

Abstract
The cysteine protease cruzipain is considered to be a validated target for therapeutic intervention in the treatment of Chagas disease. Hence, peptidomimetic cruzipain inhibitors having a reactive group (known as warhead) are subject to continuous studies to discover novel antichagasic compounds. Here, we evaluated how different warheads for a set of structurally similar related compounds could inhibit the activity of cruzipain and, ultimately, their trypanocidal effect. We first investigated in silico the intrinsic reactivity of these compounds by applying the Fukui index to correlate it with the enzymatic affinity. Then, we evaluated their potency against T. cruzi (Y and Tulahuen strains), which revealed the reversible cruzain inhibitor Neq0656 as a better trypanocidal agent (ECY.strain50 = 0.1 μM; SI = 58.4) than the current drug benznidazole (ECY.strain50 = 5.1 μM; SI > 19.6). We also measured the half-life time by HPLC analysis of three lead compounds in the presence of glutathione and cysteine to experimentally assess their intrinsic reactivity. Results clearly illustrated the reactivity trend for the warheads (azanitrile > aldehyde > nitrile), where the aldehyde displayed an intermediate intrinsic reactivity. Therefore, the aldehyde bearing peptidomimetic compounds should be subject for in-depth evaluation in the drug discovery process.
Aldehyde peptide like compounds display a bivalent reactive profile and improved antichagasic potency.
1. Introduction
Chagas disease, whose etiological agent is the protozoan parasite Trypanosoma cruzi, is a serious health and social problem for people living in Latin America and areas previously considered non-endemic such as Japan, East Europe and North America.1 More than 300 000 new cases are reported every year in 21 countries around the world, with an average of one million people currently infected with T. cruzi.2–4 The only two existing drugs available for the treatment of this unmet medical need, benznidazole and nifurtimox, show many side effects and high inefficiency in the chronic stage of the disease.5 Despite this, benznidazole has recently been approved for therapeutic use in children under 12 in the USA. Nonetheless and beyond doubt, new drugs that are safe and efficacious are therefore critically needed. One approach consists in the discovery and development of cruzain (Cz) inhibitors, which is the major T. cruzi cysteine protease responsible for the survival and propagation of the protozoan parasite.6 Recently, we have reported different covalent reversible inhibitors of cruzain as potent trypanocidal agents.7–10 Moreover, we explored the effects on the affinity of cruzain inhibition by replacing a nitrile group using alternative warheads.11
The active site of cruzain is V-shaped containing a Cys25, His162, and Asn182 catalytic triad. In general, the catalytic cysteine is deprotonated by the histidine, which is stabilized by the Asn175 and a Trp184 residue shielding the thiolate–imidazolium ion-pair from the solvent. The stabilized negative charge renders the active site cysteine capable of attacking the warhead of certain types of covalent inhibitors.12 The same mechanism is present for different mammalian and protozoan cysteine proteases. In general, 19 different types of warheads have been applied for cysteine protease inhibition. However, just a few of them inhibit cruzain.11,12 K777, the leader of the first generation of irreversible Cz inhibitors, was withdrawn from the preclinical phase due to substantial side effects caused by its irreversible mode of action.12–14 The main concern is to consider that warheads require a balanced reactivity profile. They should be sufficiently reactive to form a covalent bond with the cysteine protease in the active site. Their inherent reactivity should be reduced to a necessary minimum to prevent nonspecific off-target labeling.15 As a result of the time-dependent nature of covalent inhibition, even optimized compounds with sufficient selectivity in screening panels may exert significant off-target reactivity after extended exposure times.12 The quantification of the half-life (t1/2) for the reaction of compounds with glutathione (GSH) or cysteine provides information about warhead electrophilicity and liability toward the putative off-target reactivity.16 A comparison of t1/2 data across a range of compounds with different warheads provides useful information for the design of new compounds within a desired reactivity range.17 Recently, Balogh and coworkers have used 137 chemical probes with 36 different warheads in order to investigate the impact of the warheads in the reactivity and specificity of a given covalent fragment.18 In another contribution, Martin and coworkers have demonstrated through an NMR based assay that the reactivity of a covalent modifier is dependent on the amino acid residue such as cysteine, serine, tyrosine, and threonine as a nucleophile in aqueous solution.19 McGregor and coworkers have used experimental techniques such as crystallography, hydrogen/deuterium exchange, and differential scanning fluorimetry to study electrophiles that are useful for targeting oncogenic K-Ras mutant proteins.20 Computational tools can also be employed to understand the reactivity of compounds, through quantum mechanics (QM) reactivity indices.21,22 These indices can help to reduce the synthetic effort required during a drug discovery endeavor while increasing knowledge of how substitutions affect warhead reactivity. Herein, we have studied the intrinsic reactivity of a set of Cz inhibitors and their trypanocidal activities on two different T. cruzi strains resulting in four trypanocidal agents equipotent to benznidazole and one nanomolar T. cruzi killer acting on the Y strain. Also, we have measured the t1/2 by HPLC for key compounds to evaluate their intrinsic reactivity in water in the presence of the cysteine nucleophilic thiol. We have found that the local electrophilicity index correlates with the Cz affinity for a set of selected pairs of compounds.
2. Results
2.1. Design, synthesis and kinetic characterization
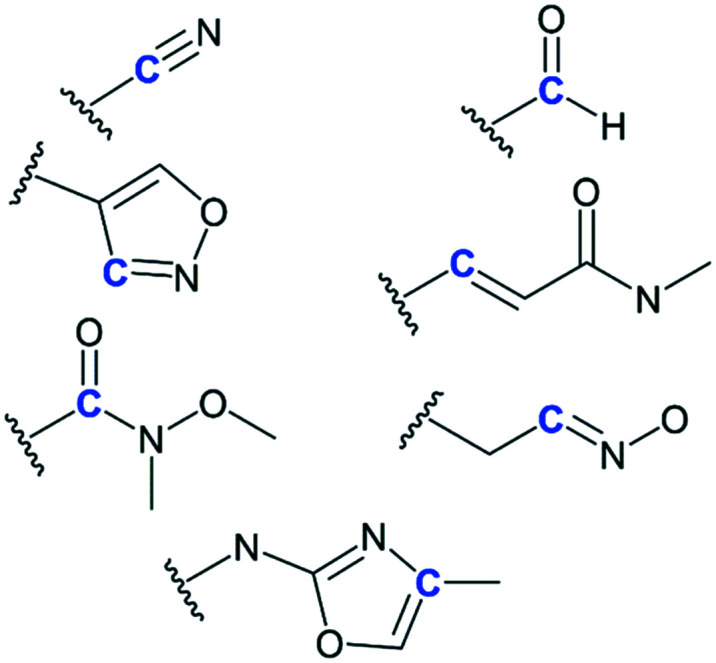
In our previous study, we reported a dipeptidyl nitrile compound, Neq0409, as a reversible covalent inhibitor of Cz. The crystal structure (PDB: 4QH6) displayed the covalent bond between the residue Cys25 at the nitrile group. Kinetic characterization reveals a fast reversible mode of binding.7 In the second step, we explored the effects on potency of cruzain inhibition by replacing a nitrile group with alternative warheads.11 Thanks to the knowledge gained in our recent SARs, we have herein designed and synthesized compounds Neq0673 and Neq0646. The other compounds present in this study have already been reported in our previous work.11Neq0673 and Neq0646 bear isoxazole as a warhead, which can be seen as prodrugs for the oxime group, which has displayed an improvement in reactivity toward Cz compared with nitrile-based inhibitors. Neq0673 and Neq0646 contain Trp and Phe moieties in P2, respectively, which are privileged substructures for inhibition of Cz and trypanocidal activity.7,9 Modifications in P3 were accomplished by exchanging a carbamate (Cbz protecting group) for an amide bond with benzoic acid to increase their metabolic stability (Fig. 1).
Fig. 1. 2D schematic representation of the compounds presented in this study. Blue color depicts the difference in warhead structures.

An overview of the synthesis of compounds Neq0646 and Neq0673 is presented in Fig. 2. First, the 4-amine-isoxazole (2) was synthesized by selective nitration of isoxazole followed by reduction with SnCl2 under acidic conditions to give the desired product. Then, the warhead (2) was coupled to the corresponding compound (3 or 4) with EDC and HOBt to afford the desired dipeptidyl isoxazole compounds. The final compounds were then purified using a HPLC equipped with a chiral column to achieve a purity higher than 95% (Fig. 1 and Scheme 1).
Fig. 2. Schematic representation of the warheads investigated using electrophilicity indices. Carbon (in blue) reacts with the thiolate nucleophile of cysteine. The Fukui index of electrophilicity was calculated for the carbon represented in blue.
Scheme 1. Synthetic scheme for the synthesis of compounds Neq0646 and Neq0673. Conditions: (a) TFAA, NH4NO3, rt, 2 h; (b) 6 M HCl, SnCl2, 1.5 h, rt; (c) Trp-OH or Phe-OH, 1 M NaOH, 0 °C, 0.5 h; (d) EDC, HOBt, THF, rt, 18 h.

Cz inhibition was evaluated by fluorometric assays. The results reported in Table 1 (see below) show a wide range of affinity, spanning from high micromolar to one-digit nanomolar inhibition. As already described in our previous work,11 substitution of the nitrile warhead for the azanitrile leads to a strong increase in potency (ΔpKi > 2.0) while all the other modifications resulted in a decrement in the reactivity profile for Cz inhibition.
The Mulliken charge (Qc) of neutral and anionic forms, electrophilic Fukui function (fc+), global electrophilicity (ω) and local electrophilicity (ωc+) were computed for the compound set from which the respective pKi against Cz was measured.
| Compound (Neq) | Q C,Neutral | Q C,anion | f c + | ω (eV) | ω c + (eV) | pKiCz |
|---|---|---|---|---|---|---|
| 500 | 0.321 | 0.332 | 0.0108 | 0.183 | 0.0020 | 6.3 |
| 539 | 0.045 | 0.050 | 0.0048 | 0.136 | 0.0006 | 5 |
| 615 | 0.835 | 0.833 | −0.0015 | 0.199 | −0.0003 | 4 |
| 646 | 0.045 | 0.049 | 0.0040 | 0.130 | 0.0005 | 4.7 |
| 652 | −0.027 | −0.044 | −0.0179 | 0.131 | −0.0023 | 3.7 |
| 653 | 0.081 | 0.071 | −0.0105 | 0.135 | −0.0014 | 4.6 |
| 654 | −0.023 | −0.026 | −0.0025 | 0.148 | −0.0004 | 3.9 |
| 655 | 0.315 | 0.333 | 0.0183 | 0.087 | 0.0016 | 5.2 |
| 656 | 0.574 | 0.580 | 0.0051 | 0.177 | 0.0009 | 5.4 |
| 657 | −0.036 | −0.063 | −0.0270 | 0.164 | −0.0044 | 3.4 |
| 673 | 0.045 | 0.049 | 0.0040 | 0.128 | 0.0005 | 5 |
| 675 | 0.125 | 0.127 | 0.0030 | 0.238 | 0.0007 | 5 |
| 677 | 0.515 | 0.537 | 0.0221 | 0.183 | 0.0041 | 8.7 |
| 690 | 0.518 | 0.536 | 0.0181 | 0.189 | 0.0034 | 8.8 |
2.2. Local electrophilicity index
One of the key aspects of the optimization process of covalent drugs is the modulation of reactivity of the warhead. Recently, Palazzesi and co-workers have used the electrophilicity index to estimate the absolute covalent warhead reactivity of acrylamides in aqueous solution.23 In another contribution, Berteotti and coworkers have investigated the mechanistic cysteine nucleophilic attack on nitrile-carrying compounds using DFT calculations and kinetic measurements in a water environment. Note that the reactivity of the inhibitor can be influenced by the presence of specific interactions in the active site of the protein. However, in this work, the reactivity of compounds is explored in a water environment following the same approach of previous studies.23,24 The advantage of this approach is that QM calculations can estimate the reactivity only based on the electrophilicity, without including protein–inhibitor specific interaction effects, which would require long computational times. Herein, the local electrophilicity was used to predict the reactivity of warheads. We have used the Fukui function, in order to estimate the electrophilic character of the carbon (see Fig. 2) involved in the nucleophilic attack of the Cys25. Overall, we make use of the electrophilic Fukui function (fc+), global electrophilicity (ω) and local electrophilicity (ωc+) to estimate the covalent inhibitor reactivity (see Table 1). Then, we have evaluated the correlation of this parameter with the ligand binding affinity. Other electronic parameters calculated for the compounds can be found in the ESI.†
From the data shown in Table 3, we have found a strong correlation between the local electrophilicity (ωc+) and the ligand binding affinity, as shown in Fig. 3. The correlation between the other parameters and pKi can be found in the ESI.†
Half-life measurement and decay constant by HPLC with glutathione and cysteine.
| Neq ID | pKi | Warhead | Half-life with glutathione | Half-life with cysteine | Decay constant with glutathione | Decay constant with cysteine |
|---|---|---|---|---|---|---|
| 409 | 6.3a | Methylene-nitrile | >6000 min | 74.3 min | — | 9.33 × 10−3 min−1 |
| 570 | 6.6a | Cyclopropane-nitrile | >6000 min | 159.0 min | — | 4.36 × 10−3 min−1 |
| 656 | 5.4 | Aldehyde | 40.0 min | 22.2 min | 1.73 × 10−2 min−1 | 3.12 × 10−2 min−1 |
| 690 b | 8.8 | Aza-nitrile | <5 min | <5 min | — | — |
| Nilvadipine | — | — | >5000 min | 49.5 min | — | 1.41 × 10−2 min−1 |
Values retrieved from the recent literature.7
When the concentrations of the compound Neq0690 and the thiol are half of the initial values, the kobs is 1.01 × 10−2 min−1 for the glutathione assay and 3.15 × 10−2 min−1 for the cysteine assay (t1/2 = 68.45 and 21.98 min respectively). The standard deviation is lower than 10% for all experiments.
Fig. 3. The putative linear equation and the coefficient of determination obtained through the linear correlation between ωc+ and pKiCz.

Fig. 3 shows that a higher value of the pKi against Cz is related to a higher value of ωc+. This result is interesting because a single descriptor obtained in the aqueous phase presented a good correlation with the inhibition of the enzyme, as other authors have done in order to obtain a descriptor correlated with the inhibition of cysteine proteases,24,25 even knowing that it is difficult to find a single property to describe the biochemical activity.21,26–28
Compounds Neq0539 (pKi = 5.0) and Neq0653 (pKi = 4.6) are over an order of magnitude less potent than the reference nitrile Neq0500 (pKi = 6.3). A ΔpKi value of 1.3 for the transformation is sufficiently small to be consistent with covalent bond formation. In the same direction, we observed a loss in potency (ΔpKi = 2.3) when exchanging the nitrile warhead to an amide group (Neq0615) which can be considered as a negative control for the covalent bond formation. The difference in potency for these compounds is nicely related to the local electrophilicity. On the other hand, the functional behaviour of warheads is sometimes assumed to be determined entirely by electrophilicity; however, the product of the covalent bond can form different non-covalent interactions with the target protein that also modulate the affinity (e.g. difference between nitrile and ketoamide dipeptidyl nitriles for cathepsin inhibition, PDB IDs: 3HHA and 1TU6).
Overall, the higher the value of local electrophilicity index for the carbon to be attacked by cysteine, the greater the stabilization of the addition of electrons; thus, this model can also be used for choosing warheads or groups that can influence the electronic properties of warheads in the design of new compounds.
2.3. Biological activities
The trypanocidal activity was then assessed for all compounds using two T. cruzi strains (Y and Tulahuen) as reported in Table 2, along with the cytotoxicity against the mammalian host cells (U2OS and LLC-MK2) and the respective selectivity indices (SIs). In general, biological activities show that Cz inhibitors are more potent against the T. cruzi Y strain than the Tulahuen one. This effect could arise from a different level of expression of Cz and its isoforms for the different strains.29 The difference in potency for different strains is already known, so it is vital to emphasize the importance of testing the same set of compounds in different strains.30Neq0500, Neq0539, and Neq0646, the dipeptidyl nitrile based compounds, appear to be trypanocidal agents equipotent to Bz, showing a low micromolar potency against the parasite (Y strain) and no cytotoxicity to the host cell. On the other hand, these dipeptidyl nitriles are ineffective against the Tulahuen strain, as also underlined in our previous work.7
Trypanocidal activity (EC50), cytotoxicity (CC50) and selectivity index (SI) for the set of compounds using the amastigote form of two strains (Y and Tulahuen) of T. cruzi in the mammalian host cells (U2OS and LLC-MK2).
| Neq ID | EC50 Tc Y (μM) | CC50 U2OS (μM) | SI | EC50 Tc Tula (μM) | CC50 LLC-MK2 (μM) | SI |
|---|---|---|---|---|---|---|
| 409 a | — | — | — | Inact | >100 | ND |
| 500 | 3.6 | >100 | >27.8 | Inact | >100 | 1.1 |
| 539 | 5.1 | >100 | >19.6 | 25.8 | >100 | >3.9 |
| 570 a | — | — | — | Inact | >100 | ND |
| 615 | ND | ND | ND | Inact | >100 | ND |
| 646 | 6.0 | >100 | >16.6 | ND | ND | ND |
| 652 | ND | ND | ND | Inact | >100 | ND |
| 653 | ND | ND | ND | Inact | >100 | ND |
| 654 | ND | ND | ND | 79.6 | >100 | >1.2 |
| 655 | 1.0 | 6.0 | 4.4 | Inact | >100 | ND |
| 656 | 0.1 | 3.0 | 58.4 | Inact | >100 | ND |
| 657 | 12.1 | >100 | >8.2 | ND | ND | ND |
| 673 | 22.4 | >100 | >8.2 | ND | ND | ND |
| 675 | Inact | >100 | ND | ND | ND | ND |
| 677 | 3.2 | 10.2 | 3.2 | 21.7 | 42.0 | 1.9 |
| 690 | 8.4 | 26.9 | 3.2 | 8.4 | 26.9 | 3.2 |
| Bz | 5.1 | >100 | >19.6 | 4.3 | >100 | >23.3 |
Values retrieved from the recent literature.7
Compound Neq0655 has a higher cytotoxic profile and low selectivity index when tested on the U2OS cell line. Azadipeptidyl nitriles display the same trend being equipotent to Bz as trypanocidal agents for both strains but with a selectivity index lower than 10. Neq0675 bearing an isoxazole as warhead is inactive for the Y strain of T. cruzi, while its analogue (Neq0646) with a Phe in P2 shows mild potency and no cytotoxicity to the same strain. Interestingly, compound Neq0656 bearing an aldehyde as a warhead is a potent trypanocidal agent for the Y strain with a high selectivity in relation to that for the mammalian cell. Although the activity of Neq0656 is not transferable to the Tulahuen strain, we believe that this result is fundamental in the ongoing research for small molecules as potent trypanocidal agents due to the high infectivity of the Y strain.31 It is noteworthy that because most of the reported compounds are covalent inhibitors of Cz, their potency against the parasite is also time-dependent; hence, future studies should be focused on the time-dependency of the provided activity.
2.4. Study of the intrinsic reactivity
Covalent inhibitors binding normally involve a two-step process in which an initial reversible binding event takes place, followed by the covalent bond-forming reaction. In the first stage, enzyme E and inhibitor I form an enzyme inhibitor complex (E⋯I) whose binding energy (ΔGbind) depends only on non-covalent interactions and is related to the inhibition constant Ki. The subsequent binding step (kinact) involves the formation of a covalent bond between the inhibitor and target. The latter step will be partly governed by the intrinsic reactivity of the warhead.15,32 In our case, the measured Ki values mostly reflect the binding free energy for non-covalent interactions and the covalent bond formation. It is difficult to define the contribution of the covalent bond formation without the kinact values. Indeed, these values are influenced by several factors other than just the chemical nature of the warhead. Also, depending on the magnitude of the non-covalent interactions with respect to the total potency outcome, we should not expect a direct correlation between the intrinsic reactivity, measured in the absence of the target enzyme, and the Ki.
However, to better evaluate the promiscuity of the different warheads independent of their biological target, the local electrophilicity (ωc+) and intrinsic reactivity are still pivotal parameters in drug discovery.
The correlation achieved between the local electrophilicity (ωc+) and the enzyme inhibition led us to investigate whether these compounds could be promiscuous electrophiles due to the high reactivity. Hence, the intrinsic reactivity (given as the half-life – t1/2) was assessed using a method described recently,17 where cysteine and glutathione were the non-specific nucleophiles. Glutathione is present in high concentration (1–10 mM) in the cell.33 It works as an important component of the intracellular redox machinery, while cysteine is a building block amino acid for many proteins, and it is the reactive amino acid in the active pocket of cysteine proteases. We measured the half-life of four compounds by HPLC using two different aqueous systems, with the results displayed in Table 3. As a model for nitrile warheads, we used Neq0409, whose crystallographic structure was previously resolved, and Neq0570, for which the reversible mode of action has also been investigated.7 In general, we can identify a trend for the intrinsic reactivity for both nucleophiles where the reactivity was azanitrile ≫ aldehyde > nitrile. In particular, the azanitrile compound Neq0690 displayed such strong reactivity with both glutathione and cysteine that it was not possible to quantify it. This high intrinsic reactivity may explain the low selectivity between the parasite and the host cell given by the low SI values (Table 2). Nitrile based compounds have low intrinsic reactivity, which was possible to measure when cysteine is present in solution as the nucleophile. As expected, the cyclopropane moiety (as in compound Neq0570) led to a small decrease in reactivity when compared with the methylene-nitrile pair (Neq0409). On the other hand, for the Cz inhibition, we observed an opposite effect thanks to the ability of the cyclopropane moiety to drive the warhead in the active site.34 Also, dipeptidyl nitriles (Neq0409 and Neq0570) are ten-times more potent inhibitors of Cz than the aldehyde (Neq0656) but less reactive to nucleophilic attack in water solution. This difference in reactivity/potency can arise from the non-covalent interaction formed by the thiomidate with the catalytic site of the enzyme.7,32
The aldehyde Neq0656 displays a balanced reactivity for both systems under study. According to Macfaul,17 compounds should have t1/2 at least equal to that of nilvadipine to be used in further assays. Nonetheless, Neq0656, with half of the t1/2 value of nilvadipine, was still selective toward the parasite, making it a good lead structure. These results are in agreement with the cytotoxicity observed in the biological studies. Therefore, we can assume that the substitution of the nitrile warhead for the aldehyde can provide a balanced reactivity, which leads to an increase in the trypanocidal activity with a high selectivity index against the host cell.
3. Material and methods
3.1. Computational details
Herein, all calculations were performed with Gaussian09.35 Geometry optimization and vibrational analysis were performed at the density functional theory (DFT) level, using the B3LYP functional36,37 and 6-311G(d,p) basis set38 in the gas phase. After confirming that the structure was at the minimum through frequency analysis, a single point calculation at the perturbation theory of Møller–Plesset (MP2)39 level and the diffuse basis set 6-311++G(d,p)38 was performed in the water phase using the PCM40 for neutral, anionic and cationic systems, keeping the external potential constant. Then, the global electrophilicity index41 (ω) was obtained by:
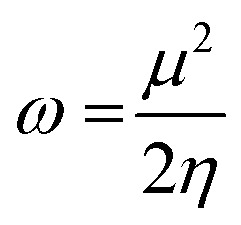 |
1 |
where μ and η are the chemical potential and chemical hardness, respectively.
In the present study, we have used the Fukui index42–45 (fk+) as a parameter for the evaluation of electrophilicity of the inhibitor. The Fukui index was obtained from the population analysis46 and charges through NBO analysis47 at the MP2 level using the diffuse basis set 6-311++G(d,p). The fk+ was calculated through finite difference, as represented below:
| fk+ = qk(N + 1) − qk(N) | 2 |
where qk(N) represents the Mulliken charge of atom k in the neutral molecule, and qk(N + 1) corresponds to the Mulliken charge of atom k in the same geometry of the neutral molecule, but in the anionic form. Since the covalent inhibition of Cz involves the nucleophilic attack of the negatively charged Cys25 (S atom) on the carbon atom of the warhead (see Fig. 2), it is important to have a parameter capable of estimating the reactivity of the warhead. Therefore, using electrophilicity and the Fukui index, it is possible to describe the electrophilic character of a reactive site within a molecule, through the local electrophilicity index41ω(r):
| ω(r) = ωfk+(r) | 3 |
3.2. Synthetic chemistry
Melting points were determined on a Büchi 510 oil bath apparatus and are uncorrected. Infrared spectra were obtained from a FT-IR Thermo Scientific Nicolet 380. The reagents, starting materials and solvents were of commercial quality and were used without further purification unless otherwise stated. All syntheses started with enantiopure amino acids. TLC analysis was carried out on Merck 60 F254 silica gel plates and visualized under UV light at 254 nm and 365 nm or by using a ninhydrin staining solution.
Purity was determined with an LC-MS instrument (AmaZon SL ESI-MS, Shimadzu LC) with a cellulose-2 Phenomenex column (250 × 4.6 mm, 5 μm) or a Daicel column (IC-chiralpak, 250 × 4.6 mm, 5 μm). Isocratic elution with MeCN and water was applied as specified (stop time 60 min, flow 0.5 mL min−1). NMR spectra were recorded on Bruker Avance 400 MHz and Bruker Avance DRX 500 MHz NMR spectrometers. Chemical shifts are reported in ppm relative to TMS or the residual proton peak of the re-protonated deuterated solvent, and the spectra were calibrated against the residual proton peak of the used deuterated solvent. The following symbols indicate spin multiplicities: s (singlet), s br (broad singlet), d (doublet), dd (doublet of doublet), t (triplet), tt (triplet of triplet), q (quartet), sept (septet), and m (multiplet). HRMS spectra were recorded on a Thermo Scientific LTQ Velos Orbitrap, in electrospray ionization (ESI) mode by direct injection.
Synthesis of 4-nitroisoxazole (1): isoxazole (15 mmol, 960 μL) was dissolved in TFAA (7.3 mL); then, NH4NO3 (22.5 mmol, 1.81 g) was added in 0.3 g portions each 15 min, keeping the reaction mixture at 25–30 °C. After complete addition, the mixture was kept at room temperature for 2 h. After that ice water (30 mL) was poured and this aqueous washing was extracted with CHCl3 (3 × 15 mL); the combined organic extracts were dried over Na2SO4 and evaporated (bath at room temperature) to give an oil that was triturated with n-hexane to give a yellow solid (50% yield). 1H-NMR (CDCl3) δ = 9.29 (s, 1H), 8.83 (s, 1H) ppm.48
Synthesis of 4-aminoisoxazole (2): to a yellow solution of 4-nitroisoxazole (1, 160 mg, 1.4 mmol) in 6 M HCl (7 mL), SnCl2 (1.327 g, 7 mmol) was added in one portion. After 1.5 h at room temperature, the resulting orange solution was treated with a saturated solution of Na2CO3 until the pH was 9. The formed solid was removed by filtration, and the aqueous solution was extracted with ethylacetate (5 × 50 mL); the organic phase, dried over MgSO4, was evaporated to give a brown oil (Rf = 0.64, ethylacetate 100%/silica) and stored at 4 °C in an inert atmosphere (65% yield). 1H-NMR (DMSO-d6) δ = 8.16 (s, 1H), 8.13 (s, 1H), 4.26 (s br, 2H) ppm.
Synthesis of 2-benzamido-3-phenylpropanoic acid (3) and 3-(1H-indol-3-yl)-2-(phenylformamido)propanoic acid (4): 2.75 mmol of the corresponding amino acid was dissolved in 1 M NaOH (6 mL) in an ice-bath. Benzoyl chloride (261 μL, 2.25 mmol) was added. After 5 min, the reaction mixture was allowed to stand at room temperature. After 20 min, the solution was cooled in ice and 1 M KHSO4 (16 mL) was added slowly. The obtained white solid was washed with 1 M KHSO4 (3 × 5 mL), H2O (10 × 3 mL), and 9 : 1 EtOH : H2O (3 × 3 mL) and dried under vacuum on P2O5 (yield 88%). For compound 3: 1H NMR (500 MHz, DMSO-d6): 12.62 (s br, 1H, D2O exchange), 10.75 (s, 1H, D2O exchange), 8.56 (m, 1H, D2O exchange), 7.28 (m, 8H), 7.13 (m, 2H), 4.54 (m, 1H), 3.29 (m, 1H), 3.19 (m, 1H). For compound 4: 1H NMR (500 MHz, DMSO-d6): 12.69 (s br, 1H, D2O exchange), 10.80 (s, 1H, D2O exchange), 8.61 (d, J = 8.0 Hz, 1H, D2O exchange), 7.81 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8 Hz, 1H), 7.51 (t, J = 7.0 Hz, 1H), 7.44 (t, J = 7.5 Hz, 2H), 7.31 (d, J = 8.0 Hz, 1H), 7.20 (s, 1H), 7.05 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 4.65 (m, 1H), 3.30 (m, 1H, H2O overlapping), 3.19 (m, 1H).
Synthesis of N-(1-(isoxazol-4-ylamino)-1-oxo-3-phenylpropan-2-yl)benzamide (Neq0646): to a suspension of (±)-2-benzamido-3-phenylpropanoic acid (3, 216 mg, 0.70 mmol), HOBt (123 mg, 0.91 mmol) and EDC (175 mg, 0.91 mmol) in CH2Cl2 (8 mL) were added under argon at 0 °C. After stirring for 1 hour at room temperature, the mixture was kept in an ice-bath, and a solution of 4-aminoisoxazole (235 mg, 2.80 mmol) in dry CH2Cl2 (2 mL) was added. The resulting mixture was kept overnight at room temperature, then the solvent was evaporated, and the residue was treated with AcOEt (30 mL) and washed with H2O (2 × 20 mL) and brine (2 × 20 mL). The organic phase was dried over MgSO4 and evaporated to give a crude residue which was purified by silica column chromatography using CHCl3/AcOEt (1 : 1) as the mobile phase, to give a solid (Rf = 0.4) crystallized from AcOEt (36% yield).
Secondary purification was carried out on a cellulose-2 Phenomenex column, in isocratic elution mode with a flow rate of 2.36 mL min−1, at 32 °C; the mobile phase composition was n-hexane/ethanol (70 : 30) (v/v) to give Neq0646. 1H-NMR (500 MHz, DMSO-d6) δ = 10.80 (s br, 1H), 10.51 (s, 1H), 9.13 (s, 1H), 8.72 (d, J = 8.0 Hz, 1H), 8.63 (s, 1H), 7.84 (m, 2H), 7.69 (d, J = 8.0 Hz, 1H), 7.52 (tt, J = 7.5 Hz, J = 1.5 Hz, 1H), 7.44 (m, 2H), 7.31 (dt, J = 8.0 Hz, J = 1.0 Hz, 1H), 7.23 (d, J = 2.0 Hz, 1H), 7.05 (m, 1H), 6.98 (m, 1H), 4.83 (qd, J = 9.5 Hz, J = 8.0 Hz, J = 5.0 Hz, 1H), 3.30 (dd, J = 14.5 Hz, J = 5.0 Hz, 1H), 3.22 (dd, J = 14.5 Hz, J = 9.5 Hz, 1H) ppm. 13C NMR (DMSO-d6): 170.72, 166.90, 147.56, 144.85, 136.52, 134.23, 131.86, 128.63, 127.97, 127.59, 124.23, 121.41, 120.17, 118.89, 118.73, 111.83, 110.55, 54.89, 27.74 ppm. HRMS (+) calc. for [C19H17N3O3]+ 335.12699, found: 336.12663 [M + H]+.
Synthesis of N-(3-(1H-indol-3-yl)-1-(isoxazol-4-ylamino)-1-oxopropan-2-yl)benzamide (Neq0673): to a suspension of (±)-2-benzamido-3-phenylpropanoic acid (216 mg, 0.70 mmol), HOBt (124 mg, 0.91 mmol) and EDC (175 mg, 0.91 mmol) in CH2Cl2 (8 mL) were added under argon at 0 °C. After stirring for 1 hour at room temperature, the mixture was kept in an ice-bath and a solution of 4-aminoisoxazole (235 mg, 2.80 mmol) in dry CH2Cl2 (2 mL) was added. The resulting mixture was kept overnight at RT, then the solvent was evaporated, and the residue was treated with AcOEt (30 mL) and washed with H2O (2 × 20 mL) and brine (2 × 20 mL). The organic phase was dried over MgSO4 and evaporated to give a crude residue that was purified by column chromatography on silica using CHCl3/AcOEt (1 : 1) as the mobile phase, to give a solid (Rf = 0.4) crystallized from AcOEt (36% yield).
Secondary purification was carried out on a cellulose-2 Phenomenex column, in isocratic elution mode with a flow rate of 2.36 mL min−1, at 32 °C; the mobile phase composition was n-hexane/ethanol (70 : 30) (v/v) to give Neq0673. 1H-NMR (500 MHz, DMSO-d6) δ = 10.80 (s br, 1H), 10.51 (s, 1H), 9.13 (s, 1H), 8.72 (d, J = 8.0 Hz, 1H), 8.63 (s, 1H), 7.84 (m, 2H), 7.69 (d, J = 8.0 Hz, 1H), 7.52 (tt, J = 7.5 Hz, J = 1.5 Hz, 1H), 7.44 (m, 2H), 7.31 (dt, J = 8.0 Hz, J = 1.0 Hz, 1H), 7.23 (d, J = 2.0 Hz, 1H), 7.05 (m, 1H), 6.98 (m, 1H), 4.83 (qd, J = 9.5 Hz, J = 8.0 Hz, J = 5.0 Hz, 1H), 3.30 (dd, J = 14.5 Hz, J = 5.0 Hz, 1H), 3.22 (dd, J = 14.5 Hz, J = 9.5 Hz, 1H) ppm. 13C NMR (DMSO-d6): 170.72, 166.90, 147.56, 144.85, 136.52, 134.23, 131.86, 128.63, 127.97, 127.59, 124.23, 121.41, 120.17, 118.89, 118.73, 111.83, 110.55, 54.89, 27.74 ppm. HRMS (+) calc. for [C21H18N4O3]+ 374.13789, found: 375.13895 [M + H]+.
3.3. Enzyme inhibition study
Recombinant cruzain, consisting of the catalytic domain of cruzipain but excluding the carboxy-terminal extension, was expressed and purified as previously described.11 The inhibitors were assayed fluorometrically (Biotek® SynergyTM HT), monitoring the hydrolysis rate of the fluorogenic substrate Z-Phe-Arg-7-amido-4-methylcoumarin (Z-FR-AMC, Sigma-Aldrich) by the enzyme cruzain with fluorescence emission at 460 nm (excitation at 355 nm) and at 37 °C. The reactions were followed over 5 min for all compounds (fast-binding, irreversible and non-covalent inhibitors) excluding Neq0690 and Neq0677 (slow-binding behaviour) for which the reaction was followed over 30 min. Enzyme kinetic assays were carried out in 200 mL of a solution containing 100 mM acetate buffer pH 5.5, 300 mM NaCl, 5 mM DTT (dithiothreitol), 5% v/v DMSO (dimethyl sulfoxide), 0.01% v/v Triton X-100 and 0.15 nM cruzain, using Corning® 96-well black flat bottom microplates. The enzyme stock aliquot was rapidly thawed at 37 °C and kept on ice until activation, in which it was incubated for 20 min in the assay buffer (100 mM acetate pH 5.5, 300 mM NaCl, 5 mM DTT) followed by an additional 2 min with inhibitors before the reaction was started by the addition of the substrate.
Visual inspection and a pre-reading of plate wells were performed to check respectively for possible precipitation and background fluorescence. None of the compounds displayed a significant fluorescence signal around 460 nm (the emission wavelength used to monitor reaction kinetics).
3.4. Mammalian cytotoxicity assay
LLC-MK2 cells were cultured in 96-well plates at a concentration of 5 × 104 cells per mL. After 48 h, the plates were washed twice with PBS, and 200 μL RPMI medium was added with serial dilutions of the compounds and benznidazole (1.95 μM to 250 μM) in triplicate. After 72 h at 37 °C, the cytotoxic activity of the compounds was determined by the classical MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] method. Briefly, 50 μL MTT dissolved in PBS (2.0 mg mL−1) was added to each well, and the plates were incubated for 4 h at 37 °C. The formed formazan crystals were dissolved with DMSO (50 μL per well), and the absorbance of the samples was measured using a spectrophotometer at 570 nm after 30 min. The cytotoxicity results (CC50) were calculated as a percentage by the formula {[(ABScontrol − ABSsample)/ABScontrol] × 100}, where ABScontrol represents the mean absorbance of the untreated control (viable cells) and ABSsample, the absorbance in each cellular treatment.
The U2OS cells were kept in high glucose DMEM media, and the culture conditions and assays were the same for LLC-MK2.
3.5. In vitro trypanocidal activity evaluation on intracellular amastigote forms (Tulahuen strain)
Cells were evaluated in 96-well plates. LLC-MK2 cells were plated at a concentration of 5 × 104 cells per mL. Trypomastigote forms of the Tulahuen LacZ strain were added at a concentration of 5 × 105 parasites per mL and placed in an incubator at 37 °C with 5% CO2 for 24 h. After the incubation period, the trypomastigote forms present were removed by successive washes with PBS, remaining only as intracellular amastigote forms. The compounds were added at different concentrations (1.95 μM to 250 μM serial dilutions) and incubated for 72 h. At the end of this period, the substrate CPRG (chlorophenol red β-d-galactopyranoside, 400 μM in 0.3% Triton X-100, pH 7.4) was added. After 4 h of incubation at 37 °C, the plates were analyzed on a spectrophotometer at 570 nm to obtain the effective concentration (EC50) to reduce the parasitemia inside the host cell. Benznidazole was used as a positive control at the same concentrations as the substances, and DMSO as a negative control. The compounds were solubilized in DMSO. The selectivity index (SI) was calculated using the formula: SI = EC50/CC50. All statistical analyses were done with the program GraphPad Prism v.5.
3.6. In vitro trypanocidal activity evaluation on intracellular amastigote forms (Y strain)
The T. cruzi Y strain was donated by A. Avila (Instituto Carlos Chagas, Fiocruz, Curitiba, Brazil). Trypomastigote forms were obtained from the supernatant of LLC-MK2 tissue cultures infected with the Trypanosoma cruzi Y. Infected cultures were maintained in low glucose DMEM media (Vitrocell) supplemented with 2% FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (all from Life Technologies), henceforth described as “Low DMEM Media”. The experiment was performed as previously described.11
3.7. Intrinsic reactivity
Liquid chromatography assay measured the half-life in a gradient mode (5–100% of B in 10 minutes) with multi-channel PDA detection (210–400 nm). The assay medium was 0.05 M phosphate buffer (pH 7.4), with 1.0 mM EDTA, 5.0 mM cysteine solution and 5% acetonitrile. Initially, 80 μL of inhibitor solution (2.5 mM) was added in the assay medium and injected in the HPLC. After that, the final solution was kept in thermal equilibrium at 37 °C. Each aliquot was taken only at the time of injection. The half-life and K were determined with GraphPad® software for a pseudo-first-order reaction. The chromatographic analysis was performed with a Shimadzu Prominence HPLC system. The HPLC system was equipped with an LC-20AT/AD ternary pumping system, SIL-20A autosampler, and CTO-20A column oven (Shimadzu Corp). The column used was a Phenomenex Gemini® 5.0 μm (4.6 × 150 mm, Phenomenex, Torrance, CA). The gradient elution, used for separation, was performed with a mobile phase composed of water as solvent A, and acetonitrile, as solvent B, at a flow rate of 1.0 mL min−1. The gradient set is as follows: 0.0–10.00 min phase B increased from 5% to 100%, 10.01–15.0 min phase B remained at 100%, 15.01 min phase B decreased to 5%, and 15.01–25.0 min phase B remained at 5%. The column oven was set at 37 °C, with the injection of 5 μL aliquots. The cysteine employed was of the levogyre form (l-cysteine), MW: 121.16 g mol−1 (C3H17NO2S) sold by the brand Sigma-Aldrich (code C7352-25G), ≥98% from a non-animal source. The glutathione used was purchased from Sigma-Aldrich (PHR1359-690MG).
The glutathione and cysteine solutions were prepared with a 50 mM final concentration in phosphate buffer with pH 7.4 and 50 mM concentration. Data have been processed with GraphPad Prism® using the one phase decay model: Y = (Y0 − plateau) × exp(−K × X) + plateau. For each experiment, a negative control without glutathione or cysteine was assessed to measure the stability of the compounds and their warheads in the time frame of the experiment.
4. Conclusions
In this study, we evaluated how the intrinsic reactivity of a set of structurally similar related compounds influenced the biological outcome of trypanocidal agents. Currently, computational methods are vital in the design of new chemical entities, but there is still little effort to estimate the intrinsic reactivity of warheads. Here, we report a successful approach to using the Fukui index to correlate anti-T. cruzi action of trypanocidal agents with different warheads. It is worth mentioning that we have discovered a new compound, Neq0656, which is 10 times more potent than the control drug, benznidazole, as a trypanocidal agent. Neq0656 also shows a higher selectivity index than benznidazole. This result underlines that warhead replacement of nitrile to aldehyde is an innovative strategy in the research of potent trypanocidal agents against the Y strain of T. cruzi. Indeed, most of the compounds here tested are also selective toward the Y strain, in comparison with the Tulahuen strain.30 This discrepancy may arise from the difference in the expression of Cz and its isoforms. We also investigated the intrinsic reactivity by HPLC to clearly illustrate that azanitrile warheads are highly reactive moieties. The higher reactivity with respect to that of nitrile can be explained by the presence of nitrogen in the alpha position, as already mentioned by Gütschow and co-workers.49 Most of them, aldehydes, which have an intermediate intrinsic reactivity between nitriles (low reactivity) and azanitriles (high reactivity), display the best biological outcome against the parasite. Although certain questions concerning the toxicity and metabolic stability of the aldehyde moiety remained unclear,50 this compound class was selected in the optimization studies of different covalent inhibitors due to its excellent potency and selectivity profile12,15 with low cytotoxicity herein observed. Outstandingly, this warhead has been selected in the structure-based design optimization processes of covalent inhibitors of the SARS-CoV-2 main protease (Mpro); the dipeptidyl aldehyde inhibitors displayed good pharmacokinetic properties and low toxicity in vivo, leading to promising drug candidates for the treatment of COVID-19.51 However, aldehydes remain relatively unpopular in drug discovery for their metabolic liability, as these chemical functionalities are prone to reduction by aldo–keto reductases, and oxidation to the corresponding acids by aldehyde dehydrogenases.52
Overall, we can elect a bivalent behaviour of compound Neq0656. On the one side, based merely on the enzymatic activity and the intrinsic reactivity, the nitrile based-compound is more efficient as it shows less intrinsic reactivity while presenting roughly the same apparent potency. On the other side, the exchange of the nitrile warhead for an aldehyde upraises the trypanocidal activity/selectivity.
Conflicts of interest
The authors declare no conflict of interest.
Supplementary Material
Acknowledgments
The authors acknowledge Fundação de Amparo à Pesquisa do Estado de São Paulo – FAPESP (grant #2013/18009-4, #2018/15904-6 and #2018/21749-3) for financing this project, and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for their financial support. JL also gives thanks for the access to the computational resources of the Supercomputer Santos Dumont (SDumont) provided by the Laboratório de Computação Científica (LNCC).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00097c
References
- Coura J. R. Viñas P. A. Nature. 2010;465:S6–S7. doi: 10.1038/nature09221. [DOI] [PubMed] [Google Scholar]
- Bern C. Kjos S. Yabsley M. J. Montgomery S. P. Clin. Microbiol. Rev. 2011;24:655–681. doi: 10.1128/CMR.00005-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelain E. J. Biomol. Screening. 2015;20:22–35. doi: 10.1177/1087057114550585. [DOI] [PubMed] [Google Scholar]
- Jackson Y. Pinto A. Pett S. Trop. Med. Int. Health. 2014;19:212–218. doi: 10.1111/tmi.12235. [DOI] [PubMed] [Google Scholar]
- Pereira P. C. Navarro E. J. Venomous Anim. Toxins Incl. Trop. Dis. 2013;19:34. doi: 10.1186/1678-9199-19-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath M. E. Eakin A. E. Engel J. C. McKerrow J. H. Craik C. S. Fletterick R. J. J. Mol. Biol. 1995;247:251–259. doi: 10.1006/jmbi.1994.0137. [DOI] [PubMed] [Google Scholar]
- Avelar L. A. A. Camilo C. D. de Albuquerque S. Fernandes W. B. Gonçalez C. Kenny P. W. Leitão A. McKerrow J. H. Montanari C. A. Orozco E. V. M. Ribeiro J. F. R. Rocha J. R. Rosini F. Saidel M. E. PLoS Neglected Trop. Dis. 2015;9:e0003916. doi: 10.1371/journal.pntd.0003916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burtoloso A. C. B. de Albuquerque S. Furber M. Gomes J. C. Gonçalez C. Kenny P. W. Leitão A. Montanari C. A. Quilles J. C. Ribeiro J. F. R. Rocha J. R. PLoS Neglected Trop. Dis. 2017;11:e0005343. doi: 10.1371/journal.pntd.0005343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianni L. Sartori G. Rosini F. De D. V. Pires G. Lopes B. R. Leitão A. Burtoloso A. C. B. Montanari C. A. Bioorg. Chem. 2018;79:285–292. doi: 10.1016/j.bioorg.2018.04.006. [DOI] [PubMed] [Google Scholar]
- Gomes J. C. Cianni L. Ribeiro J. dos Reis Rocho F. da Costa Martins Silva S. Batista P. H. J. Moraes C. B. Franco C. H. Freitas-Junior L. H. G. Kenny P. W. Leitão A. Burtoloso A. C. B. de Vita D. Montanari C. A. Bioorg. Med. Chem. 2019;27:115083. doi: 10.1016/j.bmc.2019.115083. [DOI] [PubMed] [Google Scholar]
- Silva D. G. Ribeiro J. F. R. De Vita D. Cianni L. Franco C. H. Freitas-Junior L. H. Moraes C. B. Rocha J. R. Burtoloso A. C. B. Kenny P. W. Leitão A. Montanari C. A. Bioorg. Med. Chem. Lett. 2017;27:5031–5035. doi: 10.1016/j.bmcl.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Cianni L. Feldmann C. W. Gilberg E. Gütschow M. Juliano L. Leitão A. Bajorath J. Montanari C. A. J. Med. Chem. 2019;62:10497–10525. doi: 10.1021/acs.jmedchem.9b00683. [DOI] [PubMed] [Google Scholar]
- K777 (Chagas)|DNDi, https://www.dndi.org/diseases-projects/portfolio/completed-projects/k777/, (accessed 17 July 2019)
- Jones B. D. Tochowicz A. Tang Y. Cameron M. D. McCall L.-I. Hirata K. Siqueira-Neto J. L. Reed S. L. McKerrow J. H. Roush W. R. ACS Med. Chem. Lett. 2016;7:77–82. doi: 10.1021/acsmedchemlett.5b00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehringer M. Laufer S. A. J. Med. Chem. 2019;62:5673–5724. doi: 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Lonsdale R. Burgess J. Colclough N. Davies N. L. Lenz E. M. Orton A. L. Ward R. A. J. Chem. Inf. Model. 2017;57:3124–3137. doi: 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- MacFaul P. A. Morley A. D. Crawford J. J. Bioorg. Med. Chem. Lett. 2009;19:1136–1138. doi: 10.1016/j.bmcl.2008.12.105. [DOI] [PubMed] [Google Scholar]
- Ábrányi-Balogh P. Petri L. Imre T. Szijj P. Scarpino A. Hrast M. Mitrović A. Fonovič U. P. Németh K. Barreteau H. Roper D. I. Horváti K. Ferenczy G. G. Kos J. Ilaš J. Gobec S. Keserű G. M. Eur. J. Med. Chem. 2018;160:94–107. doi: 10.1016/j.ejmech.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Martin J. S. MacKenzie C. J. Fletcher D. Gilbert I. H. Bioorg. Med. Chem. 2019;27:2066–2074. doi: 10.1016/j.bmc.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor L. M. Jenkins M. L. Kerwin C. Burke J. E. Shokat K. M. Biochemistry. 2017;56:3178–3183. doi: 10.1021/acs.biochem.7b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin K. Karakaş D. Spectrochim. Acta, Part A. 2018;188:537–546. doi: 10.1016/j.saa.2017.07.036. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Suarez M. Aizman A. Soto-Delgado J. Contreras R. J. Org. Chem. 2012;77:90–95. doi: 10.1021/jo201465g. [DOI] [PubMed] [Google Scholar]
- Palazzesi F. Grundl M. A. Pautsch A. Weber A. Tautermann C. S. J. Chem. Inf. Model. 2019;59:3565–3571. doi: 10.1021/acs.jcim.9b00316. [DOI] [PubMed] [Google Scholar]
- Ehmke V. Quinsaat J. E. Q. Rivera-Fuentes P. Heindl C. Freymond C. Rottmann M. Brun R. Schirmeister T. Diederich F. Org. Biomol. Chem. 2012;10:5764. doi: 10.1039/C2OB00034B. [DOI] [PubMed] [Google Scholar]
- Oballa R. M. Truchon J.-F. Bayly C. I. Chauret N. Day S. Crane S. Berthelette C. Bioorg. Med. Chem. Lett. 2007;17:998–1002. doi: 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- Cherkasov A. Muratov E. N. Fourches D. Varnek A. Baskin I. I. Cronin M. Dearden J. Gramatica P. Martin Y. C. Todeschini R. Consonni V. Kuz'min V. E. Cramer R. Benigni R. Yang C. Rathman J. Terfloth L. Gasteiger J. Richard A. Tropsha A. J. Med. Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal Roy S. Comput. Biol. Chem. 2018;75:91–100. doi: 10.1016/j.compbiolchem.2018.04.015. [DOI] [PubMed] [Google Scholar]
- Parthasarathi R. Subramanian V. Roy D. R. Chattaraj P. K. Bioorg. Med. Chem. 2004;12:5533–5543. doi: 10.1016/j.bmc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Lima A. P. C. A. dos Reis F. C. G. Serveau C. Lalmanach G. Juliano L. Ménard R. Vernet T. Thomas D. Y. Storer A. C. Scharfstein J. Mol. Biochem. Parasitol. 2001;114:41–52. doi: 10.1016/S0166-6851(01)00236-5. [DOI] [PubMed] [Google Scholar]
- Zingales B. Miles M. A. Moraes C. B. Luquetti A. Guhl F. Schijman A. G. Ribeiro I. Mem. Inst. Oswaldo Cruz. 2014;109:828–833. doi: 10.1590/0074-0276140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez H. O. Guerrero N. A. Fortes A. Santi-Rocca J. Gironès N. Fresno M. Acta Trop. 2014;139:57–66. doi: 10.1016/j.actatropica.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Silva J. R. A. Cianni L. Araujo D. Batista P. H. J. de Vita D. Rosini F. Leitão A. Lameira J. Montanari C. A. J. Chem. Inf. Model. 2020;60:1666–1677. doi: 10.1021/acs.jcim.9b01138. [DOI] [PubMed] [Google Scholar]
- Meister A. J. Biol. Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- Dos Santos A. M. Cianni L. De Vita D. Rosini F. Leitão A. Laughton C. A. Lameira J. Montanari C. A. Phys. Chem. Chem. Phys. 2018;20:24317–24328. doi: 10.1039/C8CP03320J. [DOI] [PubMed] [Google Scholar]
- Gaus M. Cui Q. Elstner M. J. Chem. Theory Comput. 2011;7:931–948. doi: 10.1021/ct100684s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C. Yang W. Parr R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J. Pople J. A. Binkley J. S. J. Chem. Phys. 1984;80:3265–3269. doi: 10.1063/1.447079. [DOI] [Google Scholar]
- Møller Chr. Plesset M. S. Phys. Rev. 1934;46:618–622. doi: 10.1103/PhysRev.46.618. [DOI] [Google Scholar]
- Miertuš S. Scrocco E. Tomasi J. Chem. Phys. 1981;55:117–129. doi: 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]
- Chattaraj P. K. Sarkar U. Roy D. R. Chem. Rev. 2006;106:2065–2091. doi: 10.1021/cr040109f. [DOI] [PubMed] [Google Scholar]
- Ayers P. W. Parr R. G. J. Am. Chem. Soc. 2000;122:2010–2018. doi: 10.1021/ja9924039. [DOI] [Google Scholar]
- Lee C. Yang W. Parr R. G. J. Mol. Struct.: THEOCHEM. 1988;163:305–313. doi: 10.1016/0166-1280(88)80397-X. [DOI] [Google Scholar]
- Liu S. Parr R. G. J. Chem. Phys. 1997;106:5578–5586. doi: 10.1063/1.473580. [DOI] [Google Scholar]
- Pérez P. J. Org. Chem. 2003;68:5886–5889. doi: 10.1021/jo030125x. [DOI] [PubMed] [Google Scholar]
- Yang W. Mortier W. J. J. Am. Chem. Soc. 1986;108:5708–5711. doi: 10.1021/ja00279a008. [DOI] [PubMed] [Google Scholar]
- Reed A. E. Curtiss L. A. Weinhold F. Chem. Rev. 1988;88:899–926. doi: 10.1021/cr00088a005. [DOI] [Google Scholar]
- Wriede U. Fernandez M. West K. F. Harcour D. Moore H. W. J. Org. Chem. 1987;52:4485–4489. doi: 10.1021/jo00229a011. [DOI] [Google Scholar]
- Löser R. Frizler M. Schilling K. Gütschow M. Angew. Chem., Int. Ed. 2008;47:4331–4334. doi: 10.1002/anie.200705858. [DOI] [PubMed] [Google Scholar]
- LoPachin R. M. Gavin T. Chem. Res. Toxicol. 2014;27:1081–1091. doi: 10.1021/tx5001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W. Zhang B. Jiang X.-M. Su H. Li J. Zhao Y. Xie X. Jin Z. Peng J. Liu F. Li C. Li Y. Bai F. Wang H. Cheng X. Cen X. Hu S. Yang X. Wang J. Liu X. Xiao G. Jiang H. Rao Z. Zhang L.-K. Xu Y. Yang H. Liu H. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malátková P. Wsól V. Drug Metab. Rev. 2014;46:96–123. doi: 10.3109/03602532.2013.853078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.