

Abstract
Eukaryotic transcription factors recognize specific DNA sequence motifs, but are also endowed with generic, non‐specific DNA‐binding activity. How these binding modes are integrated to determine select transcriptional outputs remains unresolved. We addressed this question by site‐directed mutagenesis of the Myc transcription factor. Impairment of non‐specific DNA backbone contacts caused pervasive loss of genome interactions and gene regulation, associated with increased intra‐nuclear mobility of the Myc protein in murine cells. In contrast, a mutant lacking base‐specific contacts retained DNA‐binding and mobility profiles comparable to those of the wild‐type protein, but failed to recognize its consensus binding motif (E‐box) and could not activate Myc‐target genes. Incidentally, this mutant gained weak affinity for an alternative motif, driving aberrant activation of different genes. Altogether, our data show that non‐specific DNA binding is required to engage onto genomic regulatory regions; sequence recognition in turn contributes to transcriptional activation, acting at distinct levels: stabilization and positioning of Myc onto DNA, and—unexpectedly—promotion of its transcriptional activity. Hence, seemingly pervasive genome interaction profiles, as detected by ChIP‐seq, actually encompass diverse DNA‐binding modalities, driving defined, sequence‐dependent transcriptional responses.
Keywords: DNA binding, E‐box, Myc, promoter, transcription
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics
Sequence‐independent interaction of transcription factor Myc with the DNA backbone is required for its chromatin association and subsequent site‐specific binding as well as transcriptional activation.
Introduction
The transcription factor Myc orchestrates complex gene expression programs that foster cell growth and proliferation, in both normal and cancer cells (e.g., Perna et al, 2012; Sabò et al, 2014; Walz et al, 2014; Kress et al, 2015; Kress et al, 2016; Muhar et al, 2018; Tesi et al, 2019). Myc dimerizes with Max (Blackwood & Eisenman, 1991) to bind DNA with a preference for the E‐box consensus sequence CACGTG (Blackwell et al, 1993; Solomon et al, 1993), through which it activates transcription (Amati et al, 1992; Kretzner et al, 1992). Within cells, however, Myc promiscuously associates with active chromatin (Guccione et al, 2006; Kim et al, 2008; Soufi et al, 2012), owing most likely to a combination of general accessibility (Sabò et al, 2014), protein–protein interactions (Thomas et al, 2015; Richart et al, 2016; Thomas et al, 2019), and non‐specific DNA binding (Ferre‐D'Amare et al, 1993; Brownlie et al, 1997; Nair & Burley, 2003; Sauvé et al, 2007): consequently, when expressed at high levels, Myc can be detected on virtually all active promoters and enhancers in the genome (Lin et al, 2012; Nie et al, 2012; Guo et al, 2014; Sabò & Amati, 2014; Sabò et al, 2014; Walz et al, 2014; Kress et al, 2016; Bywater et al, 2020). Hence, while Myc‐activated promoters tend to show overrepresentation of E‐box motifs and stronger Myc recruitment (Walz et al, 2014; Lorenzin et al, 2016; de Pretis et al, 2017; Tesi et al, 2019), the role of DNA sequence recognition in Myc activity remains to be clarified.
Here, we exploited the structure of the DNA‐bound Myc/Max dimer (Nair & Burley, 2003) to design Myc mutants bearing substitutions in residues that contact either the DNA phosphodiester backbone or specific bases within the consensus binding motif (E‐box), and profiled the DNA‐binding and gene‐regulatory activities of these mutants in murine cells. Our data reveal that non‐specific DNA binding is required for Myc to engage onto active regulatory elements in the genome, preceding sequence recognition; beyond merely stabilizing Myc onto select target loci, sequence‐specific binding contributes to its precise positioning and transcriptional activity.
Results
Structure‐based mutagenesis of the Myc DNA‐binding domain
Myc/Max dimerization depends upon the contiguous helix‐loop‐helix and leucine zipper domains of each protein (HLH‐LZ: ca. 70 amino acids) and is a strict prerequisite for DNA binding, mediated by the short basic region (15 a.a.) that precedes the HLH (Blackwood & Eisenman, 1991; Amati et al, 1992). In line with those biochemical findings, structural studies on DNA‐bound or free dimers, including Myc/Max (Nair & Burley, 2003; Sammak et al, 2019), Max/Max (Ferre‐D'Amare et al, 1993; Brownlie et al, 1997; Sauvé et al, 2004; Sauvé et al, 2007), and other bHLH proteins (Murre, 2019), showed that dimerization allows positioning of the basic regions for insertion into the DNA major groove (Fig 1A).
Figure 1. Structure‐based design and biochemical characterization of the MycHEA and MycRA mutants.

- Structure of the DNA‐bound Myc/Max dimer (Nair & Burley, 2003), with an alignment of the Myc and Max basic regions (numbering based on the 439 a.a. human Myc protein (GenBank nr. AAA36340.1).
- H359 and E363 in Myc establish H‐bonds with the complementary G6 and C1’ bases, respectively, E363 forming an additional bond with A2. R364, R366, and R367 contact the phosphodiester backbone, R367 interacting also with G4. Note that, in keeping with the symmetric configuration of the Myc/Max dimer (panel A) and with the similar structure of the Max homodimer (Ferre‐D'Amare et al, 1993), the corresponding residues in Max (R33/35/36, and H28/E32) form equivalent contacts with the other half of the E‐box palindrome.
- Far‐UV CD spectra at 20°C (top) and thermal denaturation monitored at 222 nm (bottom) for Myc (red) and Max (black) bHLH‐LZ constructs (at 16 and 8 µM, respectively), alone or in combination (Myc + Max), as indicated. SUM Myc + Max: theoretical sum of the individual Myc and Max curves. The Myc variant used (WT, RA, or HEA) is indicated at the top. The increased ellipticity signals at 222 nm and increased melting temperatures of the experimental Myc + Max mixtures (blue) compared to the theoretical sums (gray) demonstrate that MycWT, MycHEA, and MycRA heterodimerize with Max to comparable extents.
- EMSA of the Max bHLH‐LZ construct alone (at 2 µM) or in the presence of either Myc construct (MycWT, MycHEA, or MycRA, all at 6 µM). The polypeptides were incubated in the presence of 500 nM of the fluorescently labeled IRD‐CACGTG probe and the complexes separated on a native polyacrylamide gel, as described (Beaulieu et al, 2012).
- EMSA titration experiment, with increasing concentrations of either Myc bHLH‐LZ variant (WT or HEA) in the presence of a fixed amount of the Max bHLH‐LZ (2 µM) and of the fluorescently labeled IRD‐CACGTG probe (500 nM). The plot shows the mean and standard deviation of the quantification of the shifted Myc/Max bands as a function of the Myc concentration (the experiment was performed in triplicate). % Bound probe: fraction of the IRD‐CACGTG probe associated with the Myc/Max complex. HEA/WT Kd ratio: Kd value for MycHEA/Max/CACGTG divided by the Kd value for MycWT/Max/CACGTG. The apparent Kd values were 3.78 µM for MycWT/Max and 5.57 µM for MycHEA/Max.
- EMSA competition of the complex between Myc/Max and the fluorescently labeled IRD‐CACGTG probe with increasing concentrations of an unlabeled CACGTG probe (0.195, 0.390, 0.781, 1.563, 3.125, 6.25, 12.50, and 25 µM). The plot at the bottom shows the mean and standard deviation of the quantification of the shifted Myc/Max bands as a function of unlabeled probe concentration (the experiment was performed in triplicate). HEA/WT Kd ratio: as in (E). The apparent Kd values were 387 nM for MycWT/Max and 622 nM for MycHEA/Max.
- Same as (F), with an unlabeled GGATCC probe as competitor. The apparent Kd values were > 25 µM for MycWT/Max and 14.5 µM for MycHEA/Max.
- Same as (F), with an unlabeled CACGTC probe as competitor. The apparent Kd values were 674 nM for MycWT/Max and 280 nM for MycHEA/Max. Note that last two lanes of the upper gel were loaded in inverted order and for this reason were cropped and flipped horizontally in the final layout.
Data information: note that the differences in apparent Kd values between panels E and F–H can be explained by the different thresholds required to detect quantifiable complexes in the protein titration experiment (E), as opposed to a loss of signal intensity from an already detectable complex in the presence of unlabeled competitor (F–H): as such, competition experiments provide a better approximation of actual binding affinities. (F–H): The plots show the mean and standard deviation. The experiments were performed in triplicate. Unpaired Student’s t‐test was used to compare IC50 values and expressed as P‐values.
Source data are available online for this figure.
We targeted two groups of residues involved in DNA contacts within the Myc basic region (Fig 1B). First, R364, R366, and R367 interact with the phosphodiester backbone. Early data showed that a mutant with the triple alanine substitution (hereafter MycRA) was proficient in dimerization with Max, but could not activate an E‐box‐driven reporter gene (Amati et al, 1992). Here, we further characterized this mutant as a candidate for loss of generic (non‐sequence‐specific) DNA binding. Second, H359 and E363 form H‐bonds with the invariant bases of the E‐box consensus (CANNTG): with the intent to impair sequence‐specific recognition, we substituted these residues with alanine (MycHEA). Most noteworthy here, binding to the E‐box should be supported by the sum of base‐ and backbone‐directed interactions (Fig 1B). Hence, while MycHEA might be predicted to retain non‐specific binding, MycRA should lose all binding modalities: as shown below, both of these predictions were confirmed experimentally.
In order to characterize the dimerization and DNA‐binding activities of the MycRA and MycHEA mutants, we used recombinant His6‐tagged polypeptides spanning the Myc and Max bHLH‐LZ domains. Circular dichroism (CD) analysis revealed similar helicoidal content and thermal denaturation profiles for all the heterodimeric Myc/Max complexes (Fig 1C) indicating equivalent dimerization properties of MycWT, MycRA, and MycHEA with Max. In an electrophoretic mobility shift assay (EMSA) with a fluorescently labeled canonical E‐box probe (CACGTG), either MycWT or MycHEA, but not MycRA, generated a specific DNA‐bound complex when combined with Max (Fig 1D): as expected, these heterodimeric forms prevailed over the Max homodimer, observable when incubating Max alone with DNA. Two experiments were performed to assess the relative DNA‐binding efficiencies of MycWT/Max and MycHEA/Max dimers: first, we incubated a fixed amount of the DNA probe with increasing amounts of recombinant proteins (Fig 1E); second, increasing amounts of unlabeled E‐box probe were used as competitor in the binding reaction (Fig 1F). These experiments yielded consistent results, revealing a reduced binding capacity of MycHEA/Max relative to MycWT/Max, with an estimated drop in affinity of ~1.5‐ and ~1.6‐fold, respectively. Note that MycHEA/Max retained higher relative affinity for the E‐box compared to an unrelated sequence (GGATCC) (Fig 1G), owing most likely to recognition of the intact CAC half‐site by Max (Fig 1A and B).
Altogether, at this level of resolution, MycRA/Max could form in solution, but showed no DNA‐binding activity, while MycHEA/Max still bound the E‐box probe, but with reduced affinity. As will be developed below, characterization of the Myc mutants in cells confirmed these alterations and shed further light on impaired sequence recognition by MycHEA, with a secondary gain in affinity for the aberrant motif CACGTC, a feature that we also confirmed by EMSA (Fig 1H).
MycRA and MycHEA are unable to sustain cell proliferation
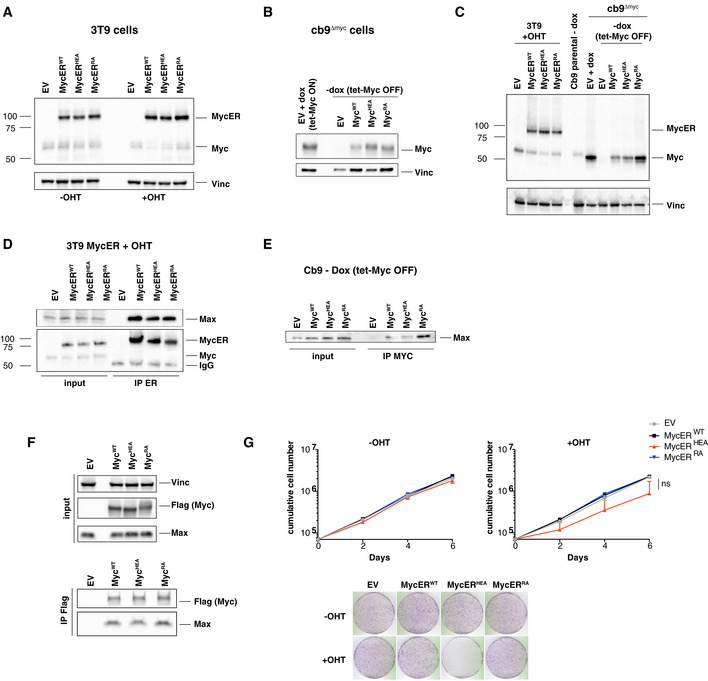
To address the activities of the above Myc mutants in vivo, we engineered two cellular models: first, we derived MycERT2 chimeras (henceforth MycERWT, MycERRA, MycERHEA) allowing post‐translational activation by 4‐hydroxytamoxifen (OHT) (Littlewood et al, 1995), and expressed these in mouse 3T9 fibroblasts (Fig EV1A); second, we used fibroblasts expressing a doxycycline‐dependent tet‐Myc transgene (cb9), inactivated the endogenous c‐myc locus by genome editing (cb9Δmyc; Fig EV2A) and transduced the cells with cDNAs encoding MycWT, MycHEA, or MycRA, thus leaving only the latter proteins upon tet‐Myc shutdown (Fig EV1B). Joint immunoblot analysis revealed comparable expression levels of the wild‐type and mutant proteins, in both the MycER and full‐length forms, slightly above endogenous Myc levels in parental 3T9 or cb9 cells (Fig EV1C). All of these Myc variants also co‐immunoprecipitated endogenous Max to comparable levels (Fig EV1D and E), as also observed in transiently transfected HEK‐293T cells (Fig EV1F), thus confirming the proficiency of MycRA and MycHEA for dimerization with Max also in live cells.
Figure EV1. Expression and characterization of the Myc mutants in cells.
-
A–CImmunoblot analysis of 3T9 or cb9Δmyc cells infected with retroviral vectors expressing the indicated MycER or Myc proteins. 3T9MycER cells were treated or not with OHT (4 h), as indicated; cb9Δmyc cells were cultured without doxycycline (−dox) for 24 h to switch off the tet‐Myc transgene, except where indicated (+dox: tet‐Myc ON). EV: empty vector. Both Myc and MycER were detected with the Y69 antibody. Note that the higher levels of MycRA seen in panel C were not observed in other experiments (panel B).
-
D, ELysates from the above 3T9 and cb9Δmyc cells were immunoprecipitated (IP) with anti‐ER and anti‐Myc (Sigma), respectively, and the precipitates analyzed by immunoblotting with the indicated antibodies. Note the proportional amounts of MycER and Max in immunoprecipitates from 3T9 cells; the amounts of the full‐length Myc proteins immunoprecipitated from cb9Δmyc cells could not be determined, due their migration close to the Ig heavy chain. One representative experiment out of 3 is shown.
-
FTop: immunoblot analysis of 293T cells transfected with plasmids expressing the indicated Flag‐tagged Myc proteins. Bottom: 293T cell lysates were immunoprecipitated with anti‐Flag, followed by immunoblot analysis. One representative experiment out of 3 is shown.
-
GGrowth curves and colony forming assays for 3T9 cells expressing the indicated MycER proteins, in the presence or absence of OHT. Data are presented as mean ± SD; n = 3 (biological replicates).
Source data are available online for this figure.
Figure EV2. Additional characterization of 3T9MycER and cb9Δmyc cells.
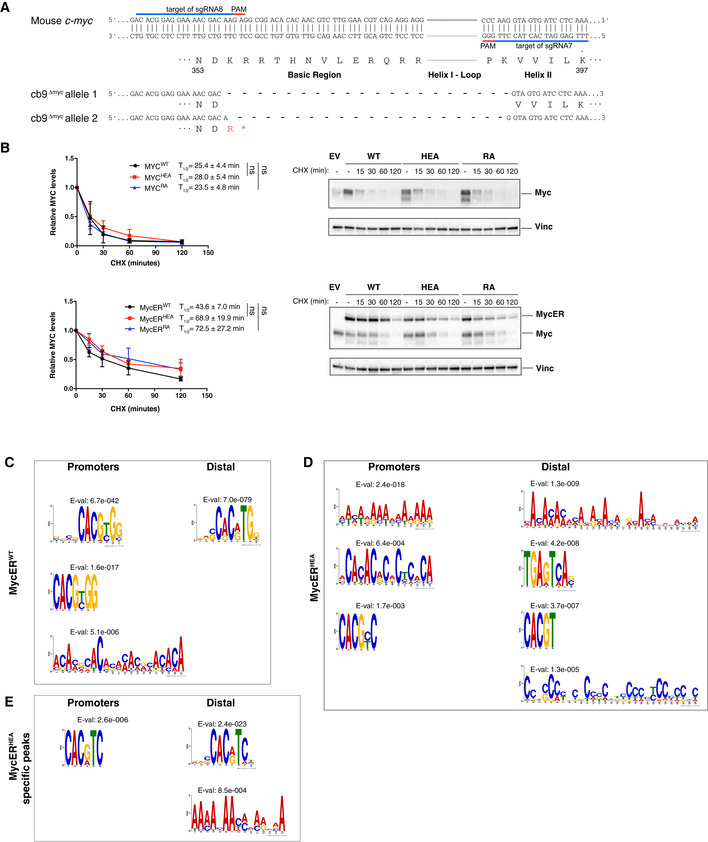
- Schematic representation of the mutant c‐myc alleles in cb9Δmyc cells (numbering based on the 439 a.a. mouse Myc protein: NCBI nr. NP 001170823.1).
- Assessment of protein stability in cb9Δmyc and 3T9 cells expressing the WT or mutant forms of Myc and MycER, respectively. cb9Δmyc cells (top) were plated without doxycycline for 24 h before cycloheximide (CHX) treatment, while 3T9 cells (bottom) were treated with OHT for 4 h before CHX treatment, followed by cell lysis at the indicated time‐points and immunoblot analysis. The quantification of three independent experiments is shown on the left as average (each normalized to time 0) and standard deviation. A representative blot for each model is shown on the right. No statistically significant differences were observed for any of the mutant proteins relative to their WT counterpart at any of the time‐points analyzed, the only points reaching P < 0.1 being MycERHEA at 15 min (P = 0.042) and MycERRA at 120 min (P = 0.075).
- De novo motif discovery analysis performed underneath the summit of the top 200 promoter‐associated or distal MycERWT peaks, as indicated. The position weight matrixes of predicted DNA‐binding motifs are shown together with their E‐values.
- As in (C) for the top 200 MycERHEA peaks.
- As in (C) for MycERHEA‐specific peaks (i.e., not bound by MycERWT).
Source data are available online for this figure.
We then took advantage of 3T9 and cb9Δmyc cells to assess the relative half‐lives of the Myc variants, by blockade of protein synthesis with cycloheximide followed by immunoblot analysis: as expected, MycERWT and MycWT showed short half‐lives in vivo, with no significant alterations for the mutant forms (Fig EV2B). Hence, at this level of resolution, neither MycHEA nor MycRA showed altered protein stability.
Finally, we addressed the ability of the Myc mutants to substitute for endogenous Myc in sustaining cell proliferation. As expected, cb9Δmyc cells infected with an empty vector (EV) arrested upon tet‐Myc shutdown, as assayed by colony formation, cell counts, and DNA synthesis (Fig 2A–C; −dox); expression of MycWT, but neither MycRA nor MycHEA, rescued proliferation in those conditions. When the tet‐Myc transgene was maintained active, MycHEA‐expressing cells showed reduced proliferative activity (Fig 2A–C; +dox), with similar effects upon activation of MycERHEA in 3T9 cells (Fig EV1G), pointing to a possible dominant‐negative effect of MycHEA. Consistent with this notion, the growth‐inhibitory action of MycHEA was manifest only in the presence of MycWT: when transduced in the c‐myc‐null rat fibroblast cell line HO15.19 (Mateyak et al, 1997), achieving mild over‐expression relative to endogenous Myc levels in parental TGR1 cells (Fig 2D), neither MycHEA nor MycRA impacted on proliferation, while MycWT strongly increased it (Fig 2E and F). In summary, MycRA and MycHEA were biologically inactive, being unable to compensate for the loss of endogenous Myc; in addition, MycHEA had dominant‐negative activity over MycWT. The molecular basis for this effect will be clarified further below.
Figure 2. MycHEA and MycRA are defective in sustaining cell proliferation.
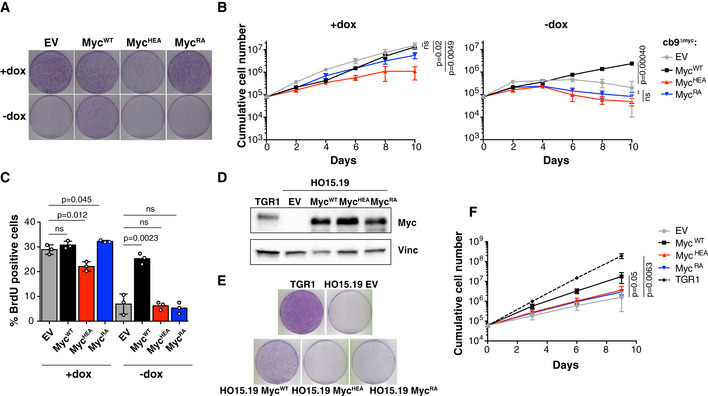
- Colony formation for cb9Δmyc cells infected with retroviral vectors expressing the indicated Myc proteins; all cells were expanded with doxycycline prior to the final plating step, upon which the compound was either maintained (+dox) or removed (−dox) to switch off the tet‐Myc transgene. One representative experiment out of 3 is shown.
- Cumulative cell counts for cb9Δmyc cells upon serial passaging with or without dox (removed at day 2). The data are presented as mean ± SD; n = 3 (biological replicates).
- Percentage of BrdU‐positive cb9Δmyc cells, in the presence or absence (24h after removal) of doxycycline. Data are presented as mean ± SD; n = 3. Two‐tailed Student’s t‐test was used to compare between two groups and expressed as P‐values.
- Immunoblot analysis of c‐myc −/− HO15.19 rat fibroblasts infected with retroviral vectors expressing the indicated Myc proteins. Parental TGR1 cells serve as control for endogenous levels of the MycWT protein.
- Colony formation for the same cells as in (D). One representative experiment out of 3 is shown.
- Cumulative cell counts for the same cells as in (D). Data are presented as mean ± SD; n = 3 (biological replicates). Two‐tailed Student’s t‐test was used to compare between two groups and expressed as P‐values.
Source data are available online for this figure.
Differential impairment in genome recognition by MycHEA and MycRA
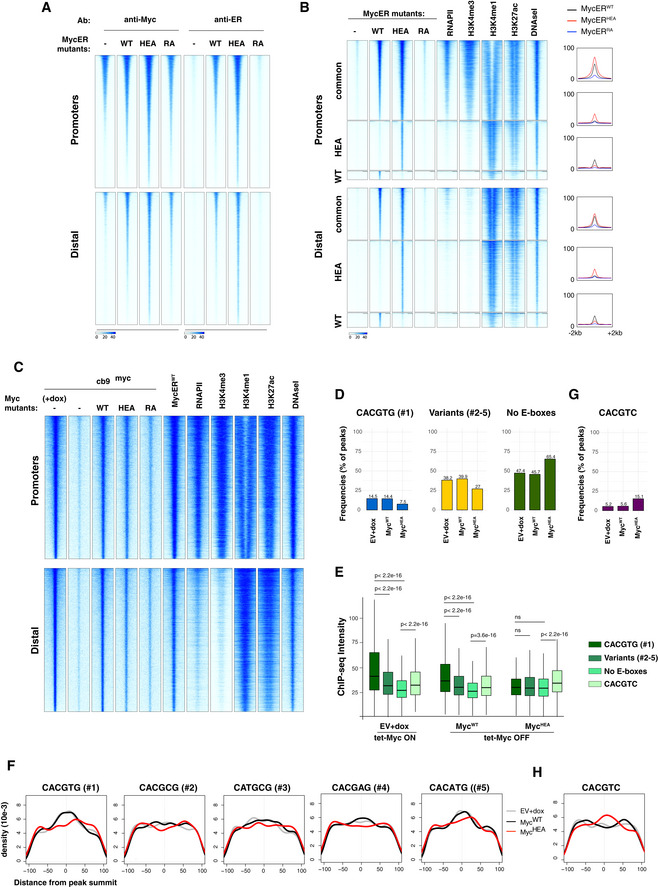
To address the ability of our Myc mutants to interact with genomic DNA in vivo, 3T9 fibroblasts expressing MycERWT, MycERRA, or MycERHEA were treated with OHT for 4h and profiled by ChIP‐seq with antibodies recognizing either Myc or the ER moiety: while the former could not discriminate between the endogenous and exogenous forms, the latter detected only the MycER variants, allowing us to specifically follow the mutated proteins (Fig EV3A). As previously reported (Sabò et al, 2014; de Pretis et al, 2017), MycERWT showed widespread association with active regulatory elements (i.e., promoters and enhancers) throughout the genome of 3T9 cells, as defined by the presence of active histone marks (H3K4me1, H3K4me3, H3K27ac), RNA polymerase II (RNAPII), and DNAseI hypersensitivity (Fig EV3B). This effect—sometimes termed “invasion” (Lin et al, 2012; Sabò et al, 2014)—was greatly attenuated with MycERRA and instead strengthened with MycERHEA, which showed even wider spreading than MycERWT onto active chromatin, in particular at gene‐distal regions. Remarkably, the regions bound the most efficiently by either MycERWT or MycERHEA were largely common to both proteins and were the most active/accessible, while those called with only one of the MycER forms were the least enriched for all of the probed features, including MycER itself (Fig EV3B). Consistent with these profiles, peak calling identified 16,762 peaks for MycERWT, 5,615 (33%) for MycERRA, and 23,873 (142%) for MycERHEA.
Figure EV3. MycHEA and MycRA show differentially altered genome‐binding profiles.
-
AHeatmaps representing normalized ChIP‐seq intensities at Myc or MycER‐associated promoters or distal sites in 3T9 cells, as indicated. Each row represents a genomic site called in at least one of the experimental samples, with each column spanning a 4 kb‐wide genomic interval centered on the union of MycER peaks (called either with anti‐Myc or anti‐ER antibody). All rows were ranked on the basis of the signal intensity of the MycERWT sample.
-
BHeatmaps representing normalized ChIP‐seq intensities at MycERWT‐ and/or MycERHEA‐associated promoters or distal sites. Each row represents a 4 kb‐wide genomic interval centered on the peak summit in either the MycERWT sample (for either the common or the WT‐only sites, as indicated on the left) or the MycERHEA sample (for HEA‐only sites). All rows were ranked on the basis of the signal intensity of the MycERWT sample (considering all signals, regardless of peak calling). The plots on the right show average ChIP‐seq intensity profiles for the 6 groups of peaks defined in the heatmap.
-
CHeatmaps representing normalized Myc ChIP‐seq intensities in cb9Δmyc cells expressing the various forms of Myc (WT, HEA, RA), as indicated. As a reference, we include the MycERWT profile (obtained with anti‐Myc antibodies) after 4h of OHT treatment. The data for RNAPII, histone marks (H3K4me3, H3K4me1, H3K27ac), and DNAseI hypersensitivity are from 3T9‐MycERWT fibroblasts without OHT (Sabò et al, 2014). Each row represents a genomic site called in at least one of the experimental cb9Δmyc samples, with each column spanning a 4 kb‐wide genomic interval centered on the union of the Myc peaks. All sites are ranked according to the intensity of the ChIP signal in the first column (cb9Δmyc cells + dox).
-
DAs Fig 3B, for cb9Δmyc cells: frequency of peaks (as %) that contain the indicated motif (within ± 100 bp from the peak summit) in each ChIP‐seq sample (EV, WT, HEA, and RA).
-
EAs Fig 3C, for cb9Δmyc cells: ChIP‐seq intensities for peaks containing the indicated motifs. The central band in the boxplot represents the median of the data, boxes the lower and upper quartiles (25 and 75%), and whiskers the minimum and maximum values. P‐values were calculated using Wilcoxon’s test.
-
FAs Fig 3D, for cb9Δmyc cells: density plots showing the distribution of the indicated motifs in a region of ± 100 bp from peak summit.
-
G, HAs in (D) and (F), respectively, for the CACGTC motif in cb9Δmyc cells.
Source data are available online for this figure.
To better characterize the effects of the mutations on DNA binding, we focused our attention on the occurrence of consensus elements, including the canonical E‐box CACGTG (or #1) and four permissive variants (#2–5: CACGCG, CATGCG, CACGAG, CACATG) (Blackwell et al, 1993; Grandori et al, 1996; Perna et al, 2012; Guo et al, 2014; Allevato et al, 2017). As previously observed (Guccione et al, 2006; Kim et al, 2008; Soufi et al, 2012; Guo et al, 2014; Sabò & Amati, 2014; Kress et al, 2016; Xin & Rohs, 2018; Bywater et al, 2020), peak intensity at MycER‐binding sites correlated primarily with chromatin and RNAPII (Fig EV3B), rather than with the presence of these consensus motifs (Fig 3A). This notwithstanding, the motifs significantly contributed to the MycERWT profiles, as evidenced by three distinctive features: first, the percentage of MycERWT peaks containing at least one motif within ± 100 bp from the peak summit was significantly above the background frequency seen with the empty vector (EV; Fig 3B); second, MycERWT peaks with canonical E‐boxes showed stronger average intensities, followed by those with variant motifs and ultimately by motif‐free peaks (Fig 3C); and third, the DNA motifs were most frequently centered under the peak summit, implying that they contributed to the precise positioning of MycERWT (Fig 3D). Most importantly, both of the MycER mutants showed substantial loss of those sequence‐associated features (Fig 3B–D)—note that while MycERHEA retained slightly higher average intensities in the presence of E‐boxes (Fig 3C), this might be due to residual recognition of half‐sites by MycHEA/Max dimers (see below).
Figure 3. Differential impact of the HEA and RA mutations on genome‐wide MycER‐binding profiles.
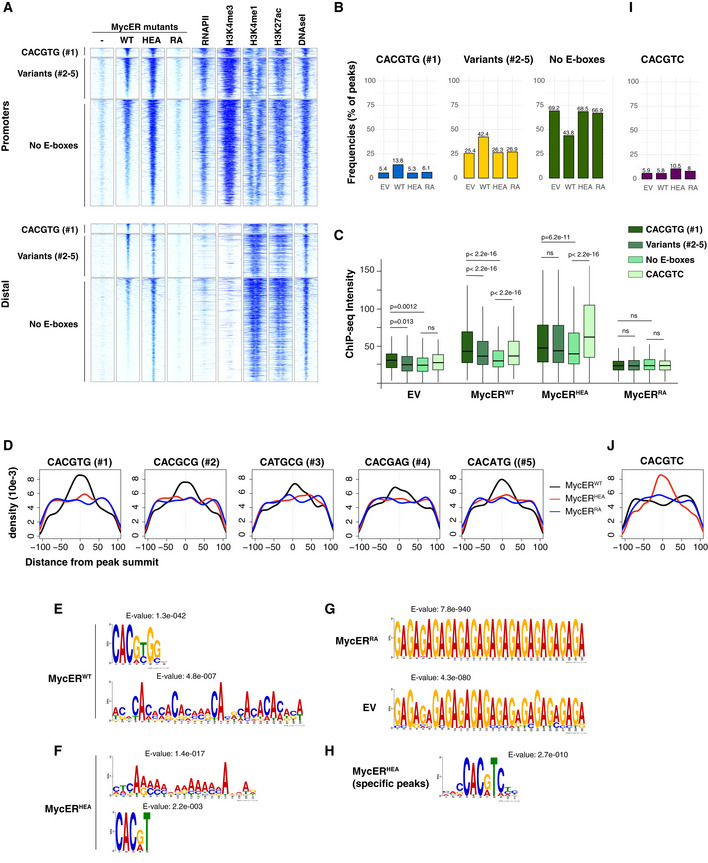
3T9 fibroblasts transduced with retroviral vectors expressing either MycER variant (WT, HEA, RA) or with the control empty vector (EV or ‐) were treated with OHT (4 h) and profiled by ChIP‐seq with anti‐ER antibodies.
-
AHeatmaps representing normalized ChIP‐seq intensities at MycER‐associated promoters or distal sites, as indicated. Each row represents a genomic site called in at least one of the experimental samples, with each column spanning a 4 kb‐wide genomic interval centered on the union of MycER peaks. All sites are ranked according to the intensity of the MycERWT signal and divided based on the presence of the indicated DNA motifs (#1: canonical CACGTG; variants #2‐5: CACGCG, CATGCG, CACGAG, CACATG; no E‐boxes: none of the above) in an interval of ± 100 bp around their peak summit. The data for RNAPII, histone marks (H3K4me3, H3K4me1, H3K27ac), and DNAseI hypersensitivity are from 3T9‐MycERWT fibroblasts without OHT (Sabò et al, 2014).
-
BFrequency of peaks (as %) that contain the indicated motif within ± 100 bp from the peak summit in each ChIP‐seq sample (EV, WT, HEA, and RA).
-
CChIP‐seq intensities for peaks (promoters + distal) containing the indicated motifs in an interval of ± 100 bp around their summit. For most accurate quantitation, intensities were computed within ± 50 bp from the summit. P‐values were calculated using Wilcoxon’s test. The central band in the boxplot represents the median of the data, boxes the lower and upper quartiles (25 and 75%), and whiskers the minimum and maximum values.
-
DDensity plots showing the distribution of the indicated motifs in a ± 100 bp interval from the peak summit.
-
E–GDe novo motif discovery analysis performed underneath the summit of the top 200 peaks called with (E) MycERWT, (F) MycERHEA, (G) MycERRA, and empty vector (EV) infected cells. The position weight matrixes of predicted DNA‐binding motifs are shown together with their E‐values.
-
HAs in (F) for the top 200 MycERHEA‐specific peaks (i.e. not bound by MycERWT).
-
I, JAs in (B) and (D), respectively, for the CACGTC motif.
The above results were obtained with MycER fusion proteins. In order to characterize the mutations in the context of full‐length Myc, we used cb9Δmyc cells expressing only MycWT, MycHEA, or MycRA (−dox, Fig EV1B and C) for ChIP‐seq profiling with Myc antibodies (Fig EV3C). As seen with the MycER chimaeras, MycRA showed substantial loss of DNA binding, while MycHEA and MycWT showed widespread binding, with distributions that largely paralleled those of MycERWT, RNAPII, and general chromatin accessibility in 3T9 cells (Fig EV3C). However, while MycERHEA showed “invasion” levels above those of MycERWT, the opposite was true for MycHEA versus MycWT (compare Fig EV3B and C): this might be due to several experimental variables, including the slightly higher levels of the MycER versus Myc proteins (Fig EV1C) and their distinct activation modes (OHT‐induced versus steady‐state). This notwithstanding, MycHEA showed a loss of E‐box selectivity analogous to that described above for MycERHEA (compare Fig 3B–D with Fig EV3, EV4, EV5).
Figure EV4. Single‐molecule microscopy analysis of Myc mutants.

- Immunoblots on subcellular fractions, used for the quantifications shown in Fig 4A. Cyt, Nuc, and Chr indicate the cytoplasmic, nucleoplasmic, and chromatin‐associated fractions, respectively, all loaded with the same cell equivalents as the total. Histone H3 and Vinculin were used as markers of the Chr and Cyt fractions, respectively. Note that the spread pattern of H3 in the central blot is most likely caused by its presence close to the front of migration in SDS–PAGE. In the first gel, the loading of the last two lanes was inverted, as indicated by the names in red. Saturated: over‐exposure of the blot, shown in order to visualize weak bands.
- Schematic representation of the illumination protocol for the SMT acquisitions to quantify the fraction of bound Myc molecules and exemplary acquisitions. The maximal projection of a representative MycWT movie is shown, displaying the nucleus boundaries (cyan dotted line) and a representative region (yellow square), for which exemplary frames are displayed on the right. The blue and red arrows highlight a bound and a diffusing molecule, respectively. Scale‐bar: 5 µm.
- Tracking the single Myc molecules allows estimating the distribution of displacements which is fit with a three‐component diffusion model to extract the fraction of bound molecules as well as the fractions and the diffusion coefficients of the diffusing molecules (see Materials and Methods).
- Schematic of the illumination protocol for the SMT movies to quantify the residence times of bound Myc molecules. Acquisition at different frame rates (ttl ranging between 200 ms and 2s) is performed to measure the residence times of bound Myc molecules at multiple time‐scales and to correct for photobleaching.
- The cumulative distributions of residence times are analyzed together using a global model accounting for photobleaching.
- The model allows estimating three characteristic times τ 1,τ 2,τ 3— inverse of the three characteristic reaction rates, whose weighted average provides an estimate of the mean residence time of the various Myc proteins on chromatin (See Materials and Methods and Fig 4C).
Figure EV5. Transcriptional analysis of MycER proteins.

- Scatter plots showing the fold change of each mRNA after 8h of OHT treatment (log2FC; y‐axis) against the intensity of Myc binding to the corresponding promoter (x‐axis) for all DEGs (defined as qval < 0.05) with a peak on the promoter in 3T9 cells expressing either MycERWT (left) or MycERHEA (right).
- As in (A), with black dots indicating the presence of the indicated motif within ± 100bp from the peak summit.
- Gene set enrichment plots for 5 custom Myc‐dependent signatures (Perna et al, 2012; Lorenzin et al, 2016; Muhar et al, 2018; Tesi et al, 2019). Normalized enrichment score (NES) and false discovery rate (FDR) values are reported for each dataset. Within each plot, genes were sorted from left to right according to the log2FC in their expression when comparing MycERWT or MycERHEA versus empty vector control cells, all treated with OHT.
MycHEA gains weak recognition of an alternative non‐E‐box motif
To better characterize the alterations in DNA sequence recognition caused by the HEA and RA mutations, we performed de novo motif analysis on the top 200 sites bound by each MycER variant, considering either all ChIP‐seq peaks (Fig 3E–H) or promoter‐associated and distal peaks separately, to account for differences in base composition (Fig EV2, EV3, EV4, EV5). As expected, MycERWT peaks enriched with high statistical significance for position weight matrices (PWMs) matching either the canonical E‐box CACGTG or variants #2–5 (Figs 3E and EV2C), but also for degenerate AC‐rich motifs, which incidentally included several CAC half‐sites. Remarkably, MycERHEA lost the main PWM of MycERWT but still enriched for the AC‐rich motifs, and secondly—i.e., with lower significance—for the partial E‐box motifs CAC(G/A)TN or CACG(C/T)C (Figs 3F and EV2D), consistent with the intact CAC half‐site being contacted by the wild‐type Max moiety (Fig 1A and B). MycERRA peaks instead were not enriched for specific motifs over the non‐specific background detected in control cells infected with the empty vector (EV; Fig 3G).
As noted above, MycERWT and MycERHEA showed largely superimposable DNA‐binding profiles, owing most likely to general chromatin accessibility. Restricting our motif analysis to sites bound only by MycERHEA (Fig EV3B) led to improved definition of the aforementioned partial motifs to CAC(G/A)TC (Figs 3H and EV2E). Thus, consistent with the contacts established by residues H359 and E363 with the conserved G6‐C1’ base pair (CACGTG: Fig 1B), substitution of these amino acids not only impaired E‐box recognition, but also altered the specificity to CACGTC: this new motif was selectively enriched (ca. 10‐15% of the HEA‐associated peaks: Figs 3I and EV3G) and correlated with stronger binding and precise positioning of the HEA, but not the WT proteins (Figs 3C and J, and EV3E and H). This change in binding specificity was confirmed in a competitive EMSA, where an unlabeled CACGTC oligonucleotide reduced binding of the MycHEA/Max dimer to the fluorescent CACGTG E‐box probe more efficiently than that of MycWT/Max (Fig 1H), opposite to what seen with the CACGTG competitor (Fig 1F). Nonetheless, it is important to note that recognition of the variant motif by MycHEA/Max did not amount to a full subversion of DNA‐binding specificity in vivo, since (i) MycERHEA‐only sites (which allowed the most stringent definition of the alternative motif) were bound at weaker levels than those shared with MycERWT (Fig EV3B) and (ii) when considering all sites, MycERWT enriched primarily for the canonical E‐box, while MycERHEA enriched for CACGTC with lower significance, and only after the degenerate AC‐rich motifs (Figs 3E and F, and EV2C and D).
In summary, the data presented so far show that MycHEA/Max dimers retain non‐specific DNA binding, while failing to recognize the canonical E‐box CACGTG and gaining weaker affinity for the alternative site CACGTC. Most importantly, MycHEA retains the propensity to distribute along active chromatin—a phenomenon that is most evident with overexpressed proteins—consistent with the notion that chromatin features (i.e., accessibility, composition, and protein–protein interactions) rather than DNA sequence are the primary determinants of Myc binding (Guccione et al, 2006; Kim et al, 2008; Soufi et al, 2012; Sabò & Amati, 2014; Thomas et al, 2015; Richart et al, 2016; Thomas et al, 2019). However, chromatin association must also rely upon close contacts with the DNA backbone, as demonstrated by the broad loss of interaction seen with the MycRA mutant. We conclude that Myc‐binding profiles, as assessed by ChIP‐seq, are determined largely by non‐specific binding events, predominating over sequence‐specific interactions.
Non‐specific DNA binding restrains free diffusion of Myc in the nucleoplasm
The above observations imply that non‐specific DNA binding may contribute to tether Myc onto active regulatory regions (promoters and enhancers), as a prerequisite for sequence‐specific binding (Sabò & Amati, 2014). As a corollary, loss of this initial tethering step in MycRA—but not MycHEA—would be expected to cause decreased chromatin association and increased protein mobility relative to MycWT. Two experiments were performed to address this issue. First, 3T9‐MycER cells (grown with OHT) were used to prepare three subcellular fractions (cytoplasm, nucleoplasm, and chromatin) and protein distribution analyzed by immunoblotting: MycERWT and MycERHEA showed roughly equal proportions of the protein in the three fractions, while MycERRA was essentially lost from chromatin (Figs 4A and EV4A). Second, we transduced 3T9 fibroblasts with vectors expressing the WT and mutant variants as Myc‐HaloTag fusion proteins, and confronted their mobility features by single‐molecule tracking microscopy (Mazza et al, 2012; Gebhardt et al, 2013). Relative to MycWT, the MycRA mutant showed reduced proportions of immobilized molecules (Figs 4B and EV4B and C) and, most importantly, close to halving of its average residence times on chromatin (Figs 4C and EV4D–F). While also showing a slight reduction in the fraction of immobilized molecules, MycHEA showed no significant alteration in residence times.
Figure 4. MycRA shows reduced chromatin association and increased nuclear mobility.

- 3T9 fibroblasts expressing the indicated MycER proteins and control cells (EV) were treated with OHT (4 h) and subjected to biochemical fractionation in three independent experiments. The quantity of MycER protein in the different fractions was quantified by Western blotting (shown in Fig EV4A) and the relative proportion in the various fractions plotted as average and standard deviation.
- Single‐molecule tracking at high frame rate: the time of ttl = 10 ms between two images allows to estimate the distribution of displacements, that is then fit by a three‐component diffusion model to estimate the fraction of molecules immobilized on chromatin. Inset: average bound fraction (see Fig EV4C and Materials and Methods). Note that MycRA displays a significantly lower bound fraction than MycWT and MycHEA (n cells = 34, 30, and 35, and n displacements = 78,550, 59,783, and 83,801 for MycWT, MycRA, and MycHEA, respectively). Error bar: SD. Statistical significance evaluated by permutation tests.
- Single‐molecule tracking at lower frame rate (ttl spanning between 200 ms and 2 s) allows to quantify the distribution of residence times (i.e. the duration of binding events). Inset: average residence times (see Fig EV4E and F and Materials and Methods). The data reveal a significantly shorter average for MycRA, relative to either MycWT or MycHEA (n cells = 35, 35, and 31, and n bound–molecules = 2,452, 2,084, and 2,171 for MycWT, MycRA, and MycHEA, respectively). Error Bar: SD. Statistical test: ANOVA–Tukey.
Data information: in (B and C), the statistical significances are indicated relative to the WT control (or RA versus HEA, as indicated): *P < 10−4; # P < 10−3; + P = 3.1 × 10−3; ns not significant.
Altogether, while defective E‐box recognition (as in MycHEA) impacted neither general protein distribution, nor mobility, loss of DNA backbone contacts (as in MycRA) caused a major decrease in chromatin retention. Together with the above ChIP‐seq profiles, we conclude that non‐specific DNA binding is required for the initial engagement of Myc onto accessible genomic regions, restricting its free diffusion in the nucleoplasm and potentially allowing localized, linear scanning of the DNA sequence.
Sequence recognition determines transcriptional activation
To address the impact of DNA‐binding alterations on transcriptional activity, we established RNA‐seq profiles following OHT treatment of 3T9‐MycERWT, MycERHEA, and MycERRA cells. As previously observed (Sabò et al, 2014; de Pretis et al, 2017), MycERWT elicited the up‐ and down‐regulation of equivalent numbers of genes (ca. 1,000 at 4 h, 2,000 at 8 h): while this effect was largely lost with the MycERRA mutant, MycERHEA mobilized even more mRNAs (Fig 5A). However, closer scrutiny revealed that the gene expression profiles elicited by MycERWT and MycERHEA were totally unrelated (Fig 5B and C). In particular, focusing on MycERWT‐responsive genes revealed that MycERHEA modulated their expression inconsistently, with a continuum of effects ranging from activation to repression (Fig 5D). Moreover, MycERHEA regulated additional genes, not modulated by MycERWT (Fig 5E). Most importantly, the differences between MycERWT‐ and MycERHEA‐driven transcriptional programs were attributable to DNA sequence, as activation correlated with enrichment of the cognate consensus motifs under the corresponding ChIP‐seq peak in the promoter (i.e., CACGTG plus variants #2‐5 for MycERWT and CACGTC for MycERHEA: Fig 5F).
Figure 5. DNA sequence recognition determines transcriptional regulation.
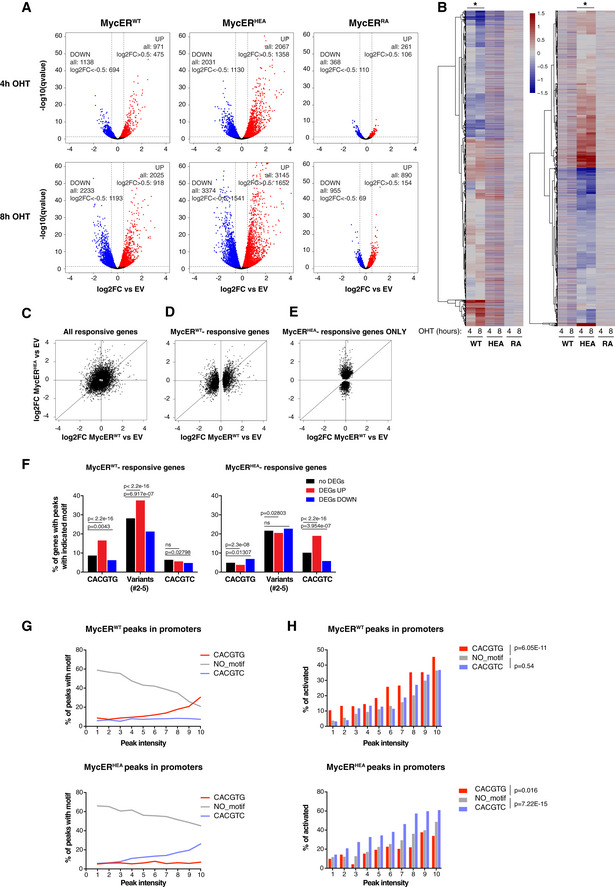
3T9 fibroblasts expressing the indicated MycER proteins and control empty vector (EV)‐transduced cells were treated with OHT (4 h, 8 h) and profiled by RNA‐seq.
-
AFold change of each annotated mRNA (log2FC, relative to the EV control), plotted against its q‐value (−log10). mRNAs showing significant up‐ and down‐regulation (qval < 0.05) are marked in red and blue, respectively. The values reported in the graphs indicate the total numbers of up‐ and down‐regulated genes (qval < 0.05) and of the subset regulated above a defined threshold of |log2FC| > 0.5.
-
BHeatmaps representing the same log2FC values as in (A) (restricted to those mRNAs with qval < 0.05 in at least one of the MycER samples). The two heatmaps differ in the samples driving the clustering, indicated by the asterisks at the top.
-
C–EScatter plots confronting fold‐change values (defined as in A) in response to MycERWT (x‐axis) and MycERHEA (y‐axis), showing the following groups of mRNAs: (C) all of the mRNAs called as DEGs (qval < 0.05) in at least one of the samples; (D) MycERWT‐regulated DEGs (whether regulated or not by MycERHEA); (E) MycERHEA‐specific DEGs (excluding those regulated by MycERWT).
-
FPercentage of promoters with the indicated DNA motifs under the ChIP‐seq peak (±100 bp from the peak summit) within each regulatory class (no DEG, UP, or DOWN) for either MycERWT (left) or MycERHEA (right). P‐values calculated with Fisher’s exact test.
-
GPercentage of promoter‐associated ChIP‐seq peaks with the indicated motifs, as a function of peak intensity (binned in deciles: 1–10) for either MycERWT (top) or MycERHEA (bottom).
-
HPercentage of DEG UP genes (qval < 0.05) as a function of peak intensity (binned as in G). Statistical test: Chi‐squared against the “no motif” condition, performed on the entire series.
Two non‐mutually exclusive mechanisms may underlie the connection between sequence recognition and transcriptional activation. First, the presence of the cognate consensus motif stabilizes DNA binding by either MycERWT or MycERHEA, as evidenced by peak intensities in ChIP‐seq profiles (Fig 5G): as a consequence, the extended residence time of the transcription factor on DNA may increase the probability of activation. In line with this scenario, Myc‐induced transcriptional programs correlated with the relative gain in Myc binding at promoters in diverse cell types (Walz et al, 2014; Lorenzin et al, 2016; de Pretis et al, 2017; Tesi et al, 2019). Likewise, up‐regulated loci showed the strongest MycER‐binding intensities in our experiments (Fig EV5A), associated with enrichment of the cognate DNA motif (i. e. CACGTG for MycERWT and CACGTC for MycERHEA; Figs 5F and EV5B). Second, beyond residence time, sequence recognition may directly contribute to the molecular activity of the transcription factor. Remarkably, our data also provided support for this scenario: indeed, at any given binding intensity (bins 1–10), loci targeted via the cognate DNA motif were more frequently activated by either MycERWT or MycERHEA, the opposite motif serving as negative control (Fig 5H).
Altogether, the above data show that sequence recognition is essential to establish adequate Myc‐activated programs. Most importantly, this step is subsequent to engagement of the factor on active chromatin, mediated by non‐specific DNA binding.
Finally, unlike activated genes, those down‐regulated by either MycERWT or MycERHEA recruited the transcription factor with the lowest efficiency and lacked enrichment of the cognate binding motif (Figs 5F and EV5A and B). Hence, as previously proposed (Kaur & Cole, 2013; de Pretis et al, 2017; Baluapuri et al, 2019), repression by either MycERWT or MycERHEA may be largely indirect. Of particular notice here, while MycERHEA did not bind the canonical CACGTG E‐box, MycERHEA‐repressed genes enriched for this motif (Fig 5F) as well as for known Myc‐dependent gene signatures (Fig EV5C), in line with the dominant‐negative action of this mutant over endogenous MycWT (Figs 2A–C and EV1G).
Discussion
Unlike pioneer factors that can access DNA in closed chromatin (Kim et al, 2008; Soufi et al, 2012), Myc and other bHLH proteins depend upon a pre‐existing active chromatin state (Guccione et al, 2006; Kim et al, 2008; Soufi et al, 2012; Guo et al, 2014; Sabò et al, 2014; Kress et al, 2016; Xin & Rohs, 2018; Bywater et al, 2020). While recognizing specific DNA sequence motifs, all of these transcription factors are also endowed with generic, non‐specific DNA‐binding activities, but how these features are integrated to determine genomic binding profiles and transcriptional outputs remains largely unresolved. Here, we addressed this question for Myc by mutating residues that contact either the DNA backbone (MycRA: R364/366/367‐A) or specific bases within the E‐box consensus motif (MycHEA: H359/E363‐A) (Fig 1A and B). Expression of these mutants in cultured mouse fibroblasts allowed us to dissect their DNA‐binding and gene‐regulatory properties (with ChIP‐seq and RNA‐seq profiling, respectively) as well as their intra‐nuclear mobility (with single‐molecule tracking microscopy), unraveling several key principles.
First, besides an open, active chromatin conformation, non‐specific DNA binding is required for Myc to engage on genomic regulatory regions, as a prerequisite for sequence‐specific recognition. In fact, this initial step underlies the majority of the cross‐linking events detected in cells with ectopic expression of Myc, as shown by the similar ChIP‐seq profiles of MycWT and MycHEA (which retains DNA backbone interactions) and the overall loss of DNA binding by MycRA. Subcellular fractionation and single‐molecule tracking experiments yielded a consistent scenario, with MycRA showing loss of chromatin association, decreased proportions of immobilized molecules, and shorter residence times, while MycHEA showed unaltered dynamics relative to MycWT. Altogether, these data imply that besides low‐affinity protein–protein interactions (Thomas et al, 2015; Richart et al, 2016; Thomas et al, 2019), DNA backbone contacts allow tethering the transcription factor onto active regulatory regions (promoters and enhancers), restricting its free diffusion in the nucleoplasm, and most likely allowing local scanning of the DNA sequence (Sabò & Amati, 2014). In this context, we note that the widespread targeting or “invasion” of active chromatin by Myc is unlikely to reflect a true change in binding kinetics, since the non‐specific DNA‐binding events detected in over‐expressing cells should also occur—albeit below experimental background—at physiological protein levels.
The second and most unexpected principle lies in the finding that DNA sequence recognition contributes not only to the stabilization and positioning of Myc onto select DNA motifs, but also to its transcriptional activity per se, implying some form of communication between the C‐terminal bHLH‐LZ and the N‐terminal transactivation domain (Amati et al, 1992; Barrett et al, 1992). For example, as reported for other transcription factors such as the glucocorticoid receptor (Watson et al, 2013) or the bHLH protein MyoD (Huang et al, 1998), sequence‐specific binding may elicit allosteric changes that modulate transcriptional activity; in line with this concept, Max bHLH‐LZ homodimers showed subtle structural differences when bound to specific versus non‐specific DNA (Sauvé et al, 2007), but whether the same occurs with Myc/Max remains unknown. Notwithstanding its molecular underpinning, this unexpected connection unravels a key specificity determinant in gene regulation by Myc, with E‐boxes contributing not merely to localization, but also to the activity of the transcription factor at select genomic loci.
The specificity of Myc‐dependent transcription became the subject of an active debate in the field in the past few years, since two initial studies (Lin et al, 2012; Nie et al, 2012) and others in their wake (Porter et al, 2017; Zeid et al, 2018; Nie et al, 2020) posited that rather than regulating select genes, Myc augments RNA synthesis at all active loci, thus acting as global “transcriptional amplifier”. This model rested largely on the coincidence between the “invasion” of active chromatin and the elevated RNA contents seen in cells with high Myc levels. However, as shown here, a sizeable fraction—and in some instances the majority—of Myc peaks in ChIP‐seq profiles reflect non‐specific DNA binding. The same applies to other transcription factors: p53, for example, also showed promiscuous chromatin association when acutely induced, yet regulated a subset of the bound loci in a sequence‐specific manner (Tonelli et al, 2015; Tonelli et al, 2017). Hence, association of a transcription factor with a given genomic element cannot be systematically equated to a productive regulatory interaction. Another key aspect to be considered here is timing: Myc is rapidly activated by mitogenic stimuli and modulates distinct gene expression programs, in either fibroblasts (Perna et al, 2012) or B‐cells (Tesi et al, 2019). This happens hours before RNA amplification—if any—or other events such as genome re‐organization, metabolic reprogramming, cell growth, or DNA replication, all of which occur in a Myc‐dependent manner (Wang et al, 2011; Nie et al, 2012; Sabò et al, 2014; Kieffer‐Kwon et al, 2017). Moreover, acute modulation of Myc levels and/or activity in proliferating cells elicited rapid transcriptional responses without changes in global RNA production (Sabò et al, 2014; Walz et al, 2014; Muhar et al, 2018).
Considering the above altogether, an objective scrutiny of matched RNA and chromatin profiles (Lin et al, 2012; Nie et al, 2012; Perna et al, 2012; Sabò et al, 2014; Walz et al, 2014; Kress et al, 2016; Lorenzin et al, 2016; de Pretis et al, 2017; Kieffer‐Kwon et al, 2017; Porter et al, 2017; Muhar et al, 2018; Zeid et al, 2018; Bywater et al, 2020) lends no formal support to the amplifier model. Instead, and as previously discussed (Kress et al, 2015; Sabò & Amati, 2018), the data are most consistent with the direct regulation of defined, yet complex, sets of Myc‐target genes, which in turn drive the wealth of secondary changes—among which RNA amplification—that contribute to cellular activation and/or transformation. In particular, Myc‐activated genes enrich for functional categories including nucleotide biosynthesis, RNA catabolism, processing, or ribosome biogenesis (e.g., Sabò et al, 2014; Muhar et al, 2018; Tesi et al, 2019), all of which globally impact RNA production (Kress et al, 2015).
Most noteworthy here, MycHEA acted as a dominant‐negative (DN) mutant, as indicated by the down‐regulation of known Myc signatures and the suppression of cell proliferation in Myc‐proficient cells. As MycHEA does not recognize the E‐box, this DN activity is unlikely to stem from direct competition with MycWT (or other bHLH proteins) for target sites. Our data show that MycHEA still dimerizes with Max, binds DNA non‐specifically, and engages on open chromatin like MycWT; however, this mutant factor drives aberrant transcriptional programs, including not only the induction of new genes, but also interference with E‐box‐containing Myc‐activated genes. Based on these results, a series of mechanisms might contribute to the DN activity of MycHEA, such as (i) titration of endogenous Max, (ii) local hindrance of endogenous Myc/Max activity at promoters through non‐specific DNA binding, and (iii) unproductive interaction with basal components or co‐factors in the transcriptional machinery, or other mechanisms yet to be determined. Most intriguingly, mutations in the residue equivalent to Myc‐E363 also conferred DN activity and altered DNA binding in other bHLH subclasses and species (from C. elegans to human) (Boisson et al, 2013; Marchegiani et al, 2015; Luchtel et al, 2019). Altogether, we surmise that the build‐up of dysfunctional bHLH dimers may be detrimental within—and perhaps across—multiple bHLH families, warranting detailed dissection of their mechanisms of action.
Finally, a key consideration here regards the multi‐faceted mechanisms by which Myc regulates gene expression: as other transcription factors, Myc interacts with a variety of core components and co‐factors in the transcriptional machinery (Tu et al, 2015; Kalkat et al, 2018) and can impact upon multiple steps, such as RNA PolII recruitment (de Pretis et al, 2017), pause release (Rahl et al, 2010), or histone modifications (Frank et al, 2001; Zippo et al, 2007). By recruiting the kinase complexes TFIIH (Cowling & Cole, 2007) and P‐TEFb (Eberhardy & Farnham, 2002), Myc may modulate phosphorylation of the RNA PolII C‐terminal domain (CTD), thus regulating not only transcription per se, but also co‐ and post‐transcriptional processes such as mRNA capping (Cowling & Cole, 2010; Lombardi et al, 2016; Posternak et al, 2017), splicing, export, or translation (reviewed in Harlen & Churchman, 2017; Herzel et al, 2017). Our data indicate that at least some of the aforementioned processes must be modulated by sequence‐specific DNA binding, warranting further mechanistic dissection of this unexpected connection.
Materials and Methods
Myc mutagenesis and subcloning
Mutagenesis of His 359 and Glu 363 to Alanine in human Myc (MycHEA mutant) was performed using the QuikChange Site‐Directed Mutagenesis Kit (Agilent Technologies # 200519), according to the manufacturer’s instructions, with a pBabe‐hygro plasmid (BH) (Morgenstern & Land, 1990) containing the human MYC cDNA (Amati et al, 1992) as template. The resulting pBH‐MycHEA plasmid was then sequenced to verify the correctness of the whole mutated cDNA (codon 359: CAC to GCC; codon 363: GAG to GCG). The resulting mutant cDNA and that encoding the Arg 364/366/367 to Ala mutant (Amati et al, 1992) (here MycRA) were subcloned in the retroviral vector pQCXIH (Clontech) or in the transfection vector pCMV‐FLAG. For expression as 4‐hydroxytamoxifen (OHT)‐dependent MycERT2 chimeras (Littlewood et al, 1995), the full‐length MYC cDNAs were fused in‐frame upstream of the variant estrogen receptor hormone‐binding domain (ERT2) into the retroviral vector pBabe‐puro (BP) (Morgenstern & Land, 1990).
Myc‐HaloTag vectors were generated using the Gibson Assembly Protocol (Gibson et al, 2009): HaloTag and MYC cDNAs (encoding MycWT, MycHEA, or MycRA) were PCR‐amplified with primers containing overlapping sequences and combined with the BP vector (digested with BamHI and EcoRI) in the Gibson Assembly Reaction, to create the final BP‐Myc‐HaloTag plasmids with in‐frame Myc‐HaloTag fusions.
Cell lines
The cb9 tet‐Myc cell line was produced through the 3T3‐immortalization protocol starting from mouse embryonic fibroblasts (E14.5) obtained from Rosa26‐rtTA/tet‐Myc mice (Croci et al, 2017) and used to derive the cb9Δmyc and the cb9‐myc HEA cell lines (see below). All the cell lines used in this work were grown in DMEM, supplemented with 10% fetal bovine serum, 2 mM l‐glutamine, and 1% penicillin/streptomycin. For the cb9Δmyc, medium was also complemented with doxycycline (1 µg/ml) to keep the tet‐Myc transgene expressed, unless otherwise specified. Mouse 3T9 fibroblasts were infected with BP‐MycERWT, MycERHEA, or MycERRA retroviruses, and selected for 2 days with puromycin (1.5 μg/ml); activation of the MycER fusion proteins was achieved addition of OHT to the culture medium (400 nM). Rat HO15.19 cells (Mateyak et al, 1997) and mouse cb9Δmyc cells were infected with BH‐ and QCXIH‐based recombinant retroviruses, respectively, expressing either MycWT, MycHEA, or MycRA; infected cells were selected with hygromycin (150 µg/ml) for 4 days. Where indicated, cells were treated with cycloheximide (Sigma, C7698‐1G; 50 mg/ml). All cell lines were routinely tested for mycoplasma contamination.
Western blot and co‐immunoprecipitation
For Western blot, protein extraction was performed by resuspending the cells in lysis buffer (300 mM NaCl, 1% NP‐40, 50 mM Tris–HCl pH 8.0, 1 mM EDTA) freshly supplemented with protease inhibitors (cOmplete™, Mini Protease Inhibitor Cocktail, Roche‐Merck, #11836153001), followed by brief sonication. After centrifugation at 16,000 g for 15 min at 4°C, cell extracts were quantified with the Bradford‐based Protein Assay kit (Bio‐Rad Protein Assay, #5000006). After addition of 6× Laemmli buffer (375 mM Tris–HCl, 9% SDS, 50% glycerol, 9% beta‐mercaptoethanol and 0.03% bromophenol blue), lysates were boiled for 5 min, electrophoresed on SDS–PAGE gels (7.5% polyacrylamide), transferred onto nitrocellulose membranes and protein expression detected with the indicated primary antibodies (see below). Chemiluminescence was detected using a CCD camera (ChemiDoc XRS + System, Bio‐Rad). Quantification of protein levels was performed using the Image Lab software (Bio‐Rad, version 4.0).
For co‐immunoprecipitation experiments, 293T cells were transfected overnight with calcium phosphate with 5 µg of plasmids encoding FLAG‐tagged MycWT, MycHEA, MycRA, or EV (empty vector) and collected 48h after transfection. After two washes in ice‐cold PBS, cells were scraped in 4 ml of ice‐cold NHEN buffer (20 mM Hepes pH 7.5, 150 nM NaCl, 0.5% NP‐40, 10% glycerol, 1 mM EDTA) freshly supplemented with protease inhibitors (cOmplete™, Mini Protease Inhibitor Cocktail, Roche‐Merck, #11836153001) and lysed for 20 min on a rotating wheel at 4°C. Complete cell disruption and DNA fragmentation was performed with three cycles of sonication (30 s on, 30 s off) with a Branson Sonifier 250 (Output Control = 2) equipped with a 3.2 mm Tip (Branson, #101‐148‐063). Lysates were cleared by centrifugation at 16,000 g for 15 min at 4°C, and protein concentration determined with the Bradford‐based Protein Assay kit (Bio‐Rad Protein Assay, #5000006). The immunoprecipitation of FLAG‐Myc was performed by incubating 2 mg of cell lysate with 40 µl of Anti‐FLAG M2 affinity gel (Sigma‐Aldrich #A2220) for 3 h in a final volume of 1 ml of NHEN buffer with agitation at 4°C. The beads were then washed five times with 1 ml of wash buffer (20 mM Hepes pH 7.5, 150 nM NaCl, 0.1% Tween, 10% glycerol, 1 mM EDTA), resuspended in 1× Laemmli buffer, and boiled for 10 min. In parallel, 2.5% of the material used for the IP was collected to be loaded as input. For co‐immunoprecipitation experiments in 3T9 and cb9Δmyc cells, the cells were lysed in a buffer containing 50 mM Tris pH 8, 1 mM EDTA, 150 mM NaCl, and 1% NP‐40; the lysates were incubated with anti‐ER (Sigma, #06‐395) or anti‐Myc (Sigma, #06‐340) antibodies, followed by protein G Sepharose beads.
3T9 MycER cells were biochemically fractionated according to the following protocol. Cell pellets were resuspended in 10 volumes of buffer RSBB (10 mM Tris–HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, Protease and phosphatase inhibitors) and incubated for 15 min at 4°C. NP‐40 was then added to a final concentration of 0.2%. Cells were lysed by douncing and the lysate centrifuged at 1,200 g for 10 min at 4°C. The supernatant was kept as the cytoplasmic fraction. Nuclei were resuspended in 20 volumes of Washing Buffer (0.88 M sucrose, 3 mM MgCl2, Protease and phosphatase inhibitors) and centrifuged for 5 min at 2,000 g at 4°C. The resulting pellet was resuspended in a 1:1 mix of glycerol buffer (20 mM Tris–HCl pH8.0, 75 mM NaCl, 0.5 mM EDTA, 50% (v/v) glycerol, 0.85 mM DTT) and nuclei lysis buffer (20 mM HEPES pH 7.6, 7.5 mM MgCl2, 0.2 mM EDTA, 300 mM NaCl, 1 M Urea, 1% v/v NP‐40, 0.1 mM DTT). After gentle vortexing and incubation (5 min on ice), the samples were pelleted at top speed in a benchtop centrifuge (16,000 g, 2 min, 4°C) and supernatants were retained as the nucleoplasmic, soluble nuclear fraction. The pellet was washed in glycerol/lysis buffer, pelleted, resuspended in Laemmli buffer, and sonicated to obtain the chromatin‐associated fraction. Protein fractions were loaded per cell equivalents and subjected to SDS–PAGE, followed by Western blot analysis with the indicated antibodies.
Production and purification of Myc and Max bHLH‐LZ peptides, heterodimerization, and DNA‐binding assay
Peptides spanning the bHLH‐LZ domains of Max (p21 isoform; sequence: MADKRAHHNA LERKRRDHIK DSFHSLRDSV PSLQGEKASR AQILDKATEY IQYMRRKNHT HQQDIDDLKR QNALLEQQVR ALEGSGC) was expressed in E. coli and purified as previously described (McDuff et al, 2009). The MycWT (MTEENVKRRT HNVLERQRR NELKRSFFAL RDQIPELENN EKAPKVVILK KATAYILSVQ AEEQKLISEE DLLRKRREQL KHKLEQLRNS CAHHHHHHHH), MycHEA, and MycRA His‐tagged variants were expressed in the pET‐3a plasmid (Genscript) and purified from the BL21 (DE3) arabinose‐inducible bacterial strain (Invitrogen) under denaturing conditions on HisTrapFF (Cytiva). Identity and purity of each purified construct was confirmed by mass spectrometry, Western blot analysis, SDS–PAGE, and UV spectroscopy.
Heterodimerization of the Myc variants with Max was assayed by circular dichroism (CD). CD measurements were performed on a Jasco J‐810 spectropolarimeter equipped with a Jasco Peltier‐type thermostat. Protein samples were prepared in 50 mM KH2PO4, 50 mM KCl, 1 mM tris(2‐carboxyethyl)phosphine (TCEP) pH 6.8 and loaded into quartz cuvettes of 0.1 cm pathlength. Far‐UV spectra were recorded at 20°C by averaging 5 scans at 0.1‐nm intervals. Thermal denaturations were recorded at 222 nm from 20 to 90°C with a heating rate of 1°C/min and a bandwidth of 2.0 nm.
The double‐stranded DNA probe (labeled with the IRD700 fluorophore) and the unlabeled non‐specific competitor probes were ordered from Integrated DNA Technologies (Coralville, IA) and resuspended in DNAse‐free water. CACGTG probe: 5′‐d(GCG CGG GCA CGT GGG CCG GGG)‐3′; CACGTC probe: 5′‐d(GCG CGG GCA CGT CGG CCG GGG)‐3′; non‐specific (NS) probe: 5′‐d(GCG CGG GGG ATC CGG CCG GGG)‐3′. The concentration of the annealed oligonucleotides was confirmed by measuring absorbance at 260 nm. Myc/Max DNA binding was assayed in an electrophoretic mobility shift assay, as previously described (Beaulieu et al, 2012); briefly, for the titration experiments, the fluorescently labeled IRD‐CACGTG probe was incubated with a fixed amount of Max bHLH‐LZ (2 µM) and increasing concentrations of either Myc construct (WT or HEA): 1, 2, 3, 4, 5, 6, 9, or 12 µM. For the competition experiments, the labeled IRD‐CACGTG probe was mixed with varying concentrations of the unlabeled competitors (0.195, 0.390, 0.781, 1.563, 3.125, 6.25, 12.50, and 25 µM), followed by addition of the proteins (2 μM Max and 6 μM Myc) in a final volume of 20 μl. Samples were incubated for 20 min prior to loading on the native PAGE in 20 mM Tris‐acetate buffer, pH 8.0. Electrophoresis conditions were 100 V for 40 min. The bands intensities were quantified using ImageJ (https://imagej.nih.gov/ij/) and analyzed using GraphPad Prism version 9.0.0 (GraphPad Software, San Diego, California USA, www.graphpad.com). Nonlinear regression was applied to determine affinities, and the fitted curves were compared using the software‐embedded comparison function.
Proliferation assays
For growth curve experiments, 70,000 Rat HO15.19 cells were plated in triplicate in 6‐well plates and counted every 3 days for 9 days. Similarly, 70,000 3T9 cells expressing the various forms of MycER were plated in the presence or absence of 400 nM OHT and counted every 2 days up to day 6. In the experiments performed with the cb9Δmyc cells, 80,000 cells per well were plated in the presence of doxycycline for 2 days, then counted, and re‐plated with or without doxycycline, every 2 days for the following 10 days. For colony forming assays (CFA), for all cell lines, 10,000 cells were plated in 10 cm dishes, let grow for 6–11 days, and stained with crystal violet.
For cell cycle analysis, cells were incubated with 33 μM BrdU for 20 min, harvested and washed in PBS, and fixed in ice‐cold ethanol. Upon DNA denaturation with 2N HCl for 25 min, cells were stained with an anti‐BrdU primary antibody (BD Biosciences, #347580) and an FITC‐conjugated anti‐mouse secondary antibody (Jackson ImmunoResearch, # 715‐545‐150). DNA was stained by resuspending the cells in 2.5 μg/ml propidium iodide (Sigma) overnight at 4°C before acquisition with a MACSQuant® Analyzer.
Antibodies
The following antibodies were used for immunoblotting: c‐Myc Y69 (Abcam ab32072), MAX (Santa Cruz sc‐197), histone H3 (Abcam, ab1791), Vinculin (Sigma, V9264), FLAG (Abcam, ab1162), HRP‐conjugated goat‐anti‐rabbit (Bio‐Rad 170‐6515), and HRP‐conjugated goat‐anti‐mouse (Bio‐Rad 170‐6516), for immunoprecipitation: c‐Myc (Sigma 06‐340) and ER‐alpha (Sigma 06‐935), for ChIP: c‐Myc N262 (Santa Cruz sc‐764) and ER‐alpha (Sigma 06‐935), and for FACS: BrdU (Becton Dickinson 347580).
Genome editing
Bi‐allelic deletion of the Myc basic region (BR) was performed exploiting the type II CRISPR‐Cas tool (Ran et al, 2013). Single guide RNA (sgRNA) sequences to target the MYC gene in the proximity of the BR‐coding region were designed using the online software CRISPR Design Tool (http://crispr.mit.edu/) and cloned as DNA inserts into pSpCas9 (BB)‐2A‐GFP (PX458) (Addgene plasmid # 48138; a gift from Feng Zhang) (Ran et al, 2013), encoding also the Cas9 protein and GFP. Out of ten tested sgRNAs, we picked the two with the highest cutting efficiency in a surveyor nuclease assay (Ran et al, 2013) (sgRNA7: ACTCCTAGTGATGGAACCC; sgRNA8: ACACGGAGGAAAACGACAAGAGG; Fig EV2A). We then transfected cb9 cells with sgRNA7 and sgRNA8 together (0.5 µg each), sorted single GFP‐positive cells on a 96‐well plate, allowed the cells to expand in culture, and screened the resulting cell clones by PCR and Sanger sequencing. We thus obtained one clone, named cb9Δmyc, in which both c‐myc alleles underwent inactivating deletions, although in different ways: one allele encoded a protein missing the BR, Helix I and the loop, and the second a truncated protein lacking the whole C‐terminal bHLH‐LZ domain (Fig EV2A).
Single‐molecule tracking (SMT) acquisition
The day before single‐molecule tracking experiments, we plated 3T9 cells infected with plasmids expressing HaloTag versions of MycWT, MycHEA, or MycRA on 4‐well LabTek covergrass chambers. One hour before imaging, cells were labeled with 1nM JF549 ligand (Grimm et al, 2016) (Janelia Farm, Ashborn, Virginia, USA), incubated for 30 min at 37°C, and extensively washed (two rounds of three washes in PBS followed by 15 min incubation at 37°C in phenol‐red free DMEM).
Imaging was carried out on a custom‐built microscope capable of inclined illumination (Tokunaga et al, 2008), based on a Olympus IX‐73 microscope frame (Olympus Life Science, Segrate, IT), equipped with a stage incubator to control temperature (37°C) and CO2 concentration (5%) and a 561 nm diode laser (100 mW Cobolt 06‐01 Series, Cobolt AB, Solna, Sweden), that is synchronized to the camera to achieve stroboscopic illumination. For fast frame‐rate acquisitions, we used an Evolve 512 EM‐CCD camera (Photometrics, Tucson, AZ, USA), in combination with a ×100, 1.49 NA oil immersion objective (Olympus Life science), resulting in a pixel size of 158 nm. In this case, we set the laser exposure to 2 ms, the time between consecutive images ttl to 10 ms, and the laser power to ~1 kW/cm2 and we collected movies composed by 1,000 frames. For slow frame‐rate acquisitions, we used a Hamamatsu Orca Fusion sCMOS camera (Hamamatsu Photonics Italia S.r.l, Arese, Italy), combined with a ×60, 1.49 NA oil immersion objective (Olympus Life Science), resulting in a pixel size of 108 nm. In this case, we set the laser exposure to 50 ms—that results in isolating bound molecules, by motion blurring of the diffusing ones (Chen et al, 2014; Hipp et al, 2019)—the laser power to 100 W/cm2 and we collected movies composed by up to 200 frames, varying the time between consecutive images ttl between 200 and 2,000 ms. For every experimental condition, we acquired at least 30 cells on two experimental days.
Analysis of the SMT movies—measurement of the bound fraction
The SMT movies collected at fast frame rate were processed using custom‐written MATLAB routines, in order to identify and track individual molecules, as previously described (Mazza et al, 2012; Loffreda et al, 2017). A maximum single‐molecule displacement of 1.2 μm was allowed between consecutive frames. The resulting tracks were analyzed to quantify the bound fraction, by populating a histogram of single‐molecule displacements with bin‐size Δr, equal to 20nm. The histogram was then normalized in order to provide the probability p(r)Δr to observe a molecule jumping a distance between r−Δr/2 and r+Δr/2 in the time between two consecutive frames ttl, which was then fit with a three‐component diffusion model, as previously described (Speil et al, 2011; Loffreda et al, 2017; Hipp et al, 2019):
where fi is the fraction of molecules moving with a diffusion coefficient equal to Di. Of note, for the slowest D 1 we measure diffusion coefficients < 0.1 μm2/s, typical of chromatin‐bound nuclear proteins at these frame rates (Hansen et al, 2017; Loffreda et al, 2017). f 1 thereby represents the average fraction of bound molecules. To provide standard deviations the fitting parameters, a bootstrapping procedure was adopted as described (Hipp et al, 2019). Briefly, we performed multiple fitting iterations, each of them after dropping 20% of the data for each of the data set. Errors are provided as standard deviations of the obtained distribution of parameters following 2,000 individual fitting iterations.
Analysis of the SMT movies—measurement of the residence times
The SMT movies collected at slow frame rate were processed using the ImageJ plug‐in TrackMate (Tinevez et al, 2017). To isolate the bound molecules, we allowed a maximum displacement of 220 nm, that allows counting 99% of chromatin‐bound molecules (Mazza et al, 2012), and we automatically filled‐in gaps of up to three consecutive frames in the tracks. We then computed the cumulative distribution of bound‐molecule residence times and we extracted kinetic parameters on the unbinding process using a global fitting procedure, that allows to minimize the artifacts due to photobleaching, as described (Gebhardt et al, 2013; Hipp et al, 2019). The data were best described by a three‐component exponential decay, providing three dissociation constants k 1,k 2,k 3, and the respective weights F 1,F 2,F 3. The average residence time was then calculated as the weighted average: . Errors were calculated as SDs from a bootstrapping procedure, as described above.
Next‐generation sequencing data filtering and quality assessment
RNA‐seq reads were filtered using the fastq_quality_trimmer and fastq_masker tools of the FASTX‐Toolkit suite (http://hannonlab.cshl.edu/fastx_toolkit/). Their quality was evaluated and confirmed using the FastQC application (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Pipelines for primary analysis (filtering and alignment to the reference genome of the raw reads) and secondary analysis (expression quantification, differential gene expression) have been integrated in the HTS‐flow system (Bianchi et al, 2016). Bioinformatic and statistical analyses were performed using R with Bioconductor and comEpiTools packages (Gentleman et al, 2004; Kishore et al, 2015). All R scripts used in data analysis and generation of figures are available upon request.
RNA‐seq
RNA extraction, processing, and sequencing, as well as the filtering of RNA‐seq reads and bioinformatic and statistical analyses, were performed as previously described (Tesi et al, 2019; Bisso et al, 2020). In particular, absolute gene expression was defined as reads per kilobase per million mapped reads, defining total library size as the number of reads mapping to exons only (eRPKM). Differentially expressed genes (DEGs) were identified using the Bioconductor Deseq2 package (Love et al, 2014) as genes whose q‐value is lower than 0.05. Functional annotation analysis to determine enriched Gene Ontology categories was performed using the online tool at http://software.broadinstitute.org/gsea/index.jsp. Gene set enrichment analysis (GSEA) was performed using the Desktop tool of the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp) with custom gene lists (Fig EV5C).
ChIP‐seq
The preparation, processing, and sequencing of ChIP‐seq samples were performed as previously described (Sabò et al, 2014). ChIP‐seq NGS reads were aligned to the mouse reference genome mm9 through the BWA aligner (http://bio‐bwa.sourceforge.net) using default settings. Peaks were called using the MACS2 software (v2.0.10) with the option “– mfold = 7,30 ‐p 0.00001 ‐f BAMPE”, thus outputting only enriched regions with P‐value < 10−5. Promoter peaks were defined as all peaks with at least one base pair overlapping with the interval between −2 kb to +2 kb from the nearest TSS. The presence of canonical and variant E‐boxes (CACGCG, CATGCG, CACGAG, CATGTG) (Blackwell et al, 1993; Grandori et al, 1996; Perna et al, 2012; Guo et al, 2014; Allevato et al, 2017) in Myc ChIP‐seq peaks was scored in a region of 100 bp around the peak summit. When comparing Myc ChIP‐seq with H3K4me3, H3K4me1, or H3K27ac histone marks to define peaks in active promoter or enhancers (Zhou et al, 2011; Calo & Wysocka, 2013), we considered two peaks as overlapping when sharing at least one base pair (findOverlaps tool of the comEpiTools R package). Motif discovery was performed using MEME‐ChIP suite (Bailey et al, 2009) with default parameters using as input the regions ± 100 bp around peak summits reported by MACS2. For heatmap and intensity plots, we used bamCoverage from deepTools 3.3.1 (Ramirez et al, 2016) to calculate read coverage per 10‐bp bin using RPKM normalization option. Heatmaps were performed through the functions computeMatrix followed by plotHeatmap from deepTools using the normalized bigwig files.
Statistical analysis
All experiments were performed at least in biological triplicates. Sample size was not predetermined, but is reported in the respective figure legends. Wilcoxon's test is used for non‐normal distributions. Fisher's exact test is used for categorical data. Chi‐squared test is used for categorical data where the contingency table is 2xN where N > 2. In the remaining cases, two‐tailed Student’s t‐test was used to compare between two groups and expressed as P‐values. Each test was used when data met their assumptions of validity.
Author contributions
PP, TK, and AS designed and performed most of the experiments described in this work. AV and MDo provided technical support. MDa, MF, MJM, and AS performed bioinformatic data analysis. AL and DM produced and analyzed data with single‐molecule tracking microscopy. M‐EB, VCC, HT, and LS produced the in vitro binding data. MM contributed to the structure‐based design of the Myc mutants. BA and AS conceived the project, co‐supervised the work, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4A
Acknowledgements
We thank Stefano Campaner, Gioacchino Natoli, Sebastiano Pasqualato, Matteo J. Marzi, Andrew Wilkie, and colleagues in our group for discussion, insight, and suggestions. We also thank L. Rotta, S. Bianchi, T. Capra, and L. Massimiliano for assistance with Illumina sequencing and L. Lavis for providing the JF549 HaloTag ligand. LS, VCC, HT, and MEB acknowledge the Cellex Foundation for providing research facilities and equipment. AL and DM were supported by Fondazione Cariplo (GR: 2014‐1157) and by the Italian Association for Cancer Research (AIRC, IG 2018‐21897). Work in the Amati laboratory was supported by funds from the European Research Council (grant agreement no. 268671‐MycNEXT), the Italian Health Ministry (RF‐2011‐ 02346976), and AIRC (IG 2015‐16768 and IG 2018‐21594).
The EMBO Journal (2021) 40: e105464.
Contributor Information
Bruno Amati, Email: bruno.amati@ieo.it.
Arianna Sabò, Email: arianna.sabo77@gmail.com.
Data availability
The RNA‐seq and ChIP‐seq data described in this work are accessible through Gene Expression Omnibus (GEO) series accession number GSE147639 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147639); previously described ChIP‐seq data (H3K4me3, H3K4me1, H3K27ac, Dnase I, RNAPII) (Sabò et al, 2014) are accessible through the accession number GSE51011 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51011).
References
- Allevato M, Bolotin E, Grossman M, Mane‐Padros D, Sladek FM, Martinez E (2017) Sequence‐specific DNA binding by MYC/MAX to low‐affinity non‐E‐box motifs. PLoS One 12: e0180147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H (1992) Transcriptional activation by the human c‐Myc oncoprotein in yeast requires interaction with Max. Nature 359: 423–426 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluapuri A, Hofstetter J, Dudvarski Stankovic N, Endres T, Bhandare P, Vos SM, Adhikari B, Schwarz JD, Narain A, Vogt M et al (2019) MYC recruits SPT5 to RNA polymerase II to promote processive transcription elongation. Mol Cell 74: 674–687.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett J, Birrer MJ, Kato GJ, Dosaka‐Akita H, Dang CV (1992) Activation domains of L‐Myc and c‐Myc determine their transforming potencies in rat embryo cells. Mol Cell Biol 12: 3130–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu ME, McDuff FO, Frappier V, Montagne M, Naud JF, Lavigne P (2012) New structural determinants for c‐Myc specific heterodimerization with Max and development of a novel homodimeric c‐Myc b‐HLH‐LZ. J Mol Recognit 25: 414–426 [DOI] [PubMed] [Google Scholar]
- Bianchi V, Ceol A, Ogier AG, de Pretis S, Galeota E, Kishore K, Bora P, Croci O, Campaner S, Amati B et al (2016) Integrated systems for NGS data management and analysis: open issues and available solutions. Front Genet 7: 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisso A, Filipuzzi M, Gamarra Figueroa GP, Brumana G, Biagioni F, Doni M, Ceccotti G, Tanaskovic N, Morelli MJ, Pendino V et al (2020) Cooperation between MYC and beta‐catenin in liver tumorigenesis requires Yap/Taz. Hepatology 72: 1430–1443 [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H (1993) Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol 13: 5216–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN (1991) Max: a helix‐loop‐helix zipper protein that forms a sequence‐specific DNA‐binding complex with Myc. Science 251: 1211–1217 [DOI] [PubMed] [Google Scholar]
- Boisson B, Wang YD, Bosompem A, Ma CS, Lim A, Kochetkov T, Tangye SG, Casanova JL, Conley ME (2013) A recurrent dominant negative E47 mutation causes agammaglobulinemia and BCR(‐) B cells. J Clin Invest 123: 4781–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlie P, Ceska T, Lamers M, Romier C, Stier G, Teo H, Suck D (1997) The crystal structure of an intact human Max‐DNA complex: new insights into mechanisms of transcriptional control. Structure 5: 509–520 [DOI] [PubMed] [Google Scholar]
- Bywater MJ, Burkhart DL, Straube J, Sabo A, Pendino V, Hudson JE, Quaife‐Ryan GA, Porrello ER, Rae J, Parton RG et al (2020) Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat Commun 11: 1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E et al (2014) Single‐molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156: 1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Cole MD (2007) The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy‐terminal domain independently of direct DNA binding. Mol Cell Biol 27: 2059–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Cole MD (2010) Myc regulation of mRNA cap methylation. Genes Cancer 1: 576–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croci O, De Fazio S, Biagioni F, Donato E, Caganova M, Curti L, Doni M, Sberna S, Aldeghi D, Biancotto C et al (2017) Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev 31: 2017–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ (2002) Myc recruits P‐TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 9: 9 [DOI] [PubMed] [Google Scholar]
- Ferre‐D'Amare AR, Prendergast GC, Ziff EB, Burley SK (1993) Recognition by Max of its cognate DNA through a dimeric b/HLH/Z domain. Nature 363: 38–45 [DOI] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c‐Myc to chromatin mediates mitogen‐induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt JC, Suter DM, Roy R, Zhao ZW, Chapman AR, Basu S, Maniatis T, Xie XS (2013) Single‐molecule imaging of transcription factor binding to DNA in live mammalian cells. Nat Methods 10: 421–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345 [DOI] [PubMed] [Google Scholar]
- Grandori C, Mac J, Siebelt F, Ayer DE, Eisenman RN (1996) Myc‐Max heterodimers activate a DEAD box gene and interact with multiple E box‐related sites in vivo. Embo J 15: 4344–4357 [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott‐Schwartz J, Liu Z, Lionnet T et al (2016) Bright photoactivatable fluorophores for single‐molecule imaging. Nat Methods 13: 985–988 [DOI] [PubMed] [Google Scholar]
- Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall' Olio V, Zardo G, Nervi C, Bernard L, Amati B (2006) Myc‐binding‐site recognition in the human genome is determined by chromatin context. Nat Cell Biol 8: 764–770 [DOI] [PubMed] [Google Scholar]
- Guo J, Li T, Schipper J, Nilson KA, Fordjour FK, Cooper JJ, Gordan R, Price DH (2014) Sequence specificity incompletely defines the genome‐wide occupancy of Myc. Genome Biol 15: 482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X (2017) CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Churchman LS (2017) The code and beyond: transcription regulation by the RNA polymerase II carboxy‐terminal domain. Nat Rev Mol Cell Biol 18: 263–273 [DOI] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, Neugebauer KM (2017) Splicing and transcription touch base: co‐transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol 18: 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp L, Beer J, Kuchler O, Reisser M, Sinske D, Michaelis J, Gebhardt JCM, Knoll B (2019) Single‐molecule imaging of the transcription factor SRF reveals prolonged chromatin‐binding kinetics upon cell stimulation. Proc Natl Acad Sci USA 116: 880–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Weintraub H, Kedes L (1998) Intramolecular regulation of MyoD activation domain conformation and function. Mol Cell Biol 18: 5478–5484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, Vitkin N, Tong Y, Sunnerhagen M, Done SJ et al (2018) MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol Cell 72: 836–848.e7 [DOI] [PubMed] [Google Scholar]
- Kaur M, Cole MD (2013) MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome‐wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res 73: 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer‐Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P et al (2017) Myc Regulates chromatin decompaction and nuclear architecture during B cell activation. Mol Cell 67: 566–578.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH (2008) An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132: 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore K, de Pretis S, Lister R, Morelli MJ, Bianchi V, Amati B, Ecker JR, Pelizzola M (2015) methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics 16: 313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M, Recordati C, Bianchi S, Rotta L, Capra T et al (2016) Identification of MYC‐dependent transcriptional programs in oncogene‐addicted liver tumors. Cancer Res 76: 3463–3472 [DOI] [PubMed] [Google Scholar]
- Kress TR, Sabò A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15: 593–607 [DOI] [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, Eisenman RN (1992) Myc and Max proteins possess distinct transcriptional activities. Nature 359: 426–429 [DOI] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI (1995) A modified oestrogen receptor ligand‐binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res 23: 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffreda A, Jacchetti E, Antunes S, Rainone P, Daniele T, Morisaki T, Bianchi ME, Tacchetti C, Mazza D (2017) Live‐cell p53 single‐molecule binding is modulated by C‐terminal acetylation and correlates with transcriptional activity. Nat Commun 8: 313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi O, Varshney D, Phillips NM, Cowling VH (2016) c‐Myc deregulation induces mRNA capping enzyme dependency. Oncotarget 7: 82273–82288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M, Wolf E (2016) Different promoter affinities account for specificity in MYC‐dependent gene regulation. Elife 5: e15161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtel RA, Zimmermann MT, Hu G, Dasari S, Jiang M, Oishi N, Jacobs HK, Zeng Y, Hundal T, Rech KL et al (2019) Recurrent MSC (E116K) mutations in ALK‐negative anaplastic large cell lymphoma. Blood 133: 2776–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchegiani S, Davis T, Tessadori F, van Haaften G, Brancati F, Hoischen A, Huang H, Valkanas E, Pusey B, Schanze D et al (2015) Recurrent mutations in the basic Domain of TWIST2 cause ablepharon macrostomia and barber‐say syndromes. Am J Hum Genet 97: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM (1997) Phenotypes of c‐myc‐deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 8: 1039–1048 [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG (2012) A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res 40: e119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDuff FO, Naud JF, Montagne M, Sauvé S, Lavigne P (2009) The Max homodimeric b‐HLH‐LZ significantly interferes with the specific heterodimerization between the c‐Myc and Max b‐HLH‐LZ in absence of DNA: a quantitative analysis. J Mol Recognit 22: 261–269 [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H (1990) Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper‐free packaging cell line. Nucleic Acids Res 18: 3587–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhar M, Ebert A, Neumann T, Umkehrer C, Jude J, Wieshofer C, Rescheneder P, Lipp JJ, Herzog VA, Reichholf B et al (2018) SLAM‐seq defines direct gene‐regulatory functions of the BRD4‐MYC axis. Science 360: 800–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C (2019) Helix‐loop‐helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev 33: 6–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SK, Burley SK (2003) X‐ray structures of Myc‐Max and Mad‐Max recognizing DNA. Molecular bases of regulation by proto‐oncogenic transcription factors. Cell 112: 193–205 [DOI] [PubMed] [Google Scholar]
- Nie Z, Guo C, Das SK, Chow CC, Batchelor E, Simons SSJ, Levens D (2020) Dissecting transcriptional amplification by MYC. Elife 9: e52483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R et al (2012) Genome‐wide mapping of Myc binding and gene regulation in serum‐stimulated fibroblasts. Oncogene 31: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JR, Fisher BE, Baranello L, Liu JC, Kambach DM, Nie Z, Koh WS, Luo J, Stommel JM, Levens D et al (2017) Global inhibition with specific activation: how p53 and MYC redistribute the transcriptome in the DNA double‐strand break response. Mol Cell 67: 1013–1025.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posternak V, Ung MH, Cheng C, Cole MD (2017) MYC mediates mRNA cap methylation of canonical Wnt/beta‐catenin signaling transcripts by recruiting CDK7 and RNA methyltransferase. Mol Cancer Res 15: 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pretis S, Kress TR, Morelli MJ, Sabò A, Locarno C, Verrecchia A, Doni M, Campaner S, Amati B, Pelizzola M (2017) Integrative analysis of RNA polymerase II and transcriptional dynamics upon MYC activation. Genome Res 27: 1658–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA (2010) c‐Myc regulates transcriptional pause release. Cell 141: 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T (2016) deepTools2: a next generation web server for deep‐sequencing data analysis. Nucleic Acids Res 44: W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richart L, Carrillo‐de Santa Pau E, Rio‐Machin A, de Andres MP, Cigudosa JC, Lobo VJ, Real FX (2016) BPTF is required for c‐MYC transcriptional activity and in vivo tumorigenesis. Nat Commun 7: 10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabò A, Amati B (2014) Genome recognition by MYC. Cold Spring Harb Perspect Med 4: a014191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabò A, Amati B (2018) BRD4 and MYC‐clarifying regulatory specificity. Science 360: 713–714 [DOI] [PubMed] [Google Scholar]
- Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A et al (2014) Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511: 488–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammak S, Hamdani N, Gorrec F, Allen MD, Freund SMV, Bycroft M, Zinzalla G (2019) Crystal Structures and nuclear magnetic resonance studies of the apo form of the c‐MYC:MAX bHLHZip complex reveal a helical basic region in the absence of DNA. Biochemistry 58: 3144–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvé S, Naud JF, Lavigne P (2007) The mechanism of discrimination between cognate and non‐specific DNA by dimeric b/HLH/LZ transcription factors. J Mol Biol 365: 1163–1175 [DOI] [PubMed] [Google Scholar]
- Sauvé S, Tremblay L, Lavigne P (2004) The NMR solution structure of a mutant of the Max b/HLH/LZ free of DNA: insights into the specific and reversible DNA binding mechanism of dimeric transcription factors. J Mol Biol 342: 813–832 [DOI] [PubMed] [Google Scholar]
- Solomon DL, Amati B, Land H (1993) Distinct DNA binding preferences for the c‐Myc/Max and Max/Max dimers. Nucleic Acids Res 21: 5372–5376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS (2012) Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell 151: 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speil J, Baumgart E, Siebrasse JP, Veith R, Vinkemeier U, Kubitscheck U (2011) Activated STAT1 transcription factors conduct distinct saltatory movements in the cell nucleus. Biophys J 101: 2592–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesi A, de Pretis S, Furlan M, Filipuzzi M, Morelli MJ, Andronache A, Doni M, Verrecchia A, Pelizzola M, Amati B et al (2019) An early Myc‐dependent transcriptional program orchestrates cell growth during B‐cell activation. EMBO Rep 20: e47987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Adams CM, Wang J, Weissmiller AM, Creighton J, Lorey SL, Liu Q, Fesik SW, Eischen CM, Tansey WP (2019) Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc Natl Acad Sci USA 116: 25260–25268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S et al (2015) Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 58: 440–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single‐particle tracking. Methods 115: 80–90 [DOI] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, Sakata‐Sogawa K (2008) Highly inclined thin illumination enables clear single‐molecule imaging in cells. Nat Methods 5: 159–161 [DOI] [PubMed] [Google Scholar]
- Tonelli C, Morelli MJ, Bianchi S, Rotta L, Capra T, Sabò A, Campaner S, Amati B (2015) Genome‐wide analysis of p53 transcriptional programs in B cells upon exposure to genotoxic stress in vivo. Oncotarget 6: 24611–24626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Morelli MJ, Sabò A, Verrecchia A, Rotta L, Capra T, Bianchi S, Campaner S, Amati B (2017) Genome‐wide analysis of p53‐regulated transcription in Myc‐driven lymphomas. Oncogene 36: 2921–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu WB, Helander S, Pilstal R, Hickman KA, Lourenco C, Jurisica I, Raught B, Wallner B, Sunnerhagen M, Penn LZ (2015) Myc and its interactors take shape. Biochim Biophys Acta 1849: 469–483 [DOI] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay‐Odelot H, Karim S, Bartkuhn M et al (2014) Activation and repression by oncogenic MYC shape tumour‐specific gene expression profiles. Nature 511: 483–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J et al (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR (2013) The glucocorticoid receptor dimer interface allosterically transmits sequence‐specific DNA signals. Nat Struct Mol Biol 20: 876–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin B, Rohs R (2018) Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res 28: 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, Scott TG, Nabet B, Erb MA, Winter GE et al (2018) Enhancer invasion shapes MYCN‐dependent transcriptional amplification in neuroblastoma. Nat Genet 50: 515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE (2011) Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 12: 7–18 [DOI] [PubMed] [Google Scholar]
- Zippo A, De Robertis A, Serafini R, Oliviero S (2007) PIM1‐dependent phosphorylation of histone H3 at serine 10 is required for MYC‐dependent transcriptional activation and oncogenic transformation. Nat Cell Biol 9: 932–944 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4A
Data Availability Statement
The RNA‐seq and ChIP‐seq data described in this work are accessible through Gene Expression Omnibus (GEO) series accession number GSE147639 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147639); previously described ChIP‐seq data (H3K4me3, H3K4me1, H3K27ac, Dnase I, RNAPII) (Sabò et al, 2014) are accessible through the accession number GSE51011 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51011).