV) medium, then fixed, permeabilized and stained with ATG13 (red) and ULK1 (green). Hoechst was used to stain nuclei. Representative micrograph and fluorescence plot are reported. Scale bar 5 μm.
-
FHeLa cells were co‐transfected with control siRNA and an empty vector (EV), or 3’UTR TFG siRNA together with an EV or HA‐TFG plasmids. Cells were grown for 1 h in starvation medium, then fixed, permeabilized and labelled with anti‐ATG13 (green) antibody. Hoechst was used to stain nuclei. The average of ATG13 puncta number per cell is reported (bottom). All data are expressed as mean ± SEM. Statistical analysis was obtained by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 (n = 3 independent experiments, n ≥ 13 fields analysed). Scale bars 5 μm.
-
G, HHeLa cells were transfected with SEC23A and SEC16 siRNAs (siSEC23A) and (siSEC16), respectively. Cells were cultured with nutrient complete medium (FED) or starvation medium (STV) for 1 h. HeLa cells were fixed, permeabilized and stained with ULK1 (red) antibody. Hoechst was used to stain nuclei. Graphs reporting the average of ULK1 puncta number per cell for the indicated conditions are shown (bottom) (n = 3 independent experiments, n ≥ 6 fields analysed for G; n = 3 independent experiments, n ≥ 9 fields analysed for H). Scale bars 5 μm. All values are expressed as the mean ± SEM. Statistics were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. ns, not significant.
-
IHeLa cells were transfected with indicated siRNAs. Protein extracts were analysed by WB to detect SEC16, ULK1, SEC23, TFG and ACTIN protein levels. Quantification of ULK1 over ACTIN is reported in the graph (right). Data are mean ± SEM. Statistics were performed by one‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01 and ns, not significant (n = 3 independent experiments).
-
JHeLa cells were transiently transfected with the indicated siRNAs and grown in complete or starvation (STV) media, in the presence or absence of CLQ for 1 h. Protein extracts were analysed by WB to detect LC3B‐II and ACTIN protein levels and SEC23A downregulation as indicated. Densitometry analysis of LC3B‐II normalized over ACTIN is reported (right). All data are reported as mean ± SEM. Significance was assigned by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 (n = 3 independent experiments).
-
KHeLa cells were transfected with indicated siRNAs, fixed and immunolabelled with ULK1 (red) antibody. Hoechst was used to stain nuclei. ULK1 puncta number was quantitated and reported in the graph (bottom). All data are mean ± SEM. Statistics were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01; ***P < 0.001 (n = 3 independent experiments, n ≥ 7 fields analysed). Scale bar 5 μm.
-
LHeLa cells were transfected with indicated siRNAs. Protein extracts were analysed by WB for indicated antibodies. ULK1 over ACTIN protein levels is reported in the graph (right). All values are expressed as the mean ± SEM. Statistics were performed by one‐way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.05 (n = 3 independent experiments).
Third, since once activated, the ULK1 complex translocates to puncta to prime autophagosome biogenesis (Karanasios et al, 2013), we assessed whether TFG could influence ULK1 puncta formation and, to this aim, measured ULK1 puncta number after TFG downregulation. Indeed, there is a significant increase in ULK1 puncta number in fed conditions, and this effect is exacerbated during autophagy induction by starvation (Figs 1F and EV1D). Also, since ATG13 puncta may often recapitulate ULK1 puncta localization during starvation (Karanasios et al, 2013 and Fig EV1E), we evaluated whether or not TFG also affected ATG13 puncta formation in our system. As shown in Figs 1G and EV1F, ATG13 puncta also increase in TFG‐depleted cells with a significant spread upon starvation, and TFG overexpression is able per se to rescue this phenotype.
Last, since it is well established that TFG has a direct role in COPII vesicle localization (Johnson et al, 2015), we decided to investigate whether TFG‐dependent effect on ULK1 could be related to TFG role in the COPII pathway. To this aim, we analysed ULK1 puncta number formation upon depletion of both SEC23A and SEC16, two important components of the COPII pathway. As shown in Fig EV1G and H, there are no significant differences in ULK1 puncta number in the conditions analysed, when compared to the control. To support this result, we analysed ULK1 protein levels in both SEC23A‐ and SEC16‐depleted cells, respectively, and found no relevant changes when compared to TFG‐depleted cells, at variance with the effect of SEC23A downregulation on autophagy flux (Fig EV1, EV2, EV3, EV4, EV5J) (Ge et al, 2014). In order to exclude that such an effect of TFG on ULK1 could be a consequence of TFG deficiency on COPII vesicles, we analysed both ULK1 puncta formation and protein levels after TFG‐SEC23A or TFG‐SEC16 depletion. As shown in Fig EV1K and L, ULK1 puncta number and protein levels are still increased in both conditions analysed, indicating that the increment of ULK1 does not depend on the presence of COPII vesicles.
Figure EV2. TFG downregulation affects ULK1‐ATG9 co‐localization.
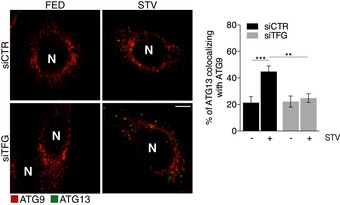
HEK293 cells stably expressing GFP‐ATG13 were interfered or not with TFG siRNA (siTFG) and starved for 1 h. Cells were fixed and immunolabelled with ATG9 (red) antibody. Co‐localization analysis was performed by Jacop plugin. Values of Mander’s coefficient for ULK1 are reported as percentage and represented as mean ± SEM. Significance was assigned by two‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01 and ***P < 0.001. Scale bar 5 μm. N, nucleus (n = 3 independent experiments, n ≥ 20 fields analysed).
Figure EV3. ATG16L1 puncta accumulation depends on ULK1 in TFG‐depleted cells.
-
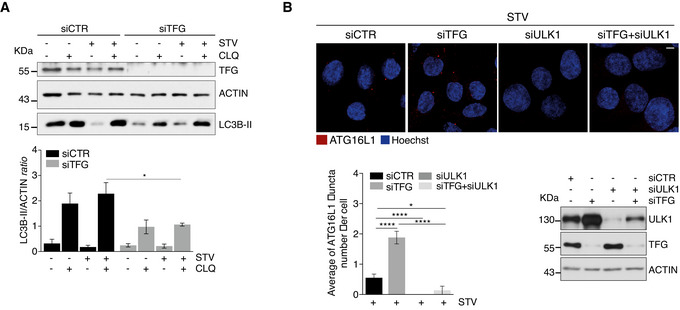
AHeLa cells were transfected with indicated siRNAs and cultured in fed or starvation conditions (STV) in the presence or absence of CLQ for 1 h. Protein extracts were analysed by WB for the expression of TFG, ACTIN and LC3B‐II. Densitometry analysis of LC3B‐II over ACTIN is shown (bottom). Values are expressed as mean ± SEM. Statistical analysis was performed by Tukey’s multiple comparison test. *P < 0.05 (n = 3 independent experiments).
-
BHeLa cells were transfected with indicated siRNAs. Cells were then cultured in starvation medium for 1 h, fixed and immunolabelled with ATG16L1 (red) antibody. Hoechst was used to stain nuclei. Analysis of ATG16L1 puncta is reported in the graph (bottom left). Values are expressed as mean ± SEM. Statistical analysis was performed by one‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 and ****P < 0.0001. Scale bar 5 μm (n = 5 independent experiments). WB analysis to detect ULK1 and TFG downregulations is also shown (bottom right).
Figure EV4. Both TFG depletion and TFGR106C mutation induce autophagy impairment.
-
AHeLa cells were co‐transfected with TFG or control siRNA together with vectors encoding for Myc‐ULK1 and HA‐LC3B. After 30’ of starvation, cells were lysed, and protein extracts were immunoprecipitated by using an anti‐HA antibody or IgG as negative control. ULK1 LC3B and TFG protein levels were analysed by WB.
-
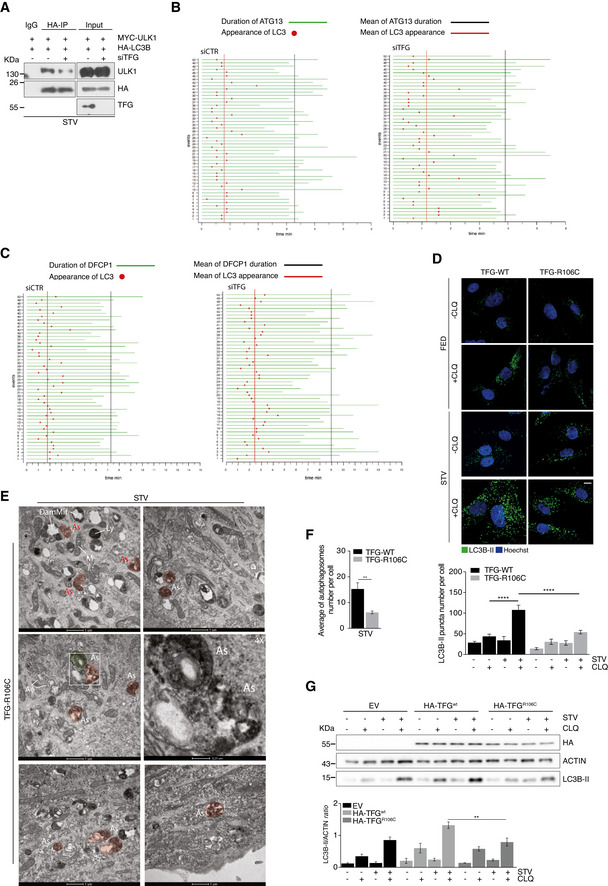
B, CTemporal relationship of LC3B and ATG13 (B) or LC3B and DFCP1 (C). Starved HEK293 cells stably expressing GFP‐ATG13 or GFP‐DFCP1 together with CFP‐LC3B were analysed by live‐cell imaging. 50 unique events showing the moment at which LC3 appears on ATG13 (B) or DFCP1 (C) (red point) are reported; green lines depict the lifespan of ATG13 particles (B) or DFCP1 structures (C), respectively (n = 3 independent experiments, n = 50 events analysed).
-
DControl (TFG‐WT) and patient’s fibroblasts (TFG‐R106C) were grown in fed or starvation conditions in the presence or absence of CLQ for 1 h. Cells were fixed permeabilized and labelled with LC3B‐II antibody. Hoechst was used to stain nuclei. LC3B‐II puncta number was analysed (bottom). Data are expressed as the mean value ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. ****P < 0.0001 (n = 5 independent experiments). Scale bar 5 μm.
-
EPatient’s fibroblasts (TFG‐R106C) were cultured in STV medium for 1 h. Cells were processed and imaged by TEM. Representative electron micrographs are reported. AΦ, autophagosome; As, abnormal structures; M, mitochondrion; DamMit, damaged mitochondrion; and Ly, lysosome. Scale bar 1 μm or 0,25 μm as indicated.
-
FQuantification of the autophagosomes number per cell in control (TFG‐WT) and patient’s fibroblasts (TFG‐R106C) grown in starvation (STV) conditions is shown. Data are expressed as the mean value ± SEM. Statistical analysis were performed by unpaired Student’s t‐test. **P < 0.01. A minimum of 10 cells for condition were observed (n = 3 independent experiments, n ≥ 10 cells/condition analysed).
-
GHeLa cells were transfected with an empty vector (EV) or HA‐TFGwt, or HA‐TFGR106C and grown in fed or starvation conditions (STV) in the presence or absence of CLQ for 1 h. Protein extracts were analysed by WB to detect HA‐TFG ACTIN and LC3B‐II as indicated. Densitometry analysis of LC3B‐II over ACTIN is shown (bottom). All data are expressed as the mean ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01 (n = 3 independent experiments).
Figure EV5. TFG‐LC3C interaction is important for ULK1 activity in autophagy induction.
-
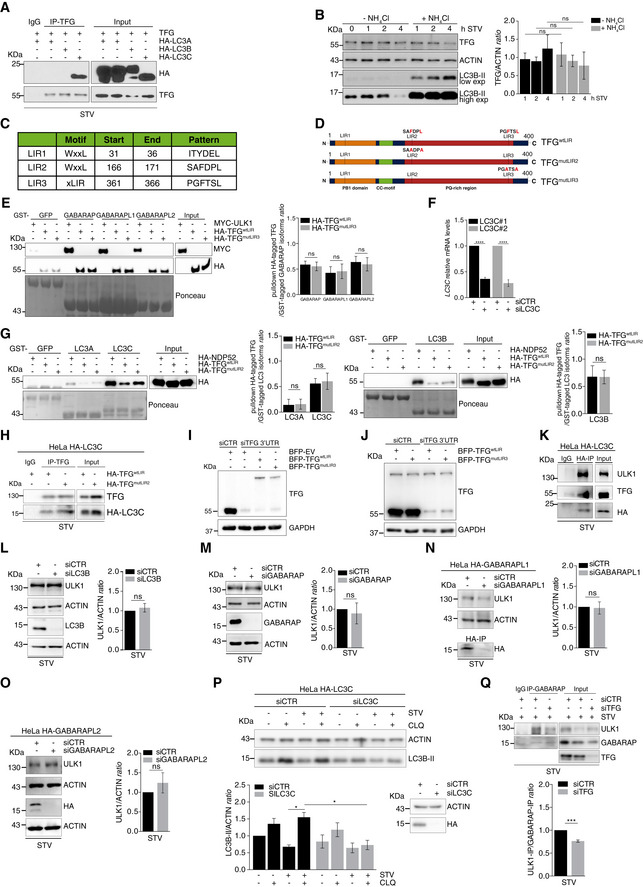
AHeLa cells were co‐transfected with untagged TFG and HA‐tagged LC3A, LC3B or LC3C. After 1 h of starvation, protein extracts were immunoprecipitated by using an anti‐TFG antibody or IgG as negative control. LC3 subfamily and TFG were analysed by WB.
-
BHeLa cells were nutrient‐starved for the indicated time periods in the presence or absence of NH4Cl for the indicated timepoints of starvation. TFG, ACTIN and LC3B‐II (as indicator of NH4Cl activity) were shown in the WB. Densitometry analysis of TFG over ACTIN protein levels was analysed in the graph (right). Data are expressed by mean ± SEM. Statistical analysis was obtained by two‐way ANOVA followed by Tukey’s multiple comparison test. ns, not significant (n = 3 independent experiments).
-
CTFG LIR motives, obtained from the autophagy database iLIR with their sequence and position, are shown.
-
DSchematic illustration of TFG wild type (TFGwtLIR) and TFG mutants (TFGmutLIR2 and TFGmutLIR3) is shown. The aromatic F and the hydrophobic L residues of the TFGwtLIR were changed into two alanine residues for both the TFG mutants.
-
EPulldown assays were performed by using purified GST‐GABARAP, GST‐GABARAPL1 and GST‐GABARAPL2 isoforms, each incubated together with HA‐TFGwtLIR‐ or HA‐TFGmutLIR3‐ or Myc‐ULK1‐transfected HeLa cell lysates, respectively. Myc‐ULK1 was used as positive control. Ponceau staining reports GST‐GABARAP isoforms and GST‐GFP used as negative control. WB analyses show Myc‐ULK1, HA‐TFGwtLIR and HA‐TFGmutLIR3. Densitometry analysis of HA‐TFG normalized over GST‐tagged GABARAP isoforms (Ponceau) was performed and reported (right). Data are mean ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. ns, not significant (n = 3 independent experiments).
-
FHeLa cells were transfected with the indicated siRNA, and LC3C mRNA relative levels were analysed by qPCR. Values are expressed as mean ± SEM. Statistical analysis was performed by unpaired t‐test. ****P < 0.0001 (n = 3 independent experiments).
-
GPulldown assays were performed by using purified GST‐LC3A, GST‐LC3B and GST‐LC3C isoforms, each incubated together with HA‐TFGwtLIR‐ or HA‐TFGmutLIR2‐ or HA‐NDP52‐transfected HeLa cell lysates, respectively. HA‐NDP52 was used as positive control. Ponceau staining reports the indicated GST‐LC3 isoforms and GST‐GFP used as negative control. WB analyses show HA‐NDP52, HA‐TFGwtLIR and HA‐TFGmutLIR2 in association with LC3A and LC3C (left) and HA‐NDP52, HA‐TFGwtLIR and HA‐TFGmutLIR2 in association with LC3B (right) are shown. Densitometry analysis of HA‐TFG normalized over GST‐tagged LC3 isoforms (Ponceau) was performed and reported as indicated (right). All values are expressed as mean ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. ns, not significant (n = 3 independent experiments).
-
HHeLa cells harbouring endogenously HA‐tagged LC3C were transfected with HA‐TFGwtLIR or HA‐TFGmutLIR2. After 30’ of starvation, cells were lysed, and protein extracts were immunoprecipitated by using an anti‐TFG antibody or IgG as negative control. LC3C and TFG were analysed by WB.
-
IHeLa cells stably expressing GFP‐ATG13 together with BFP‐empty vector (EV) or BFP‐TFGwtLIR or BFP‐TFGmutLIR3 were co‐transfected with mCherry‐SEC16 together with siRNA targeting endogenous TFG (siTFG 3’UTR) or with a non‐targeting control (siCTR). Endogenously interfered cells were used to perform live‐cell imaging analysis (Fig 5I). WB analysis to detect endogenous or BFP‐TFG protein levels is reported.
-
JHeLa cells stably expressing GFP‐ATG13 together with BFP‐TFGwtLIR or BFP‐TFGmutLIR3 were transfected with siRNA targeting endogenous TFG (siTFG 3’UTR) or with a non‐targeting control (siCTR) as indicated. Endogenously interfered cells were used to perform live‐cell imaging analysis (Fig 5J). WB analysis to detect endogenous or BFP‐TFG protein levels is reported.
-
KHeLa cells harbouring endogenously HA‐tagged LC3C were starved for 30’. Protein extracts were immunoprecipitated by using an anti‐HA antibody or IgG as negative control. ULK1, TFG and HA‐LC3C protein levels were analysed by WB.
-
L, MHeLa cells were transfected with unrelated siRNA (siCTR) or siRNA targeting LC3B (siLC3B) (L) or GABARAP (siGABARAP) (M). (L‐M) After 30’ of starvation, cells were lysed and protein extracts were analysed by WB for indicated markers. ULK1 over actin is reported in the graphs (right). Data are shown as mean value ± SEM. Statistical analysis was performed by unpaired Student’s t‐test. ns, not significant (n = 3 independent experiments).
-
NHeLa cells harbouring endogenously HA‐tagged GABARAPL1 were transfected with siRNA targeting GABARAPL1 or unrelated siRNA (siCTR). After 30’ of starvation, cells were lysed. Part of protein extract was immunoprecipitated with anti‐HA antibody to evaluate the downregulation of GABARAPL1. Remaining protein extract was analysed by WB as shown. ULK1 over ACTIN is also reported (right). Data are shown as mean value ± SEM. Statistical analysis was performed by unpaired Student’s t‐test. ns, not significant (n = 3 independent experiments).
-
OHeLa cells harbouring endogenously HA‐tagged GABARAPL2 were transfected with siRNA targeting GABARAPL2 or unrelated siRNA (siCTR). Cells were starved for 30’ and lysed. Protein extract was analysed by WB to detect the indicated markers. ULK1 over ACTIN is also reported (right). Data are as mean ± SEM. Statistical analysis was performed by unpaired Student’s t‐test. ns, not significant (n = 3 independent experiments).
-
PHeLa cells harbouring endogenously HA‐tagged LC3C were transiently transfected with siLC3C or siCTR and grown in complete or starved (STV) media, in the presence or absence of CLQ for 1 h. LC3B‐II and ACTIN were analysed by WB. Densitometry analysis of LC3B‐II normalized over ACTIN is reported (bottom left). LC3C downregulation is also shown (bottom right). All data are reported as mean ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 (n = 3 independent experiments).
-
QHeLa cells were co‐transfected with indicated siRNAs and cultured in starvation medium for 30’. Cells were lysed, and protein extracts were immunoprecipitated by using an anti‐GABARAP antibody or IgG as negative control. ULK1, GABARAP and TFG protein levels were analysed by WB. ULK1‐IP over GABARAP‐IP is reported in the graph (bottom). Data are mean ± SEM. Statistical analysis was performed by unpaired Student’s t‐test. ***P < 0.001 (n = 3 independent experiments).
Altogether, these results show that TFG i) controls ULK1 protein levels at a steady state, ii) regulates ULK1‐ATG13 puncta number and size, and iii) acts on ULK1 puncta formation independently of its function in the COPII pathway.
TFG is essential for proper ULK1 complex distribution during autophagy
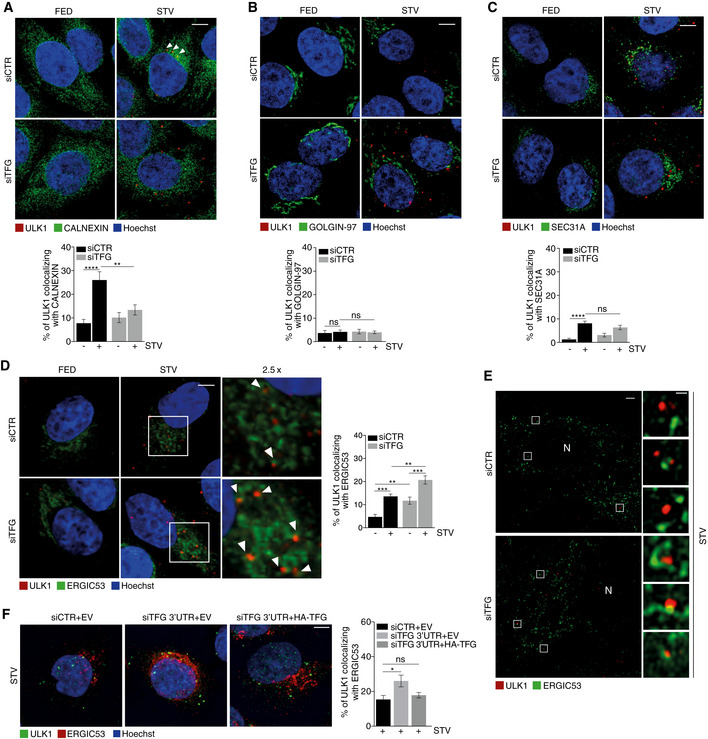
Based on these data, we set up experiments in order to verify ULK1 puncta distribution upon TFG depletion. To this aim, we evaluated ULK1 puncta co‐localization by IF with several intracellular structures: ER, ATG9‐positive vesicles, Golgi complex, COPII vesicles and ERGIC, respectively. In control conditions, upon autophagy induction, there is an increase in ULK1 puncta that co‐localize with both ER and ATG9 (Figs 2A and EV2A); this increase does not occur when TFG is depleted and autophagy is induced. About ULK1–Golgi complex association, we observed no differences in all conditions analysed (Fig 2B). Of note, the ERGIC compartment has been proposed as an important membrane source for autophagosome formation, and it was suggested that upon autophagy induction, ERGIC‐derived COPII vesicles act as template for LC3 lipidation (Ge et al, 2014). Therefore, we investigated whether TFG depletion could affect or not ULK1 puncta localization to both COPII vesicles and ERGIC compartments, by analysing the percentage of ULK1 puncta co‐localizing with SEC31A and ERGIC53, respectively. Strikingly, while TFG depletion upon autophagy induction does not induce significant variation in the association between ULK1 and COPII (Fig 2C), the percentage of ULK1 puncta juxtaposed to the ERGIC structures significantly increased in the same condition (Fig 2D). Moreover, we evaluated the association between ULK1‐ERGIC upon TFG depletion and autophagy induction through a super‐resolution analysis by structured illumination microscopy (SIM). As shown in Fig 2E, we found that, upon TFG downregulation, a portion of ULK1 puncta is ERGIC‐associated, with this defining TFG as an important factor for a close juxtaposition between ULK1 and ERGIC. Indeed, we also succeeded to restore canonical ULK1‐ERGIC co‐localization by simply re‐expressing TFG in a classical rescue experiment (Fig 2F).
Figure 2. TFG is essential for proper ULK1 complex distribution during autophagy.
-
A–DHeLa cells were transfected with specific RNAi oligonucleotides (siTFG) or unrelated oligonucleotides as a negative control (siCTR) and grown in fed or starved conditions for 1 h. Cells were fixed and co‐labelled with ULK1 (red) antibody and (A) anti‐CALNEXIN (green) antibody to highlight ER structures (n = 3 independent experiments, n ≥ 16 fields analysed), (B) GOLGIN‐97 (green) antibody to label GOLGI complex (n = 3 independent experiments, n = 12 fields analysed), (C) SEC31A (green) to point out COPII vesicles (n = 3 independent experiments, n ≥ 16 fields analysed) and (D) ERGIC53 (green) to mark ERGIC compartment (n = 3 independent experiments, n ≥ 14 fields analysed). Analysis of ULK1 localization with each compartment is reported in each graph. Co‐localization analyses were performed by ImageJ plugin Jacop. Values of Mander’s coefficient for ULK1 are expressed in percentage as mean ± SEM. Statistical analyses were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01; ***P < 0.001; ****P < 0.0001; and ns, not significant. Scale bars 5 μm. White arrowheads point at ULK1 puncta associated with the analysed markers.
-
EHeLa cells were transfected as in A), labelled with the indicated antibodies and imaged by SIM microscope. Scale bar in large panels, 5 μm; in small panels, 1 μm. N, nucleus.
-
FHeLa cells were co‐transfected with control siRNA and an empty vector (EV), or 3’UTR TFG siRNA together with an EV or HA‐TFG plasmids. Cells grown for 1 h in starvation medium then fixed, permeabilized and labelled with anti‐ULK1 (green) and anti‐ERGIC53 (red) antibodies, respectively. Hoechst was used to stain nuclei. Co‐localization analysis was performed as in D). Values of Mander’s coefficient for ULK1, expressed as percentage, are mean ± SEM. Statistical analyses were performed by one‐way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.05; ns, not significant (n = 3) (n = 3 independent experiments, n ≥ 9 fields analysed). Scale bar 5 μm.
Source data are available online for this figure.
In sum, this set of results highlights a role for TFG in controlling ULK1 puncta subcellular distribution during autophagy induction.
Inhibition of TFG results in impaired autophagy flux as a consequence of stalled omegasomes
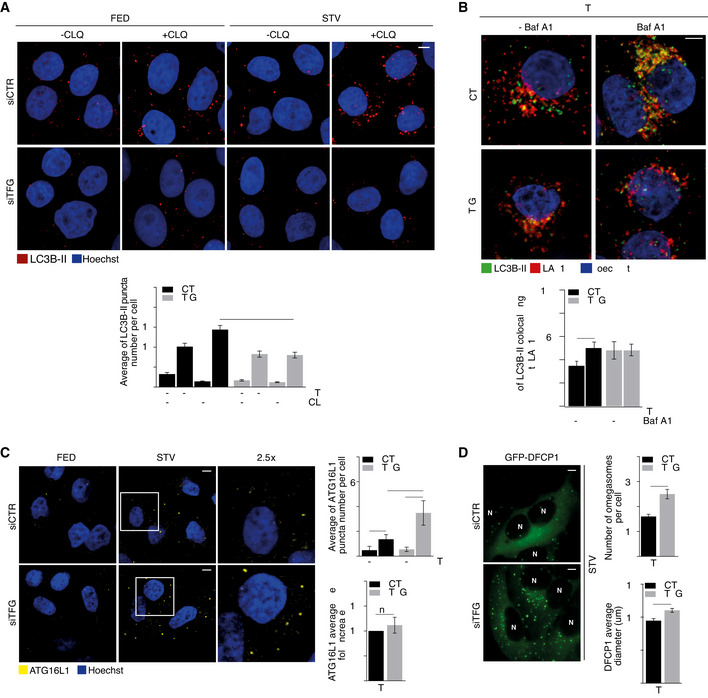
In order to evaluate whether or not TFG knockdown had an impact on the autophagy flux, we analysed the LC3B status by different approaches. First, a significant reduction in the number of LC3B puncta, which measure early LC3B‐II recruitment to autophagosomes (Itakura & Mizushima, 2010), is visible only upon starvation (+STV/+CLQ), when comparing control with TFG‐depleted cells (Fig 3A). Second, Western Blot (WB) analyses reveal a significant decrease in LC3B‐II protein levels after both starvation and TFG depletion, in the presence of CLQ, when compared with controls (Fig EV3A). Third, the co‐localization between LC3B‐II puncta and LAMP1, an endosomal and lysosomal marker, shows that TFG depletion impacts on autophagosome clearance, when compared with control cells (Fig 3B). Altogether, these results support the concept of a global impairment of the autophagy process after TFG depletion.
Figure 3. Inhibition of TFG results in impaired autophagy flux.
-
AHeLa cells were transfected with specific RNAi oligonucleotides (siTFG) or unrelated oligonucleotides as a negative control (siCTR) and grown in fed (FED) or starvation conditions (STV) in the presence or absence of CLQ for 1 h. Cells were then fixed and stained using LC3B‐II (red) antibody. Analysis of LC3B‐II puncta number is reported (bottom). All data are expressed as the mean value ± SEM. Statistical analysis were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. ****P < 0.0001 (n = 4 independent experiments, n ≥ 25 fields analysed). Scale bar 5 μm.
-
BHeLa cells were transfected as in A) and cultured in starvation conditions (STV) in the presence or absence of bafilomycin A1 (Baf A1) for 1 h. Cells were then fixed and stained using LC3B‐II (green) and LAMP1 (red) antibodies. Hoechst was used to mark nuclei. Co‐localization analyses were performed by ImageJ plugin Jacop. Values of Mander’s coefficient for LC3B‐II are expressed as mean ± SEM. Statistical analysis were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 (n = 3 independent experiments, n ≥ 13 fields analysed). Scale bar 5 μm.
-
CHeLa cells were transfected as in A) and grown in fed (FED) or starvation conditions (STV) for 1 h. Cells were then fixed and stained using ATG16L1 (yellow) antibody. ATG16L1 puncta number (upper right) and the average size of ATG16L1 as fold increase (bottom right) were reported. All values are expressed as mean ± SEM. Statistical analyses were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. **P < 0.01 and ****P < 0.0001 or by unpaired Student’s t‐test when two groups were compared. ns, not significant (n = 3 independent experiments, n ≥ 9 fields analysed). Scale bar 5 μm.
-
DHEK293 cells stably expressing GFP‐DFCP1 were transfected as in A) and cultured in starved conditions for 40’. Cells were visualized with cellSens microscope for live‐cell imaging. Pictures show the images taken at the same number of frames for each video. Analyses of the number of DFCP1 structures per cell (upper right) and the mean of omegasomes structures diameter (μm) (bottom right) were reported. N, nucleus. All values are expressed as mean ± SEM. Statistical analyses were performed by unpaired Student’s t‐test. *P < 0.05 and ***P < 0.001 (n = 3 independent experiments). Scale bar 5 μm.
Source data are available online for this figure.
Next, given the impact of TFG on the ULK1 complex, we decided to evaluate whether loss of TFG also affected the number and size of other early autophagy proteins: ATG16L1, a FIP200‐interacting protein (Gammoh et al, 2013; Nishimura et al, 2013), belonging to the ATG12–5/16 complex, and DFCP1, a marker of omegasomes (Karanasios et al, 2013). As shown in Fig 3C, we found a significant increase in ATG16L1 puncta formation after TFG downregulation upon starvation. Omegasome structures were then analysed by using an HEK293 cell line stably expressing green fluorescence protein (GFP)‐DFCP1 (Axe et al, 2008). Interestingly, DFCP1‐labelled structures are significantly increased in both number and size upon TFG depletion, as shown in Fig 3D. Our data thus suggest that depletion of TFG causes a significant increase of early pro‐autophagy complexes, most likely by inducing omegasome stalling. This is due to the effect of TFG on ULK1 since, as shown in Fig EV3B, the increment of ATG16L1 puncta is overturned by ULK1 depletion.
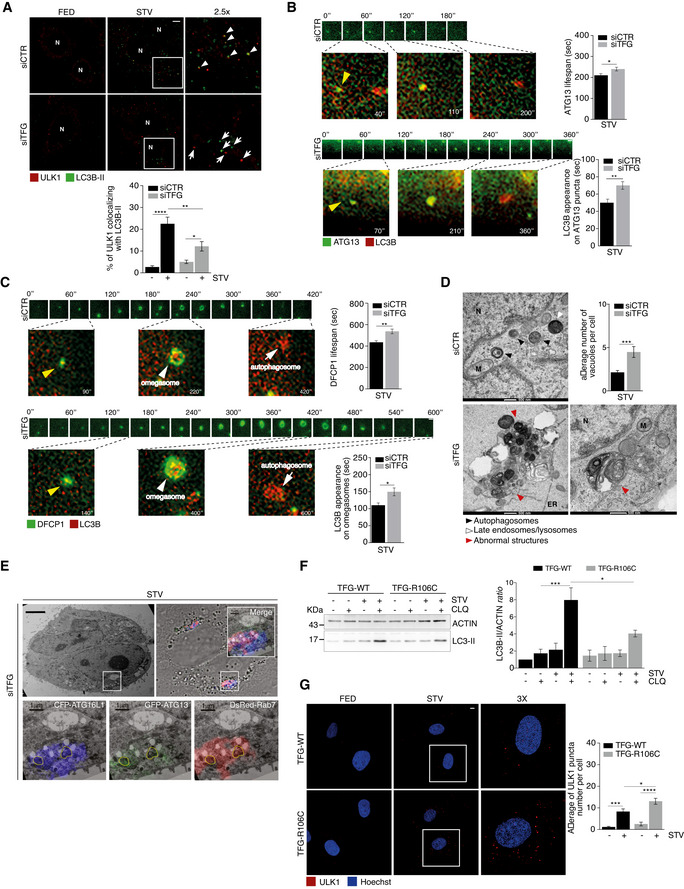
Further, we evaluated the association and the dynamics between IM components (ULK1 complex, DFCP1) and LC3B, since DFCP1 marks a transient omegasome, whereas LC3B is recruited shortly after DFCP1 at the omegasome and becomes then trapped on the inner membrane of a complete autophagosome until its degradation. Thus, we proved that starvation is able to promote a significant reduction in endogenous ULK1‐LC3B association after TFG depletion in HeLa cells (Fig 4A). Moreover, by a co‐IP assay, we found a decrease in ULK1‐LC3B‐II interaction in the same conditions (Fig EV4A). Next, we overexpressed ATG13, a reliable marker (and less cytotoxic than ULK1 itself) of the ULK1 complex, and DFCP1, as a marker of the omegasome, to examine in depth the dynamics of their association with LC3B by means of a live imaging‐based approach. To this aim, we employed HEK293 cells stably expressing GFP‐ATG13 or GFP‐DFCP1, respectively, in which we co‐transfected a plasmid encoding for cyan fluorescence protein (CFP)‐LC3B, in the presence or absence of TFG. In Fig 4B‐C (and Fig EV4, EV5; Movies [Link], [Link]) is shown that, upon TFG downregulation, the lifespan of both ATG13 (340 s vs. 200 s in control conditions) and DFCP1 (580 s vs. 400 s in control conditions) is significantly extended. Also, in the same condition, i.e. upon TFG depletion, the appearance of LC3B on ATG13 particles (70 s vs. 40 s in control conditions) or DFCP1 structures (140 s vs. 90 s in control conditions) shows a significant delay. In sum, these results indicate that depletion of TFG perturbs the correct dynamic relationships between pro‐autophagic proteins.
Figure 4. Inhibition of TFG results in stalled omegasomes and accumulation of abnormal structures.
-
AHeLa cells were transfected with specific RNAi oligonucleotides (siTFG) or unrelated oligonucleotides as a negative control (siCTR); after culturing cells in fed (FED) or starvation conditions (STV) for 1 h, cells were fixed and labelled with the indicated antibodies. Co‐localization analysis was performed by using ImageJ Jacop plugin and mean ± SEM of Mander’s coefficients of ULK1, expressed as percentage, is reported (bottom). White arrowheads point at co‐localization events between ULK1 and LC3B‐II; white arrows track puncta not co‐localizing; N, nucleus. Statistical analyses were performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05; **P < 0.01; and ****P < 0.0001 (n = 3 independent experiments, n ≥ 16 fields analysed). Scale bar 5 μm.
-
B, CHEK293 cells stably expressing either GFP‐ATG13 (B) or GFP‐DFCP1 (C) were transiently transfected with specific RNAi oligonucleotides (siTFG) or unrelated oligonucleotides as a negative control (siCTR), together with CFP‐LC3B plasmid (visualized in red) and cultured in starved conditions for 40’. Wide‐field live‐cell imaging of starved cells was taken by cellSens microscope. Representative images of lifespan of both ATG13 (B) and (C) are reported. Images of both ATG13 (B) and DFCP1 (C) forming association with LC3B are also shown. The appearance of LC3B on ATG13 (B) (bottom right) and on DFCP1 (bottom right) (C) puncta was quantified and reported. All values are expressed in seconds (s) as mean ± SEM. Yellow arrowheads point at the DFCP1 and LC3B particles in the first frame from their onset, and white arrowheads and white arrows show omegasomes and autophagosomes as indicated. Statistical analyses were performed by unpaired Student’s t‐test. *P < 0.05 and **P < 0.01 (n = 3 independent experiments, n = 50 events/condition analysed). Scale bar 1 μm.
-
DHeLa cells were transfected as in A). Cells were imaged by TEM. Representative electron micrographs (x26500) are reported. The average number of abnormal structures per cell is shown (bottom graph). N, nucleus; M, mitochondria; and ER, endoplasmic reticulum. The average number of abnormal vacuoles per cell was evaluated (upper right). Statistical analysis was performed by unpaired Student’s t‐test. ***P < 0.001. A minimum of 50 cells were observed. Cell count was performed (n = 3 independent experiments, n ≥ 51 cells analysed). Data are presented as mean ± SEM. Scale bar 500 nm as indicated.
-
EHeLa cells stably expressing GFP‐ATG13 and CFP‐ATG16 were transiently co‐transfected with DsRed‐Rab7 and TFG siRNA (siTFG). Cells were cultured in starved conditions for 1 h. Representative electron micrographs (x3900) and merge of the three fluorophores CLEM are shown. Micrographs at higher magnification (x13500) of a region showing the three fluorophores split and merged are reported. Scale bars: 5 μm upper panels, 1 μm bottom panels.
-
FControl (TFG‐WT) and patient’s (TFG‐R106C) fibroblasts were grown in fed or starvation conditions in the presence or absence of CLQ for 1 h and then lysed. Protein extracts were analysed by WB to detect ACTIN and LC3B‐II as indicated. Densitometry analysis of LC3B‐II over ACTIN is shown (right). All data are expressed as the mean ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05 and ***P < 0.001 (n = 3 independent experiments).
-
GControl (TFG‐WT) and patient’s (TFG‐R106C) fibroblasts were grown in fed or starvation conditions for 1 h. Cells were fixed, permeabilized and labelled with anti‐ULK (red) antibody. Hoechst was used to stain nuclei. The average of ULK1 puncta number per cell is shown (right). All data are expressed as the mean value ± SEM. Statistical analysis was performed by two‐way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05; ***P < 0.001; and ****P < 0.0001 (n = 3 independent experiments, n ≥ 14 fields analysed). Scale bar 5 μm.
Source data are available online for this figure.
Next, to monitor at an ultrastructural level the appearance of disturbed autophagosomes in TFG‐depleted conditions, we performed a detailed transmission electron microscopy (TEM) analysis (Fig 4D). Indeed, upon TFG depletion and autophagy induction, we observed the occurrence of what we termed "Abnormal structures" (As), containing multiple smaller compartments, multilamellar structures and single‐membrane vesicles including electron‐dense intraluminal material; by contrast, in control starved cells no significant accumulation of waste materials, a sign of proper execution of autophagy, was detectable. Indeed, a cytosolic accumulation of such As is compatible with an impairment of autophagy, and these structures were in fact positive for ATG13, ATG16L and RAB7, as shown by correlative EM (CLEM) experiments (Fig 4E).
Of the highest interest in biomedicine, mutations within the TFG coiled‐coil domain (amino acids 97–124) have been proposed to impair its function and underlie early‐onset forms of HSP (Beetz et al, 2013; Harlalka et al, 2016). Thus, we tested fibroblasts from a patient with homozygous variant p.R106C in TFG (Catania et al, 2018) and observed a reduction in autophagy flux (Figs 4F and EV4D and F), a significant accumulation of ULK1 puncta when compared with controls (Fig 4G), together with a massive presence of As (Fig EV4E). Additionally, decreased autophagy flux was observed by exogenously expressing an HA‐TFGR106C mutant construct in HeLa cells (Fig EV4G).
Altogether, these results clearly indicate that TFG is important for a proper autophagy progression and more precisely in the transition phase between autophagosome initiation and elongation. Also, this role may be relevant in HSP.
The role of TFG in autophagy requires its LIR‐dependent association with LC3C
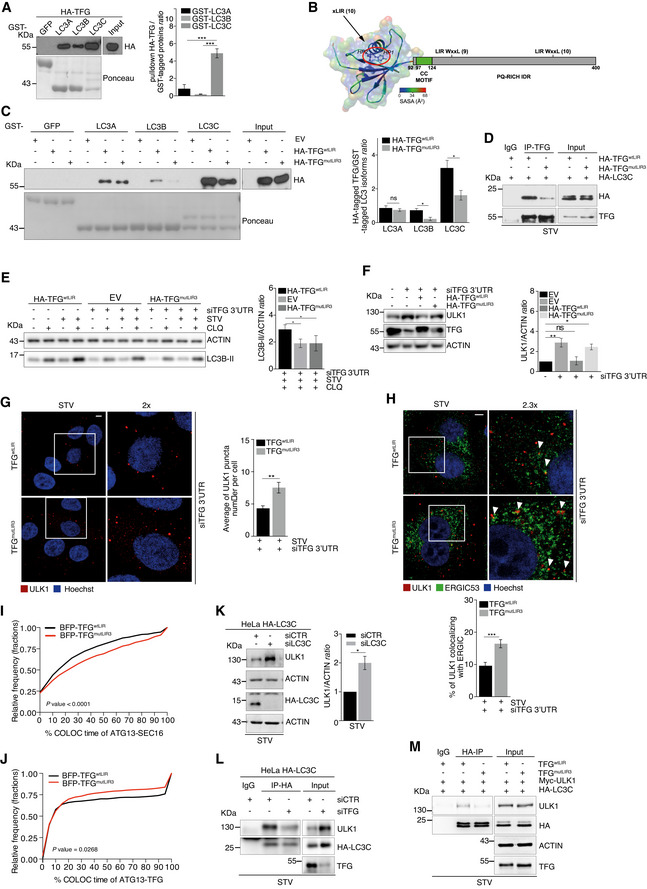
Given that TFG is necessary for the recruitment of LC3B to IM components, and since ERGIC/ERES compartments represent a primary membrane source necessary to trigger LC3B lipidation (Ge et al, 2013; Ge et al, 2014), we investigated TFG interaction with individual members of the LC3 family. First, we performed a GST pulldown assay by using human LC3 isoforms (LC3A, LC3B and LC3C) and found that TFG is able to bind all LC3 members with different strength and with a clear preference for LC3C (Fig 5A). Also, LC3C, at variance with LC3A‐B, clearly co‐immunoprecipitates with TFG (Fig EV5A). Of note, since most proteins that interact with LC3 members are themselves degraded by autophagy, we induced autophagy by starvation and treated cells with ammonium chloride (NH4Cl), a well‐known lysosome inhibitor, in a time course. As shown in Fig EV5B, no changes in TFG protein levels are detected, suggesting that TFG is not merely an autophagy substrate but may act as a regulatory ATG8‐interacting protein. Indeed, ATG8‐interacting proteins bind non‐covalently both LC3 and GABARAP proteins via a short linear sequence named LIR (Rogov et al, 2014). We identified three possible LIR motifs of TFG with similar position‐specific scoring matrix (PSSM) (Fig EV5C) located in the regions 31–36 (LIR1), 166–171 (LIR2) and 361–366 (LIR3), respectively. We then analysed their propensity to disorder or their capability to be located in folded protein domains. We thus noticed that the LIR 31–36 is located within a predicted PB1 domain for TFG, a domain also found in another LIR‐containing protein, p62, and known to be involved in protein–protein interactions (Kirkham, 1974). The region 166–171, which has the lowest PSSM score, is not predicted to be fully disordered according to the consensus in MobiDB, and it has small propensity for helical structures according to an analysis with FELLS. The LIR in the region 361–366 thus seems, in principle, to be the most suitable candidate due to its localization in a disordered region of the protein with also “anchor” motifs, which are common to LIRs (Jacomin, et al, 2016). Moreover, we obtained a model of the TFG PB1 domain (residues 10–91) by using as a template the PB1 domain from protein kinase C (Fig 5B and Table EV1) and used this model to rule out that the LIR residues 31–36 could in fact be accessible for interaction with the LC3s. The two key residues for interaction with the hydrophobic grooves of LC3 proteins, Y33 and L36, featured a solvent accessibility of their side chain atoms of 26 and 6.2%, respectively (Birgisdottir, et al, 2013; Johansen & Lamark, 2020). This confirms that the region 31–36, despite containing a sequence similar to a LIR motif, is almost buried in the PB1 domain and not available for interaction with LC3 proteins. Our analyses thus point to LIR3, located in the C‐terminal region 361–366 (361‐PGFTSL‐366), as a bona fide functional LIR for interaction between TFG and the LC3s, with 363F and 366L occupying the hydrophobic HP1 and HP2 pockets, respectively. To next verify whether TFG binds human ATG8 proteins in a LIR‐dependent manner, we generated two HA‐tagged TFG LIR mutant constructs (HA‐TFGmutLIR2 and HA‐TFGmutLIR3). In particular, the aromatic residue phenylalanine (F) at position 1 and the hydrophobic residue leucine (L) at position 4 were changed into alanine (A) in both mutant constructs (Fig EV5D). By GST pulldown assay, we next tested whether each TFG‐LIR3 mutant construct was able or not to abrogate the capability of TFG to interact with LC3 and GABARAP family members. As shown in Figs 5C and EV5E, mutations of the LIR3 motif decrease significantly the affinity of TFG for both LC3B and LC3C, but not for LC3A or any GABARAP family members. Taking into consideration that i) LC3C recruits the secretion‐autophagy co‐regulating protein TECPR2 to ERES (Stadel et al, 2015), ii) LC3C is required for recruiting other ATG8 orthologues to the site of autophagosome formation (Von Muhlinen et al, 2012), iii) LC3C shows the strongest affinity for TFG and iv) no obvious co‐immunoprecipitation of LC3A and LC3B with TFG was detected (Fig EV5A), we analysed more in depth the involvement of LIR3 in the TFG‐LC3C binding. First, we confirmed the expression of endogenous LC3C in HeLa cells, by analysing its mRNA levels (Fig EV5F); next, we took advantage from a HeLa cell line harbouring endogenously expressed HA‐tagged LC3C. As shown in Fig 5D, we confirmed by co‐IP that the LC3C‐TFG interaction is dramatically decreased in the presence of TFGmutLIR3, at variance with HA‐TFGmutLIR2 (Fig EV5G and H).
Figure 5. The role of TFG in autophagy requires its LIR‐dependent association with LC3C.
-
APulldown assay was performed incubating purified GST‐tagged LC3 (A, B and C) isoforms and HA‐TFG‐transfected HeLa cell lysates. HA‐TFG protein levels were analysed by WB; LC3 isoforms and GFP are shown in the Ponceau staining. Densitometry analysis of TFG over GST‐tagged LC3 isoforms is reported in the graph (right). Data are expressed as mean ± SEM. Statistical analysis was performed by one‐way ANOVA followed by Tukey’s multiple comparison test. ***P < 0.001 (n = 3 independent experiments).
-
BThe model illustrates the domain composition of TFG, including a PB1 domain (residues 10–91), a coiled‐coil motif (CC, residues 97–124) and a C‐terminal region including a proline–glutamine‐rich mostly disordered region. The location of the predicted LIR motifs is also reported, along with the associated PSSM scores from iLIR. The model of the 3D structure of the PB1 domain is coloured according to the solvent accessibility of each residue as derived by Chimera, and the residues for the predicted LIR motif 31–36 for HP1 and HP2 hydrophobic pockets of LC3 proteins are highlighted as sticks.
-
CPulldown assays were performed by using purified GST‐LC3 (A, B and C) isoforms, each incubated together with empty vector (EV) or HA‐TFGwtLIR‐ or HA‐TFGmutLIR3‐transfected HeLa cell lysates, respectively. Ponceau staining reports GST‐LC3 isoforms and GST‐GFP used as a negative control. WB analyses show HA‐TFGwtLIR or HA‐TFGmutLIR3 in the pulldown and in total extracts (Input). Densitometry analyses of HA‐tagged TFG normalized over GST‐tagged LC3 isoforms (Ponceau) were performed and reported (right). Data are mean value ± SEM. Statistical analysis was performed by multiple t‐test. *P < 0.05 and ns, not significant (n = 3 independent experiments).
-
DHeLa cells were co‐transfected with HA‐LC3C together with HA‐TFGwtLIR or HA‐TFGmutLIR3 plasmids. After 30’ of starvation, protein extracts were immunoprecipitated using an anti‐TFG antibody or IgG as a negative control. LC3C and TFG were analysed by WB.
-
EHeLa cells were co‐transfected with 3’UTR TFG siRNA together with HA‐TFGwtLIR or an empty vector (EV) or TFGmutLIR3 plasmids, expressed at near endogenous levels. Cells were cultured with complete or starvation (STV) media and grown in the presence or absence of CLQ for 1 h. ACTIN and LC3B‐II protein levels are shown. Densitometry analysis of LC3B‐II over actin for indicated conditions is shown in the graph (right). Data are mean value ± SEM. Statistical analysis was performed one‐way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.05 (n = 3 independent experiments).
-
FHeLa cells were co‐transfected with unrelated oligonucleotides as a negative control (siCTR) and empty vector (EV) or 3’UTR siRNA TFG siRNA together with EV or HA‐TFGwtLIR or TFGmutLIR3 plasmids, expressed at near endogenous levels. Protein extracts were analysed by WB to detect ULK1, TFG and ACTIN as indicated. Densitometry analysis of ULK1 over ACTIN is shown (right). Data are mean value ± SEM. Statistical analysis was performed by one‐way ANOVA followed by Dunnett’s multiple comparison test. *P < 0.05; **P < 0.01; and ns, not significant (n = 4 independent experiments).
-
G, HHeLa cells stably expressing untagged TFGwtLIR or TFGmutLIR3 were transfected with 3’UTR TFG siRNA, starved for 1 h, fixed, permeabilized and immunolabelled with ULK1 (red) alone or ULK1 (red) and ERGIC53 (green) (H). Hoechst was used to stain nuclei. ULK1 puncta number (G) was analysed and reported as mean ± SEM in the graph (right) (n = 3 independent experiments, n = 24 fields analysed). Co‐localization analyses between ULK1 (red) and ERGIC53 (green) (H) were performed by Jacop plugin. Values of Mander’s coefficient for ULK1, expressed as percentage, are reported as mean ± SEM (bottom graph) (n = 3 independent experiments, n = 19 fields analysed). White arrowheads point at co‐localization events between ULK1 and ERGIC53. Statistical analyses were performed by unpaired Student’s t‐test. **P < 0.01 and ***P < 0.001. Scale bar 5 μm.
-
IHeLa cells stably expressing GFP‐ATG13 together with BFP‐TFGwtLIR or BFP‐TFGmutLIR3 were co‐transfected with a siRNA targeting endogenous TFG (siTFG 3’UTR) and mCherry‐SEC16 plasmid and then cultured for 40’ with starvation medium. Cells were visualized with cellSens microscope for live‐cell imaging. Analysis of the percentage of GFP‐ATG13 lifetime co‐localizing with mCherry‐SEC16 in the presence of TFGwtLIR or TFGmutLIR3 is reported in the cumulative relative frequency plot. Statistical analysis was performed by Kolmogorov–Smirnov test. P < 0.0001 (n = 3 independent experiments, n ≥ 995 number of co‐localization events analysed).
-
JHeLa cells stably expressing GFP‐ATG13 together with BFP‐TFGwtLIR or BFP‐TFGmutLIR3 were transfected with 3’UTR TFG siRNA to downregulate endogenous TFG. Cells were cultured for 40’ with starvation medium and visualized with cellSens microscope for live‐cell imaging. Analysis of the percentage of GFP‐ATG13 lifetime co‐localizing with BFP‐TFGwtLIR or BFP‐TFGmutLIR3 is reported in the cumulative relative frequency plot. Statistical analysis was performed by Kolmogorov–Smirnov test. P = 0.0268 (n = 3 independent experiments, n ≥ 379 number of co‐localization events analysed).
-
KHeLa cells harbouring endogenously HA‐tagged LC3C were transfected with specific RNAi oligonucleotides (siLC3C) or unrelated oligonucleotides as a negative control (siCTR) and cultured in starvation medium for 30’. Protein extracts were analysed by WB to detect ULK1, ACTIN and HA‐LC3C as indicated. Densitometry analysis of ULK1 over ACTIN is reported (right). Data are expressed as mean ± SEM. Statistical analysis was performed by unpaired Student’s t‐test. *P < 0.05 (n = 3 independent experiments).
-
LHeLa cells harbouring endogenously HA‐tagged LC3C were transfected with a siRNA targeting TFG (siTFG) or with a non‐targeting control (siCTR). After 30’ of starvation, cells were lysed, and protein extracts were immunoprecipitated by using an anti‐HA antibody or IgG as negative control. Protein extracts were analysed by WB to detect ULK1 and HA‐LC3C as indicated.
-
MHeLa cells stably expressing untagged TFGwtLIR or TFGmutLIR3 were co‐transfected with HA‐LC3C and Myc‐ULK1 plasmids as indicated. Cells were starved for 30’, and protein extracts were immunoprecipitated using an anti‐HA antibody or IgG as negative control. Protein extracts were analysed by WB to detect ULK1, HA‐LC3C, TFG and ACTIN as indicated.
Source data are available online for this figure.
Next, to assess the functional importance of the TFG‐LC3C interaction in autophagy, we evaluated the autophagy flux after complementing TFG‐deficient cells with TFGwtLIR or TFGmutLIR3, respectively. As shown in Fig 5E, LC3B‐II levels are reduced by the TFG mutant construct, when compared with the WT form. Consistent with this observation, ULK1 canonical protein levels are recovered upon complementation with TFGwtLIR but not with TFGmutLIR3 (Fig 5F). Next, we performed an IF analysis to test the importance of TFG‐LC3C interaction in both ULK1 puncta formation and intracellular localization. As shown in Fig 5G and H, the integrity of the LIR motif is critical for both formation and distribution of ULK1 complex puncta. Indeed, in live imaging experiments, by analysing co‐tracking percentage of ATG13 puncta with both ERES/ERGIC (labelled by SEC16, Figs 5I and EV5I) and TFGwtLIR or TFGmutLIR3, respectively (Figs 5J and EV5J), we found that the TFG‐LC3C interaction is functional to ULK1 complex distribution and to the TFG‐ULK1 association. In line with this, we were able to detect both ULK1 and TFG in complex with LC3C at an endogenous level (Fig EV5K).
Next, we asked whether or not depletion of ATG8 proteins resulted in a similar phenotype as that detected upon TFGmutLIR3 expression. Intriguingly, only knockdown of LC3C (Figs 5K and EV5L–O) significantly increases ULK1 protein levels. Importantly, siLC3C is sufficient per se to impair the autophagy flux (Fig EV5P).
Finally, since ULK1 function in autophagy depends on its interaction with ATG8 proteins (Alemu et al, 2012; Kraft et al, 2012; Wirth et al, 2019), we investigated whether TFG could favour ULK1‐ATG8s interaction. By co‐IP experiments, we found that knockdown of TFG strongly decreases ULK1 complex association with both LC3C and GABARAP during starvation (Figs 5L and EV5Q). This function also depends on the TFG LIR motif, since TFGmutLIR3 induces a similar effect (Fig 5M).
Altogether, these findings indicate that the TFG‐LC3C interaction is crucial for autophagy progression by regulating ULK1 accumulation, ULK1 puncta localization and ULK1‐LC3C/GABARAP binding.
Discussion
Emerging evidence highlights the important crosstalk existing between autophagy and the early secretory pathway, thus emphasizing the multitasking role that proteins involved in a specific process could play in the other one, in response to cellular conditions. In recent years, it became quite apparent that COPII vesicles are important for autophagosome biogenesis (Ge et al, 2014). At the interface between ER(ES) and ERGIC, TFG promotes uncoating of COPII vesicles after their release from the ER, with this providing a platform that facilitates fusion between these vesicles and the ERGIC compartment (Johnson et al, 2015; Hanna et al, 2017). In this study, we identified TFG as a positive regulator of autophagosome formation during autophagy induction by nutrient deprivation. Indeed, depletion of TFG leads to several consequences for functional autophagy. First, we found TFG as an ULK1‐interacting protein and that its absence promotes higher levels of ULK1, ATG13 and DFCP1 proteins, suggesting a role for this factor in the transition phase between autophagosome initiation and elongation. In line with the increase of early autophagy markers, ULK1 activity is not impaired upon TFG depletion, and it is still able to recruit proteins that may act at the early steps of autophagosome formation. Of the highest importance, here we show that the effect of TFG depletion on ULK1 is not accounted for by the COPII pathway. In fact, under starvation conditions, at variance with depletion of SEC23 and SEC16, the absence of TFG revealed an increase in puncta number and size of ULK1, ATG13 and DFCP1.
It is well known that ULK1 protein levels are finely regulated upon autophagy induction. In particular, ULK1 is targeted by several E3 ubiquitin ligases, such as NEDD4L (Nazio et al, 2016) and CUL3‐KLHL20 (Liu et al, 2016), which lead to ULK1 degradation by the ubiquitin–proteasome system. Interestingly, ultrastructural data show that, at variance with control conditions, the absence of TFG triggers accumulation of abnormal structures (As) containing multiple smaller compartments and membranes that are compatible with immature and aborted autophagosomes. These structures, positive for the autophagosome markers ATG13 and ATG16L, are quite large in size, possibly because they recruit membranes that are not pre‐loaded with mATG8 orthologs, generating as a consequence an expanded isolation membrane that cannot progress into a complete and fully functional autophagosome. Of note, the structures we found by EM analyses are reminiscent of characteristic multilamellar vesicles called MVBs (multivesicular bodies), and they are also labelled by the MVB marker Rab7. However, since TFG has been identified as a potential interactor of some proteins involved in sorting and formation of MVB (Schlundt, et al, 2009; Hein, et al, 2015), we cannot exclude that the absence of TFG could also have an effect on this pathway.
In addition, loss of TFG reduces the association between LC3B and the IM components, leading to a delay of dissociation of early autophagy proteins from membranes and an impairment in the autophagosome transition phase between initiation and elongation.
Interestingly, by dissecting the molecular details of the TFG role in autophagy, we identified TFG as novel and unexplored LC3C‐binding protein. Indeed, our data argue for a direct close interaction between LC3C and TFG, through a canonical LIR motif that we have mapped in a P/Q rich TFG region. At variance with the established importance of LC3B in autophagy, only a few studies have focused on LC3C in terms of its subcellular localization and regulation (Muhlinen et al, 2012; Stadel et al, 2015; Liu et al, 2017; Le Guerroué et al, 2017; Wetzel et al, 2020). LC3C is described in such reports as a key factor in regulating various selective autophagy processes. Here we demonstrate a role for the axis TFG‐LC3C in regulating starvation‐dependent autophagy. Indeed, disruption of the TFG‐LC3C interaction influences ULK1 protein levels, puncta formation and distribution and consequently autophagy progression, similarly to TFG depletion. Most likely, this interaction is also functional for a proper localization of early autophagy proteins and for the correct recruitment of LC3B to the IM. Another interesting finding is that TFG favours both ULK1‐LC3C and ULK1‐GABARAP interaction. ULK1 complex members (ULK1, ATG13 and FIP200) bind strongly GABARAP, GABARAP‐L1, GABARAP‐L2 and LC3C, and weakly LC3A and LC3B (Alemu et al, 2012). We could thus speculate that LC3C may cooperate with TFG as a recruiting anchor at the omegasome sites for autophagosome formation and that the TFG‐LC3C interaction could be crucial in globally regulating ULK1 during autophagy.
Interestingly, from a biomedical viewpoint, emerging evidence shows a tight correlation between inherited neuropathy‐associated genes and autophagy alteration. In particular, mutations in RAB7 (protein required for autophagosome and lysosome fusion) cause CMT2B neuropathy (Verhoeven et al, 2003), and CMT2B patients carrying the V162M RAB7A mutation show alteration in autophagy flux. In addition, the function of TECPR2 in maintaining ER export and the formation of early autophagosome intermediates is disrupted by HSP‐causing mutations and this, in turn, causes an autophagy impairment (Stadel et al, 2015). Of note, several mutations in TFG (such as R106C, G269V and P285L) have been demonstrated to cause different neuropathologies (HSP, CMT2 and HMSN‐P). The R106C TFG mutant induces defects in the octamer assembly, impairing the TFG structure; the G269V TFG mutant produces cytoplasmic aggregates, which would trap wild‐type TFG. Last, Ishiura and colleagues demonstrated that motor neurons in HSMN‐P of patients show TFG/ubiquitin‐ and/or TDP‐43 (TAR DNA‐binding protein 43 kDa) immunopositive cytoplasmic inclusions (Ishiura et al, 2012). The finding that HSP patient fibroblasts carrying mutated TFG display reduced autophagy levels, defective ULK1 puncta turnover and abnormal structures in the cytoplasm highlights the importance of our discovery in biomedicine. In the case investigated, defective autophagy could be postulated as a possible contributor to ontogenesis or progression of the HSP disease; in particular, neurons are sensitive to accumulation of protein aggregates or damaged organelles and altered autophagy is now recognized as an important cause of neuronal degeneration in both central and peripheral nervous system. In sum, the role of TFG in autophagy could thus be key in a novel pathogenic mechanism for neuropathy associated with its mutation, and our study supports the idea that targeting autophagy may represent a potential approach to improve patients conditions.
Finally, TFG has been found to play a role in oncogenesis, especially as a fusion protein; moreover, several reports suggest that TFG alone could act both as a tumour suppressor and an oncogene, although its role in this context remains unclear (Chen & Tseng, 2014). The fusion partners of TFG include NTRK1 or ALK, and this results in the generation of an oncogene. Interestingly, in both instances, a role for TFG in targeting kinase activities of NTRK1 and ALK to ER exit sites has been found and this, in turn, causes tumour transformation. It would be thus of the highest interest to study a role for TFG‐fusion proteins in autophagy mis‐regulation, a condition well known to affect cancer cell survival.
Materials and Methods
Human subjects
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Skin biopsies were performed after obtaining informed consent approved by the Ethics Committee of Fondazione IRCCS Istituto Neurologico Carlo Besta.
Cell culture and treatments
HeLa and HEK293 cells were cultured in DMEM (EuroClone, ECB7501L) supplemented with 10% foetal bovine serum (FBS) (GIBCO, 10270106), 2 mM L‐glutamine (EuroClone, ECB3000D) and 1% penicillin/streptomycin (EuroClone, ECB3001B) solution. HEK293 cells stably expressing GFP‐ATG13 or GFP‐DFCP1 were also selected in 800 mg/ml Geneticin (GIBCO, 11811031). HeLa cells harbouring endogenously HA‐tagged LC3C, GABARAP, GABARAPL1 and GABARAPL2 were cultured in DMEM with 10% FBS, 5% glutamine and selected in 4 μg/ml of blasticidin (GIBCO, A1113903). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. The induction of autophagy by nutrients starvation was obtained washing the cells with phosphate‐buffered saline (PBS) (EuroClone, ECB4053) and incubating with Earle’s balanced salt solution (EBSS; Sigma‐Aldrich, 254134) for the indicated time periods. Autophagy induction of HEK293 or HeLa cells for live imaging experiments was accomplished washing three times with pre‐warmed starvation medium (140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 1% bovine serum albumin (BSA; Sigma‐Aldrich A9647) and 20 mM Hepes pH 7.4) and incubating at 37°C under 5% CO2 for 40 min. Lysosome activity was inhibited for 1 h with 20 μM CQ (Sigma‐Aldrich, C6628) or with 10 mM NH4Cl (Sigma‐Aldrich, 254134) or with 100 nM of bafilomycin A1 (Baf A1) (Selleck chemicals, S1413) for the indicated time periods. Protein translation was inhibited using 50 μg/μl cycloheximide (CHX; Sigma‐Aldrich, C4859). All used cell lines were transiently transfected with Lipofectamine 2000 (Thermo Fisher Scientific, 11668‐019) according to the manufacturer’s instructions. Patient fibroblasts and control were obtained from skin biopsy as previously reported (Catania et al, 2018). All experiments were performed on cells at the same passage number. Cell lines were regularly tested and verified to be mycoplasma negative by PCR; all cell lines tested negative for mycoplasma.
Plasmids and retroviral transduction
Plasmid encoding for wild‐type Myc‐ULK1 was provided by S.A. Tooze (The Francis Crick Institute, Lincoln's Inn Fields Laboratories, London, England, UK). DsRed‐rab7WT plasmid was by Addgene (#12661). Vectors encoding for GST‐GFP, GST‐LC3A, GST‐LC3B, GST‐LC3C, HA‐LC3A, HA‐LC3B and HA‐LC3C were a kind gift from C. Behrends. HeLa cell lines harbouring endogenously HA‐tagged LC3C, or GABARAP, or GABARAPL1, or GABARAPL2 were obtained as previously described (Le Guerroué et al, 2017). GST‐GABARAP, GST‐GABARAPL1 and GST‐GABARAPL2 were a gift from I. Dikic (Institute of Biochemistry II, Goethe University, Frankfurt Germany). Plasmid encoding for CFP‐LC3B, mCerry‐SEC16 together with HEK293 cells stably transfected with GFP‐ATG13 or GFP‐DFCP1 vectors were provided by N.T. Ktistakis (Babraham Institute, Cambridge, UK). To obtain pCI‐HA‐TFG plasmid, TFG was cleaved from pcDNA3‐TFG by KpnI/NotI and was sub‐cloned to the pCI‐HA vector purchased from Addgene. TFG mutant constructs were generated by using the site‐directed mutagenesis kit (Agilent Technologies, 200518). The sequences used are as follows: pCI HA‐TFGR106C R to C: 5' CAAGTCAGGTGAAATATCTC TGT CGAGAACTGATAGAACT 3'. pCI HA‐TFGmutLIR2: F to A, 5′‐GGCAGCAAGTATGTCTGCTGCTGATCCTTTAAAAAACCAAG‐3′; L to A, 5′‐GGCAGCAAGTATGTCTGCTGCTGATCCTGCAAAAAACCAAG‐3′. pCI HA‐TFGmutLIR3: F to A, 5′‐ATCAACCAAGACCAGGTGCTACTTCACTTCCTGGAAG‐3′; L to A, 5′‐CCAGGTGCTACTTCAGCTCCTGGAAGTACCATG‐3′. To obtain stable cell lines, TFGwtLIR and TFGmutLIR3 were sub‐cloned into the SFG (Quintarelli et al, 2018) retrovirus vector in frame with the 2A sequence and the truncated cell‐surface CD19 selectable marker. Then, the retroviral supernatants were produced in 293T cells, as previously described (Caruana et al, 2015). For live‐cell imaging or CLEM analysis, stable cell lines were obtained using the following vectors: TFGwtLIR and TFGmutLIR3 sub‐cloned into pMSCV‐IRES‐Blue FP (Addgene #52115), pMRX‐IP/SECFP‐ATG16A1 (Addgene #58994) and pMXs‐IP‐EGFP‐ATG13 (Addgene #38191), as described in the figure legends.
RNA Interference (RNAi)
RNAi was achieved by RNA oligonucleotide duplex listed in Table 1.
Table 1.
Oligonucleotides related to the experimental procedures
| Thermo Fisher Scientific | TFG | 5′‐ACAGCAGUACCAGGCGAGCAAUUAU‐3′ |
| 5′‐AUAAUUGCUCGCCUGGUACUGCUGU‐3′ | ||
| Sigma‐Aldrich | TFG‐3′UTR | 5′‐CCAAAAGACUCCAGUACUA‐3′ |
| 5′‐UAGUACUGGAGUCUUUUGG‐3′ | ||
| Thermo Fisher Scientific | SEC23A | 5′‐GGGUGAUUCUUUCAAUACU‐3′ |
| 5′‐AGUAUUGAAAGAAUCACCC‐3′ | ||
| Sigma‐Aldrich | SEC16 | 5′‐CCCAAGACUGCAGAACCCAGCUAUU‐3′ |
| 5′‐GCAGCUCUGGAACUUAGUA‐3′ | ||
| Sigma‐Aldrich | ULK1 | 5′‐GAGAACGUCACCAAGUGCAAGCUGU‐3’ |
| 5′‐ACAGCUUGCACUUGGUGACGUUCUC‐3′ | ||
| Thermo Fisher Scientific | MAP1LC3C | 5′‐GCUUGGCAAUCAGACAAGAGGAAGU‐3′ |
| 5′‐ACUUCCUCUUGUCUGAUUGCCAAGC‐3′ | ||
| Sigma‐Aldrich | MAP1LC3B | 5′‐CUCCCUAAGAGGAUCUUUAUU‐3′ |
| 5′‐AAUAAAGAUCCUCUUAGGGAG‐3′ | ||
| Sigma‐Aldrich | GABARAP | 5’‐GGUCAGUUCUACUUCUUGA‐3’ |
| 5′‐UCAAGAAGUAGAACUGACC‐3′ | ||
| Horizon/Dharmacon | GABARAPL1 | 5′‐CUAGUGCCCUCUGACCUUA‐3′ |
| 5′‐UAAGGUCAGAGGGCACUAG‐3′ | ||
| Sigma‐Aldrich | GABARAPL2 | 5′‐UGGGCUAGGUGCACCGUAA‐3′ |
| 5′‐UUACGGUGCACCUAGCCCA‐3′, |
2.5 × 105 cells per well were transfected with 100 pmol siRNA oligos in six‐well plates using Lipofectamine 2000 following the manufacturer’s instructions
Western blot analysis
Cells were rinsed in PBS on ice and lysed in RIPA buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl2 1% Triton X‐100, 0,25% sodium deoxycholate and 0,1% SDS) plus protease inhibitor cocktail (Sigma‐Aldrich, P8340), Na4VO3 0.1 mM (Sigma‐Aldrich, S6508), NaF 1 mM (Sigma‐Aldrich 201154) and β‐glycerophosphate 5 mM (Sigma‐Aldrich G9422).
Cell extracts were centrifuged at 13,000 rpm for 10 min at 4°C. Protein concentrations were determined with the Bio‐Rad Protein Assay Kit (Bio‐Rad, 5000113 and 5000114). Cell extracts or immunoprecipitates were denatured by adding 1 volume of 4 × Laemmli SDS sample loading buffer with β‐mercaptoethanol and boiled at 95°C for 10 min. Then, proteins were separated by SDS–PAGE. PVDF (Merck Millipore, IPVH00010) membranes were incubated with primary antibodies followed by horseradish peroxidase‐conjugated secondary antibody (Bio‐Rad, rabbit 1706515, mouse 1706516) and visualized with ECL (Merck Millipore, WPKLS0500). Densitometry analysis was performed using ImageJ software. Comparison between control and sample in the WB intensity measurement was made from the same WB. Of note, in some cases, WB was captured using digital cameras (Alpha Innotech FluorChem SP, or iBright FL1500), whereas others were captured on film (Aurogene, AU1102). Backgrounds in WB images from different experimental approaches were then equalized by changing their exposition post‐acquisition.
Antibodies
The primary antibodies used in this study were as follows: mouse anti‐HA tag (Santa Cruz Biotechnology, sc‐7392) and rabbit anti‐HA tag antibody (Santa Cruz Biotechnology, sc‐805), rabbit anti‐HA (Cell Signaling Technology, #3724), mouse anti‐TFG (Santa Cruz Biotechnology, sc‐515054) and rabbit anti‐TFG (Santa Cruz Biotechnology, sc‐98969), rabbit anti‐TFG (Abcam, ab156866), mouse anti‐TFG (Novus Biological, NBP2‐62212), mouse anti‐Myc (9E10) (Santa Cruz Biotechnology, sc‐40), rabbit anti‐LC3‐XP (D11) (Cell Signaling Technology, #3868), mouse LC3B (5F10) (Nanotools, 0231‐100), rabbit anti‐ULK1 (D8H5) (Cell Signaling Technology, #8054), rabbit anti‐ULK1 (N‐17) (Santa Cruz Biotechnology, sc‐10900), mouse anti‐GOLGIN‐97 (CDF4) (Thermo Fisher Scientific, A‐21270), rabbit anti‐ATG16L1 (D6D5) (Cell Signaling Technology, #8089), rabbit ATG9A (D4O9D) (Cell Signaling Technology, #13509) rabbit anti‐ATG13 (E1Y9V) (Cell Signaling Technology, #13468), mouse anti‐ATG13 (2H4.2) (Merck Millipore, MABC46), rabbit anti‐pS318‐ATG13 (Rockland, 600‐401‐C49), rabbit anti‐ATG14 (Cell Signaling Technology, #5504) rabbit pS29‐ATG14 (Cell Signaling Technology, #13155), rabbit anti‐Actin (Sigma‐Aldrich, A2066), rabbit anti‐GADPH (Sigma‐Aldrich, G9545), mouse anti‐Calnexin (AF18) (Santa Cruz Biotechnology, sc‐23954), mouse anti‐SEC31A (BD, 612350) rabbit anti‐SEC23A (Cell Signaling Technology, #8162) and mouse anti‐ERGIC53 (Enzo Life Science, ABS300‐0100). All antibodies are listed in Table EV2.
Immunoprecipitation assay
HeLa cells were rinsed with ice PBS and lysed in NP‐40 lysis buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% NP‐40 and protease inhibitor cocktail (Sigma‐Aldrich, P8340)) or with a buffer used for co‐IP with ATG8 proteins (10 mM Tris–HCl pH 7.4, 100 mM NaCl, 2 mM EDTA, 1% NP‐40). For endogenous co‐IP, was used a buffer containing 40 mM Hepes pH 7.4, 2 mM EDTA and 0,3% chaps, and for IP washes was added 150 mM of NaCl. IP assays were performed with 0.5 mg of lysates for co‐IP in overexpression, 1.5 mg of lysates for co‐IP in semi‐endogenous conditions and 2 mg of lysate in endogenous conditions. Lysates were then incubated with 1 μg, 1.5 μg or 2 μg, respectively, of indicated primary antibody at 4°C with rotation overnight (O/N) before addiction of 15 μl dynabeads protein G (Invitrogen, 10003D) and then incubated at 4°C for 1 h. Immunoprecipitates were then washed 5 × 5 min with its own lysis buffer or wash buffer and then denatured by adding 1 volume of 4 × Laemmli SDS sample buffer (Thermo Fisher Scientific, NP0007) with β‐mercaptoethanol and incubated at 95° for 10 min.
Pulldown assay
Recombinant GST‐fusion proteins were expressed in E. coli BL21 (DE3) (Promega, L1195) using 0.5 mM IPTG at 25°C O/N. Bacteria were lysed in NET‐N buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P‐40) containing EDTA‐free protease inhibitor mixture (Sigma‐Aldrich, P8465) and subjected to sonication. GST proteins were purified and immobilized on glutathione agarose (Thermo Scientific, 16100). HA‐TFGwtLIR or HA‐TFGmutLIR3 or positive control (supplementary Fig 5E and F) vectors were transiently transfected in HeLa cells as indicated. Then, cells were lysed with NET‐N buffer. GST pulldown assays were performed by incubating immobilized GST‐fusion proteins with protein extract derived from HeLa cells for 2 h at 4°C with gentle agitation. Unbound proteins were removed by washing the resins five times with NET‐N buffer. GST proteins were eluted from the beads with 10 mM reduced glutathione in 50 mM Tris–HCl (pH 8.0), for 30 min with gentle agitation at room temperature. The eluted samples were boiled for 5 min in sample loading buffer and separated by SDS–PAGE.
Immunofluorescence analysis
Cells were washed in PBS and fixed with 4% paraformaldehyde in PBS for 10 min. After permeabilization with 0.4% Triton X‐100 (Sigma‐Aldrich, X‐100, T9284) in PBS for 5 min. For LC3 staining, cells were fixed and permeabilized with cold methanol for 10’ at −20°. Coverslips were incubated for 1 h with 3% normal goat serum (Sigma‐Aldrich, NS02L) for blocking non‐specific signals. Cells were incubated (O/N) at 4°C with primary antibodies and 1% normal goat serum. Cells were then washed with PBS and incubated for 1 h and labelled with anti‐mouse (Thermo Fisher Scientific, A11017, A21425) or anti‐rabbit (Thermo Fisher Scientific A21430, A11070) secondary antibodies. Nuclei were stained with 1 μg/ml Hoechst for 5 min at room temperature. Images were obtained by several confocal microscopes, depending of hosting laboratory: Olympus FV1000 laser‐scanning confocal microscope, equipped with a 60x (NA 1.35) oil immersion objective (Tor Vergata University, Rome), Leica TCS‐SP8X laser‐scanning confocal microscope equipped with a 60x (NA 1.4) oil immersion objective (Bambino Gesù Pediatric Hospital, Rome) and Carl Zeiss LSM 780 Confocal 60x (NA 1.4) oil immersion objective and N‐Nikon Structured Illumination Microscopy (N‐SIM) with 100x (NA 1.49) oil immersion objective with lateral resolution of 120 nm (Babraham Institute, Cambridge). Image analysis has been performed using “Fiji” (Fiji Is Just ImageJ) open‐source software. Co‐localization analysis was performed by using “Jacop plugin”. For all co‐localization analyses, to evaluate the percentage of ULK1 puncta localized to several intracellular structures or particles, Mander’s coefficient was used. Images were processed to equally remove the background without affecting the original signal. Co‐localization values were represented as a percentage of pixels of a channel overlapping with the other channel. All acquisitions were performed in non‐saturated single focal central planes, and images were captured from randomly selected fields of view.
Live‐cell imaging and analysis of live‐cell imaging movies
Olympus cellSens, a wide‐field imaging system, with a 100x oil immersion objective and a sCMOS camera was used to capture images of live cells in Babraham Institute, Cambridge, for Fig 4B and C. Olympus cellSens with a 100x oil immersion objective and a CCD camera was used to capture images of live cells in the University of Ferrara, Italy, for the analysis in Fig 5I and J. HEK293 cells stably expressing GFP‐ATG13 or GFP‐DFCP1, or HeLa cells stably expressing GFP‐ATG13 and BFP‐TFGwtLIR or GFP‐ATG13 and BFP‐TFGmutLIR3 were plated onto 60‐mm dishes and transiently co‐transfected with the indicated plasmids together with indicated siRNAs. Transfected cells were transferred onto 22‐mm‐diameter glass coverslips (BDH) and on the third day, the individual coverslips were secured in an imaging chamber with 2 ml of starvation medium. The assembled imaging chamber was secured onto the microscope stage, and cells were maintained at 37°C in the incubation system of cellSens microscope. Each movie is composed of 240 pictures (frames) each taken every 10 s. Live‐cell imaging movies were analysed by Fiji, a Java‐based image processing program.
Autophagy dynamics analysis
Analysis of dynamics between autophagy proteins was performed by the examination of 50 events deriving from three independent experiments. The events analysed are characterized by the number of frames, expressed in seconds, from the moment at which the analysed particles (GFP‐ATG13 and GFP‐DFCP1) are clearly emerging to the collapse. Events corresponding to particles moving out of focus and then back in were excluded from the analysis. Same type of analysis was performed for the CFP‐LC3 (visualized in red); in this case, we analysed just the moment in which CFP‐LC3 structures appeared on GFP‐ATG13 or GFP‐DFCP1 particles taking advantage of coordinates of these particles.
ATG13 tracking and particles‐proximity analysis
Time‐lapse images of GFP‐ATP13 and BFP‐TFG or mCherry‐SEC16 were processed and quantified using open‐source software Fiji (Fiji – ImageJ). Images were first corrected for noise suppression and background subtraction using a median filter (kernel size 2 pixels) and a rolling ball algorithm (kernel size 150 pixels), respectively. As no significant changes in total fluorescence intensity per field of view were expect apart from fluorescence degradation, time‐lapse stacks were corrected for photobleaching using the simple‐ratio method. Eventual lateral shifts were corrected using the MultiStackReg plugin, and the shift was detected as translation of the GFP‐ATP13 channel and then re‐applied to the other channels. Finally, dot‐shaped structures were emphasized using a 2D top‐hat algorithm (kernel size 7x7). After image processing, GFP‐ATP13 puncta were tracked using the TrackMate plugin. LoG detector and simple LAP tracer were used for spot detection and tracking, respectively. Of all detected tracks, those starting at timepoint < 2 or > 238 were discarded to ensure that all particles were tracked in the entirety of their lifetime. All tracks with lifetime shorter than nine frames were considered non‐specific or partial detection then discarded. At the end of the detection, gaps were filled with spots and spot size was automatically re‐calculated. Fluorescence intensity for each fluorescence channel on each track was exported and analysed with Excel. To identify frames were GFP‐ATP13 co‐localized with BFP‐TFG or mCherry‐Sec16, maximum fluorescence intensity of the non‐GFP channel from all the timepoints of all the tracks was summarized. Then considering that pixel size was set below optical resolution, a timepoint where the maximum fluorescence intensity doubled the median of the distribution was considered as co‐localized.
Correlative Light Electron Microscopy (CLEM) and Transmission Electron Microscopy (TEM)
Cells stably expressing CFP‐ATG16L1 and GFP‐ATG13 were transfected as indicated. Cells were plated in 30‐μm Grid‐500 glass bottom dish (ibidi #81168) (18 × 103 cells / dish) and after 24 h were starved (EBSS) or not for 1 h. After treatment, cells were fixed with 3,7% paraformaldehyde (PFA) in PBS for 1 h at RT. Cell orientation and position in the 30‐μm Grid‐500 glass bottom dish (ibidi #81168) were identified and images acquired by using a confocal inverted microscope (Leica) TCS SP5 II, HC PL FLUOTAR 20X NA 0.50 or 63X NA 0.80 oil objective. After image acquisition, cells for CLEM and TEM were processed in parallel by fixation with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4, 1 h at RT. After washing with 0.1 M cacodylate buffer, cells were treated with 1% osmium tetroxide plus potassium ferrocyanide 1% in 0.1 M sodium cacodylate buffer for 1 h at 4°C. The samples were then rinsed, dehydrated in a series of graded ethanol and embedded in an epoxy resin (Sigma). Ultrathin sections of the embedded samples (60–70 nm) were obtained by using an Ultrotome V (LKB) ultramicrotome and were counterstained with uranyl acetate and lead citrate. The sections were finally analysed using a Tecnai G2 (FEI) transmission electron microscope operated at 100 kV. The images were captured with a Veleta (Olympus Soft Imaging System) using 2.800X, 3.900X or 13.500X magnification of the sections.
The images obtained by fluorescence and electron microscopy were aligned using ImageJ. For overlapping, the nucleus and reference structures were taken as reference and different magnifications adapted to the scale bars.
qPCR
Total RNA was isolated by using RNeasy mini kit (QIAGEN, 217004). One µg RNA was retrotranscribed using M‐MLV enzyme and oligodT (Promega, C1101). qPCR was performed using SYBR Green PCR Master Mix Applied Biosystems (Thermo Fisher Scientific, 4309155) with the QuantStudio 12K Flex Applied Biosystems. The following oligonucleotide primers were used for LC3C: (LC3C#1: 5’‐TGCCGGTGATAGTAGAAA‐3’; 5’‐GGTGTTCCTGGTACAGCT‐3’ and LC3C#2: 5'‐TGACCATGACCCAGTTCC‐3'; 5'‐TCCTCATCCTTGTAGTCTCT‐3'), and for β2m: (5’‐CTCCGTGGCCTTAGCTGTC‐3’; 5’‐TCTCTGCTGGATGACGTGAG‐3'). The mRNA expression of the gene of interest was normalized to β2m expression, and the ΔΔCt method was used; mRNA levels were then normalized to the control conditions.
LIR prediction
We predicted the possible LIRs of TFG with iLIR and associated PSSM scores (Jacomin et al, 2016). For each LIR, we assessed the propensity to be located in disordered regions with MobiDB (Piovesan et al, 2018) and other structural properties with FELLS (Piovesan et al, 2017).
Modelling of PB1 domain of TFG
We used HHPred (Söding, et al, 2005) to search for templates and the optimal alignment, along with Modeller (Webb, & Sali, 2017) to obtain a three‐dimensional (3D) model for the TFG PB1 domain. We identified only templates with a sequence identity lower than 20% with respect to the PB1 domain of TFG, which indicates a remote homology with known structures. Among the matches identified by HHPRED, we focused our attention on the PB1 domains from the protein kinase C (PDB entry 1WMH, chain B, Hirano et al, 2005) for which we obtained a sequence identity of 19% according to the HHPRED alignment (Table S1). The predicted secondary structures for the PB1 domain of TFG using the DSSP algorithm (Zacharias & Knapp, 2014), which is based on intra backbone hydrogen bond networks, matched properly with the known secondary structures of the templates and the gaps in the alignment where less than three residues long and located in disordered regions of the PB1 fold. The quality of the model was evaluated with VADAR (Volume Area Dihedral Angle Reporter) (Willard et al, 2003) with particular attention to the region where the first predicted LIR motif is located. We estimated the relative solvent accessible surface area of the residues 31–36 using NACESS (http://wolf.bms.umist.ac.uk/naccess/).
Statistical analysis
Densitometry analysis was performed using ImageJ software. All statistical calculations were performed and graphed using GraphPad Prism. Each point value represents the mean ± SEM (as reported in the figure legends). Number of independent experiments and number of replicates used to perform statistics are reported in the figure legends. Statistical significance was measured using an unpaired Student's t‐test, assuming a two‐tailed distribution, one‐way and two‐way analysis of variance (ANOVA) followed by Tukey’s post hoc test or followed by Dunnett’s multiple comparison test, or multiple t‐test, or Kolmogorov–Smirnov test using GraphPad Prism program as reported in the figure legends. Significance is defined as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; and ns, not significant. The source data are available in the supplementary information page.
Author contributions
FN and FC conceived the presented idea, developed the theory and, together with MCari, designed the work plan and wrote the paper. FL supervised and supported this work besides providing key intellectual advice. MCari. performed most of the experiments. MB helped with IFs. GM, BT, CF and DC helped with the biochemical assays. LA and IC generated both TFGwtLIR and TFGmutLIR3 constructs and produced viruses for all stable cell lines used. RN performed TEM experiments and analyses. MES and MCarr performed TEM and CLEM assays. NTK supervised high‐resolution imaging approaches. MBon performed live‐cell imaging analysis. FE generated HeLa cell lines harbouring endogenously expresses ATG8 proteins. SP and VD performed confocal images acquisition. MK, ML and EP developed the PB1 model and carried out the bioinformatics analyses. CP and VT provided control’ and TFG mutated patient’s fibroblasts. PP, CG, CB, FLG, NTK, MM and FL provided critical reagents.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We are grateful to S. Walker for the technical assistance. We thank the "Cell lines and DNA Bank of Genetic Movement Disorders and Mitochondrial Diseases” of the Telethon Network of Genetic Biobanks (grant GTB12001J). Francesco Cecconi’s laboratory is supported by grants from the Danish Cancer Society (KBVU R72‐A4408, R146‐A9364, R231‐A14034), the Novo Nordisk Foundation (NNF13OC0007559, NNF16OC0022544), the Lundbeckfonden (R233‐2016‐3360), the LEO Foundation (LF17024), “Associazione Italiana per la Ricerca sul Cancro” (AIRC project IG 2019 #23543) and The Italian Ministry of Research (MIUR, project PRIN 2017 FS5SHL Radius). F. Cecconi’s and E. Papaleo’s laboratories in Copenhagen are part of the Center of Excellence for Autophagy, Recycling and Disease (CARD), funded by the Danmarks Grundforskningsfond (DNRF125), Carlsberg fondet Distinguished Fellowship (CF18‐0314) to E.P. and a Netaji Subhash ICAR international fellowship (Govt. of India) to M.K. This research was also supported in part by grants from the Italian Ministry of Health (Ricerca corrente) to F. Nazio; the Italian Ministry of Education, University and Research (Research Project of National Relevance 2017 ID 2017WC8499) to F. Locatelli; and AIRC (Associazione Italiana Ricerca sul Cancro, 5x1000 ID 9962 and AIRC IG 2018 ID 21724) to F. Locatelli. This work is in part supported by European Research Council Grant 853057‐InflaPML (to C.G.) and EMBO Short‐Term Fellowship (number 7463) to M. Carinci. We give a special thanks to Vanda Turcanova for her help with cloning and mutagenesis, and Dr. Cristina Valacca, (Copenhagen) Prof. Silvia Campello, Dr. Valentina Cianfanelli and Dr. Carlo Rodolfo (Rome) for their strenuous help and support. C.B. was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the frameworks of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198) and of the Collaborative Research Center 1177 (ID 259130777).
The EMBO Journal (2021) 40: e103563.
Contributor Information
Francesca Nazio, Email: francesca.nazio@opbg.net.
Francesco Cecconi, Email: cecconi@cancer.dk.
Data availability
All the data on models and predictions are freely available in the GitHub repository https://github.com/ELELAB/LIR_TFG. All other data required to evaluate the conclusions here presented are in the paper or in Supplementary Materials.
References
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T (2012) ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3‐interacting region (LIR) motifs. J Biol Chem 287: 39275–39290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT (2008) Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3‐phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182: 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beetz C, Johnson A, Schuh AL, Thakur S, Varga R‐E, Fothergill T, Hertel N, Bomba‐Warczak E, Thiele H, Nürnberg G et al (2013) Inhibition of TFG function causes hereditary axon degeneration by impairing endoplasmic reticulum structure. Proc Natl Acad Sci USA 110: 5091–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgisdottir ÅB, Lamark T, Johansen T (2013) The LIR motif ‐ crucial for selective autophagy. J Cell Sci 126: 3237–3247 [DOI] [PubMed] [Google Scholar]
- Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, Ittmann MM, Marchetti D, Dotti G (2015) Heparanase promotes tumor infiltration and antitumor activity of CAR‐redirected T lymphocytes. Nat Med 21: 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catania A, Battini R, Pippucci T, Pasquariello R, Chiapparini ML, Seri M, Garavaglia B, Zorzi G, Nardocci N, Ghezzi D et al (2018) R106C TFG variant causes infantile neuroaxonal dystrophy “plus” syndrome. Neurogenetics 19: 179–187 [DOI] [PubMed] [Google Scholar]
- Chen Y, Tseng S‐H (2014) Targeting tropomyosin‐receptor kinase fused gene in cancer. Anticancer Res 34: 1595–1600 [PubMed] [Google Scholar]
- Colecchia D, Stasi M, Leonardi M, Manganelli F, Nolano M, Veneziani BM, Santoro L, Eskelinen EL, Chiariello M, Bucci C (2018) Alterations of autophagy in the peripheral neuropathy Charcot‐Marie‐Tooth type 2B. Autophagy 14: 930–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Wang J, Zhu M, Stahmer K, Lakshminarayan R, Ghassemian M, Jiang Y, Miller EA, Ferro‐Novick S (2016) Sec24 phosphorylation regulates autophagosome abundance during nutrient deprivation. eLife 5: e21167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funderburk, S.F., Wang, Q.J., Yue, Z (2010) The Beclin 1‐VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol 20: 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Florey O, Overholtzer M, Jiang X (2013) Interaction Between FIP200 and ATG16L1 Distinguishes ULK1 Complex‐Dependent and ‐Independent Autophagy. Nat Struct Mol Biol 20: 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan W, Zhang C, Siu KY, Satoh A, Tanner JA, Yu S (2017) ULK1 phosphorylates Sec23A and mediates autophagy‐induced inhibition of ER‐to‐Golgi traffic. BMC Cell Biol 18: 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Melville D, Zhang M, Schekman R (2013) The ER‐Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. eLife 2: e00947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Zhang M, Schekman R (2014) Phosphatidylinositol 3‐kinase and COPII generate LC3 lipidation vesicles from the ER‐Golgi intermediate compartment. eLife 3: e04135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guerroué F, Eck F, Jung J, Starzetz T, Mittelbronn M, Kaulich M, Behrends C (2017) Autophagosomal content profiling reveals an LC3C‐dependent piecemeal mitophagy pathway. Mol Cell 68: 786–796 [DOI] [PubMed] [Google Scholar]
- Hanna MG, Block S, Frankel EB, Hou F, Johnson A, Yuan L, Knight G, Moresco JJ, Yates JR, Ashton R et al (2017) TFG facilitates outer coat disassembly on COPII transport carriers to promote tethering and fusion with ER–Golgi intermediate compartments. Proc Natl Acad Sci USA 114: E7707–E7716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlalka GV, McEntagart ME, Gupta N, Skrzypiec AE, Mucha MW, Chioza BA, Simpson MA, Sreekantan‐Nair A, Pereira A, Günther S et al (2016) Novel genetic, clinical, and pathomechanistic insights into TFG‐associated hereditary spastic paraplegia. Hum Mutat 37: 1157–1161 [DOI] [PubMed] [Google Scholar]
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F et al (2015) A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723 [DOI] [PubMed] [Google Scholar]
- Hirano Y, Yoshinaga S, Takeya R, Suzuki NN, Horiuchi M, Kohjima M, Sumimoto H, Inagaki F (2005) Structure of a cell polarity regulator, a complex between atypical PKC and Par6 PB1 domains. J Biol Chem 280: 9653–9661 [DOI] [PubMed] [Google Scholar]
- Ishihara N, Hamasaki M, Yokota S, Suzuki K, Kamada Y, Kihara A, Yoshimori T, Noda T, Ohsumi Y (2001) Autophagosome requires specific early Sec proteins for its formation and NSF/SNARE for vacuolar fusion. Mol Biol Cell 12: 3690–3702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiura H, Sako W, Yoshida M, Kawarai T, Tanabe O, Goto J, Takahashi Y, Date H, Mitsui J, Ahsan B et al (2012) The TRK‐fused gene is mutated in hereditary motor and sensory neuropathy with proximal dominant involvement. Am J Hum Genet 91: 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6: 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomin AC, Samavedam S, Promponas V, Nezis IP (2016) iLIR database: A web resource for LIR motif‐containing proteins in eukaryotes. Autophagy 12: 1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong YT, Simoneschi D, Keegan S, Melville D, Adler NS, Saraf A, Florens WMP, Cavasotto CN, Fenyö D, Cuervo AM et al (2018) The ULK1‐FBXW5‐SEC23B nexus controls autophagy. eLife 7: e42253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, Lamark T (2020) Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol 432: 80–103 [DOI] [PubMed] [Google Scholar]
- Johnson A, Bhattacharya N, Hanna M, Pennington JG, Schuh AL, Wang L, Otegui MS, Stagg SM, Audhya A (2015) TFG clusters COPII‐coated transport carriers and promotes early secretory pathway organization. EMBO J 34: 811–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, Ktistakis NT (2013) Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 126: 5224–5238 [DOI] [PubMed] [Google Scholar]
- Karanasios E, Walker SA, Okkenhaug H, Manifava M, Hummel E, Zimmermann H, Ahmed Q, Domart M‐C, Collinson L, Ktistakis NT (2016) Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun 7: 12420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkham MB (1974) Trace elements in sludge. Science 184: 1030 [DOI] [PubMed] [Google Scholar]
- Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I et al (2012) Binding of the Atg1/ULK1 kinase to the ubiquitin‐like protein Atg8 regulates autophagy. EMBO J. 31: 3691–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C‐C, Lin Y‐C, Chen Y‐H, Chen C‐M, Pang L‐Y, Chen H‐A, Wu P‐R, Lin M‐Y, Jiang S‐T, Tsai T‐F et al (2016) Cul3‐KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell 61: 84–97 [DOI] [PubMed] [Google Scholar]
- Liu X, Li Y, Wang X, Xing R, Liu K, Gan Q, Tang C, Gao Z, Jian Y, Luo S et al (2017) The BEACH‐containing protein WDR81 coordinates p62 and LC3C to promote aggrephagy. J Cell Biol 216: 1301–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro‐Novick S (2011) Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 473: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T (2003) Mouse Apg16L, a novel WD‐repeat protein, targets to the autophagic isolation membrane with the Apg12‐Apg5 conjugate. J Cell Sci 116: 1679–1688 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Ohsumi Y (2011) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27: 107–132 [DOI] [PubMed] [Google Scholar]
- Moyer BD, Allan BB, Balch WE (2001) Rab1 interaction with a GM130 effector complex regulates COPII vesicle cis–Golgi tethering. Traffic 2: 268–276 [DOI] [PubMed] [Google Scholar]
- von Muhlinen N, Akutsu M, Ravenhill BJ, Foeglein Á, Bloor S, Rutherford TJ, Freund SMV, Komander D, Randow F (2012) LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol Cell 48: 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazio F, Carinci M, Valacca C, Bielli P, Strappazzon F, Antonioli M, Ciccosanti F, Rodolfo C, Campello S, Fimia GM et al (2016) Fine‐tuning of ULK1 mRNA and protein levels is required for autophagy oscillation. J Cell Biol 215: 841–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N (2013) FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 14: 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Tamura N, Kono N, Shimanaka Y, Arai H, Yamamoto H, Mizushima N (2017) Autophagosome formation is initiated at phosphatidylinositol synthase‐enriched ER subdomains. EMBO J 36: 1719–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan D, Tabaro F, Paladin L, Necci M, Micetic I, Camilloni C, Davey N, Dosztányi Z, Mészáros B, Monzon AM et al (2018) MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res 46: 471–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan D, Walsh I, Minervini G, Tosatto SCE (2017) FELLS: fast estimator of latent local structure. Bioinformatics 33: 1889–1891 [DOI] [PubMed] [Google Scholar]
- Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, Lavarello C, Sinibaldi M, Weber G, Del Bufalo F et al (2018) Choice of costimulatory domains and of cytokines determines CAR T‐cell activity in neuroblastoma. Oncoimmunology 7: e1433518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dötsch V, Johansen T, Kirkin V (2014) Interactions between autophagy receptors and ubiquitin‐like proteins form the molecular basis for selective autophagy. Mol Cell 53: 167–178 [DOI] [PubMed] [Google Scholar]
- Schlundt A, Sticht J, Piotukh K, Kosslick D, Jahnke N, Keller S, Schuemann M, Krause E, Freund C (2009) Proline‐rich Sequence Recognition. Mol Cell Proteomics 8: 2474–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodkin MR, Elazar Z (2013) The Atg8 family: multifunctional ubiquitin‐like key regulators of autophagy. Essays Biochem 55: 51–64 [DOI] [PubMed] [Google Scholar]
- Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33: 244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadel D, Millarte V, Tillmann KD, Huber J, Tamin‐Yecheskel B‐C, Akutsu M, Demishtein A, Ben‐Zeev B, Anikster Y, Perez F et al (2015) TECPR2 cooperates with LC3C to regulate COPII‐dependent ER export. Mol Cell 60: 89–104 [DOI] [PubMed] [Google Scholar]
- Steinmetz TD, Schlötzer‐Schrehardt U, Hearne A, Schuh W, Wittner J, Schulz SR, Winkler TH, Jäck H‐M, Mielenz D (2020) TFG is required for autophagy flux and to prevent endoplasmic reticulum stress in CH12 B lymphoma cells. Autophagy 22: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Nakagawa M, Nakahara K, Suehara M, Matsuzaki T, Higuchi I, Higa H, Arimura K, Iwamasa T, Izumo S et al (1997) A new type of hereditary motor and sensory neuropathy linked to chromosome 3. Ann Neurol 41: 771–780 [DOI] [PubMed] [Google Scholar]
- Tsai P‐C, Huang Y‐H, Guo Y‐C, Wu H‐T, Lin K‐P, Tsai Y‐S, Liao Y‐C, Liu Y‐T, Liu T‐T, Kao L‐S et al (2014) A novel TFG mutation causes Charcot‐Marie‐Tooth disease type 2 and impairs TFG function. Neurology 83: 903–912 [DOI] [PubMed] [Google Scholar]
- Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer‐Grumbach M, Kwon JM, FitzPatrick D, Schmedding E, De Vriendt E, Jacobs A et al (2003) Mutations in the small GTP‐ase late endosomal protein RAB7 cause charcot‐marie‐tooth type 2B neuropathy. Am J Hum Genet 72: 722–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tan D, Cai Y, Reinisch KM, Walz T, Ferro‐Novick S (2014) A requirement for ER‐derived COPII vesicles in phagophore initiation. Autophagy 10: 708–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, Sali A (2017) Protein structure modeling with MODELLER. Methods Mol Biol. 1654: 39–54 [DOI] [PubMed] [Google Scholar]
- Wetzel L, Blanchard S, Rama S, Beier V, Kaufmann A, Wollert T (2020) TECPR1 promotes aggrephagy by direct recruitment of LC3C autophagosomes to lysosomes. Nat Commun 11: 2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard L, Ranjan A, Zhang H, Monzavi H, Boyko RF, Sykes BD, Wishart DS (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res 31: 3316–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Zhang W, Razi M, Nyoni L, Joshi D, O'Reilly N, Johansen T, Tooze SA, Mouilleron S (2019) Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat Commun 10: 2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR, Eimer S, Audhya A (2011) TFG‐1 function in protein secretion and oncogenesis. Nat Cell Biol 13: 550–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias J, Knapp EW (2014) Protein secondary structure classification revisited: processing DSSP information with PSSC. J Chem Inf Model 54: 2166–2179 [DOI] [PubMed] [Google Scholar]
- Zoppino FCM, Militello RD, Slavin I, Alvarez C, Colombo MI (2010) Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11: 1246–1261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
All the data on models and predictions are freely available in the GitHub repository https://github.com/ELELAB/LIR_TFG. All other data required to evaluate the conclusions here presented are in the paper or in Supplementary Materials.