

Abstract
Human T-cell lymphotropic virus type 1 (HTLV-1) is a retrovirus that can cause severe paralytic neurologic disease and immune disorders as well as cancer. An estimated 20 million people worldwide are infected with HTLV-1, with prevalence reaching 30% in some parts of the world. In stark contrast to HIV-1, no direct acting antivirals (DAAs) exist against HTLV-1. The aspartyl protease of HTLV-1 is a dimer similar to that of HIV-1 and processes the viral polyprotein to permit viral maturation. We report that the FDA-approved HIV-1 protease inhibitor darunavir (DRV) inhibits the enzyme with 0.8 µM potency and provides a scaffold for drug design against HTLV-1. Analogs of DRV that we designed and synthesized achieved sub-micromolar inhibition against HTLV-1 protease and inhibited Gag processing in viral maturation assays and in a chronically HTLV-1 infected cell line. Cocrystal structures of these inhibitors with HTLV-1 protease highlight opportunities for future inhibitor design. Our results show promise toward developing highly potent HTLV-1 protease inhibitors as therapeutic agents against HTLV-1 infections.
Graphical Abstract
Introduction
Human T-cell lymphotropic viruses type 1 and type 2 (HTLV-1/2) were the first human retroviruses described nearly 40 years ago.1–2 For decades HTLV-1 has been known to be highly carcinogenic and cause severe paralytic neurologic disease as well as immune disorders that can increase susceptibility to bacterial infections. HTLV-1 infection can lead to adult T-cell leukemia/lymphoma (ATL), HTLV-associated myelopathy/tropical spastic paraparesis (HAM/TSP), uveitis and infective dermatitis.3–4 HTLV-1 is transmitted via the same routes as HIV-1 (sexually, via blood or mother-to-child) with significant HIV/HTLV co-infections reported in Europe, America, and Africa.5 About 20 million people worldwide are infected with HTLV-1,6 reaching endemic rates of 30% in some parts of the world. According to a recent report, up to 45% of the adult population among five Aboriginal communities in central Australia tested positive for HTLV-1.7 With global human movement, HTLV-1 infections are starting to expand into previously non-endemic regions as recently reported for Spain from Latin American immigrants.8 Unfortunately, there are no vaccines or direct acting antivirals (DAAs) against HTLV-1.
In the absence of preventive vaccines and DAAs, treatment options for HTLV-1 infected patients are scarce. For the estimated 3–5% of the HTLV-1 infected patients who develop ATL, chemotherapy is ineffective with very poor survival rates and relapse leading to death.9–10 Earlier studies with HIV-1 protease inhibitor ritonavir, although not very potent against HTLV-1, were promising against HTLV-1 induced ATL in cell lines and mouse models.11 In clinic, a combination of zidovudine, an HIV-1 reverse transcriptase inhibitor, and IFN-ɑ significantly improved prognosis, indicating active HTLV-1 replication in ATL patients that can be targeted by antiretrovirals. As with HIV-1 infections, development of specific DAAs has a great potential to improve treatment outcomes. Such DAAs may also prove to be effective in all HTLV-1 associated human diseases, for pre-exposure prophylaxis (PrEP) especially in areas where the virus is endemic, and for preventing mother-to-child transmissions.12
HTLV-1 utilizes a similar viral machinery as HIV-1, and inhibiting the viral protease would prevent viral maturation. HIV-1 protease inhibitors are a success story of rational structure-based drug design, with the most recent FDA-approved drug, darunavir (DRV), having exceptional low-picomolar potency against wild type (WT) HIV-1 protease.13 As peptidomimetic transition-state analogues, protease inhibitors are the most potent among HIV-1 antivirals, as enzymes bind strongest to their transition-state as compared to substrates or products.14–15 Due to this potency and relatively high barrier to resistance, DRV has been tried as a monotherapy in patients with treatment-naïve HIV where the viral load and replication are relatively low.16 Success against HIV-1 protease suggests similar potency can be achieved against HTLV-1 protease.
HTLV-1 protease is a 28 kDa homodimeric aspartyl protease that is 28% identical to HIV-1 protease with 45% identity between active site residues,17–18 yet HTLV-1 protease has considerably distinct substrate specificity (Table S1).18–22 The crystal structure of HTLV-1 protease was determined in 200520 and early attempts at inhibitor design involved peptide scanning with non-hydrolysable substrates.23 Iterations of this process incorporated unnatural amino acids24–25 and different capping moieties26–27 leading to the identification of allophenylnorstatin-based HTLV-1 protease inhibitors with low nanomolar potency.22 This inhibitor did not progress to the clinic, likely due to high peptidic character and large molecular weight. A few peptidic inhibitors were cocrystallized28 and deposited into the Protein Data Bank (PDB)29–30 providing valuable insights into inhibitor binding at the HTLV-1 active site. HIV-1 protease inhibitor indinavir (IDV), an FDA-approved drug, was also reported to weakly inhibit HTLV-1.31 More recently, pyrrolidine-based C2-symmetric non-peptidic inhibitors were investigated and achieved low nanomolar potency.32 Despite these efforts, there is still a need for exploring chemical scaffolds that will ultimately result in a clinically relevant HTLV-1 protease inhibitor with sub-nanomolar potency.
We tested the FDA-approved HIV-1 protease inhibitor DRV and our own novel DRV analogues against HTLV-1 protease. Several modifications to the DRV scaffold improved the inhibitors to low nanomolar potency, similar to the most potent HTLV-1 protease inhibitors to date. These compounds successfully inhibited maturation of virus-like particles (VLPs) and infectious particles produced by the chronically HTLV-1 infected cell line (SP) by blocking the processing of viral Gag. Through the determination of cocrystal structures and comparison with analogous complexes in HIV-1 protease, we present additional insights into how to further increase potency and potentially develop the first antivirals against HTLV-1.
Results and Discussion
DRV and Analogs Inhibit HTLV-1 Protease
We expressed and purified the 116-amino-acid construct of HTLV-1 protease for enzyme inhibition assays and crystallization. This protease construct has 9 amino acids removed from the C-terminus to facilitate crystallization,20 which does not affect catalytic activity.33 With a FRET-based enzymatic assay, the activity of HTLV-1 protease was tested using two peptide substrates based on HTLV-1 matrix/capsid (MA/CA) and capsid/nucleocapsid (CA/NC) cleavage sites (Table S2). The Michaelis-Menten constant (KM) was measured as 101.3 ± 1.9 µM and 31.6 ± 5.9 µM, consistent with previously reported values.34 HTLV-1 catalyzed cleavage of the MA/CA substrate an order of magnitude faster than that of CA/NC, with a catalytic efficiency of 0.21± 0.02 µM−1s−1. Thus, the purified HTLV-1 protease is folded and active in our conditions.
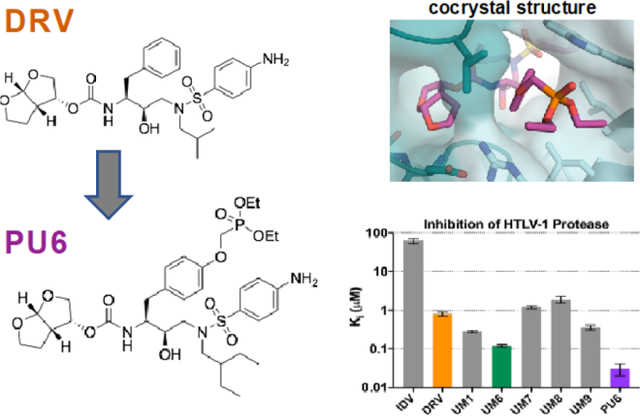
The FDA-approved HIV-1 protease inhibitors IDV and DRV were tested against HTLV-1 protease (Figure 1a,b and Table S3). IDV weakly inhibited HTLV-1 protease (Ki = 62.7 µM), while DRV was two orders of magnitude more potent with a Ki of 0.8 µM. We previously found that modifications at the P1′ and P2′ positions of DRV improved potency against resistant variants of HIV-1 protease.35 These DRV analogues with larger hydrophobic P1′ moieties, increasing from an isobutyl to sec-butyl and isohexyl, were also tested against HTLV-1 protease (Table S3 and Figure S1). Some of the analogs had significantly improved potency, the best being UM6 with a Ki of 0.12 ± 0.01 µM, which is ~7-fold lower compared to DRV. In both UM1 and UM6, increasing the hydrophobicity at P1′ improved potency. However, modifications at the P2′ position did not further increase potency, indicating the aniline moiety of DRV is better than phenoxymethyl, hydroxymethylbenzene, or benzo(1,3)dioxole. Thus we identified DRV as a viable scaffold for designing inhibitors against HTLV-1 protease.
Figure 1. Inhibitors tested against HTLV-1 protease.
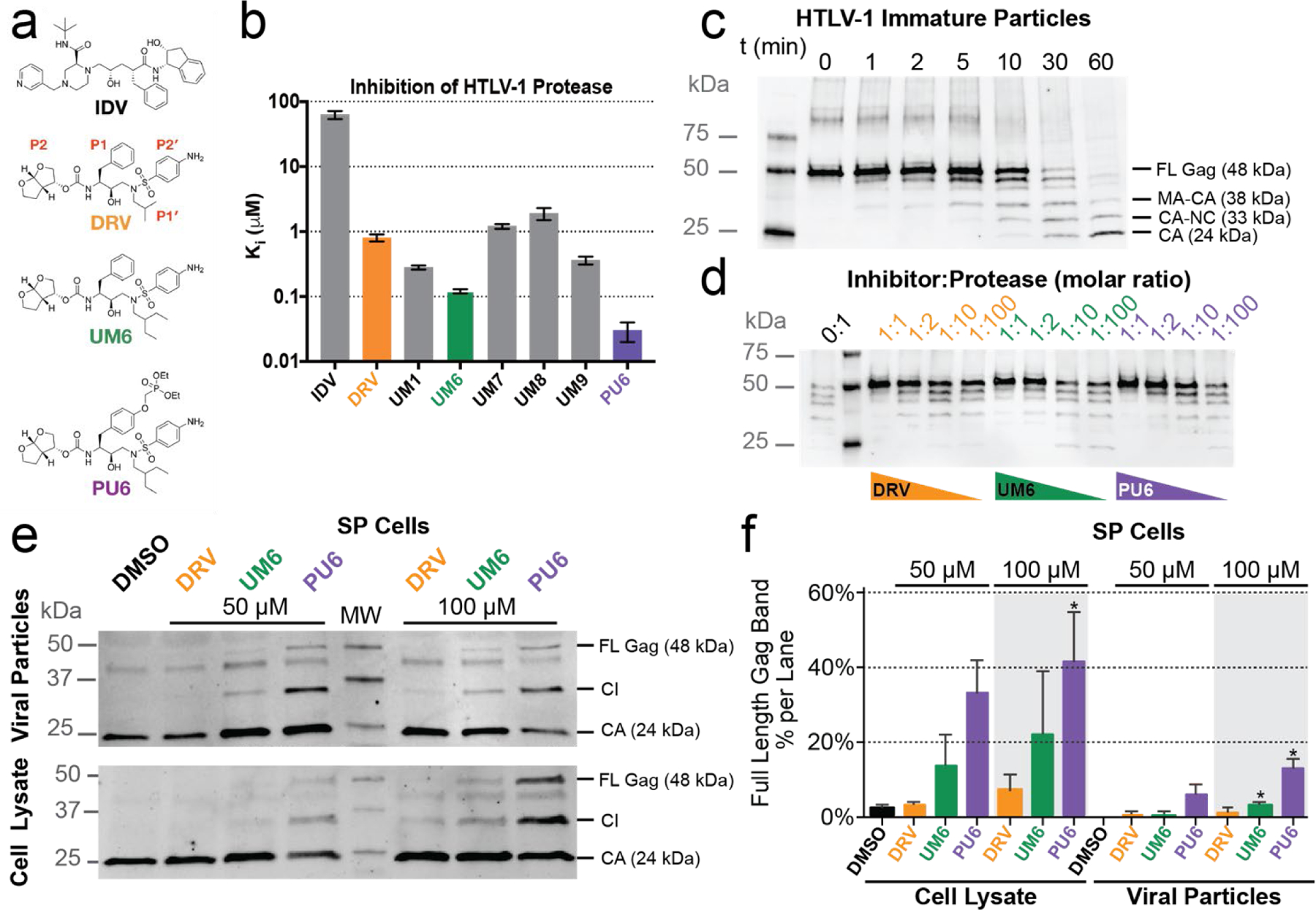
(a) Chemical structure of indinavir (IDV), darunavir (DRV) with P2–P2′ moieties labeled, and DRV analogs that exhibited improved potency. (b) Enzyme inhibition constants (Ki) for IDV, DRV, and DRV analogs against HTLV-1 protease. (c) HTLV-1 Gag cleavage by protease in immature virus particles. Gag cleavage by HTLV-1 protease (1 µM) at 37° C without inhibitor as a function of time. Cleavage products visualized with anti-HTLV-1 p24 (capsid). Expected cleavage products of full-length (FL) Gag are matrix-capsid (MA-CA), capsid-nucleocapsid (CA-NC), capsid (CA). (d) HTLV-1 protease inhibited in vitro against VLP Gag. Gag cleavage after 60 min with decreasing molar ratio of inhibitor to protease. At a 1:1 ratio (1 µM), no Gag cleavage products are observed for any inhibitor, while below 1:10 ratio (100 nM inhibitor) some p24 capsid is observed after 60 min. (e) HTLV-1 Gag cleavage from SP cell lysates and from particles released into cell culture supernatants were treated with 50 µM or 100 µM inhibitor for 48 hrs. Samples were then visualized by using an anti-HTLV-1 p24 antibody. (f) Immunoblot quantification of the FL Gag band as a percentage of total bands in each lane for the different SP cell drug treatments reported as the mean ± SEM (n=3). Band labels: full-length Gag (FL Gag), cleavage intermediates (CI), and capsid (CA). * denotes p-value < 0.05 with values compared to the DMSO control sample.
Finally, we added a diethylphosphonate group via a methyloxy (O-CH2-O) linker to UM6 at the para position of the P1 phenyl (PU6) as this moiety had improved potency in a similar scaffold against highly drug resistant HIV-1 proteases.36 With this additional phosphonate modification, the resulting novel inhibitor PU6 inhibited HTLV-1 protease 4-fold better than UM6 (Ki = 0.03 ± 0.01 µM). Therefore, through exploration of a small panel of DRV analogues, an HTLV-1 protease inhibitor with low-nM potency was identified with over 26-fold improvement over the FDA-approved inhibitor DRV.
Inhibition of HTLV-1 Gag Processing
In addition to the biochemical enzymatic assays, DRV and the two analogs UM6/PU6 were tested for their ability to prevent Gag maturation (Figure 1c–f). HTLV-1 Gag expressed in Hek293T cells produced immature particles, which were harvested, resolved by SDS-PAGE, and visualized using anti-capsid antibodies (see Methods for details) (Figure 1c–d). In the absence of any inhibitor, HTLV-1 protease rapidly processed Gag in immature particles to yield smaller fragments corresponding to cleavage products (Figure 1c). DRV and both analogs effectively inhibited Gag cleavage and particle maturation at as low as 1:1 ratio. Decreasing inhibitor concentration resulted in a dose-dependent response (Figure 1d). To further validate inhibition of HTLV-1 protease, SP cells (which are chronically infected with HTLV-1) were treated with the same inhibitors (Figure 1e). The SP cell line harbors low numbers of HTLV-1 proviruses and produces virus particles that incorporate full-length Gag,37 which makes SP an ideal cell culture system for testing the inhibition of HTLV-1 maturation. , Gag cleavage products were detected by using a CA (p24) primary antibody in both SP cell lysates and virus particles released in the cell culture supernatants. In both lysates and supernatants, amount of full-length (FL) Gag and larger size cleavage intermediates (CI) increased when the cells were treated with 50 µM or 100 µM DRV or the two analogues UM6/PU6. The PU6 analogue showed the greatest amount of FL Gag present in both concentrations of inhibitor tested: 32.4 ± 15.5% and 41.0 ± 13.7% band percentage (mean ± SEM) for 50 µM and 100 µM respectively in the cell lysate as compared to DMSO (2.0 ± 1.0%). For the PU6 treatment of the viral particles produced from the SP cells, the FL Gag band percentage was 5.8 ± 2.8% and 12.9 ± 3.1% (mean ± SEM) for 50 µM and 100 µM treatments respectively (Figure 1f, S2). In agreement with the enzyme inhibition assays, the highest amount of protease inhibition was observed for PU6 in vitro with the SP cell line. Therefore, DRV analogs inhibit HTLV-1 protease and the subsequent Gag processing, which is required for viral maturation.
Cocrystal Structures of DRV and Analogs Bound to HTLV-1 Protease
To elucidate how the DRV scaffold binds to HTLV-1 protease, high-resolution cocrystal structures with DRV, UM6, and PU6 (2.05–2.29 Å resolution) were determined (crystallography statistics in Table S4). The three complexes crystallized in the same space group (P6322), containing one homodimer in the asymmetric unit. For comparison, the structure of PU6 bound to HIV-1 protease was also determined to 1.84 Å resolution (Table S4). Following established convention, the monomer that contacts the aniline side of DRV (P2′) is denoted the prime (′) or B chain, while the A chain that interacts with the bis-THF (P2) will remain non-prime. Starting from these crystal structures, a series of molecular dynamics (MD) simulations were performed to assess the stability of the inhibitor–protease interactions observed in the crystal structures.
Interactions Lost in the Binding of DRV to HTLV-1 Versus HIV-1 Protease
In the cocrystal structure determined, DRV was bound at the active site of HTLV-1 protease with an overall conformation similar to that in HIV-1 protease (Figure 2). However, because of variations in the active site, evident in dramatically altered substrate specificity, certain key protease–inhibitor interactions were either lost or weakened in HTLV-1 protease. As with all HIV-1 protease inhibitors, DRV is a transition state analogue: the central hydroxyl moiety interacts with the side chain oxygens of both catalytic aspartates at a distance of 2.5–3.2 Å between heavy atoms. In HTLV-1 protease, these distances increased to 2.8–3.5 Å. In addition, the more open active site in HTLV-1 protease resulted limited packing of the inhibitor and the flaps, with no evidence of coordinated waters in the electron density maps. Finally, DRV is not large enough to keep optimal distance to the catalytic residues while coordinating bis-THF moiety interactions at the S2 subsite. In HIV-1 protease, the bis-THF oxygens have hydrogen bonds with the backbone nitrogen atoms of Asp29 and Asp30 [Figure 2b] within 2.8–3.2 Å while remaining 3.5 Å away from the repulsive force of the carbonyl oxygen of Asp30. This binding also allows the NH atom of the carbamate linker to interact with the carbonyl oxygen of Gly27 at a distance of 3.0 Å. In HTLV-1 protease, the bis-THF had a single hydrogen bond with the backbone nitrogen of D36 at a favorable distance of 2.9 Å. The nitrogen of Met37 and carbonyl oxygen of Gly34 were both beyond hydrogen bonding distance (3.4–3.6 Å). Thus, these three major disruptions of hydrogen bonds in HTLV-1 protease (Figure 2b) between (1) the central hydroxyl and catalytic aspartates (which centers the inhibitor within the active site); (2) the loss of the tetrahedral network coordinating the inhibitor to the flaps through a conserved water; (3) the bis-THF moiety and backbone nitrogens in the S2′ subsite, weaken the affinity with DRV as previously observed in highly DRV-resistant HIV-1 protease variants.38
Figure 2. Comparison of DRV binding to HTLV-1 versus HIV-1 protease.
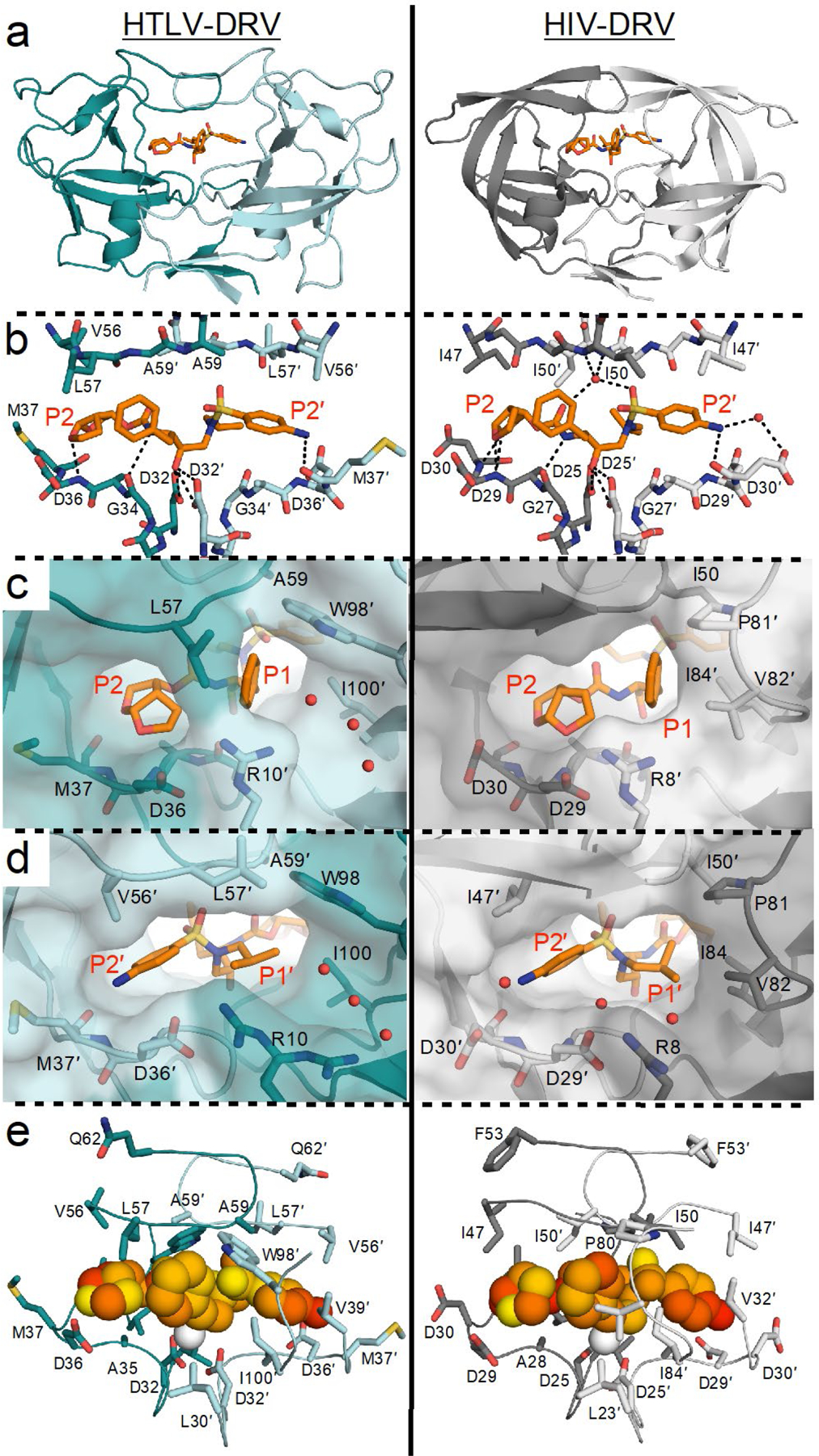
(a) Cocrystal structures of DRV bound to HTLV-1 and HIV-1 protease (PDB IDs 6W6Q and 6DGX, respectively). (b) Inter-molecular hydrogen bonds between DRV and protease active site. (c) Close-up view of aniline moiety in the S2′ subsite. (d) Close-up view of bis-THF moiety in the S2 subsite. (e) Packing around DRV at the active site visualized through mean inter-molecular vdW interactions with protease during MD simulations, where yellow to red indicates increased packing. In all panels, the prime side monomer (chain A) interacting with the bis-THF moiety is in darker shade, and DRV is depicted as orange sticks in panels A–D.
The packing of DRV is dramatically different in HTLV-1 compared with HIV-1 protease. In HTLV-1, the phenyl ring of P1 moiety does not pack well against the hydrophobic residues Leu30′, Trp98′, Ile100′, and Leu57 in the S1 subsite (Figure 2c) and the exposed ring coordinates a channel filled with water molecules. In contrast in HIV-1 protease, the phenyl ring of P1 packs against the hydrophobic side chains in the 80s loop, especially Val82 (Figure 2c). This reflects the substrate specificity of HIV protease where residues with aromatic rings (Phe and Tyr) naturally occur at the P1 position of cleavage sites [Table S1] and modifications to increase van der Waals (vdW) contacts at this site can greatly improve potency against resistant proteases.36, 39–40 The cocrystal structure with HTLV-1 protease indicates modifications to the P1 moiety to either stack against the hydrophobic side chains in the S1 subsite or exploit the available space in the channel may increase potency.
In homodimeric proteases, the S2′ subsite is symmetrically related to the S2 subsite and made up of identical residues. In the S2′ subsite of HIV-1 protease, DRV P2′ aniline makes a direct hydrogen bond with the backbone oxygen of Asp30′ and a water-mediated interaction with the side chain of Asp30′ [Figure 2d]. In our DRV analogs (UM7–9), modifications to P2′ –including phenoxymethyl, hydroxymethylbenzene, benzo(1,3)dioxole– improved potency against resistant variants by establishing more favorable interaction with the backbone nitrogen of Asp30′.35 In contrast, DRV and analogs harboring the aniline moiety had the best potency among those tested against HTLV-1 protease (Figure 1, Table S3). The cocrystal structure (Figure 2d) shows the aniline nitrogen is roughly equidistant from both the backbone oxygen and backbone nitrogen of residue Met37′ but not within hydrogen bonding distance. The aniline benzene ring had a slightly different rotation angle and S2′ subsite had a deeper hydrophobic pocket beyond Val56′ toward Phe67′. These differences may underlie why the P2′ modifications did not increase potency toward HTLV-1 protease.
When DRV bound to HTLV-1 protease, both P1 and P1′ moieties were largely exposed to channels occupied by water molecules (Figure 2c,d). These moieties are nestled in hydrophobic pockets in HIV-1 protease with favorable hydrophobic contacts. In HTLV-1 protease the favorable packing around these moieties was substantially decreased, which was also the case for the overall inhibitor due to the larger active site (Figure 2e).
Structural Basis of Improved Potency of DRV Analogs toward HTLV-1 Protease
Cocrystal structures of DRV analogs that had improved potency were determined with HTLV-1 protease. Both UM6 and PU6 have a larger isohexyl moiety at the P1′ position compared to DRV’s isobutyl. When bound to the protease, these aliphatic moieties had diverging conformations (Figure 3a) which could all be accommodated within the relatively large hydrophobic S1′ subsite (Figure 3b). The conformation of the P1′ moiety seemed to impact that of the P2′ aniline, suggesting subsite interdependence and the need to optimize these moieties simultaneously. The P1′ isohexyl moiety was not large enough to displace any water molecules in the extended S1′ channel. However, hydrophobic packing against S1′ residues (Ala59′ and Trp98) were enhanced (Figure S3), likely underlying the improved potency of analogs with the larger P1′ moiety. Even larger P1′ moieties that pack against these hydrophobic moieties while extending into the channel with a polar face to interact with Arg10 may further increase the binding affinity.
Figure 3.
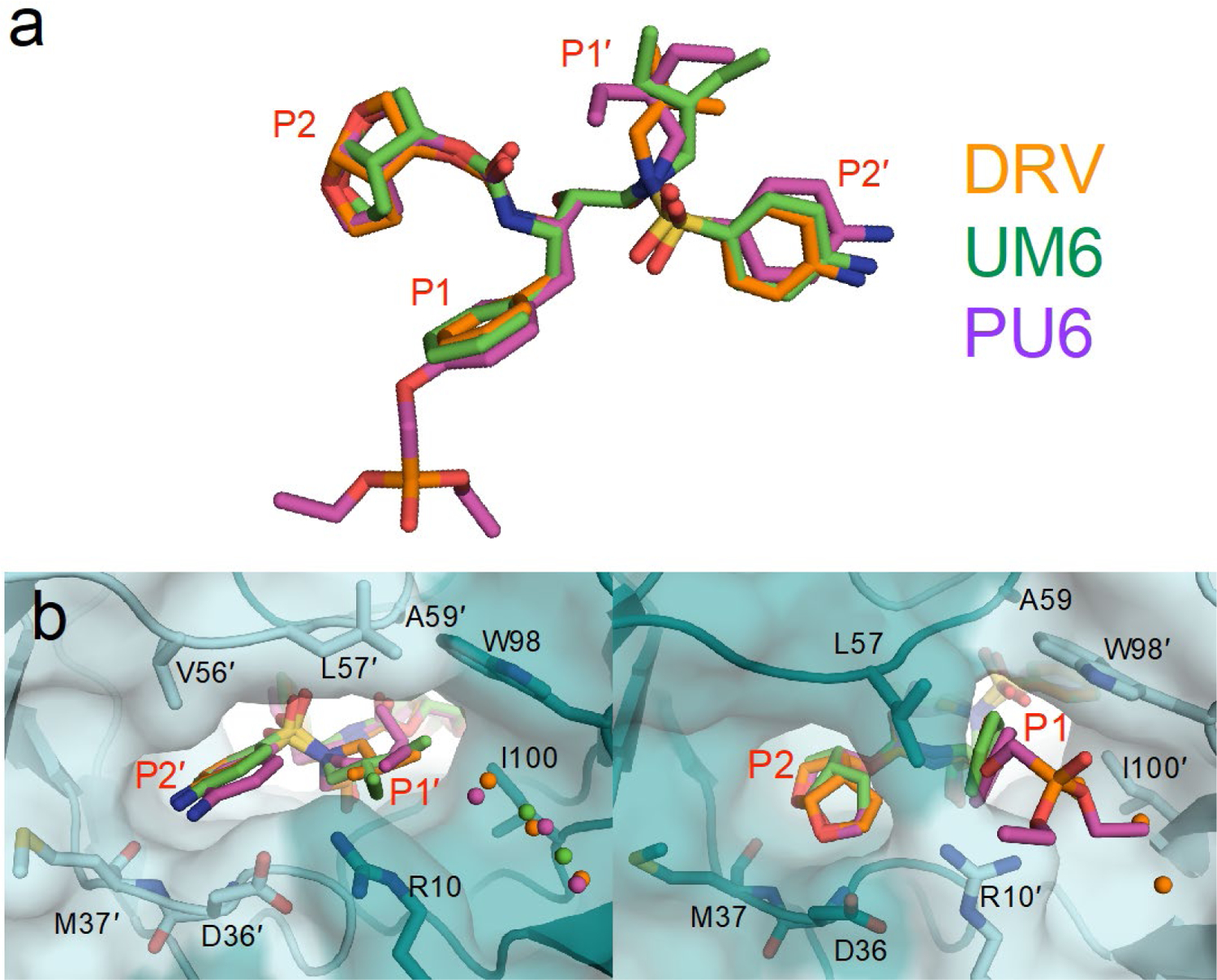
Comparison of DRV and DRV analogs when bound to HTLV-1 protease. (a) Alignment of inhibitors. (b) Close-up view of P2′–P1′ moiety in the S2′–S1′ subsite, and P2–P1 moieties in the S2–S1 subsite. The phosphonate moiety of PU6 extends into the S1 subsite, displacing conserved crystallographic waters.
On the flip side of the active site, the invariant P2 bis-THF and P1 benzene ring superimposed well with only minor conformational divergence (Figure 3a,c). The phosphonate added to the P1 ring in PU6 extended into the S1 channel, substantially increasing vdW contacts with Leu57, Trp98′, Arg10′ (Figure S3). These enhanced interactions account for the order of magnitude increase in the measured inhibition constant (Figure 1).
The phosphonate addition to the P1′ benzene ring has been reported for other HIV-1 protease inhibitor scaffolds to increase potency against resistant variants by “solvent anchoring”.36 We determined the crystal structure of our PU6 inhibitor bound to HIV-1 protease to both investigate the mechanism of increased potency and to compare with HTLV-1 protease (Figure 4). The conformation of the phosphonate moiety was completely different in the two cocrystal structures. In HIV-1 protease, one branch of the phosphonate moiety interacted with Pro81′ in the 80s loop while the other extended to the flaps. This suggests that rather than solvent interactions, the mechanism of improved potency against resistant HIV-1 protease variants is through interactions with the invariant Pro81′ and stabilization of the closed conformation of the flaps. In HTLV-1 protease, the phenyl ring intercalates between Leu57 and Trp98′, while Leu57 prevents the phosphonate group from interacting with the protease flaps. Instead, the phosphonate addition extends into the S1 channel, as detailed above.
Figure 4.
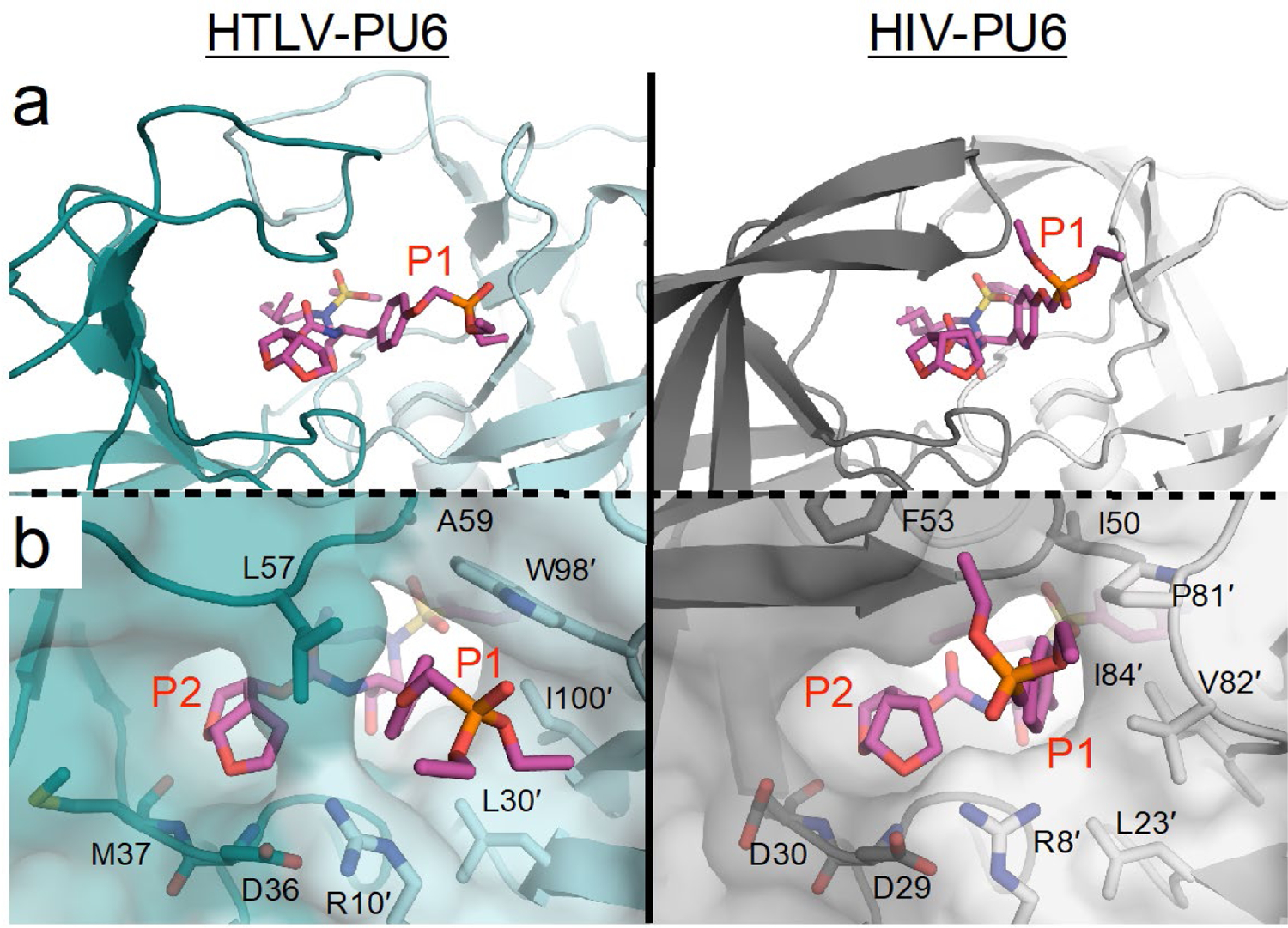
Comparison of PU6 when bound to HTLV-1 and HIV-1 protease. (a) The phosphonate moiety of PU6 sticks out into the S1 subsite in HTLV-1, whereas it binds up against the flaps in HIV-1 protease. (b) Residue L57 prevents the phosphonate from interacting with the flaps of HTLV-1 protease.
The crystal structures indicate that exploration of larger P1′ and P1 moieties to fill the water-occupied channels is highly promising. Our MD simulations indicate Ala59/59′ and Trp98/98′ to be highly flexible (Figure S4). Modifications that increase interactions with Ala59/59′ to stabilize the flaps and with Trp98/98′ to lock the side chain conformer will likely further increase inhibitor potency. These tryptophan residues and phenylalanine (Phe67/Phe67′) in the S2/S2′ subsites also provide an opportunity for π-π stacking interactions between protease and inhibitor (Figure S5). Previously reported C2-symmetric pyrrolidine-based inhibitors with P1/P1′ aromatic rings had π-π stacking interactions with Trp98, which improved potency.32 In our MD simulations, π-π stacking of P1 phenylalanine with Trp98 was maintained only about half of the time, and only PU6 formed edge-to-face π-π stacking with Phe67′ again for about half of the simulation time. Stabilizing these stacking interactions may further increase potency. Overall, analysis of crystal structures suggests that DRV offers a promising scaffold for HTLV-1 protease inhibitor design, and pinpoints interactions that can be optimized to achieve potent inhibition.
Conclusion
HTLV-1 is a growing global threat with no DAAs or vaccines to prevent the spread of the disease. We sought to inhibit HTLV-1 protease using DRV, the most potent FDA-approved HIV-1 protease inhibitor. Our results show that DRV is a promising scaffold for inhibitor design and antiviral development against HTLV-1 protease. With modifications to two moieties of DRV, the lead compound (PU6) achieved substantially (~27-fold) improved potency in vitro. Furthermore, PU6 inhibited Gag processing by the HTLV-1 protease as observed with released virus particles as well as in cell lysates from a HTLV-1-chronically infected cell line. The structural insights gained here will guide the design of highly potent HTLV-1 protease inhibitors that can prevent viral replication and infectious spread.
METHODS
Construction of the HTLV-1 Protease Expression Plasmid
The 116 amino acid HTLV-1 variant (Uniprot Accession ID: Q82134) was ordered from Genscript on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. A L40I mutation was included to prevent autoproteolysis.34
Protein expression and purification
The expression, isolation, and purification of HTLV-1 protease used for the kinetic assays and crystallization was carried out as previously described for HIV-1 protease.41–42 Briefly, the gene encoding the HTLV-1 protease was subcloned into the heat-inducible pXC35 expression vector (ATCC) and transformed into E. coli TAP-106 cells. Cells grown in 6 L of Terrific Broth were lysed with a cell disruptor and the protein was purified from inclusion bodies.43 The inclusion body centrifugation pellet was dissolved in 50% acetic acid followed by another round of centrifugation to remove impurities. Size exclusion chromatography was used to separate high molecular weight proteins from the desired protease. This was carried out on a 2.1 L Sephadex G-75 superfine column (Millipore Sigma) equilibrated with 50% acetic acid. The cleanest fractions of HTLV-1 protease were refolded into a 10-fold dilution of 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Folded protein was concentrated down to 1–2 mg/mL and stored at −80°C. Protein aliquots were thawed, concentrated to ~10 mg/mL, and a final purification was performed with a Pharmacia Superdex 75 FPLC column equilibrated with 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Protease fractions purified from the size exclusion column were concentrated to 1–2 mg/mL using an Amicon Ultra-15 10-kDa device (Millipore). Freshly purified protease was used for crystallization and the rest was stored at −80°C. This stored protease was used for KM and Ki assays.
Enzyme Binding Assays to Determine KM
To determine the enzyme activity, in a 96-well plate, purified protease was provided a 10-amino acid substrate containing the natural cleavage site (MA/CA or CA/NC) with an EDANS/DABCYL FRET pair dissolved in 8% DMSO was 2/3 serially diluted from 0 to 40 μM for MA/CA and 0 to 100 μM to CA/NC. Using a PerkinElmer Envision plate reader, 5 µL of HTLV-1 protease were added to the 96-well plate to obtain a final concentration of 1 μM. Fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts. FRET inner filter effect correction was applied as previously described.44 Data corrected for the inner filter effect was analyzed with Prism8.
Enzyme Inhibition Assays to Determine Ki
To determine the enzyme inhibition constant (Ki), in a 96-well plate, each inhibitor was 2/3 serially diluted from 400 μM to 6.9 μM for IDV, 30 μM to 0.5 μM for DRV and UM6 or 2 μM to 0.03 μM for PU6. All assays included a 0 μM control, and incubated with 1 μM protein for 1 hour. A 10-amino acid substrate containing a solubility enhanced HTLV-1 MA/CA protease cleavage site (Ac-R-E(EDANS)-APQVLPVMHPK(DABCYL)-R-NH2) with an EDANS/DABCYL FRET pair (BAChem) was dissolved in 4% DMSO. Using the Envision plate reader, 5 µL of the substrate was added to the 96-well plate to a final concentration of 10 µM. The fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts. Data was analyzed with Prism8. UM6 was used as a control in all assays.
HTLV-1 Immature Gag Virus Like Particle Production and Purification
Hek293T cells were grown to ~70% confluency and were maintained in DMEM supplemented with 10% HyClone FetalClone III (FC3) (GE Healthcare Life Sciences, USA) and penicillin/streptomycin (Invitrogen, USA) 37 °C and 5% CO2. Cells were then transfected with a human codon optimized pN3 HTLV-1 Gag and a HTLV-1 envelope (Env) expression construct at a 10:1 molar ratio (4 µg total DNA) by using GenJet Plus (SignaGen Laboratories, USA) to generate immature HTLV-1 particles.45–46 Cell culture supernatants were collected 48 h post-transfection and cellular debris was cleared by low speed centrifugation (3000 x g) for 5 min at 20 °C and then passed through a 0.22 µm filter. To purify the particles, cell culture supernatants were subjected to ultracentrifugation over an 8% Optiprep cushion (Millipore Sigma, USA) (109,000 x g for 1.5 hours at 4 °C) in a 50.1 Ti rotor (Beckman Coulter, USA), and virus particle pellets were resuspended in STE buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.4, 1 mM Ethylenediaminetetraacetic acid (EDTA)). Particles were then layered over a 10–30% Optiprep step gradient (10% steps) and subjected to ultracentrifugation for 3 h at 250,000 x g in a SW55 Ti rotor (Beckman Coulter, USA) at 4 °C. Virus particles were removed from the gradient between the 20 and 30% Optiprep layers by using a hypodermic syringe. The isolated fraction was diluted to a final volume of 5 mL in STE buffer and pelleted by ultracentrifugation (195,000 x g) for 1 h at 4 °C. Pellets were resuspended in 50 uL of STE buffer and stored at −80 °C.
In Vitro Immature Virus Particle Protease Inhibition Reactions
Purified immature HTLV-1 particles were dialyzed into protease cleavage buffer (CB) composed of 100 mM NaCl, 50 mM sodium acetate pH 5.3, 2 mM DTT, and 1% glycerol. To solubilize the virus particle membrane, Triton X-100 was added to a final concentration of 1% (v/v) 20 min prior to the HTLV-1 protease cleavage reaction. Protein concentration of the solubilized particles were determined by using a BCA assay (Thermo Fisher Scientific, USA). Time course analysis cleavage reaction of HTLV-1 protease with immature HTLV-1 particles was done over 60 min in the presence of 1 µM protease at 37 °C. For each time point, 20 µg of HTLV-1 Gag was removed and the reaction was quenched by addition of 4x SDS-PAGE loading buffer and heat denatured at 100 °C for 10 min. Each sample was analyzed by using SDS-PAGE and immunoblot analysis. Analysis of protease inhibitors was conducted using the same experimental strategy at the indicated concentration of inhibitor being added to the virus particle cleavage reaction prior to the addition of protease and samples were quenched after a 60 min cleavage reaction. Control cleavage reaction was treated with the highest concentration of DMSO (0.01 % v/v) as used for the sample with the highest inhibitor concentrations. Samples were run on Tris-glycine SDS gels with a 4–20% acrylamide gradient (Bio-Rad, USA) Primary antibody for immunoblot was a monoclonal mouse anti HTLV-1 p24 (Santa Cruz Biotechnology, USA; P/N: sc-53891) at a 1:2,000 dilution in 2.5% (w/v) non-fat milk powder diluted in tris-buffered saline with tween 20 (TBST). Secondary antibody goat anti mouse IgG StarBright Blue 700 (Bio-Rad, USA; P/N: 12004158) at a 1:1,000 dilution in 2.5% (w/v) non-fat milk powder diluted in TBST was used for detection using a ChemiDoc imager (Bio-Rad, USA).
SP Cell Culture and Protease Inhibition Treatments
SP cells were cultured in RPMI (Invitrogen, Carlsbad, CA, USA) containing 20% HyClone (FC3) from Thermo Scientific (Waltham, MA) and 1% penicillin-streptomycin from Life Technologies (Grand Island, NY, USA) and supplemented with human IL-2 (Roche Diagnostics, Indianapolis, IN, USA) at a concentration of 10 U/ml.47 Cells were grown to a density of ~5.05 cells/mL and the media was exchanged into media containing the drug treatment at the two concentrations tested (50 µM and 100 µM). DMSO was used as a treatment control at the max concentration (0.01 % v/v) as used for the sample with the highest inhibitor concentrations. Cells were treated for 48 h in an incubator at 37 °C with 5% CO2. To harvest the samples the cell culture supernatant and cells were separated with a 5 min spin at 1000 x g. The cell culture supernatant was filtered through a 0.22 µm syringe filter and ultracentrifuged over an 8% Optiprep cushion (Millipore Sigma, USA) (109,000 x g for 1.5 hours at 4 °C) in a 50.1 Ti rotor (Beckman Coulter, USA). The resulting viral pellet was resuspended in 200 µL of RIPA buffer (150 mM NaCl, 1% (v/v) Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 25 mM Tris pH 7.4) supplemented with cOmplete, mini, EDTA-free cellular protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). The SP cell pellet was washed once with phosphate buffered saline (PBS) and resuspended in a final volume of 200 µL of RIPA buffer cOmplete, mini, EDTA-free cellular protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA). Immunoblot samples were then prepared and detected as described in the previous method section. Gel band quantification analysis was performed using Fiji.48 A one-tailed T-test for two independent means (95% confidence) was used to determine if the FL Gag band from the various drug treatments in the immunoblot analysis was statistically different from the DMSO controls.
Protein Crystallization
Many crystallization conditions produced HTLV-1 protease cocrystals with a hexagonal plate morphology, but the condition most reliably producing larger crystals in three dimensions was discovered using the JCSG+ sparse screen, well C6, containing 40% (v/v) PEG 300, 0.1M Phosphate/Citrate pH 4.2. All cocrystals were grown at room temperature by hanging drop vapor diffusion method in a 24-well VDX hanging-drop trays (Hampton Research) with a protease concentration of 4.0 mg/mL with 5-fold molar excess of inhibitor and mixed with the precipitant solution at a 1:1 ratio. The precipitant solution consisted of 39–41% (v/v) PEG 300 with 0.1 M Phosphate/Citrate buffer at pH 4.2 and the crystallization drops were set with 1 µL of precipitant solution and 1 µL protein-inhibitor solution and micro-seeded with a cat whisker and dried over a well solution of 3.0–4.0 M NaCl. Diffraction quality crystals were obtained within 2 weeks. Data were collected at 100 K and due to the high percentage of low molecular weight PEG there was no need for supplemental cryoprotectant. The cocrystal of PU6 bound to HIV-1 protease was generated as previously described.38, 49–51
Data Collection and Structure Solution
The three HTLV-1 protease cocrystals were shot at the Chicago APS Synchrotron Beamline 23-ID-D using beamline control software JBluIce.52 The diffraction images were indexed, integrated, and scaled using the GM/CA autoprocessing pipeline which utilizes XDS,53 POINTLESS,54 and AIMLESS.55 X-rays diffracted through a cocrystal of PU6 bound to HIV-1 protease were collected by our in-house Rigaku Saturn944 system and intensities were indexed, integrated, and scaled using HKL3000.56
Structures were solved by molecular replacement in the program PHASER57 using an HTLV-1 protease monomer (PDB: 3WSJ) or a WT HIV-1 protease monomer (PDB: 6DGX). Model building and refinement was performed using Coot58 and Phenix.59 Ligands were designed in Maestro and the output sdf file was used in the Phenix program eLBOW60 to generate the cif file containing atomic positions and constraints necessary for ligand refinement. Iterative rounds of crystallographic refinement were carried out until convergence was achieved. To limit bias throughout the refinement process, five percent of the data were reserved for the free R-value calculation.61 MolProbity was applied to evaluate the final structures before deposition in the PDB.62 Structure analysis, superposition and figure generation was done using PyMOL.63 X-ray data collection and crystallographic refinement statistics are presented in the Supplementary Information [Table S4].
Structural Analysis
To calculate the intermolecular vdW interaction energies the crystal structures were prepared using the Schrödinger Protein Preparation Wizard.64 Hydrogen atoms were added, protonation states were determined, and the structures were minimized. Subsequently, force field parameters were assigned using the OPLS3 force field.65 Interaction energies between the inhibitor and protease were estimated using a simplified Lennard-Jones potential, as previously described in detail.66 Briefly, the vdW energy was calculated for pairwise interactions depending on the types of atoms interacting and the distance between them.
Molecular Dynamics Simulations
High resolution HTLV-1 cocrystal structures with DRV, UM6, and PU6 (PDB: 6W6Q, 6W6R, 6W6S) were used as starting coordinates for molecular dynamics simulations. All starting structures were prepared using the Protein Preparation Wizard from Schrodinger.64 Crystallographic water molecules were retained, missing atoms were added using Prime,67 and PROPKA68–69 was used to determine the protonation state of side chains at pH 7.0. The resulting structure was minimized under restraint to a convergence criterion of 0.3 Å using Impref.70
All molecular dynamics simulations were carried out using Desmond, within Schrodinger, 64 with the OPLS3e force field for the inhibitor and protein. The systems were prepared as previously discussed.71 Briefly, the cocrystal structures were placed within a cubic TIP3P water box measuring 15 Å on each side. Chloride ions were first used to neutralize the system and sodium and chloride atoms were added to reach a physiological 0.15 M salt concentration. Prior to simulation, each solvated system was relaxed using a series of restrained minimization stages as previously described.71 These stages consisted of successive minimizations with restraints on i) the heavy protein atoms, ii-iii) the protein backbone atoms and finally iv) no restraints. The restraining force constants were 1000, 1000 and 5 kcal mol−1 Å−2 for stages i), ii) and iii), respectively and the minimization was done using steepest descent followed with the limited-memory BFGS method to a tolerance of 0.5 kcal mol−1 Å−1. During unrestrained minimization, this tolerance was further reduced to 0.05 kcal mol−1 Å−1. Molecular dynamics for each system was carried out in triplicate, with each of the three 100 ns simulations starting with different randomized velocities.
No unexpected or unusually high safety hazards were encountered in any of the experiments.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health grants 1R21AI149716–01A1, P01 GM109767 and R01 GM98550. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH–Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289–01A1). We thank the beamline specialists at 23-ID-D for their help in data collection. N.T. is supported by NIH fellowship grants T32 DA007097 and F32 AI150351.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/.
• Figures depicting representative dose-response curves, immunoblot quantification of Gag processing, per-residue van der Waals contacts, root-mean-squared fluctuations of Cα atoms, and π-π stacking interactions from molecular dynamics simulations.
• Tables listing amino acid sequence of HIV-1 and HTLV-1 protease cleavage sites, kinetic parameters for HTLV-1 protease enzymatic activity, enzyme inhibition constants (Ki), X-ray data collection and crystallographic refinement statistics.
REFERENCES
- 1.Poiesz BJ; Ruscetti FW; Gazdar AF; Bunn PA; Minna JD; Gallo RC, Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 1980, 77 (12), 7415–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalyanaraman VS; Sarngadharan MG; Robert-Guroff M; Miyoshi I; Golde D; Gallo RC, A new subtype of human T-cell leukemia virus (HTLV-II) associated with a T-cell variant of hairy cell leukemia. Science 1982, 218 (4572), 571–573. [DOI] [PubMed] [Google Scholar]
- 3.Proietti FA; Carneiro-Proietti AB; Catalan-Soares BC; Murphy EL, Global epidemiology of HTLV-I infection and associated diseases. Oncogene 2005, 24 (39), 6058–6068. [DOI] [PubMed] [Google Scholar]
- 4.Goncalves DU; Proietti FA; Ribas JG; Araujo MG; Pinheiro SR; Guedes AC; Carneiro-Proietti AB, Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev 2010, 23 (3), 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beilke MA, Retroviral coinfections: HIV and HTLV: taking stock of more than a quarter century of research. AIDS Res Hum Retroviruses 2012, 28 (2), 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willems L; Hasegawa H; Accolla R; Bangham C; Bazarbachi A; Bertazzoni U; Carneiro-Proietti AB; Cheng H; Chieco-Bianchi L; Ciminale V; Coelho-Dos-Reis J; Esparza J; Gallo RC; Gessain A; Gotuzzo E; Hall W; Harford J; Hermine O; Jacobson S; Macchi B; Macpherson C; Mahieux R; Matsuoka M; Murphy E; Peloponese JM; Simon V; Tagaya Y; Taylor GP; Watanabe T; Yamano Y, Reducing the global burden of HTLV-1 infection: An agenda for research and action. Antiviral Res 2017, 137, 41–48. [DOI] [PubMed] [Google Scholar]
- 7.Einsiedel L; Woodman RJ; Flynn M; Wilson K; Cassar O; Gessain A, Human T-Lymphotropic Virus type 1 infection in an Indigenous Australian population: epidemiological insights from a hospital-based cohort study. BMC Public Health 2016, 16, 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Mendoza C; Piron M; Gonzalez R; Jimenez A; Caballero E; Roc L; Benito R; Ramos JM; Soriano V; Group HSS, Clinical Presentation of Individuals With Human T-Cell Leukemia Virus Type-1 Infection in Spain. Open Forum Infect Dis 2019, 6 (2), ofz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bazarbachi A; Hermine O, Treatment of adult T-cell leukaemia/lymphoma: current strategy and future perspectives. Virus Res 2001, 78 (1–2), 79–92. [DOI] [PubMed] [Google Scholar]
- 10.Nasr R; El-Sabban ME; Karam JA; Dbaibo G; Kfoury Y; Arnulf B; Lepelletier Y; Bex F; de The H; Hermine O; Bazarbachi A, Efficacy and mechanism of action of the proteasome inhibitor PS-341 in T-cell lymphomas and HTLV-I associated adult T-cell leukemia/lymphoma. Oncogene 2005, 24 (3), 419–430. [DOI] [PubMed] [Google Scholar]
- 11.Dewan MZ; Uchihara JN; Terashima K; Honda M; Sata T; Ito M; Fujii N; Uozumi K; Tsukasaki K; Tomonaga M; Kubuki Y; Okayama A; Toi M; Mori N; Yamamoto N, Efficient intervention of growth and infiltration of primary adult T-cell leukemia cells by an HIV protease inhibitor, ritonavir. Blood 2006, 107 (2), 716–724. [DOI] [PubMed] [Google Scholar]
- 12.Hurst SA; Appelgren KE; Kourtis AP, Prevention of mother-to-child transmission of HIV type 1: the role of neonatal and infant prophylaxis. Expert Rev Anti Infect Ther 2015, 13 (2), 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Surleraux DL; Tahri A; Verschueren WG; Pille GM; de Kock HA; Jonckers TH; Peeters A; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu-Jeyabalan M; Schiffer CA; Wigerinck PB, Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J Med Chem 2005, 48 (6), 1813–1822. [DOI] [PubMed] [Google Scholar]
- 14.Wolfenden R, Transition state analogues for enzyme catalysis. Nature 1969, 223 (5207), 704–705. [DOI] [PubMed] [Google Scholar]
- 15.Schramm VL, Enzymatic transition state theory and transition state analogue design. J Biol Chem 2007, 282 (39), 28297–28300. [DOI] [PubMed] [Google Scholar]
- 16.Girard PM; Antinori A; Arribas JR; Ripamonti D; Bicer C; Netzle-Sveine B; Hadacek B; Moecklinghoff C, Week 96 efficacy and safety of darunavir/ritonavir monotherapy vs. darunavir/ritonavir with two nucleoside reverse transcriptase inhibitors in the PROTEA trial. HIV Med 2017, 18 (1), 5–12. [DOI] [PubMed] [Google Scholar]
- 17.Tozser J; Zahuczky G; Bagossi P; Louis JM; Copeland TD; Oroszlan S; Harrison RW; Weber IT, Comparison of the substrate specificity of the human T-cell leukemia virus and human immunodeficiency virus proteinases. Eur J Biochem 2000, 267 (20), 6287–6295. [DOI] [PubMed] [Google Scholar]
- 18.Kadas J; Weber IT; Bagossi P; Miklossy G; Boross P; Oroszlan S; Tozser J, Narrow substrate specificity and sensitivity toward ligand-binding site mutations of human T-cell Leukemia virus type 1 protease. J Biol Chem 2004, 279 (26), 27148–27157. [DOI] [PubMed] [Google Scholar]
- 19.Shuker SB; Mariani VL; Herger BE; Dennison KJ, Understanding HTLV-I protease. Chem Biol 2003, 10 (5), 373–380. [DOI] [PubMed] [Google Scholar]
- 20.Li M; Laco GS; Jaskolski M; Rozycki J; Alexandratos J; Wlodawer A; Gustchina A, Crystal structure of human T cell leukemia virus protease, a novel target for anticancer drug design. Proc Natl Acad Sci U S A 2005, 102 (51), 18332–18337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naka H; Teruya K; Bang JK; Aimoto S; Tatsumi T; Konno H; Nosaka K; Akaji K, Evaluations of substrate specificity and inhibition at PR/p3 cleavage site of HTLV-1 protease. Bioorg Med Chem Lett 2006, 16 (14), 3761–3764. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen JT; Kato K; Hidaka K; Kumada HO; Kimura T; Kiso Y, Design and synthesis of several small-size HTLV-I protease inhibitors with different hydrophilicity profiles. Bioorg Med Chem Lett 2011, 21 (8), 2425–2429. [DOI] [PubMed] [Google Scholar]
- 23.Kimura T; Nguyen JT; Maegawa H; Nishiyama K; Arii Y; Matsui Y; Hayashi Y; Kiso Y, Chipping at large, potent human T-cell leukemia virus type 1 protease inhibitors to uncover smaller, equipotent inhibitors. Bioorg Med Chem Lett 2007, 17 (12), 3276–3280. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen JT; Zhang M; Kumada HO; Itami A; Nishiyama K; Kimura T; Cheng M; Hayashi Y; Kiso Y, Truncation and non-natural amino acid substitution studies on HTLV-I protease hexapeptidic inhibitors. Bioorg Med Chem Lett 2008, 18 (1), 366–370. [DOI] [PubMed] [Google Scholar]
- 25.Zhang M; Nguyen JT; Kumada HO; Kimura T; Cheng M; Hayashi Y; Kiso Y, Synthesis and activity of tetrapeptidic HTLV-I protease inhibitors possessing different P3-cap moieties. Bioorg Med Chem 2008, 16 (10), 5795–5802. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M; Nguyen JT; Kumada HO; Kimura T; Cheng M; Hayashi Y; Kiso Y, Locking the two ends of tetrapeptidic HTLV-I protease inhibitors inside the enzyme. Bioorg Med Chem 2008, 16 (14), 6880–6890. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen JT; Kato K; Kumada HO; Hidaka K; Kimura T; Kiso Y, Maintaining potent HTLV-I protease inhibition without the P3-cap moiety in small tetrapeptidic inhibitors. Bioorg Med Chem Lett 2011, 21 (6), 1832–1837. [DOI] [PubMed] [Google Scholar]
- 28.Satoh T; Li M; Nguyen JT; Kiso Y; Gustchina A; Wlodawer A, Crystal structures of inhibitor complexes of human T-cell leukemia virus (HTLV-1) protease. J Mol Biol 2010, 401 (4), 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berman H; Henrick K; Nakamura H, Announcing the worldwide Protein Data Bank. Nat Struct Biol 2003, 10 (12), 980. [DOI] [PubMed] [Google Scholar]
- 30.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhnert M; Steuber H; Diederich WE, Structural basis for HTLV-1 protease inhibition by the HIV-1 protease inhibitor indinavir. J Med Chem 2014, 57 (14), 6266–6272. [DOI] [PubMed] [Google Scholar]
- 32.Kuhnert M; Blum A; Steuber H; Diederich WE, Privileged Structures Meet Human T-Cell Leukemia Virus-1 (HTLV-1): C2-Symmetric 3,4-Disubstituted Pyrrolidines as Nonpeptidic HTLV-1 Protease Inhibitors. J Med Chem 2015, 58 (11), 4845–4850. [DOI] [PubMed] [Google Scholar]
- 33.Demir A; Oguariri RM; Magis A; Ostrov DA; Imamichi T; Dunn BM, Kinetic characterization of newly discovered inhibitors of various constructs of human T-cell leukemia virus-1 (HTLV-1) protease and their effect on HTLV-1-infected cells. Antivir Ther 2012, 17 (5), 883–892. [DOI] [PubMed] [Google Scholar]
- 34.Louis JM; Oroszlan S; Tozser J, Stabilization from autoproteolysis and kinetic characterization of the human T-cell leukemia virus type 1 proteinase. J Biol Chem 1999, 274 (10), 6660–6666. [DOI] [PubMed] [Google Scholar]
- 35.Nalam MN; Ali A; Reddy GS; Cao H; Anjum SG; Altman MD; Yilmaz NK; Tidor B; Rana TM; Schiffer CA, Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem Biol 2013, 20 (9), 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Callebaut C; Stray K; Tsai L; Williams M; Yang ZY; Cannizzaro C; Leavitt SA; Liu X; Wang K; Murray BP; Mulato A; Hatada M; Priskich T; Parkin N; Swaminathan S; Lee W; He GX; Xu L; Cihlar T, In vitro characterization of GS-8374, a novel phosphonate-containing inhibitor of HIV-1 protease with a favorable resistance profile. Antimicrob Agents Chemother 2011, 55 (4), 1366–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meissner ME; Mendonca LM; Zhang W; Mansky LM, Polymorphic Nature of Human T-Cell Leukemia Virus Type 1 Particle Cores as Revealed through Characterization of a Chronically Infected Cell Line. J Virol 2017, 91 (16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henes M; Lockbaum GJ; Kosovrasti K; Leidner F; Nachum GS; Nalivaika EA; Lee SK; Spielvogel E; Zhou S; Swanstrom R; Bolon DNA; Kurt Yilmaz N; Schiffer CA, Picomolar to Micromolar: Elucidating the Role of Distal Mutations in HIV-1 Protease in Conferring Drug Resistance. ACS Chem Biol 2019, 14 (11), 2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller JF; Andrews CW; Brieger M; Furfine ES; Hale MR; Hanlon MH; Hazen RJ; Kaldor I; McLean EW; Reynolds D; Sammond DM; Spaltenstein A; Tung R; Turner EM; Xu RX; Sherrill RG, Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg Med Chem Lett 2006, 16 (7), 1788–1794. [DOI] [PubMed] [Google Scholar]
- 40.Aoki M; Hayashi H; Rao KV; Das D; Higashi-Kuwata N; Bulut H; Aoki-Ogata H; Takamatsu Y; Yedidi RS; Davis DA; Hattori SI; Nishida N; Hasegawa K; Takamune N; Nyalapatla PR; Osswald HL; Jono H; Saito H; Yarchoan R; Misumi S; Ghosh AK; Mitsuya H, A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ozen A; Lin KH; Kurt Yilmaz N; Schiffer CA, Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc Natl Acad Sci U S A 2014, 111 (45), 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King NM; Melnick L; Prabu-Jeyabalan M; Nalivaika EA; Yang SS; Gao Y; Nie X; Zepp C; Heefner DL; Schiffer CA, Lack of synergy for inhibitors targeting a multi-drug-resistant HIV-1 protease. Protein Sci 2002, 11 (2), 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hui JO; Tomasselli AG; Reardon IM; Lull JM; Brunner DP; Tomich CS; Heinrikson RL, Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J Protein Chem 1993, 12 (3), 323–327. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y; Kati W; Chen CM; Tripathi R; Molla A; Kohlbrenner W, Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Analytical biochemistry 1999, 267 (2), 331–335. [DOI] [PubMed] [Google Scholar]
- 45.Grigsby IF; Zhang W; Johnson JL; Fogarty KH; Chen Y; Rawson JM; Crosby AJ; Mueller JD; Mansky LM, Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology 2010, 7, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maldonado JO; Cao S; Zhang W; Mansky LM, Distinct Morphology of Human T-Cell Leukemia Virus Type 1-Like Particles. Viruses 2016, 8 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rowe T; Dezzutti C; Guenthner PC; Lam L; Hodge T; Lairmore MD; Lal RB; Folks TM, Characterization of a HTLV-I-infected cell line derived from a patient with adult T-cell leukemia with stable co-expression of CD4 and CD8. Leuk Res 1995, 19 (9), 621–628. [DOI] [PubMed] [Google Scholar]
- 48.Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; Tinevez JY; White DJ; Hartenstein V; Eliceiri K; Tomancak P; Cardona A, Fiji: an open-source platform for biological-image analysis. Nat Methods 2012, 9 (7), 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lockbaum GJ; Leidner F; Rusere LN; Henes M; Kosovrasti K; Nachum GS; Nalivaika EA; Ali A; Yilmaz NK; Schiffer CA, Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease. ACS Infect Dis 2019, 5 (2), 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henes M; Kosovrasti K; Lockbaum GJ; Leidner F; Nachum GS; Nalivaika EA; Bolon DNA; Kurt Yilmaz N; Schiffer CA; Whitfield TW, Molecular Determinants of Epistasis in HIV-1 Protease: Elucidating the Interdependence of L89V and L90M Mutations in Resistance. Biochemistry 2019, 58 (35), 3711–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rusere LN; Lockbaum GJ; Lee SK; Henes M; Kosovrasti K; Spielvogel E; Nalivaika EA; Swanstrom R; Yilmaz NK; Schiffer CA; Ali A, HIV-1 Protease Inhibitors Incorporating Stereochemically Defined P2’ Ligands To Optimize Hydrogen Bonding in the Substrate Envelope. J Med Chem 2019, 62 (17), 8062–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pothineni SB; Venugopalan N; Ogata CM; Hilgart MC; Stepanov S; Sanishvili R; Becker M; Winter G; Sauter NK; Smith JL; Fischetti RF, Tightly integrated single- and multi-crystal data collection strategy calculation and parallelized data processing in JBluIce beamline control system. J Appl Crystallogr 2014, 47 (Pt 6), 1992–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans P, Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 1), 72–82. [DOI] [PubMed] [Google Scholar]
- 55.Evans PR, An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otwinowski Z; Minor W, [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 57.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J Appl Crystallogr 2007, 40 (Pt 4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 59.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moriarty NW; Grosse-Kunstleve RW; Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr 2009, 65 (Pt 10), 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brunger AT, Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355 (6359), 472–475. [DOI] [PubMed] [Google Scholar]
- 62.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB 3rd; Snoeyink J; Richardson JS; Richardson DC, MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 2007, 35 (Web Server issue), W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DeLano WL, Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40, 82–92. [Google Scholar]
- 64.Schrödinger Release 2019–2: Maestro, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]
- 65.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA, OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12 (1), 281–296. [DOI] [PubMed] [Google Scholar]
- 66.Nalam MN; Ali A; Altman MD; Reddy GS; Chellappan S; Kairys V; Ozen A; Cao H; Gilson MK; Tidor B; Rana TM; Schiffer CA, Evaluating the Substrate-Envelope Hypothesis: Structural Analysis of Novel HIV-1 Protease Inhibitors Designed to be Robust against Drug Resistance. J. Virol 2010, 84 (10), 5368–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacobson MP; Friesner RA; Xiang Z; Honig B, On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol 2002, 320 (3), 597–608. [DOI] [PubMed] [Google Scholar]
- 68.Olsson MH; Sondergaard CR; Rostkowski M; Jensen JH, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J Chem Theory Comput 2011, 7 (2), 525–537. [DOI] [PubMed] [Google Scholar]
- 69.Sondergaard CR; Olsson MH; Rostkowski M; Jensen JH, Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J Chem Theory Comput 2011, 7 (7), 2284–2295. [DOI] [PubMed] [Google Scholar]
- 70.Banks JL; Beard HS; Cao Y; Cho AE; Damm W; Farid R; Felts AK; Halgren TA; Mainz DT; Maple JR; Murphy R; Philipp DM; Repasky MP; Zhang LY; Berne BJ; Friesner RA; Gallicchio E; Levy RM, Integrated Modeling Program, Applied Chemical Theory (IMPACT). J Comput Chem 2005, 26 (16), 1752–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leidner F; Kurt Yilmaz N; Paulsen J; Muller YA; Schiffer CA, Hydration Structure and Dynamics of Inhibitor-Bound HIV-1 Protease. J Chem Theory Comput 2018, 14 (5), 2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.