

SUMMARY
We lack a mechanistic understanding of aging-mediated changes in mitochondrial bioenergetics and lipid metabolism that affect T cell function. The bioactive sphingolipid ceramide, induced by aging stress, mediates mitophagy and cell death; however, the aging-related roles of ceramide metabolism in regulating T cell function remain unknown. Here, we show that activated T cells isolated from aging mice have elevated C14/C16 ceramide accumulation in mitochondria, generated by ceramide synthase 6, leading to mitophagy/mitochondrial dysfunction. Mechanistically, aging-dependent mitochondrial ceramide inhibits protein kinase A, leading to mitophagy in activated T cells. This aging/ceramide-dependent mitophagy attenuates the antitumor functions of T cells in vitro and in vivo. Also, inhibition of ceramide metabolism or PKA activation by genetic and pharmacologic means prevents mitophagy and restores the central memory phenotype in aging T cells. Thus, these studies help explain the mechanisms behind aging-related dysregulation of T cells’ antitumor activity, which can be restored by inhibiting ceramide-dependent mitophagy.
Graphical Abstract
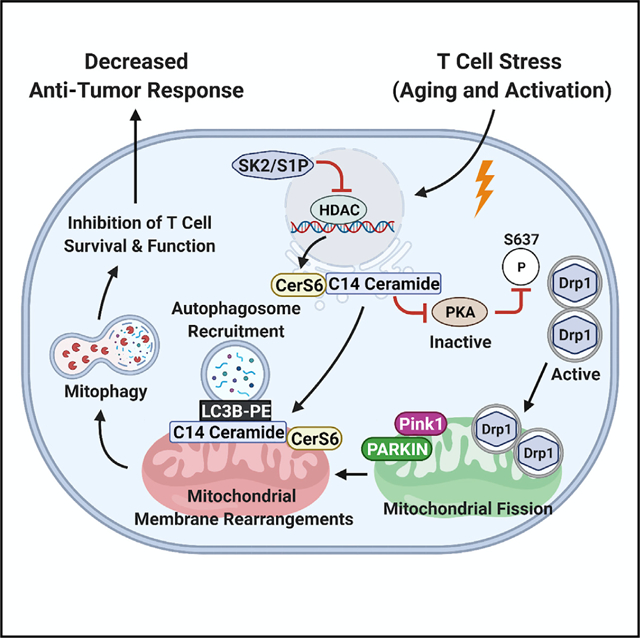
In brief
Vaena et al. define the mechanism whereby aging-mediated induction of ceramide-dependent mitophagy ameliorates antitumor functions of T cells via the inhibition of PKA. The attenuation of ceramide metabolism or activation of PKA prevents mitophagy and restores the central memory phenotype in aging T cells, improving T cell-mediated immunotherapeutic control of tumor growth.
INTRODUCTION
Memory T cells are dependent on lipid metabolism and mitochondrial function to generate energy for survival and persistence (Baazim et al., 2019; O’Sullivan et al., 2014). Components of the immune system, including CD4+ and CD8+ T cells, are vulnerable to mitochondrial dysfunction as organisms age (Bronietzki et al., 2015; Chougnet et al., 2015; Ron-Harel et al., 2015). Aging is a process characterized by a progressive loss of physiological integrity that leads to impaired tissue and organ function, increasing vulnerability to various diseases such as neurodegeneration and cancer (Bonneuil, 2007; Cantuti-Castelvetri et al., 2005; Kaur et al., 2016; Wang et al., 2017). The hallmarks of aging include mitochondrial dysfunction, telomere attrition, loss of proteostasis, and altered intercellular communication (Dodig et al., 2019; Revuelta and Matheu, 2017; Roy et al., 2002; Salminen et al., 2018; Shay and Wright, 2007). Altered mitochondrial function has been postulated as one of the central regulators of the aging process (Brown-Borg and Anderson, 2017; Campisi et al., 2019; Champagne et al., 2016; Sliter et al., 2018; Wang et al., 2010) with decreased mitochondrial respiratory capacity and ATP generation (Mellouk and Bobé, 2019; Mendelsohn et al., 2018). The protective T cell memory repertoire deteriorates with age (Giannakis et al., 2019; Minhas et al., 2019; Nicholls, 2004).
In contrast to naive T cells, the functions of resident memory T cells decline with the aging process (Kishton et al., 2017; Le Page et al., 2018; Martinez-Jimenez et al., 2017; Naumov et al., 2011; Ouyang et al., 2011), and the disproportionate activation of memory cells contributes to the failure to generate immune memory in the aging host (Pulko et al., 2016; Schenten et al., 2014; Sckisel et al., 2015). However, mechanisms involved in aging-dependent inhibition of T cell effector functions, especially for controlling cancer, are mostly unknown. Recent studies demonstrated that suppression of T cell effector function and memory phenotype is regulated, in part, by alterations in lipid metabolism and signaling (Klein Geltink et al., 2020; Pearce et al., 2009; Weinberg and Chandel, 2014).
The bioactive sphingolipid ceramide, generated by ceramide synthase, is an adaptive stress effector induced in response to various stress stimuli such as aging (Choi et al., 2015; Hannun and Obeid, 2018; Trayssac et al., 2018; Yi et al., 2016; Zelnik et al., 2020). Ceramide synthase (CerS) was first discovered in yeast as a longevity assurance gene (LAG1), as the reduced ceramide generation consequent to LAG1 deletion increased longevity (Riebeling et al., 2003; Venkataraman and Futerman, 2000). There are 6 homologs of mammalian LAG1/ceramide synthases (CerS1–6) that generate ceramides containing different fatty acid chain lengths with distinct functions (Park et al., 2014; Petrache et al., 2013; Pewzner-Jung et al., 2006). For example, while CerS1/4 mainly generates C18/C20 ceramides, CerS2 is responsible for the generation of very-long-chain C22–24 ceramides, and C14/C16 ceramides are synthesized mainly by CerS5 and CerS6. After the hydrolysis of ceramide by ceramidases to sphingosine and fatty acids, sphingosine is then phosphorylated by sphingosine kinase 1 or 2 (SphK1 or SphK2) for the generation of pro-survival sphingosine 1-phosphate (S1P), which enhances immune cell egress to the bloodstream, activating immune function (Bektas et al., 2005; Blaho et al., 2015; Hait et al., 2009; Singh and Spiegel, 2020; Watterson et al., 2007). Ceramide is known to induce mitophagy and cell death (Giannakis et al., 2019; Kishton et al., 2017; Mendelsohn et al., 2018; Minhas et al., 2019; Nicholls, 2004); however, whether it exerts negative effects on antitumor functions of T cells and immunotherapy remains unknown. One possible connection between aging, ceramide metabolism, and declined T cell effector functions is the induction of mitophagy (Dany et al., 2016; Dany and Ogretmen, 2015; Oleinik et al., 2019; Sentelle et al., 2012; Thomas et al., 2017).
Stress-induced ceramide is known to bind and recruit autophagosomes via LC3-II (through F52 interaction) to mitochondria (MITO) that then undergo Drp1-mediated fission, resulting in mitophagy, the degradation of MITO by autophagic machinery (Dany et al., 2016; Dany and Ogretmen, 2015; Oleinik et al., 2019; Sentelle et al., 2012; Thomas et al., 2017). Ceramide-mediated mitophagy limits ATP production and results in the inhibition of cancer cell proliferation and growth (Dany et al., 2016; Dany and Ogretmen, 2015; Oleinik et al., 2019; Sentelle et al., 2012; Thomas et al., 2017). However, whether this process plays any roles in immune aging has not been reported. This study aimed to determine whether aging-mediated ceramide metabolism and signaling play a role in T cell regulation via mitophagy induction. The data demonstrate that CD8+ T cells isolated from older mice (between 8 and 24 months) compared to younger counterparts (2–4 months) exhibit reduced antitumor function, regulated by CerS6-dependent C14 ceramide-induced mitophagy through the inhibition of protein kinase A (PKA) and the subsequent activation of Drp1. Genetic and pharmacologic inhibition of ceramide synthesis and Drp1-mediated mitophagy or activation of PKA restored the ability of the older T cells to improve tumor control via adoptive T cell transfer.
RESULTS
Aging decreases T cell survival and function. To delineate the role of aging in T cell biology, we isolated CD4+ and CD8+ T cells from wild-type (WT) C57BL/6 mice splenocytes at various ages between 1 and 24 months and measured their growth/survival in vitro. Although naive T cells from 1- to 10-month-old age groups showed no significant difference in their growth/survival (Figure 1A), T cells isolated from 8- to 24-month-old WT mice exhibited a significant decrease in survival after ex vivo T cell receptor (TCR) activation using anti-CD3 and anti-CD28 antibodies with interleukin-2 (IL-2) for 72 h (Figure 1B). Interestingly, alterations of survival of T cells upon TCR activation appeared to start as early as ages 8–18 months compared to 2–6 months (Figure 1B). To identify upstream mechanisms involved in limiting T cell survival upon aging stress, we mainly focused on T cells isolated from 2- to 6- (young, Y) versus 8- to 18- (aging, A) month-old mice, which exhibited significant and consistent changes in their survival in response to TCR activation, similar to aged (or old, O) mice at 20–24 months of age (Figure 1B). The data showed that the attenuation of survival was consistent with decreased interferon-γ (IFN-γ) expression in the living T cells isolated from A compared to Y mice (Figure 1C). Interestingly, analysis of T cells isolated from Y and A mice using scanning electron microscopy (SEM) demonstrated that aging stress-mediated inhibition of survival and cytokine secretion is also associated with decreased T cell size and altered surface phenotype (shape and texture) in T cells from A versus Y mice (Figure S1A) and reduced cell diameter (Figures S1B and S1C). To assess whether aging stress affects T cell function, we measured the effects of CD8+ Pmel T cells (activated using gp-100 and IL-2 for 72 h) isolated from Y or A pMel mice on B16 cell killing in co-culture studies at 1, 6, and 16 h (Chatterjee et al., 2014, 2018). CD8+ T cells from Pmel mice bear TCR transgenes (Tgs) specific to melanoma-associated antigen (gp-100), which allows them to specifically target and kill B16 melanoma cells. CD8+ T cells from Y and A Pmel mice exhibited similar expression levels of the TCR (Vβ13), and the expression of the Tg on the cells is not affected by the aging process (Figure S1D). Data demonstrated that T cells isolated from Y pMel mice kill B16 melanoma after 6–16 h of co-culture, whereas T cells isolated from A pMel mice had almost no cytotoxic function against B16 melanoma in vitro (Figure 1D). Overall, these data suggest that aging inhibits the survival and antitumor functions of ex vivo TCR-activated T cells.
Figure 1. Aging stress attenuates T cell function in part via induction of mitophagy in response to ex vivo TCR activation.
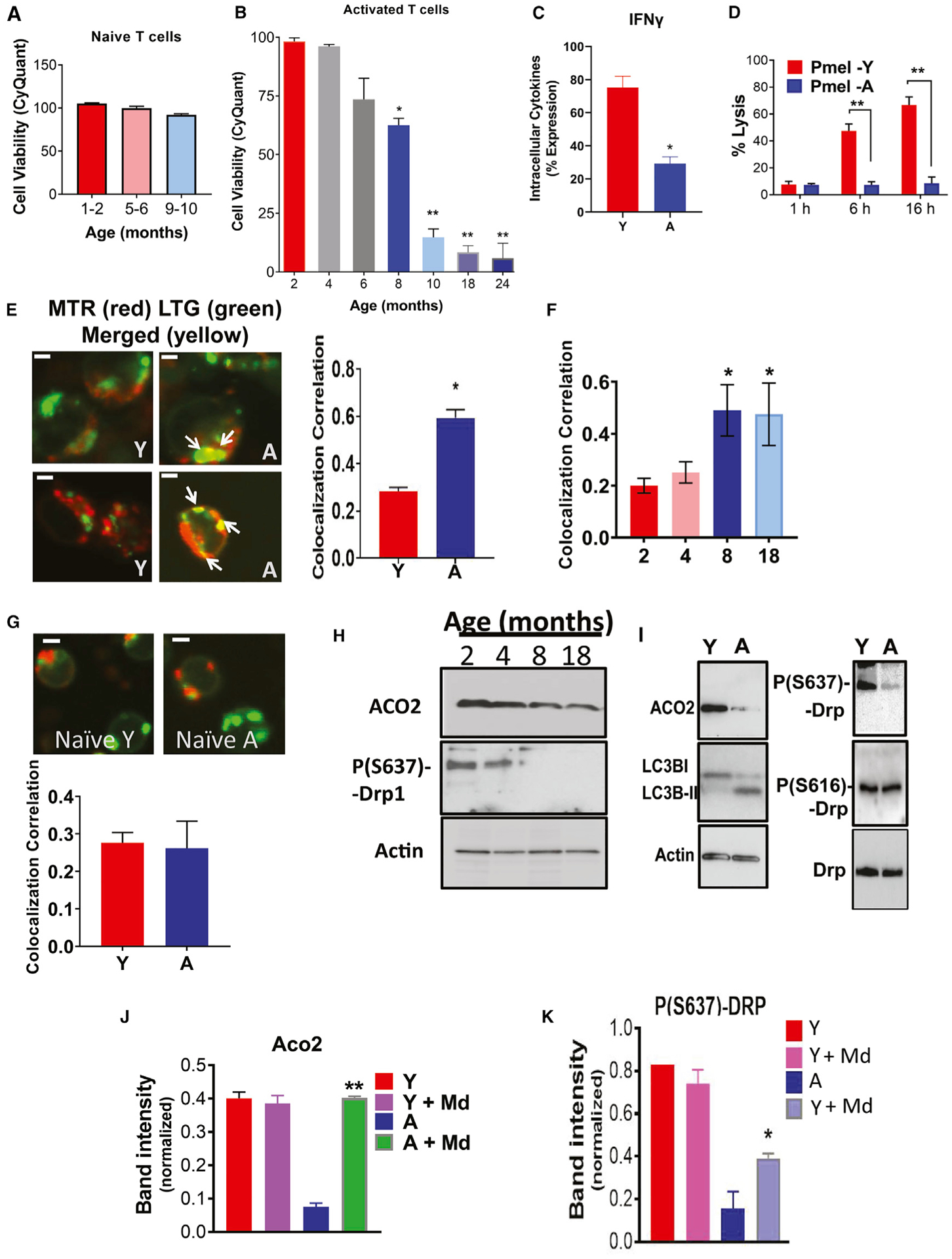
(A and B) Viability of naive (A) or TCR activated (B) T cells isolated from Y or A mice was measured by trypan blue exclusion assay.
(C) Flow cytometry analyses show IFN-γ suppression in activated T cells isolated from A compared to Y mice. Data are means ± SDs from 3 independent experiments (n = 3). *p < 0.05 as determined by Student’s t test.
(D) The ability of aging Pmel T cells to kill B16 melanoma cancer cells in co-cultures is impaired compared to T cells isolated from Y mice. Data are means ± SDs from 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test.
(E and F) Mitophagy of Y (isolated from 2- and 4-month-old mice) and A (isolated from 8- and 18-month-old mice) activated T cells were measured by live-cell imaging using confocal microscopy stained for mitochondria (MITO; MitoTracker Red [MTR]) and lysosomes (LysoTracker Green [LTG]). Scale bars, 1 μm. Micrographs represent at least 3 independent experiments of Y and A T cells (E). The right panel shows the quantification of colocalization extracted from the coefficient of colocalization (Rc). White arrows indicate merged (yellow). Scale bars, 1 μm. Quantification of colocalization extracted from the coefficient of colocalization (Rc) using the Fiji software (F). Data are means ± SDs from 3 independent experiments (n = 3). *p < 0.05 as determined by Student’s t test.
(G) Live-cell imaging of naive Y and A T cells, stained MITO (MTR) and lysosomes (LTG). Scale bars, 1 μm. Micrographs represent at least 3 independent experiments of Y and A T cells. The bottom panel shows the quantification of colocalization extracted from the coefficient of colocalization (Rc). Data are means ± SDs from 3 independent experiments (n = 3).
(H and I) Western blot measuring the levels of ACO2 and P-S637-Drp1 in activated T cells from Y (2- and 4-month-old), and A (8- and 18-month-old) mice. Protein expression was normalized to β-actin. (I) Western blot measuring cytoplasmic levels of LC3B-I, LC3B-II, and ACO2 (H), or total-Drp1, P-(S637)-Drp1, and P-(S616)-Drp1 (I), in activated T cells from Y and A mice.
(J and K) Western blot to detect P-S637-Drp1 (J) and ACO2 (K) in activated T cells with/without mitophagy inhibitor mDivi (Md). Data are means ± SDs from 3 independent experiments (n = 3). *p < 0.05, **p < 0.01 as determined by Student’s t test.
Aging stress induces mitophagy in T cells. Because aging stress is known to induce ceramide generation (Liu et al., 2013; Montoliu et al., 2014), which then activates mitophagy (Dany and Ogretmen, 2015, Sentelle et al., 2012, Dany et al., 2016, Thomas et al., 2017, Oleinik et al., 2019) to limit cell growth/survival, we next measured whether this process plays a role in the decreased function of T cells isolated from A compared to Y mice in response to ex vivo TCR activation. To detect ceramide-dependent mitophagy, we performed live-cell imaging and immunofluorescence. We measured the colocalization of MITO and lysosomes using MitoTracker Red (MTR) and LysoTracker Green (LTG) or ceramide and TOM20 (outer mitochondrial membrane protein) using anti-ceramide and anti-TOM20. Colocalization of MTR-LTG and ceramide-LC3 indicates mitophagy activation. These studies revealed higher levels of MTR-LTG and ceramide-LC3 colocalization in activated CD4+ and CD8+ T cells isolated from A compared to Y mice (Figures 1E and 1F). No mitophagy was observed in naive (non-activated) Y or A T cells, as we did not detect any MTR-LTG, ceramide-TOM20, or ceramide-LC3 colocalization (Figures 1G, S1E, and S1F). Mitophagy induction in response to the activation of T cells from A compared to Y mice was also detectable by decreased aconitase 2 (ACO2), a mitochondrial matrix protein that becomes degraded upon mitochondrial digestion by autophagosomes (Figure 1H, upper panel). This was consistent with LC3 activation, which is detectable by the presence of lipidated protein (LC3-II) in aging T cells (Figure 1I, left, center panel). Mitophagy induction was also associated with the activation of Drp1, detected by decreased levels of inhibitory Phospho-S637-Drp1 (P-S637-Drp1) in T cells isolated from A compared to Y mice (Figures 1H and 1I). There were no detectable changes in the abundance of P-S616-Drp1 or total Drp1 in these A versus Y T cells (Figure 1I, right, center panel). Also, pharmacologic inhibition of Drp1 using Mdivi largely prevented mitophagy activation and protected Drp1 activation or ACO2 degradation in T cells isolated from A compared to Y mice (Figures 1J and 1K). There were no changes in apoptosis, detected by annexin V, caspase 3, or granzyme B expression in TCR-activated T cells obtained from A and Y mice (Figures S1G–S1I). Reduction of the effector memory T cells CD44hi/CD62Llo was also detected in cells isolated from A compared to Y WT mice (Figure S1J). Analyses of cytokines/chemokines using ELISA showed decreased IFN-γ and elevated levels of IL-1α in T cells isolated from A compared to Y mice (Figures S1H, S1K, and S1L). Thus, these data support the induction of mitophagy and not apoptosis in A compared to Y T cells in response to ex vivo TCR activation.
Aging stress induces ceramide-dependent mitophagy
To determine whether the induction of mitophagy is associated with ceramide metabolism and/or signaling, we measured the effects of fumonisin B1 (FB1), a pan inhibitor of CerS1–6, on mitophagy in response to ex vivo TCR activation in T cells isolated from A and Y mice. Inhibition of CerS1–6 by FB1 prevented mitophagy induction and inhibited ceramide-TOM20 colocalization and autophagy/LC3 activation in A compared to Y T cells (Figures 2A and 2B). It should be noted that ceramide detection using anti-ceramide antibody in Figure 2A still shows substantial ceramide signal in cells treated with FB1. We believe that this is dependent on the sensitivity of the fluorescence for ceramide detection, as FB1 inhibits ceramide generation, but it does not eliminate cellular ceramides completely. We also performed mass spectrometry-based lipidomics (Gencer et al., 2017; Panneer Selvam et al., 2015) in isolated MITO of naive versus TCR-activated T cells isolated from Y and A mice. The lipidomics studies were performed in cell extracts enriched in MITO of a versus Y T cells. The data revealed that, compared to T cells from Y mice, C14 ceramide and C16 ceramide (to a minor extent) accumulation is increased selectively in MITO of T cells from A mice without any significant changes in C18 ceramides (Figure 2C), or C20–C26 ceramides, sphingosine, and S1P (Figure S2A). The measurement of total ceramides also showed a substantial increase in C14 ceramide levels in A compared to Y T cells (Figure S2B). ACO2 was present in MITO fraction but not in S or mitochondria-associated membranes (MAMs). Similarly, FACL-4 was enriched in MAMs but not in other fractions (Figure S2C).
Figure 2. Aging induces mitochondrial CerS6/C14 ceramide and mediates mitophagy in activated T cells.
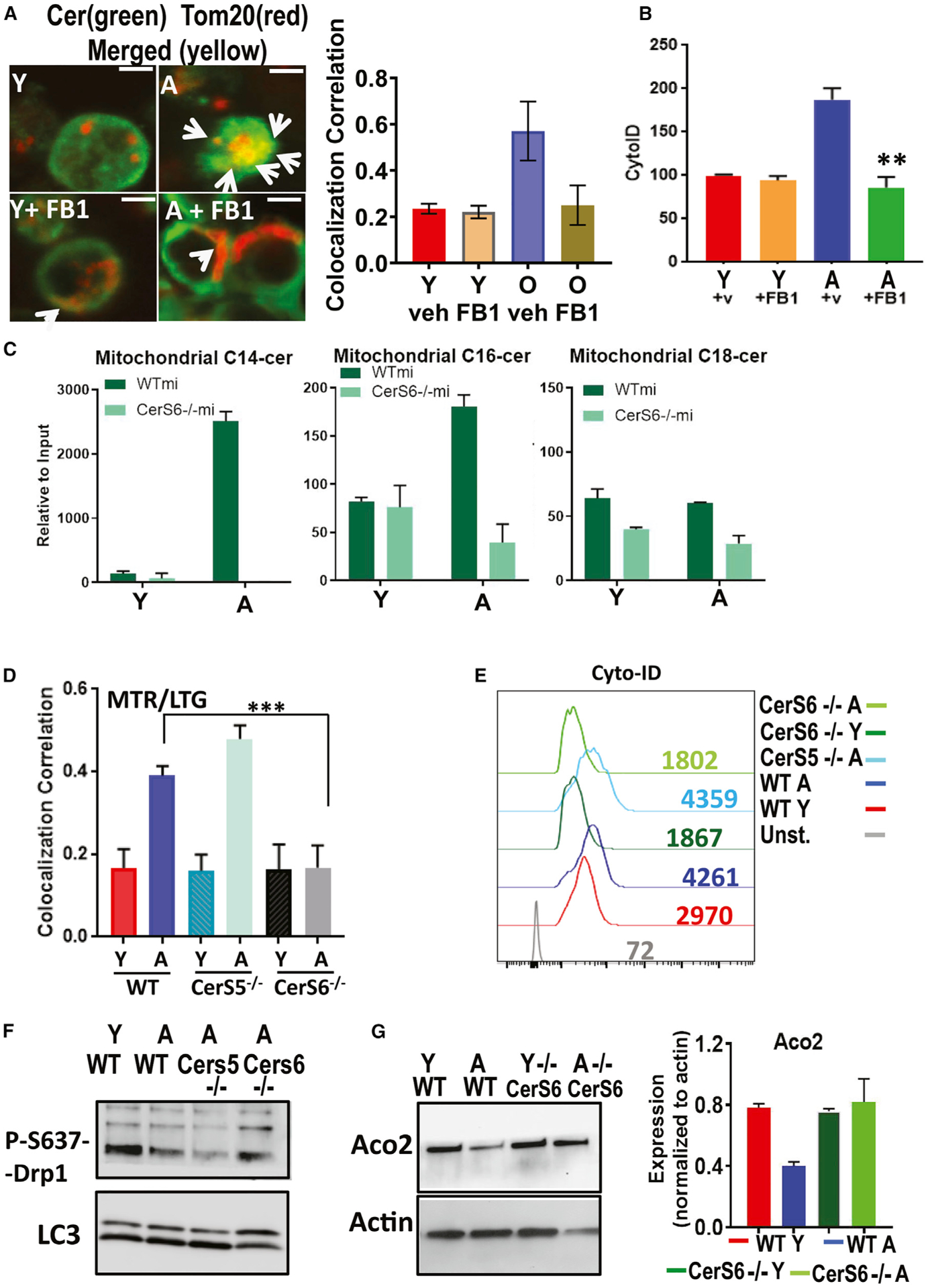
(A) Confocal microscopy for Y and A T cells with/without FB1 dual labeled with TOM20 (red, mitochondrial marker), and ceramide (green) fluorescent antibodies. White arrows indicate merged (yellow). Scale bars, 1 μm. Images represent at least 3 independent experiments. The right panel shows the quantification of colocalization extracted from the coefficient of colocalization (Rc). Data are means ± SDs from 3 independent experiments (n = 3).
(B) Autophagy was measured staining with CytoID followed by flow cytometry of 3-day activated Y and A T cells with/without FB1. Data show means ± SDs from 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test.
(C) High-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) measurements of C14 ceramide (left), C16 ceramide (center), and C18 ceramide (right) in activated T cells isolated from Y and A mice. Data are means ± SDs from 3 experiments (n = 3). *p < 0.05 as determined by Student’s t test.
(D) Mitophagy was detected by live-cell imaging for measuring the colocalization of MTR-LTG in T cells obtained from Y and A WT, CerS5−/− and CerS6−/− mice. Quantification of colocalization was performed using the coefficient of colocalization (Rc). Data are means ± SDs from at least 3 experiments (n = 3). ***p < 0.001 as determined by Student’s t test.
(E–G) LC3/autophagy activation was measured by cyto-ID (E), and protein abundance of P-S637-Drp1 and LCBI/LCBII (F) or ACO2 and actin (G) were measured using extracts of activated T cells isolated from Y and A WT, CerS5−/−, and/or CerS6−/− mice. These data represent at least 3 independent experiments (n = 3). Data are means ± SDs from 3 independent experiments (n = 3).
Unlike long-chain (C18–C20 ceramides, synthesized by CerS1 and CerS4) and very-long-chain ceramides (C22–C26 and longer ceramides, synthesized by CerS2 and CerS3), short-chain (C14/C16) ceramides are primarily synthesized by CerS5 or CerS6 (Park et al., 2014; Petrache et al., 2013; Pewzner-Jung et al., 2006). To identify the enzyme responsible for C14/C16-ceramide accumulation in MITO, we isolated CD4+/CD8+ T cells from Y and A WT, CerS5−/− and CerS6−/− (knockout) mice (Scheffel et al., 2017; Sofi et al., 2017). We then measured MITO ceramide accumulation and mitophagy upon TCR activation in these T cells. Genetic loss of CerS6, but not CerS5, completely prevented MITO-C14 ceramide accumulation (Figure 2C) mitophagy and inhibited MTR-LTG colocalization, LC3 activation, reduction of inhibitory P-S637 expression, and ACO2 degradation in T cells isolated from A CerS6−/− compared to age-matched WT mice (Figures 2D–2G). Thus, these data indicate that aging stress selectively induces CerS6-generated C14 ceramide accumulation in T cell MITO, associated with LC3 and Drp1 activation and mitophagy upon TCR activation.
CerS6/C14 ceramide-mediated mitophagy in aging compared to young T cells also resulted in reduced mitochondrial function, indicated by a decreased oxygen consumption rate (OCR) without any effects on glycolysis or extracellular acidification rate (ECAR) in response to TCR activation, which is measured by real-time flux analyses using the Seahorse platform. Loss of CerS6 largely prevented mitochondrial dysfunction and OCR in ex vivo TCR-activated T cells obtained from A compared to Y CerS6−/− mice (Figures 3A–3C). Ceramide-mediated mitophagy was also consistent with reduced ATP abundance in T cells obtained from A versus Y WT mice, which was protected in aging T cells obtained from A compared to Y CerS6−/− mice (Figures 3D and 3E). Prevention of mitophagy in T cells isolated from A CerS6−/− mice was also consistent with reduced mitochondrial C14/C16 ceramide levels compared to controls (Figures 2C and S2A). Mitochondrial and metabolic dysfunction in aging T cells was also detected in RNA sequencing studies. The expression of genes encoding for various enzymes associated with T cell metabolism, such as PLA2 (phospholipase 2), LPAR3 (lysophosphatidic acid receptor 3), GRIN2C (glutamate receptor), and ACSL6 (fatty acyl-coA synthase) was found to be highly reduced in T cells isolated from A WT compared to CerS6−/− mice (Figure S3A). This suggests that while aging stress is associated with decreased T cell survival/function, loss of CerS6 restores T cell metabolism during aging. The expression of GSTA2 (glutathione S-transferase) was highly elevated in TCR-activated aging WT T cells, suggesting the involvement of increased reactive oxygen species and/or peroxidation during aging stress-mediated mitophagy, which is also prevented in T cells obtained from A CerS6−/− mice (Figure S3A). RNA sequencing was also consistent with reduced mRNA abundance of IFN-γ in T cells isolated from A compared to Y WT mice, which is also reversed in T cells isolated from A versus Y CerS6−/− mice (Figure S3B). There was also a group of cytokines/chemokines whose expression was inversely associated with aging versus young T cells isolated from WT compared to Cer6−/− mice (Figure S3B). These cytokines/chemokines include IL-2, IL-6, CXCL3, IL-19, CCL22, CXCR2, CCL9, CCL6, CCL17, and IL-119, which are induced versus reduced in T cells isolated from A WT versus CerS6−/− compared to their young counterparts (Figure S3B). However, we observed that the expression of IFN-γ, CXCR1, IL-11, and IL-24, which are associated with T cell activation, are reduced versus increased in T cells isolated from A WT versus CerS6−/− compared to their young counterparts (Figure S3B). These studies support that aging stress-mediated CerS6/C14-ceramide induces T cell mitophagy, leading to dysfunctional mitochondria and reduced ATP, which is consistent with reduced central memory phenotype.
Figure 3. Aging attenuates mitochondrial function of activated T cells via ceramide-mediated mitophagy.
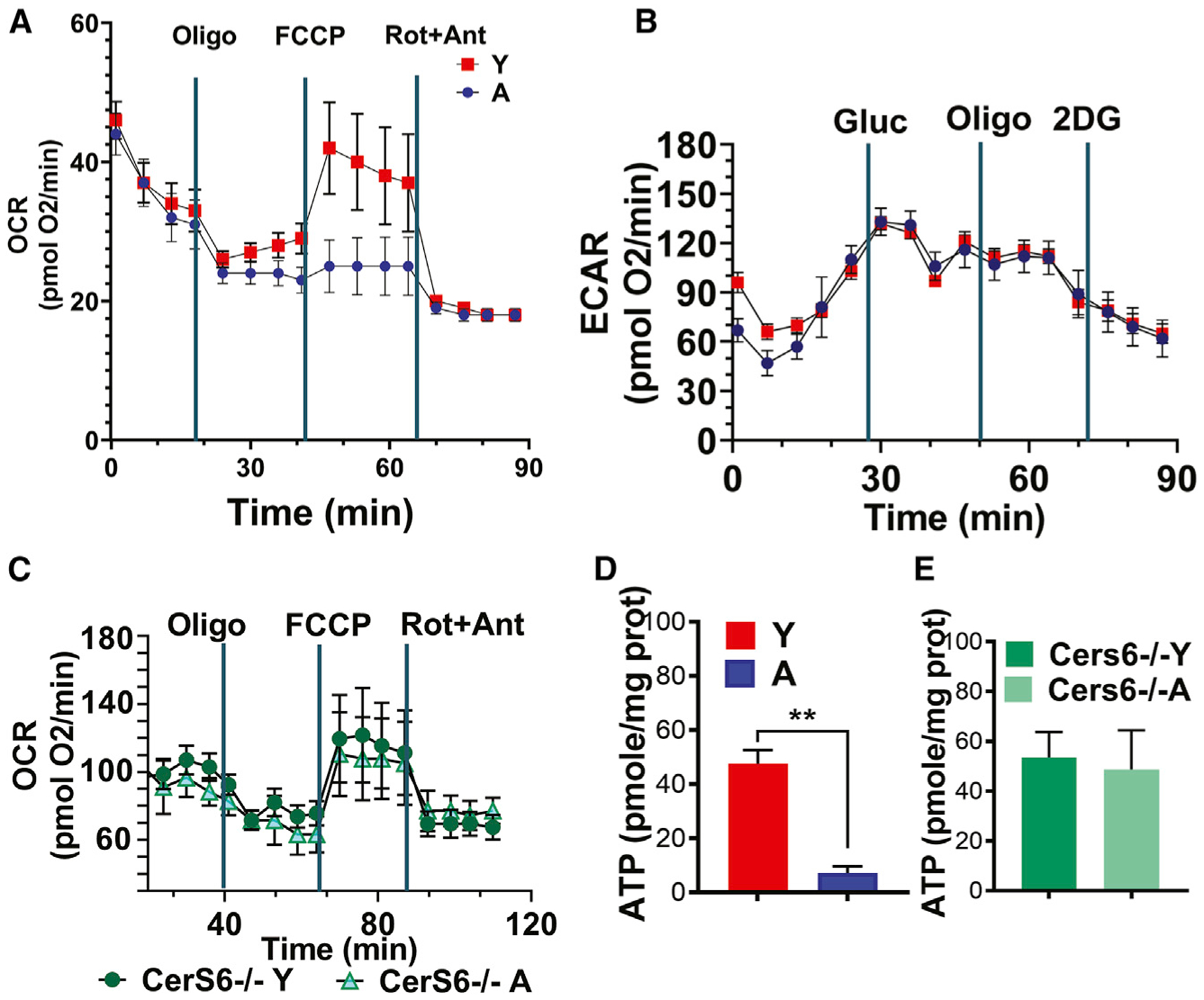
(A and B) Metabolic responses of 3-day activated T cells (0.4 × 106 cells/well) obtained from Y and A WT mice were used to determine the oxygen consumption rate (OCR) under basal conditions and in response to the electron-transport chain inhibitors oligomycin, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and rotenone/antimycin A (A). The glycolytic rates by ECAR detection in response to glucose, oligomycin, and 2-deoxyglucose were measured by the Seahorse platform (B) in T cells shown in (A).
(C) OCR of T cells isolated from Y and A CerS6−/− mice was measured by the Seahorse platform.
(D and E) ATP contents of T cells obtained from Y and A WT (D) or CerS6−/− mice were measured using the ATP detection kit. Data represent at least 3 independent experiments (n = 3). Data are means ± SDs from 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test.
CerS6/C14 ceramide generation in T cells is linked to SphK2/S1P signaling. Analyses of RNA sequencing data for the expression of genes involved in sphingolipid metabolism in T cells also revealed that the reduction of ceramide accumulation/generation was increased in aging WT T cells, while it is reduced in aging CerS6−/− T cells compared to their young counterparts (Figure S3C). Among these genes, ACER1 and ACER2, which cleave ceramide, are induced in T cells isolated from A CerS6−/− compared to WT mice, while the expression of CerS6 and SphK2 is greatly reduced in aging CerS6−/− while largely increased in aging WT T cells compared to their young counterparts (Figure S3C). These data are consistent with reduced ceramide and mitophagy in aging CerS6−/− T cells. Interestingly, these data also suggested to us a hypothesis that there may be crosstalk between CerS6 and SphK2 expression in T cells upon aging stress (Figure S3D). The loss of SphK2 almost completely prevented short-chain ceramide accumulation in the MITO of T cells isolated from A compared to Y SphK2−/− mice (Selvam and Ogretmen, 2013; Panneer Selvam et al., 2018) (Figure 4A). The loss of SphK2 expression in T cells isolated from Y SphK2−/− mice was confirmed by western blotting (Figure 4A, inset). This is consistent with the prevention of mitophagy in aging compared to young SphK2−/− T cells, which showed reduced ceramide-TOM20 colocalization, inhibition of LC3 or Drp1 activation, and protection of ACO2 degradation (Figures 4B–4E). Also, SEM studies revealed that the diameter of T cells obtained from Y and A SphK2−/− mice was similar to that of young WT T cells, which is consistent with the protection of aging SphK2−/− T cell function and morphology (Figure S1C). While the levels of IFN-γ were protected, IL-1α levels were reduced in T cells isolated from A versus Y SphK2−/− mice to similar levels observed in young WT T cells (Figures S1G–S1I). These findings indicate that aging stress induces CerS6/C14 ceramide, Drp1 activation, and mitophagy, which may be linked to SphK2/S1P signaling, and that genetic loss of SphK2 also protects against aging stress-related mitophagy and dysfunction in T cells.
Figure 4. CerS6/C14-ceramide and SphK2/S1P crosstalk induces aging-dependent mitophagy in activated T cells.
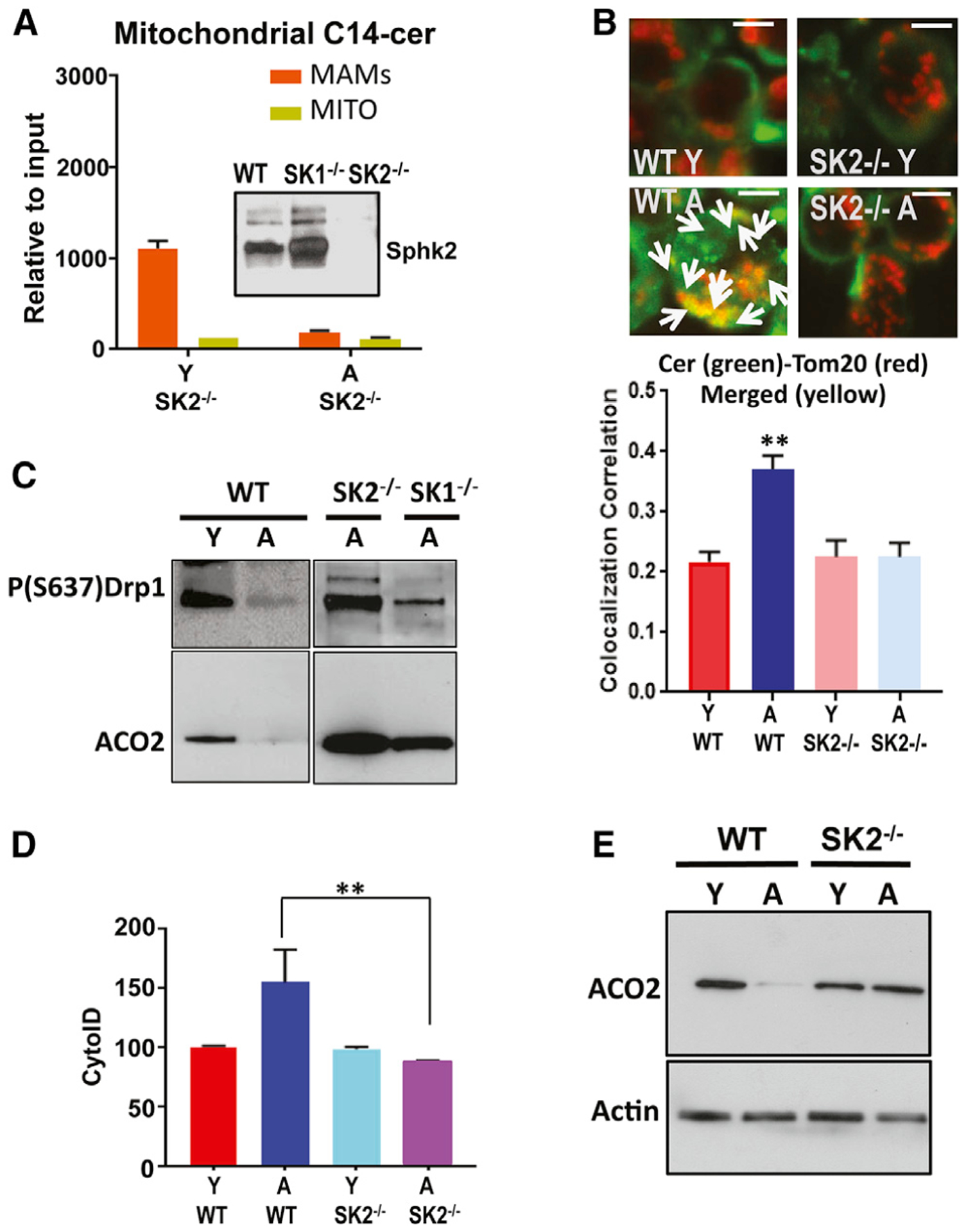
(A) Mitochondrial accumulation of C14 ceramide was measured using mass spectrometry/lipidomics in activated T cells obtained from Y and A SphK2−/− (SK2) mice in MAMs and MITO-enriched fractions.
(B) Colocalization of ceramide (green) and TOM20 (red) was measured to detect mitophagy in activated T cells isolated from Y and A WT and SphK2−/− (SK2) mice. Lower panel indicates the quantification of colocalization (Rc). Data are means ± SDs from 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test.
(C) P(S637)-Drp1 and ACO2 protein abundance was measured by western blotting in T cells isolated from Y and A WT or A SphK2−/− (SK2) and SphK1−/− (SK1) mice.
(D and E) TCR-activated T cells obtained from Y and A WT or SphK2−/− (SK2) mice were used for the detection of LC3/autophagy by cyto-ID (D) and ACO2 degradation by mitophagy using western blotting (E). Actin was used as a loading control (E). These studies represent 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test in (D).
Because SphK2/S1P signaling inhibits HDAC1/2 to regulate gene expression (Hait et al., 2009), we next assessed whether this process is involved in affecting CerS6 transcription in T cells isolated from A mice. While CerS6 mRNA was significantly increased in T cells isolated from A compared to Y WT mice, its expression was blunted in T cells isolated from A SphK2−/− mice (Figure 5A). CerS6 mRNA was not detectable by qPCR in T cells isolated from CerS6−/− mice used as a negative control (Figure 5A). Furthermore, the inhibition of HDAC1/2 using the pharmacologic inhibitor MS275 (Wooten-Blanks et al., 2007) resulted in the induction of CerS6 mRNA (Figure 5B) and mitophagy, increasing colocalization of MTR-LTG, and LC3 activation in T cells isolated from Y WT mice (Figures 5B–5D). Inhibition of HDAC1/2 using MS275 also attenuated, relative to vehicle-treated controls, the cytotoxic effects of T cells from Y Pmel WT mice against B16 melanoma cells (Figure 5E).
Figure 5. SphK2/S1P signaling via HDAC1/2 inhibition induces CerS6/ceramide-dependent mitophagy in aging T cells.
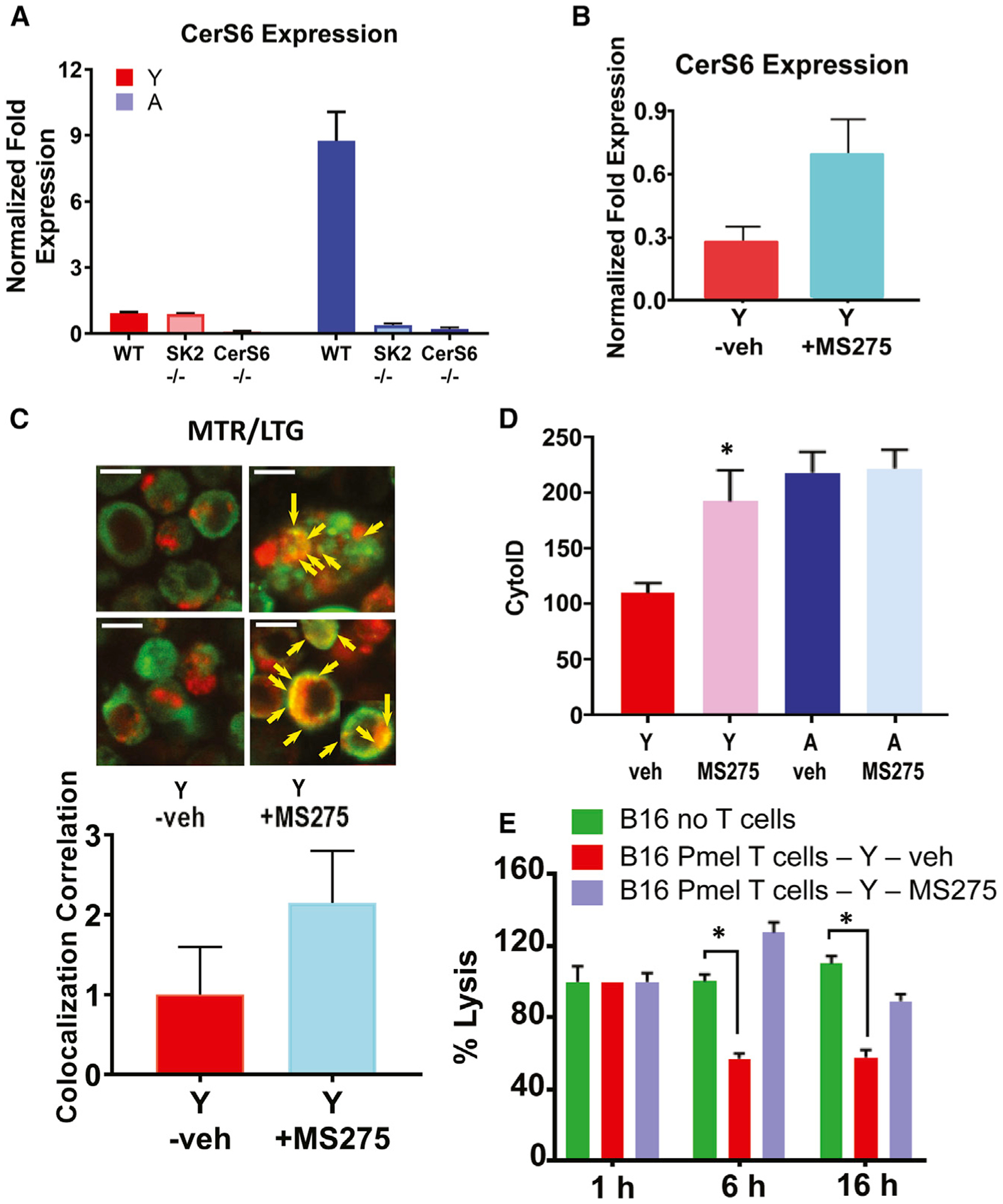
(A and B) Expression of CerS6 mRNA in the absence (A) or presence of HDAC inhibitor MS275 (B) was measured by qPCR in ex vivo TCR activated T cells isolated from Y and A WT, SphK2−/− (SK2), or CerS6−/− mice. Data are means ± SDs from 3 independent experiments (n = 3).
(C and D) Activated T cells obtained from Y and/or A mice were cultured with/without MS275, and mitophagy/autophagy was detected by live-cell imaging using MTR-LTG colocalization (arrows indicate colocalized signals), and scale bars indicate 1 μm. Images represent at least 3 independent experiments. Quantification of colocalization (Rc) (C, lower panel). LC3 activation as measured by Cyto-ID (D). Data are means ± SDs from 3 independent experiments (n = 3). *p < 0.05 as determined by Student’s t test.
(E) TCR-activated T cells in the presence of splenocytes isolated from Y Pmel mice were co-cultured with B16 melanoma cells for 1, 6, and 16 h in the absence/presence of MS275. Data are means ± SDs from at least 3 experiments (n = 3). *p < 0.05 as determined by Student’s t test.
Aging stress-mediated ceramide inhibits PKA, resulting in Drp1 activation and mitophagy in activated T cells. Ceramide-dependent mitophagy in T cells isolated from A mice appeared to be downstream of decreased Drp1 phosphorylation at S637, which leads to mitophagy by the activation of Drp1. Thus, we investigated the mechanism by which this process occurs. PKA is known to regulate inhibitory S637 phosphorylation of Drp1 (Dany et al., 2016). To determine whether PKA plays a role in ceramide/Drp1-induced mitophagy, we treated T cells isolated from Y WT mice with PKA inhibitor H89, whereas treated T cells were isolated from A WT mice with bcAMP (bromo-cyclic AMP), an activator of PKA (Dany et al., 2016). We measured their effects on the phosphorylation of Drp1 at S637 and mitophagy (Figures S4A–S4D). PKA inhibition using H89 activated Drp1 in young T cells (decreased Drp1 phosphorylation at S637) and induced mitophagy compared to vehicle-treated controls (Figures S4A–S4C), whereas PKA activation using bcAMP increased P-Drp1 at S637 and inhibited mitophagy in aging T cells (Figures S4A and S4D). Moreover, the inhibition of ceramide synthesis using FB1 largely protected mitophagy induction in response to PKA inhibitor H89 in T cells isolated from Y mice compared to vehicle-treated controls (Figure S4E). Thus, these data suggest that PKA regulation plays a critical dual role in controlling ceramide-mediated Drp1-mitophagy induction in TCR-activated T cells; inhibition of PKA induces Drp1 activation, whereas PKA activation inhibits aging/ceramide-dependent Drp1 and mitophagy in T cells. We then investigated whether the effect of ceramide on PKA is direct or indirect. Recombinant/purified PKA protein was preincubated with the ceramide analogs of different chain lengths LCL29 or LCL461 (Novgorodov et al., 2005; Szulc et al., 2006) and determined their effects on the catalytic PKA activity in vitro. These data showed that ceramide significantly inhibits PKA activity (Figure S5A), with Kd = 0.05 μM. The role of PKA in mitophagy induction in aging T cells was also consistent with decreased expression of the catalytic subunit of PKA (PKAc) in T cells isolated from A compared to Y mice (Figures S5B and S5C). Interestingly, we did not observe any detectable change in the expression of the regulatory subunit of PKA (PKAr) in young compared to aging T cells (Figure S5C). Also, the proximity ligation assay (PLA) showed that CerS6 and PKA colocalization was highly induced in ex vivo TCR-activated T cells isolated from A compared to Y mice (Figures S5D and S5E). PKA inhibitor H89 prevented the association between CerS6 and PKAc in aging T cells (Figures S5D and S5E). The decrease in the expression of the PKAc in A T cells was attenuated in the CerS6−/− T cell counterparts (Figures S5F and S5G). These data suggest that exogenous ceramide or CerS6-generated endogenous ceramide inhibits PKA activity in vitro and in TCR-activated T cells, reducing PKAc expression in aging T cells, which subsequently results in Drp1 activation and mitophagy, limiting aging T cell survival and function.
To further assess the role of ceramide and PKA in the regulation of Drp1, we measured the effects of LCL-29 (exogenous ceramide analog) on P-S637-Drp1 in the presence/absence of bcAMP. These studies were performed in TCR-activated Y T cells that were transfected with the WT versus mutant LC3 with F52A conversion, which inhibits ceramide-LC3 binding and mitophagy (Dany et al., 2016; Dany and Ogretmen, 2015; Oleinik et al., 2019; Sentelle et al., 2012; Thomas et al., 2017) (Figure S6A). Treatment of T cells with ceramide (20 μM for 6 h) activated Drp1 by the inhibition of Drp1 phosphorylation at S637 (lane 2), which was prevented by the expression of mutant LC3-F52A (lane 4) but not WT-LC3 (lane 3) compared to empty vector-transfected and vehicle-treated controls (lanes 1 and 2; Figure S6A). Moreover, the activation of PKA using bcAMP prevented ceramide-mediated Drp1 activation, increasing P-S637-Drp in cells transfected with WT-LC3 or LC3-F52A (lanes 5 and 6) compared to controls; Figure S6A). These data were also consistent with the effects of ceramide on mitophagy induction in vector- and WT-LC3-transfected young T cells, which is protected by the expression of mutant LC3-F52A or the activation of PKA by bcAMP (Figure S6B). Also, the treatment of Y T cells with LCL-29 highly induced LC3 activation, increasing lipidated LC3-II expression in vector- and WT-LC3-transfected cells (Figure S6C). However, the expression of mutant LC3 with the F52A conversion that inhibits ceramide-mediated mitophagy inhibited ceramide-induced LC3 activation compared to vehicle-treated control T cells (Figure S6C). The expression of LC3-I35A, which does not affect ceramide-LC3 binding, did not prevent ceramide-mediated LC3 activation in Y T cells compared to controls (Figure S6C). Moreover, pharmacologic inhibition of SphK2 using ABC294640 (Britten et al., 2017; Panneer Selvam et al., 2015) prevented CerS6 expression and mitophagy, decreasing Drp1 activation via inducing inhibitory P-Drp1-S637 and restoring ACO2 expression in A T cells (isolated from 18-month-old WT mice) compared to age-matched vehicle-treated controls (Figures S6D–S6G).
To investigate the role of PKAc in ceramide-mediated mitophagy in T cells, we have generated conditional knockout mice, in which the PKAc was deleted selectively in T cells by crossbreeding the floxed-PKAc floxed (Prkaa2fl/fl) with CD4Cre mice. We then measured the effects of the genetic loss of PKAc on the mitophagy of T cells isolated from Y mice. The data showed that the loss of PKAc in Prakaa2fl/fl/CD4Cre (Figures 6A–6C) both in heterozygous and homozygous mice (loss of PKAc expression in T cells obtained from homozygous Prakaa2fl/fl/CD4Cre knockout mice is confirmed by western blotting; Figure 6B) induced mitophagy in young T cells, inducing TMG-LysoTracker Red (LTR) colocalization and Drp1 activation compared to young WT T cells (Figures 6A and 6C). The inhibition of Drp1 using Mdivi prevented mitophagy and protected LC3 and Drp1 activation or inhibited ACO2 degradation in young T cells obtained from Prakaa2fl/fl/CD4Cre mice compared to young WT T cells (Figures 6D–6F). Overall, these data point to a critical role for PKA inhibition by ceramide-mediated induction of LC3/Drp1-dependent mitophagy in aging T cells.
Figure 6. Mitophagy is induced in activated T cells obtained from young T cell-specific PKAc−/− mice.
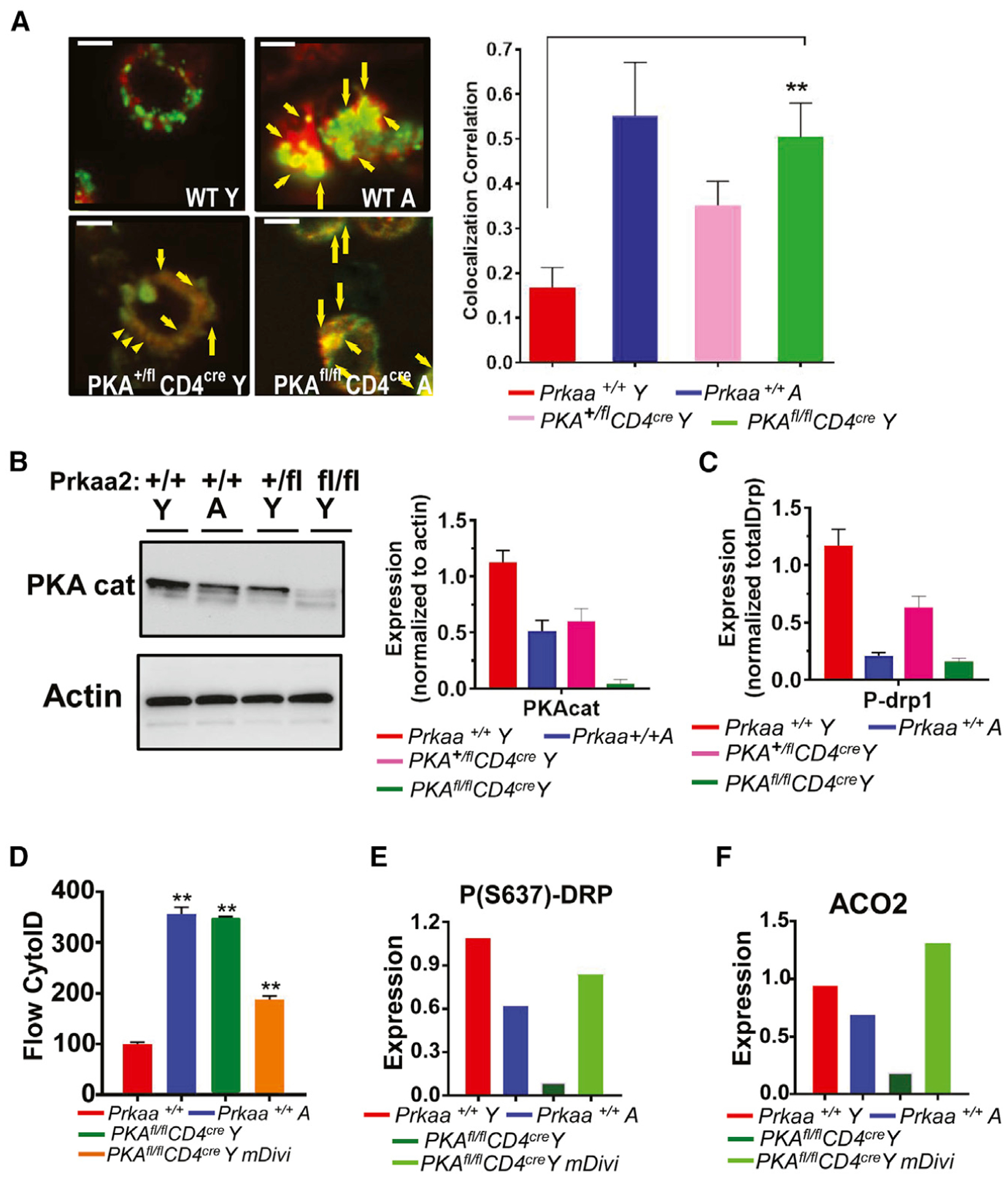
(A) T cells obtained from Y and A WT or 1 or 2 alleles targeted mutation of murine PKA (Prkaa2) PKA+/fl CD4cre and PKAfl/fl CD4cre mice were analyzed using live-cell microscopy for MTR-LTG colocalization. Arrows indicate merged (yellow); scale bars: 1 μm. Images represent at least 3 independent experiments. The right panel shows the quantification of colocalization extracted from the coefficient of colocalization (Rc). Data are means ± SDs from 3 independent experiments (n = 3). **p < 0.01 as determined by Student’s t test.
(B and C) Western blotting was performed to detect PKAc (cat) (B) or P-(S637)-Drp1 (C) in T cells obtained from Y and A WT or Y Prkaa2 knockout mice (PKA+/+ and PKA+/fl CD4cre and PKAfl/fl CD4cre). The quantification of PKAc abundance was shown in the right panel in (B). Data are means ± SDs from 3 independent experiments (n = 3).
(D–F) Mitophagy/autophagy was detected in T cells isolated from Y and A PKA+/+, and PKA+/fl CD4cre, and PKAfl/fl CD4cre mice in the absence/presence of mDivi for the measurement of cyto-ID (D) using flow cytometry, or P(S637)-Drp1 and ACO2 using western blotting. Data are means ± SDs from at least 3 experiments (n = 3). **p < 0.01 as determined by Student’s t test.
The loss of CerS6 and SphK2 improves the antitumor functions of aging T cells. To measure the effects of the CerS6-SphK2 axis on aging stress and mitophagy in T cells, we performed co-culture and adoptive T cell transfer studies in vitro and in vivo, respectively. In co-culture studies, T cells isolated from Y pMel mice were able to induce B16 cell death, whereas T cells obtained from A pMel mice were not effective in B16 cell killing (Figures S7A and S7B). However, T cells obtained from A CerS6−/−/pMel (CS6-pMel) or A SphK2−/−/pMel (pMel/SK2−/−) were as effective as their young counterparts in inducing B16 cell death in vitro (Figures S7A and S7B). Then, we isolated CD8+ T cells from Y and A WT/pMel or SphK2−/−/pMel mice, TCR activated them ex vivo, and injected them in C57BL/6 mice with subcutaneously established B16 melanoma. The effects of these activated T cells on controlling cancer growth/proliferation were measured compared to controls (mice with B16 melanoma that receive no adoptive transfer). The data revealed that T cells isolated from Y pMel mice suppressed tumor growth significantly better (p < 0.05) than T cells isolated from A mice with improved survival (Figures 7A and 7B). These data were also consistent with the protection of the effector memory T cells obtained from A versus Y CerS6−/− and SphK2−/− mice (Figure S1J). Moreover, T cells isolated from A SphK2−/−/pMel mice were equally effective as their young counterparts in suppressing tumor growth, resulting in longer survival than their aging pMel T cell counterparts (Figures 7A and 7B). These data were consistent with the reduced mitophagy of T cells isolated from A pMel/SphK2−/− mice with decreased MTR-LTG and CerS6-Tom20 colocalization (Figures 7C and 7D). After measuring the tumor size, we isolated tumor-infiltrating lymphocytes (TILs) and expanded them ex vivo to measure mitophagy. The data revealed that mitophagy was increased in TILs isolated from A pMel mice, but not in A pMel/SphK2−/− mice (Figure 7E). When we co-cultured these expanded TILs with B16 cells in vitro, we observed increased B16 cell death in response to incubation with TILs obtained from Y compared to A pMel mice (Figure 7F). However, TILs obtained from Y and A SphK2−/−/pMel mice were equally effective in inducing B16 cell death (Figure 7F). Interestingly, when we performed studies to measure the effects of naive CD4+ cells isolated from Y versus A WT and CerS6−/− mice on inducing colitis in Rag−/− recipient mice, there was no significant difference (Figures S7C and S7D). These results suggest that the genetic loss of SphK2 or CerS6 in CD8+ T cells obtained from A SphK2−/− or CerS6−/− mice prevents ceramide-dependent mitophagy and restores their antitumor functions, overcoming aging stress in T cells.
Figure 7. Genetic loss of SphK2 enhances the ability of aging T cells to kill melanoma cells in adoptive cell transfer therapy.
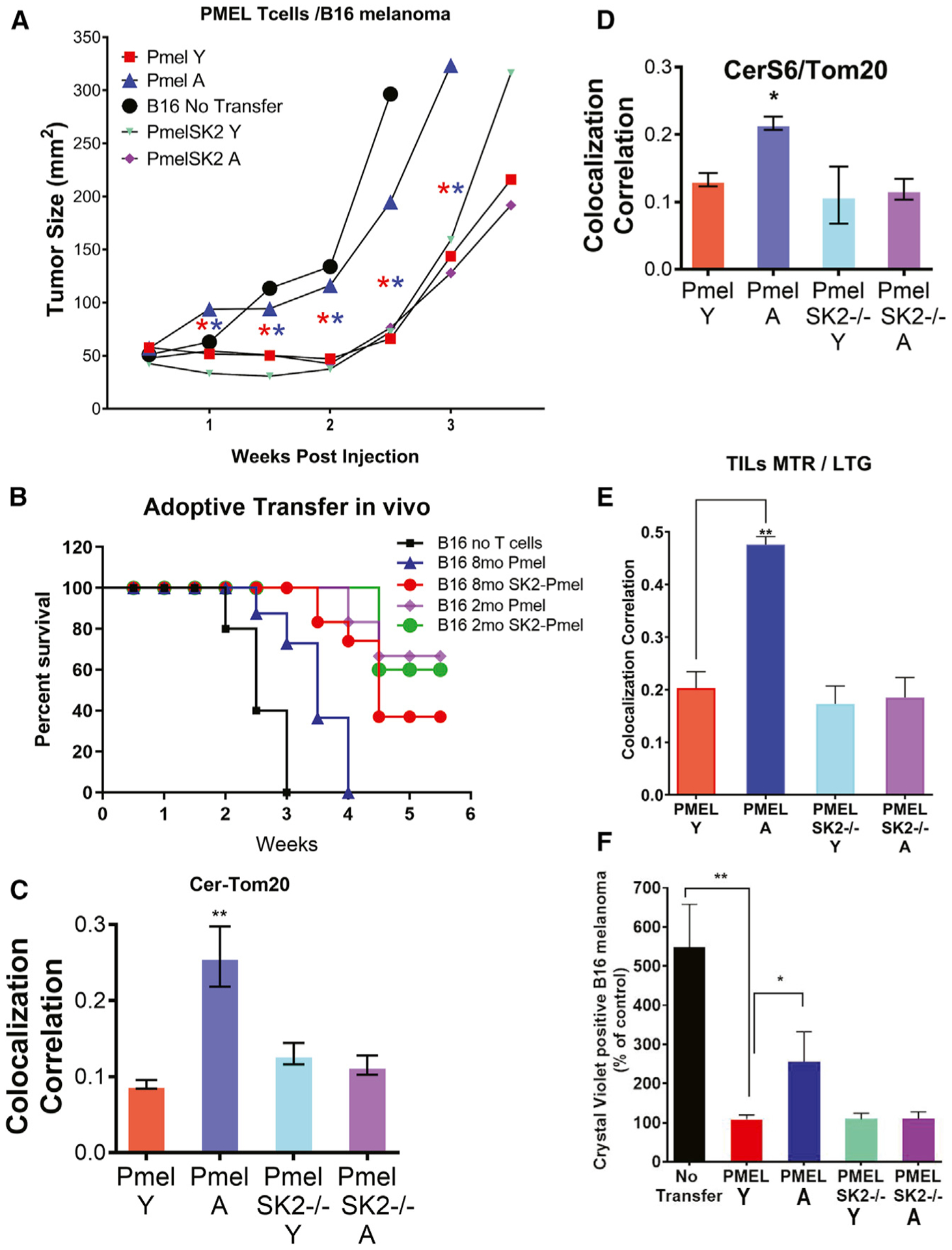
(A and B) Pmel and Pmel-Sphk2−/− splenic Y and A T cells, activated for 3 days with cognate antigen gp-100, were used in the adoptive T cell transfer experiments. C57BL/6 mice (n = 10/group) were inoculated (subcutaneously [s.c.]) with B16-F10 melanoma cells for 9 days before injecting cyclophosphamide (CTX). After 24 h of CTX injection, tumor-bearing C57BL/6 were either kept untreated (no transfer) or adoptively transferred with 1.3 × 106 gp-100-activated pMel T cells. Tumor growth was measured using digital calipers twice weekly. Tumor growth (A) and survival (B) were measured as described in Method details. Error bars represent means ± SDs. **p < 0.01 as determined by Student’s t test.
(C–E) Colocalization of ceramide (Cer) and TOM20 (C) or CerS6-TOM20 (D) in TILs obtained from Y and A pMel or pMel/SphK2−/− mice were performed by dual labeling using immunofluorescence or live-cell imaging for MTR-LTG colocalization (E). Data are means ± SDs from 3 independent experiments (n = 3). *p < 0.05, **p < 0.01 as determined by Student’s t test.
(F) TILs were isolated from tumors in gender- and age-matched tumor-bearing mice recipients for Y and A pMel or pMel/Sphk2−/− T cells. TILs were evaluated in a colony-formation assay using Crystal Violet staining. *p < 0.05 or **p < 0.01 as determined by Student’s t test.
DISCUSSION
This study’s main goal was to discover and describe the mechanisms that regulate T cell aging and antitumor functions of T cells by mitochondrial dysfunction due to alterations in lipid metabolism and signaling. Our studies demonstrated that aging-mediated ceramide stress in T cells, isolated from 8-month-old compared to younger (2-month-old) mice, induces mitochondrial dysfunction via PKA inhibition, leading to Drp1 activation and mitophagy, limiting their antitumor functions in culture and mice. These data also revealed that the inhibition of ceramide synthesis and the activation of PKA in T cells isolated from 8-month-old mice attenuated mitophagy and restored effector T cell function to inhibit tumor growth after adoptive CD8+ T cell transfer.
It is known that memory T cells are vulnerable to mitochondrial dysfunction as organisms age (Baazim et al., 2019; Bronietzki et al., 2015; Chougnet et al., 2015; O’Sullivan et al., 2014; Ron-Harel et al., 2015), but we lack a clear mechanistic understanding of T cell aging through mitochondrial dysfunction, which results in the suppression of memory phenotype and antitumor functions of effector T cells. Memory T cells use lipolysis, catalyzed in MITO, to generate energy for their survival and persistence (Klein Geltink et al., 2020; Pearce et al., 2009; Weinberg and Chandel, 2014). It has been well documented that bioactive sphingolipid ceramide, generated by CerS1–6), is induced in response to aging-associated stress signaling (Cutler and Mattson, 2001; Cutler et al., 2014; Jazwinski and Conzelmann, 2002; Szulc et al., 2006). Inhibition of ceramide generation was shown to extend lifespan (Riebeling et al., 2003; Venkataraman and Futerman, 2000). However, the roles and mechanisms whereby ceramide metabolism is involved in T cell aging regulation, leading to the suppression of T cell function and memory phenotype, have been mostly unknown. The data presented in this work revealed that aging-dependent ceramide synthesis inhibits anticancer functions of central memory T cells (TCM) by inducing mitophagy and that inhibition of ceramide-dependent mitophagy restores TCM function in aged T cells. Mechanistically, our data suggest that the aging-dependent induction of CerS6/C14 ceramide in the outer mitochondrial membrane inhibits PKA, which then results in the decreased inhibitory phosphorylation at S637 and subsequent activation of Drp1, leading to mitochondrial fission and mitophagy in activated T cells. Thus, the inhibition of CerS6 and Drp1, or activation of the PKAc subunit restores and improves the aging-related anticancer function of effector T cells. It is interesting that while our data showed the accumulation of C14/C16 ceramide in MITO leading to mitophagy, synthesized by CerS6 in older T cells, CerS6-generated C14/C16 ceramide has been implicated in inducing mitochondrial degradation and dysfunction in obesity-induced metabolic stress in adipose tissues (Hammerschmidt et al., 2019). CerS1–6 enzymes and their ceramide products with different fatty acyl chain lengths in the regulation of distinct biological processes have been studied previously. One common factor that regulates these distinct functions of ceramides appears to be the localization of CerS1–6 enzymes and their ceramide products. For example, in head and neck cancer cells, mitochondrial transport of CerS1 by p17/PERMIT was shown to induce C18 ceramide accumulation and mitophagy (Oleinik et al., 2019), which was also mediated by the C16 pyridinium-ceramide analog that accumulates selectively in MITO. Thus, these data suggest that regardless of the fatty acyl chain lengths of ceramides, their subcellular localization/transport (either ceramide or CerS1–6 enzymes) in response to different cellular stress stimuli plays essential roles to affect their downstream signaling in inducing mitophagy, which is also context dependent.
Once the immune memory repertoire is built, morbidity and mortality from infections decline. However, protective adeptness increasingly deteriorates with age (Giannakis et al., 2019; Minhas et al., 2019; Nicholls, 2004). The low proliferation rate of naive T cells in the periphery is the major adaptation to prevent immune suppression and preserve a diverse repertoire of T cells. In contrast to naive T cells, memory T cells exhibit a high degree of compartmentalization, as exemplified by resident memory T cells (Kishton et al., 2017; Le Page et al., 2018; Martinez-Jimenez et al., 2017; Naumov et al., 2011; Ouyang et al., 2011). To this end, aging memory T cells balance their maintenance and expansion while avoiding clonal dominance, because the disproportionate activation of memory cells contributes to the failure to generate immune memory in the aging host (Pulko et al., 2016; Schenten et al., 2014; Sckisel et al., 2015). Studies in human T cells have associated aging with changes in autophagy, membrane properties to form rafts, and producing reactive oxygen species. Several different models, such as yeast, worms, and flies, have shown that sphingolipids play critical roles in regulating aging and lifespan (Cutler and Mattson, 2001; Cutler et al., 2014). It is well documented that sphingolipids play vital roles in the regulation of immune and adaptive cell senescence. For example, most sphingolipids were found to be higher in T cells from A compared to Y mice, and the addition of exogenous C2 and C6 ceramide inhibited T cell proliferation (Kirk and Miller, 1999; Martinova, 1998; Sopel et al., 2019). These data are consistent with our data presented here, demonstrating that age-related ceramide increases may contribute to the proliferative and functional defects observed in aged T cells through the induction of Drp1-mediated mitochondrial fission and mitophagy. Interestingly, it is also shown that Drp1-induced mitochondrial dynamics play key roles in T cell migration, proliferation, and metabolic reprogramming (Simula et al., 2018) when aging stress and ceramide-dependent mitophagy are not invoked.
Our studies are consistent with previous studies that showed that dysfunctional MITO in T cells induce multimorbidity and senescence (Desdín-Micó et al., 2020). It is also known that aging-related dysfunctions of memory T cells are associated with increased senescence and defects in IL-7R or cytoskeleton, which affect compartmentalization and functions of memory T cells (Garcia and Miller, 2002; Kim et al., 2006; Mattoo et al., 2009; Ouyang et al., 2011; Pulko et al., 2016; Quinn et al., 2018).
These data have important biological implications in regulating T cell aging and adoptive T cell transfer for anticancer immunotherapy. For example, our data revealed that diminished antitumor functions of CD8+ T cells obtained from A mice could be restored by the inhibition of aging-mediated ceramide stress through pharmacologic inhibitors or genetic loss of CerS6, SphK2, Drp1, or PKAc. However, it was curious that the immunogenic functions of CD4+ naive cells in inducing colitis in Rag−/− recipient mice appeared to be independent of aging and ceramide synthesis. Similarly, in a previous study, it was shown that CerS6-generated C16 ceramide plays a critical role in inducing graft-versus-host disease, without affecting graft-versus-leukemia after allogeneic bone marrow transplantation (Sofi et al., 2017). These studies suggest that aging-induced ceramide stress and mitophagy in T cells may be selectively mediated upon ex vivo activation for adoptive T cell transfer, limiting antitumor functions of CD8+ T cells. In addition, our data are supported by a recent study, which demonstrated that dysfunctional MITO in autophagic vesicles were largely increased in CD4+ T cells obtained from older (≥70 years old) compared to younger (20–39 years old) healthy human participants with dysregulation of oxidative phosphorylation and energy metabolism (Bektas et al., 2019).
Although this study provides a mechanistic link between aging and alterations of antitumor functions of T cells via ceramide-dependent mitophagy, inducing dysfunctional mitochondrial metabolism in mice models, it is unknown whether a similar mechanism has any impact on aging human T cells and if there is a time point in the aging continuum when mitophagy prevention or reversal is limited. It remains unknown whether the inhibition of ceramide-dependent mitophagy could have adverse effects in aging T cells to increase autoimmunity and/or cytokine storm in response to infection in humans. A recent study (Bektas et al., 2019) demonstrated that dysfunctional MITO in autophagic vesicles, one of the indication markers of mitophagy, are highly accumulated in CD4+ T cells obtained from older compared to younger human donors. Whether this process is associated with mitochondrial ceramide signaling by the inhibition of PKA and activation of Drp1 in human T cells needs to be determined. It should be noted, however, that several studies have shown that mitophagy may be beneficial in aging tissues, and Drp1 overexpression prolongs the lifespan in model organisms (Rana et al., 2017; Yang et al., 2011). These studies support the idea that the Drp1 activity and mitophagy processes are highly complicated and context dependent.
Also, in our studies, unlike activated CD8+ T cells’ antitumor functions, naive CD4+ T cell transfer did not appear to be regulated by ceramide-mediated mitophagy and aging for inducing colitis in mice. Thus, another limitation of this study is to identify whether aging-mediated ceramide signaling regulates dysfunctional mitochondrial metabolism in specific populations of both mouse and human T cells. Moreover, it remains unknown how the metabolism of specific ceramides with different fatty acyl chain lengths, such as C18 or C14 ceramide, generated by distinct CerS enzymes CerS1 or CerS6, respectively, are regulated in mitochondrial membranes in response to aging stress in T cells versus other cell types, such as adipocytes, hepatocytes, keratinocytes, neurons, or cancer cells, which are vulnerable to aging-related disorders. To this end, if aging stress alters lipid oxidation, the cholesterol content of the mitochondrial membranes in T cells remains to be explored. Also remaining to be explored is whether the use of agents that have been shown to support steady-state mitochondrial function can help avoid this ceramide-associated mitophagy in aging T cells. One of these agents is α-lipoic acid, which plays an important role as an antioxidant as well as a cofactor for mitochondrial metabolism (Stoner et al., 2016). The therapeutic potential of such antioxidants has been examined in several diseases, and it would be interesting to explore their potential role in improving mitochondrial health in aging T cells.
Overall, this study provides a mechanistic connection between aging and the induction of ceramide-mediated mitophagy, resulting in mitochondrial dysfunction, which then limits ATP production and T cell proliferation and expansion (Giannakis et al., 2019; Kishton et al., 2017; Mendelsohn et al., 2018; Minhas et al., 2019; Nicholls, 2004). Thus, the dysregulation of effector T cell functions through the aging-ceramide-mitophagy axis may have important implications in aging-dependent impairments in the anticancer functions of T cells. This suppressive axis may also be involved in inducing aging-associated chronic inflammation or attenuation of the immune defense against bacterial or viral infections, including severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) (Braun et al., 2020; Li et al., 2020; Zheng et al., 2020), which was reported to be associated with defective T cell functions in some elderly patients with coronavirus disease 2019 (COVID-19).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Besim Ogretmen (ogretmen@musc.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The RNA sequencing data generated during this study are available at Gene Expression Omnibus (GEO) with the accession number GSE148498.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and tumor models
Vertebrate animals were maintained in pathogen-free facilities. Experimental procedures were performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina. C57BL/6, B6Rag−/−, Pmel, PKAfl/fl CD4cre mice were obtained from Jackson Laboratory (Bar Harbor, ME). Pmel-Sphk2−/−mice, and Pmel-CerS6−/− mice were developed in the lab. Pmel+ animals and Rag1−/− mice were employed for the melanoma and colitis studies, respectively. The animals were maintained in house for 2 months (young) or 8–18 months for the aging stress condition. Knockout mice were crossed with Pmel mice, and after generations F1 and F2 mice were maintained until 8–18 months. B16-F10 mouse melanoma tumor cells (0.3 × 106) were injected subcutaneously (s.c.) into the left flank of 8-week-old C57BL/6mice. After nine days of tumor establishment, recipient mice were injected (i.p) with cyclophosphamide (4 mg/mice) before adoptively transferring (i.v.) either Pmel+ (CD8+Vβ13+) or Pmel-SK2−/− or Pmel-CerS6−/− cells on 10. Recipient mice were given IL2 (50,000 U/mouse; i.p) for three consecutive days after ACT. Sex and aged-matched animals were used throughout these studies, and we did not observe any gender-specific outcomes.
Cell lines
B16-F10 (RRID: CVCL_0159) was obtained from American Type Culture Collection (ATCC), suggested to be of male origin
METHOD DETAILS
Cell lines and reagents
B16 mouse melanoma cancer cells were grown in RPMI containing 10% FBS (Cellgro) and 1% penicillin/streptomycin (Cellgro). Cells were treated with 10 uM of the following reagents for 3h: Fumonisin B1, LCL-29-ceramide, or 1μM of H89/PKAi, or bcAMP, or 5 μM ABC294640 and mDivi for 24h unless otherwise stated. YO-PRO-1 was loaded for 15 min (Oleinik et al., 2019).
T cell differentiation
Naive total T cells were purified from the total splenocytes of 8–72 week-old WT (C57BL/6) and KO (Sphk2−/−, CerS1–6, PKAfl/flCD4cre) mice, first by incubating the cells with biotinylated anti-CD19, anti-CD11b, anti-CD11c, anti-NK1.1, anti-CD25 (cell signaling technology), followed by negative selection with streptavidin magnetic particles (BD Biosciences). Total splenocytes from 6–72 week-old TCR transgenic mouse Pmel (bears class-I restricted CD8+ T cells) or Pmel knockouts: Pmel-Sphk2−/− or Pmel-CerS6−/− were also used. Within experiments, mice were age and sex-matched. In some experiments, purified naive T cells or total splenocytes were differentiated to Th17 (3 ng/ml TGFβ, 25 ng/ml IL6, 10 μg/ml anti-IL4 and 10 μg/ml anti-IFNγ) or Th0 (100 IU/ml IL2) in the presence of plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml). For Pmel TCR transgenic mice, splenocytes were stimulated with 1 μg/ml gp100 melanoma antigen in the presence of 100 IU/ml IL2. T cells were differentiated for three days in IMDM media supplemented with 10% FCS, 4mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 55 μM beta-mercaptoethanol under 5% CO2, atmospheric oxygen at 37°C in a humidified incubator. To evaluate intracellular cytokines by flow cytometry, we re-stimulated T cells with PMA/ionomycin for 4 h or with plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) in the presence of Golgi inhibitors.
Cell viability, permeability assays, and co-culture studies
Cell viability was assessed by CyQuant following manufacturer’s instructions and by trypan blue exclusion assay. According to the manufacturer’s specifications, ATP was measured with a commercially available kit (FLAA, Sigma) using bioluminescence at 560 nm. Cell permeability and co-culture experiments were performed as previously described (Chatterjee et al., 2014; Nganga et al., 2019).
Adoptive T cell protocol
B16-F10 mouse melanoma tumor cells (0.3 × 106) were injected subcutaneously (s.c.) into the left flank of 8–10 weeks old C57BL/6 mice. After nine days of tumor establishment, recipient mice were injected (i.p) with cyclophosphamide (4 mg/mice) before adoptively transferring (i.v) Pmel (CD8+Vβ13+) cells on day 10. Recipient mice were given IL2 (50,000 U/mouse; i.p) for three consecutive days after ACT.
Colitis model
Naive CD4+ T cells (CD4+CD25−CD44−CD62L+) were purified separately by negative selection. The cocktail of antibodies used to purify CD4 T cells (eBioscience) included anti-mouse CD25, anti-mouse CD49b), anti-mouse TER-119, anti-human/mouse CD45R, anti-mouse CD11b, anti-human/mouse, anti-mouse and anti-mouse CD8a anti-biotin microbeads were from Miltenyi Biotec. Naive T cells (1×106 CD4+) from WT or CerS6−/− mice were intravenously injected into lymphopenic Rag1−/− hosts to induce colitis. The loss of body weight assessed the disease progression, and the scoring analysis in the control mice injected with PBS only. Immune cells were isolated from spleens and lymph nodes. Recipient mice were monitored for weight loss and other clinical signs of colitis once per week. Clinical scores were tabulated as 4 parameters: weight loss, general health (posture, fur texture, and skin integrity), stool quality, and rectal prolapse. Individual mice were scored from 0 to 4 for stool quality and rectal prolapse and 0 to 3 for general health. Recipients at the moribund stage were euthanized and counted for lethality. Representative samples of the colon were excised from recipients 60 days after T cell transfer and subjected to pathology scoring which was tabulated as 7 parameters: a degree of inflammation, goblet cell loss, presence of abnormal crypts, presence of crypt abscesses, presence of mucosal erosion and ulceration, submucosal spread to transmural involvement, and meatuses counted at 40X magnification.
Flow cytometry
Staining for cell surface markers was performed by incubating cells with the antibody at 1:200 dilutions in FACS buffer (0.1% BSA in PBS) for 30 min at 4°C in the dark. For intracellular cytokines (IFNγ, IL4, and IL6) staining, surface markers were stained before fixation/permeabilization (BD Cytofix/Cytoperm Kit, BD Biosciences, San Jose, CA). Samples were acquired on LSR Fortessa/X-20 and analyzed with FlowJo software (Tree Star, OR).
Coating of coverslips for T cells imaging
A protocol was developed to visualize live and fixed T cells on confocal microscopes as an adaptation of steps applied to use suspension cells in instrumentation for measuring cell bioenergetics. Poly-L-lysine coated 35-mm glass-bottomed dishes (MatTek Corporation) were treated with Cell Tak compound at RT for 1h, followed by 5 washes with ddiH20 without allowing coverslips to dry until cells were loaded. The humidifier chamber was employed for image acquisition of these cells.
Detection of mitophagy by live-cell imaging
On the day of the experiment, 50,000 cells were loaded with MTR (0.5 μM) for 60 min in a humidified atmosphere at 370C in standard growth media. Afterward, the cells were washed and incubated with LTG (0.5 μM) for 20 min. After MTR and LTG had been loaded, one-third of the initial LTG concentration was kept in the media for the duration of the experiment. Cells were loaded on glass coverslips coated as described above after removing washing ddiH20, and live-cell images were acquired using a humidifier chamber on the Olympus FluoView FV10i LIV laser scanning confocal microscope, MUSC Cell, and Molecular Imaging Core Facility. Images were analyzed by Flouview Fv10i software. Quantification was performed using Fiji (ImageJ).
Detection of LC3 activation by Cyto-ID
LC3 activation was assessed with the Cyto-ID Autophagy Detection kit (Enzo) in live cells using flow cytometry according to the manufacturer’s instructions.
Immunofluorescence
Activated cells were incubated with the respective treatment, washed two times with 1XPBS Cells (50,000 per well) were fixed and permeabilized using 4% paraformaldehyde (20 min) and 0.1% Saponin in 1 × PBS (pH 7.4) for 10 min. The cells were then blocked with 1% BSA /PBS (pH 7.4) for 1 h. Cells were incubated for 18 h at 4°C with antibodies specific for Ceramide, LC3, Tom 20, FLAG, CerS1–6, (1:50) in blocking solution, followed by Alexa Fluor 488-, 594, or −633-conjugated secondary antibodies (1:1000) an additional hour. Cells were loaded on coverslips coated as described above, immediately after removing washing ddiH20 and cell images were acquired using a humidifier chamber to avoid dehydration. Immunofluorescence was performed using an Olympus FV10i microscope with the corresponding channels for visualizing fluorescence, or Zeiss LSM 880 NLO Quasar confocal/multiphoton microscope with a Fast Airyscan super-resolution detector. Images were taken at 63X magnification for at least three random fields were selected for quantification. Images were analyzed by Flouview Fv10i software. Quantification was performed using Fiji (ImageJ).
Ultra-structural analysis using transmission electron microscopy
After removal of culture medium, T cells were and rinsed with 1X PBS and fixed in freshly prepared 2% (w/v) glutaraldehyde in 0.1 M cacodylate buffer. After post-fixation in 2% (v/v) osmium tetroxide, specimens were embedded in Epon 812, and sections were cut orthogonal to the cell monolayer with a diamond knife. Thin sections were visualized in a JEOL 1010 transmission electron microscope (TEM). In immunoelectron microscopy studies, activated T cells were collected and rinsed with 1X PBS. They were fixed for 15 min with 4% paraformaldehyde, and cell permeabilization was achieved with 0.1% Saponin for 10 min, washed twice, and blocked with 1% BSA in PBS for 20 min. Primary antibodies (1:50 dilution) were added to cells and incubated overnight at 40C. After washing with 1% BSA PBS, Nanogold (1.4 nm) (Nanoprobes) conjugated donkey anti-mouse, or anti-rat Fab fragments (1:200) were incubated with cells for 1 h. After post-fixation with 1% glutaraldehyde in PBS for 10 min at RT, Li Silver enhancement was performed for 5 min. After rinsing with distilled H2O, enhanced with silver staining before specimens were embedded in Epon 812, and sections were cut orthogonal to the cell monolayer with a diamond knife. Thin sections were visualized in a JEOL 1010 TEM.
Ultra-structural analysis using scanning electron microscopy
SEM was performed as we recently described (Nganga et al., 2019) except that no trypsinization was performed, and the cells were collected by scraping on ice. Cell-cell contacts were preserved by scraping cells for collection. In short, samples were fixed with primary fixative (2% glutaraldehyde, 2% paraformaldehyde in 0.1 M cacodylate buffer pH 7.35). Secondary fixation was achieved with 1% osmium tetroxide in 0.1 M cacodylate buffer. After both fixation steps, samples were washed three times with ddH20 then dehydrated with increasing ethanol concentrations (20% to 100%). Samples were submerged in 100% ethanol, and critical point drying was achieved with Tousimis Autosamdri-931 (Rockville, MD). Before imaging, 3–6 nm iridium was applied to samples and images were acquired with a Magellan 400 field emission scanning electron microscope (FEI) (Hillsboro, OR) at Purdue Imaging Facility.
Sub-cellular fractionation and isolation of crude mitochondria
For mitochondria preparations, cells were resuspended in Buffer A and incubated for 10 min on ice, followed by Dounce homogenization. After centrifugation, the supernatant was saved and the pellet resuspended in Buffer B to the same volume as Buffer A, following the same rupturing steps. Combined supernatants were centrifuged at 12,000 g for 15 min at 4oC to obtain the mitochondrial fraction. For the membrane fraction, after obtaining the mitochondrial pellet, the supernatant was collected and centrifuged at 100,000 × g for 1 h in an ultracentrifuge. The pellet was resuspended in wash buffer and recentrifuged for 45 min. The membrane pellet was then resuspended in the lysis buffer (250 mM Sucrose, 20 mM HEPES (7.4), 10 mM KCl, 1 mM EDTA, 1mM EGTA, 10% glycerol, 0.1% SDS, 1 mM DTT and PI Cocktail). Western blotting was used to detect the purity of the fractions with Tom 20 as a mitochondrial marker, and with β-tubulin actin as a cytosolic marker.
Isolation of purified mitochondrial fractions and MAMs
In brief, T cells were activated, washed, and homogenizing in IB buffer (225 mM mannitol, 75-mM sucrose, 0.1 mM EGTA, and 30 mM Tris-HCl pH 7.4), the homogenate was centrifuged at 600 g for 5 min. After discarding debris and nuclei, the supernatant was centrifuged twice at 7,000 × g for 10 min, and once at 10,000 g for 10 min. The mitochondrial pellet was resuspended in ice-cold mitochondria resuspending buffer (MRB) (250 mM mannitol, 5 mM HEPES (pH 7.4) and 0.5 mM EGTA) and subjected to Percoll gradient (225 mM mannitol, 25 mM HEPES (pH 7.4), 1 mM EGTA, 30% v/v Percoll). After 1 h centrifugation at 95,000 g, two bands were visualized in the tube containing the Percoll gradient: a thick band containing purified mitochondria located at the bottom, and a MAM’s band located in the upper middle. The bands were individually collected and washed steps, and after centrifugation at 100, 000 × g, mitochondria (MITO) and MAMs were isolated, resuspended in small volumes of MRB and suspended using the loose glass pestle (Oleinik et al., 2019) and analyzed by Lipidomics or immunoblotting with corresponding antibodies.
Immunoblotting
Activated T cells were homogenized in RIPA buffer (25mM Tris-HCl pH7.4, 150mM NaCl, 1% Triton X-100,1% sodium deoxycholate, EDTA (20ul 0f 0.5M stock) including protease inhibitor and phosphatase inhibitor cocktails using a gauge (26.5%) syringe five times, incubated for 20 minutes on ice. The samples were centrifuged at 12000 g for 15 minutes at 4°C. The supernatants were collected, and proteins were quantified with the Bradford method. 4X loading dye was added to sample and boiled using a heating block at 95 degrees for 5 minutes before loading SDS gradient gels 4%–20% were used to run the samples using the Bio-Rad Criterion apparatus, followed by semi-dry transfer onto PVDF membrane. Blocking was done with 5% milk in 0.1%PBST. Primary antibodies were used at 1:2000 dilution overnight at 4°C. Proteins were analyzed by western blotting using β-actin (Sigma, St. Louis, MO) or other antibodies. Original blot images are provided in Data S1.
Quantitative real-time PCR
Total RNA was extracted from cell pellets of the indicated T cell subsets (2 × 106 cells) using Trizol reagent (Life Technologies, Grand Island, NY). cDNA was generated from 1 μg total RNA using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Reactions were carried out using SsoFast probes mix (Bio-Rad) and TaqMan primer probes (ThermoFisher Scientific) in the CFX96 Detection System (BioRad, Hercules, CA). The expression of different genes was quantified relative to RPLPO.
Plasmids and transfections
WT and mutant LC3B-FLAG expression vectors with F52A and G120A conversions were as we previously described (Sentelle et al., 2012). Naive T cells were isolated either from spleens, then transfected with the plasmids above using AmaxaTM mouse T cell nucleofectorTM kit from Lonza following manufacturers’ protocol. Transfected cells were cultured in Th0 differentiation media described in the corresponding section for 48 hours.
Lipidomics analyses of sphingolipids
The Analytical Unit performed lipid extraction and analysis in the Lipidomics Shared Resource at MUSC. Briefly, cells were lysed with RIPA buffer, and analyses of endogenous bioactive sphingolipids were performed on a ThermoFisher TSQ Quantum liquid chromatography/triple-stage quadrupole mass spectrometer system operating in a multiple reaction monitoring (MRM) positive ionization mode, as previously described (Dany et al., 2016; Dany and Ogretmen, 2015; Oleinik et al., 2019; Sentelle et al., 2012; Thomas et al., 2017). Lipid levels were normalized by the level of protein present in samples (pmol/mg protein).
Isolation of tumor-infiltrating T cells
To obtain tumor-infiltrating T cells (TILs) from subcutaneously established stable B16-F10 melanoma-bearing mice, tumors were excised, chopped finely using tweezers, and scissors, and then digested with 2 mg/ml collagenase type IV (Stemcell technologies, Vancouver, BC) for 45 min. Following digestion, tumors were filtered through 70μM cell strainers (BD Biosciences, San Jose, CA). The cell suspension was washed in culture medium twice by centrifugation at 1500 rpm for 10 min at 4°C. After the second wash, the cells were resuspended in 6 mL PBS and layered carefully over 3 mL Ficoll-paque (GE Healthcare) followed by centrifugation at 1500 rpm for 30 min. The enriched TILs obtained at the interface as a thin buffy layer were washed with PBS twice and finally resuspended in FACS staining buffer for further staining procedures
Multiplex analyses of cytokines
In this study, we used Luminex xMAP technology for multiplexed quantification of 45 Mouse cytokines, chemokines, and growth factors. The multiplexing analysis was performed using the Luminex 100 system (Luminex, Austin, TX, USA) by Eve Technologies Corp. (Calgary, Alberta). Forty-four markers were simultaneously measured in the supernatants from gp100 re-stimulated TILs using a MILLIPLEX Mouse Cytokine/Chemokine 32-plex kit and a MILLIPLEX Mouse Cytokine/Chemokine 13-plex kit (Millipore, St. Charles, MO, USA) according to the manufacturer’s protocol. The 45-plex consisted of Eotaxin, Erythropoietin, 6Ckine, Fractalkine, G-CSF, GM-CSF, IFNB1, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-11, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-16, IL-17, IL-20, IP-10, KC, LIF, LIX, MCP-1, MCP-5, M-CSF, MDC, MIG, MIP-1α, MIP-1β, MIP-2, MIP-3α, MIP-3B, RANTES, TARC, TIMP-1, TNFα, and VEGF. The assay sensitivities of these markers range from 0.3 to30.6 pg/mL for the 45-plex. Individual analyte values are available in the MILLIPLEX protocol.
Measurement of cellular and mitochondrial bioenergetics
Mitochondrial bioenergetics were measured by real time flux analyses using the Seahorse platform as we previously described (Chakraborty et al., 2019; Sentelle et al., 2012). In brief, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined using the Seahorse XF96 analyzer (Agilent Technologies, Santa Clara, CA). Briefly, T cells (0.5×106/well) were plated on a Cell-Tak coated Seahorse culture plate for 30 min. OCR, a measure of OXPHOS, was analyzed under basal condition, followed by 1.0 μM oligomycin, 1.0 μM fluoro-carbonyl cyanide phenylhydrazone (FCCP) and 2 μM rotenone, plus 100 nM antimycin A. ECAR, a measure of glycolysis, was measured under basal conditions and in response to glucose (5.5 mM), Oligomycin (1.0 μM), and 2-deoxyglucose (2-DG) (100 mM) (Sentelle et al., 2012). All reagents were purchased from Sigma-Aldrich, St. Louis, MO.
Proximity ligation assay
Proximity ligation assays were performed using Duolink in situ red kit (Sigma) per manufacturer’s instructions. Then, it was analyzed as described previously (Nganga et al., 2019).
Membrane permeability/dead cell apoptosis kit with YO-PRO-1 and PI
B16 melanoma cells were cultured to 60%–70% confluency and then activated T cells were 10-fold for co-cultures at 37 C.
Trypan blue exclusion assay
Cancer cells were seeded in 12-well plates and allowed to adhere for 20 hours before activated T cells or media were added. Medium containing dead cells was pelleted with trypsinized cells, then resuspended in 1X PBS, and counted in a hemocytometer using the trypan blue dye (Sigma-Aldrich) at a 1:2 dilution.
Lipidomics analysis
Cells were collected by scraping off from the dishes and washed twice with cold PBS. Further preparation of samples and measurement of endogenous ceramides by LC-MS/MS followed the protocol described previously (Oleinik et al., 2019). Briefly, samples were supplemented with internal standards, and 2 mL of isopropyl alcohol: water: ethyl acetate (30:10:60; v:v:v) was added to the extracts. Samples were subjected to two rounds of vortex and sonication followed by 10-min centrifugation at 4,000 rpm. The supernatant or top layer was used as lipid extract and subjected to LC-MS/MS for analysis of ceramide species. The MUSC Lipidomics Shared Resources performed lipid extraction and analyses. Inorganic phosphates (Pi) were used for normalization.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were reported as mean ± standard deviation. Statistical analysis was performed by ANOVA or Student’s t test using Prism/GraphPad software version 7; p < 0.05 were considered statistically significant, as described previously (Nganga et al., 2019). Descriptive statistics were used to summarize continuous variables in which the group comparisons were explored using a Mann-Whitney test. Kaplan-Meier analysis was used to estimate the overall survival probabilities. The survival curves comparisons between groups were explored using log-rank tests. Besides, the segmented mixed model was used to identify continuous measurement trends over time and the potential change point of accelerated growth for each subject.
All data reported are arithmetic means from three or four independent experiments performed with technical replicates ± standard deviation (SD) unless stated otherwise. The unpaired Student’s t test was used to evaluate the significance of the differences between individual groups, accepting p < 0.05 as a threshold of significance. Data analyses were performed using the Prism software (GraphPad, San Diego, CA). In animal studies, analyses of time-to-sacrifice and tumor size (mm2) were performed using R statistical software version 3.2.3 and SAS version 9.4. Time-to-sacrifice was censored for animals, not meeting conditions for sacrifice at the end of the experiment. Graphical displays of survival probabilities were constructed using Kaplan-Meier plots. Log-rank tests were used to compare survival probabilities between experimental groups. Tumor growth over time was modeled using linear mixed-effects regression. We used a log base 2 transformations of tumor size to achieve approximate normality. Models included fixed effects for experimental condition and time (as a continuous variable) and their two-way interaction, and mouse-specific random effects to accommodate the correlation between measures obtained from the same animal over time. In cases where data were pooled across multiple experiments, models were appropriately adjusted for experiment effects. All models further adjusted for baseline tumor size to accommodate differences in tumor size at treatment initiation. Power was calculated to detect a survival probability of 50%, 60%, or 70% versus a null survival probability of 5% at a given time point based on a log-rank test with two-sided α = 0.05. The differences between the null and alternative survival probabilities are substantial, but we expect differences that translate clinically to be significant in the in vivo models. For group sample sizes of 10, only the most substantial differences are detectable with approximately 80% power. However, with a group sample size of 14 animals, all differences considered are detectable with 80% power. The majority of our experiments were performed using between 10 and 14 animals per group. We, therefore, consider our experiments to be appropriately powered to detect clinically meaningful differences. Finally, we note that in cases where data from multiple experiments were pooled to achieve the appropriate group sample size, analyses were appropriately modified to adjust for any batch effect attributed to the experiment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3 | BioXCell | Clone: 145-2C11; Cat# BE0001-1; RRID: AB_1107634 |
| Anti-mouse CD28 | BioXCell | Clone: 37.51; Cat# BE0015-1; RRID: AB_1107624 |
| Anti-mouse IL4 | BioXCell | Clone: 11B11; Cat# BE0045; RRID: AB_1107707 |
| Anti-mouse IFNγ | BioXCell | Clone: XMG1.2; Cat# BE0055; RRID: AB_1107694 |
| Anti-mouse CD25-biotin | eBioscience | Cat# 13-0251-85; RRID: AB_466401 |
| Anti-mouse C49b-biotin | eBioscience | Cat# 13-5971-85; RRID:AB_466826 |
| Anti-mouse TER-119-biotin | eBioscience | Cat# 13-5921-85; RRID:AB_466798 |
| Anti-human/mouse CD45R-biotin | eBioscience | Cat# 13-0452-85; RRID:AB_466450 |
| Anti-mouse CD11b-biotin | eBioscience | Cat# 13-0112-85; RRID:AB_466360 |
| Anti-human/mouse CD44-biotin | eBioscience | Cat# 13-0441-85; AB_466443 |
| Anti-mouse CD8a-biotin | eBioscience | Cat# 13-0081-86; AB_466348 |
| Anti-biotin microbeads | Miltenyi Biotec | Cat# 130-090-485; AB_244365 |
| CD4-PE | eBioscience | Clone: GK5.1; Cat# 12-0041-83; RRID: AB_465506 |
| CD4-PE/Cy7 | Biolegend | Clone: GK5.1; Cat# 100422; RRID: AB_312707 |
| CD4-APC | Biolegend | Clone: GK5.1; Cat# 100412; RRID: AB_312697 |
| CD4-APC/Cy7 | Biolegend | Clone: GK5.1; Cat# 100414; RRID: AB_312699 |
| CD8-PE/Cy7 | Biolegend | Clone: 53-6.7; Cat# 100722; RRID: AB_312761 |
| CD8-APC | Biolegend | Clone: 53-6.7; Cat# 100712; RRID: AB_312751 |
| CD8-FITC | Biolegend | Clone: 53-6.7; Cat# 100706; RRID: AB_312745 |
| IL17-PE | Biolegend | Clone: TC11-18H10.1; Cat# 506904; RRID: AB_315464 |
| IL17-Pacific Blue | Biolegend | Clone: TC11-18H10.1; Cat# 506918; RRID: AB_893544 |
| IFNγ-PE | Biolegend | Clone: XMG1.2; Cat# 505808; RRID: AB_315402 |
| IFNγ-Alexa647 | Biolegend | Clone: XMG1.2; Cat# 505814; RRID: AB_493314 |
| CD25-APC | Biolegend | Cat# 102012; RRID:AB_312861 |
| IL4-PE | Biolegend | Clone 11B11 Cat# 504103 RRID AB_315317 |
| CD62L-APC | Biolegend | Clone: MEL14; Cat# 104412; RRID AB_313099 |
| CD44-PerCP/cye5.5 | Biolegend | Clone: IM7; Cat# 103032; RRID AB_2076204 |
| Vβ13-FITC | BD Biosciences | Clone: MR1 2–3; Cat# 553204; RRID: AB_394706 |
| Anti- Lass6 (CerS6) | Santa Cruz Biotechnology | sc-65127; RRID:AB_2133113 |
| Anti- Lass1 (CerS1) | Santa Cruz Biotechnology | sc-65096; RRID:AB_2132952 |
| Anti- Lass5 (CerS5) | Santa Cruz Biotechnology | sc-135038; RRID:AB_10609786 |
| Anti-Aco2 | Cell Signaling | Cat# 6922S, RRID:AB_10828218 |
| Anti-LC3B | Cell Signaling | Cat# 2775, RRID:AB_915950 |
| Anti-Tom20 | Santa Cruz Biotechnology | Cat# sc-17764, RRID:AB_628381 |
| Anti-Ceramide | Enzo | Cat# MD15B4 ALX-804–196-T050; RRID:AB_10541503 |
| Anti-p62/SQSTM1 | Cell Signaling | Cat#5114 |
| Anti-actin | Sigma-Aldrich | Cat# A2066, RRID:AB_476693 |
| Anti-FLAG | Sigma-Aldrich | Clone: 2EL-1B11 Cat# MAB3118; RRID:AB_94705 |
| Anti-Sphk1 (SK1) | Abcam | Cat# ab71700; RRID:AB_1270891 |
| Anti-Sphk2 (SK2) | Protein Tech | 17096-1-AP GenBank access. BC010671; RRID:AB_10598479 |
| Anti-Goat Alexa647 | Thermo Fisher Scientific | Clone: N/A; Cat# A21447; RRID: AB_141844 |
| Anti-DLP1 (Dynamin-like protein/Drp1) | BD Transduction | Cat# 611112; RRID:AB_398423 |
| AntI p(S637)-Drp1 | Cell Signaling Technology | Clone: N/A; Cat# 4867S; RRID:AB_10622027 |
| Anti p(S616)-Drp1 | Cell Signaling Technology | Clone:N/A; Cat#3455S; RRID:AB_2085352 |
| Anti PKA catalytic subunit Iα/β | Santa Cruz Biotechnology | Cat# sc-28315; RRID:AB_628136 |
| Anti PKA regulatory subunit Iα/β reg (B-6) | Santa Cruz Biotechnology | sc-271125; RRID:AB_10611494 |
| Anti PKA regulatory subunit IIα reg (40) | Santa Cruz Biotechnology | sc-136262; RRID:AB_2168239 |
| Anti PKA regulatory subunit IIβ reg (C-2) | Santa Cruz Biotechnology | sc-376778 |
| Anti-AKAP 149 (B-10) | Santa Cruz Biotechnology | sc-377450 |
| Anti-PINK1 (17HCLC), | ThermoFisher | Cat# 710993; RRID:AB_2633104 |
| Anti-PARKIN | ThermoFisher | Cat# 13399; RRID:AB_2159914 |
| Ac-Histone H4 (E-5) | Santa Cruz Biotechnology | sc-377520 |
| Anti-Rabbit HRP | Cell Signaling Technology | Clone: N/A; Cat# 7074S; RRID:AB_2099233 |
| Anti-Rabbit PE | Jackson ImmunoResearch Laboratories | Clone: N/A; Cat# 111-116-144; RRID: AB_2337985 |
| Anti-Rabbit Alexa647 | Jackson ImmunoResearch Laboratories | Clone: N/A; Cat# 111-607-003; RRID: AB_2338084 |
| Anti FACL-4 | Santa Cruz Biotechnology | Cat# sc365230; RRID:AB_10843105 |
| Anti VDAC | Santa Cruz Biotechnology | Cat# sc-8830 |
| Anti-cytochrome C | BD Transduction Laboratories | Cat# 556433; RRID:AB_396417 |
| Anti β-Tubulin | Cell Signaling Technology | Clone: N/A; Cat#2146S; RRID:AB_2210545 |
| Anti-IP3R3 | BD Transduction Laboratories | Cat# 610312; RRID:AB_397704 |
| inVivoMAb anti-mouse PD-1 | BioxCell | Cat# BE0146 Clone RMPI 14 RRID: RRID:AB_10949053 |
| Chemicals, peptides, and recombinant proteins | ||
| H89 | Abcam | Cat#ab120341 |
| H89 | InvivoGen | Cat# tlrl-h89 |
| PKA inhibitor peptide | Sigma-Aldrich | Cat# 12–151 |
| bcAMP | Sigma-Aldrich | Cat# B5386 |
| ABC294640 | RedHill Biopharm | Cat# ABC924640 |
| cAMP Elisa Kit | Cayman Chemical | Cat# 581001 |
| mDivi | Sigma-Aldrich | Cat#M0199 |
| Anti-rabbit Immuno-gold labeled, 1.4 nm | Nanoprobes, Inc. | #2004, #2006 |
| Anti-mouse Immuno-gold labeled 10 nm | Nanoprobes, Inc. | #2022 |
| PF-543 | Calbiochem | Cat# 567741 |
| YO-PRO®−1 iodide (491/509) | Invitrogen | Cat# Y3603 |
| MatTek | MatTek Corp. | Cat# P35GC-1.5-14-C |
| Mitotracker Red FM 581/644 | Molecular Probes | Cat# M22425 |
| Mitotracker Deep Red FM 644/665 | Molecular Probes | Cat# M22426 |
| Mitotracker Green FM 490/516 | Molecular Probes | Cat# M7514 |
| LTG 504/511 | Molecular Probes | Cat# L7526 |
| 2-Deoxy-D-glucose (2DG) | Sigma Aldrich | Cat# D6134 |
| Antimycin A | Sigma Aldrich | Cat# A8674 |
| Rotenone | Sigma Aldrich | Cat# R8875 |
| Oligomycin | Sigma Aldrich | Cat# O4876 |
| FCCP | Sigma Aldrich | Cat# C2920 |
| IMDM | GE Healthcare, HyClone | Cat# SH3022801 |
| Ficoll-paque | GE Healthcare, HyClone | Cat# 17-1440-03 |
| RPMI-1640 (Glucose free) | Thermo Fisher Scientific | Cat# 11879-020 |
| Penicillin-Streptomycin | Corning | Cat# 30-001-CI |
| Fetal Bovine Serum (FBS) | Atlanta Biologicals | Cat# S11150 |
| ACK Lysing Buffer | Thermo Fisher Scientific | A1049201 |
| Cell Strainer 40μm | Thermo Fisher Scientific | 22363547 |
| β-mercaptoethanol | Thermo Fisher Scientific | 21985023 |
| Cell-TAK | Corning | Cat# 354240 |
| rIL12 | Biolegend | Cat# 577004 |
| rIL6 | Biolegend | Cat# 575704 |
| rTGFβ | Biolegend | Cat# 580702 |
| rhIL2 | NCI, Biological Resources Branch | https://frederick.cancer.gov/Science/BrbRepository/#/preclinicalRepository |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| gp10025–33 peptide (KVPRNQDW) | Genscript | Cat# RP20344 |
| Nucleofector Kits for Mouse T Cells | Lonza | Cat# VPA-1006 |
| RIPA Lysis Buffer | Thermo Fisher Scientific | Cat# 89900 |
| Protein inhibitors | Thermo Fisher Scientific | Cat# 78430 |
| HaltTM Phosphatase inhibitors single use 100X cocktail | Thermo Fisher Scientific | Cat# 78428 |
| Critical commercial assays | ||
| CyQUANT® Direct Cell Proliferation Assay | Thermo Fisher Scientific | Cat# C35011 |
| Cyto-ID autophagy/mitophagy detection kit | Enzo | ENZ-51031-0050 |
| LIVE/DEAD | Thermo Fisher Scientific | L34967 |
| cAMP Determination | Cayman | Cat# 581001 |
| PKA Activity in vitro | Enzo | Cat# ADI-EKS-390A |
| β-galactosidase | Invitrogen | I-2904 |
| Adenosine 5’-triphosphate (ATP) Bioluminescent Assay Kit | Sigma | Cat# FLAA-1KT |
| iScript cDNA Synthesis Kit | Biorad | Cat# 1708891 |
| SsoAdvanced Universal SYBR® Green Supermix | Biorad | Cat# 1725274 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34554 |
| Deposited data | ||
| RNA sequencing data Title: Aging stress and ceramide dependent RNA sequencing expression profiling depends on ceramide synthase 6 | NCBI, Gene Expression Omnibus | Accession number: GSE148498 |
| Experimental models: cell lines | ||
| B16-F10 (validated cell line) | ATCC | CRL-6475 |
| Experimental models: organisms/strains | ||
| C57BL/6 | Jackson Laboratory | Stock# 000664 |
| B6.129S7-Rag1tm1Mom/J | Jackson Laboratory | Stock# 002216 |
| Prkaa1 PKAfl/fl | Jackson Laboratory | Stock# 014141 |
| CD4cre | Jackson Laboratory | Stock# 017336 |
| Pmel Sphk2−/− | Ogretmen’s Laboratory | Generated in this study |
| Pmel CerS6−/− | Ogretmen’s Laboratory | Generated in this study |
| Prkaa2 PKAfl/fl CD4cre | Ogretmen’s Laboratory | Generated in this study |
| Oligonucleotides | ||
| IL23r Forward Primer TTCAGATGGGCATGAATGTTTCT | IDT, Coralville | N/A |
| IL23r Reverse Primer CCAAATCCGAGCTGTTGTTCTAT | IDT, Coralville | N/A |
| IL-22 Forward Primer ATGAGTTTTTCCCTTATGGGGAC | IDT, Coralville | N/A |
| IL-22 Reverse Primer GCTGGAAGTTGGACACCTCAA | IDT, Coralville | N/A |
| IL-9 Forward Primer CATCAGTGTCTCTCCGTCCCAACTGAT | IDT, Coralville | N/A |
| IL-9 Reverse Primer GATTTCTGTGTGGCATTGGTCAG | IDT, Coralville | N/A |
| βactin Forward Primer ACGTAGCCATCCAGGCTGGTG | IDT, Coralville | N/A |
| βactin Reverse Primer TGGCGTGAGGGAGAGCAT | IDT, Coralville | N/A |
| SphK2 Probe: Mm00445021_m1 | ThermoFisher | Cat# 4331182 |
| SphK1 Probe: Mm00448841_g1 | ThermoFisher | Cat# 4331182 |
| CerS1 Probe: Mm03024093_mH | ThermoFisher | Cat# 4331182 |
| CerS4 Probe: Mm00482658_m1 | ThermoFisher | Cat# 4331182 |
| CerS5 Probe: Mm00510998_m1 | ThermoFisher | Cat# 4331182 |
| CerS6 Probe: Mm00556165_m1 | ThermoFisher | Cat# 4331182 |
| RPLP0 Probe: Mm00725448_s1 | ThermoFisher | Cat# 4331182 |
| Software and algorithms | ||
| FlowJo 10.2 | TreeStar, OR | https://www.flowjo.com/solutions/flowjo/downloads/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Agilent Seahorse Wave 2.4 | Agilent | https://www.agilent.com/en-us/products/cell-analysis-(seahorse)/seahorse-wave-software |
| CFX Manager 3.1 | Biorad | https://www.bio-rad.com/en-us/sku/soft-cfx-31-patch-cfx-manager-software-v3-1-upgrade |
| Fiji | NIH Image | https://imagej.net/Fiji |
| FV10i | Olympus Corp. | FV10-ASW https://www.grapecity.com |
Highlights.
CerS6-generated C14/C16 ceramide induces mitophagy in activated aging T cells
Aging-induced mitochondrial ceramide inhibits protein kinase A, leading to mitophagy
Aging and ceramide-dependent mitophagy attenuates the antitumor functions of T cells
Inhibiting ceramide-dependent mitophagy restores aging T cells’ antitumor activity
ACKNOWLEDGMENTS
We thank the members of the Ogretmen Laboratory for their technical support and helpful discussions. We also thank Drs. Craig Beeson, Shilpak Chatterjee, Nancy Smythe, and Zdizlaw Schulz at the Medical University of South Carolina for their helpful discussions and expertise. This work was supported by research funding from the National Institutes of Health (NIH; AG069769, CA214461, CA214461-03S1, DE016572, andP01 CA203628 to B.O.) and SmartState Endowment in Lipidomics and Drug Discovery. The core facilities utilized are supported by NIH (C06 RR015455), Hollings Cancer Center Support Grant (P30 CA138313), or the Center of Biomedical Research Excellence (Cobre) in Lipidomics and Pathobiology (P30 GM103339). The Zeiss 880 microscope was funded by a Shared Instrumentation Grant (S10 OD018113). This study was performed in strict accordance with the recommendations in the Guide for the Care and the Use of Laboratory Animals of the NIH. All of the animals were handled according to approved Institutional Animal Care and Use Committee (IACUC) protocol (2018-00412) of the Medical University of South Carolina.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109076.
DECLARATION OF INTERESTS
B.O. is an inventor on a provisional patent application covering ceramide analogs as therapeutic agents owned by the MUSC Foundation for Research Development. S.M. and B.O. are co-founders of Lipo-Immuno Tech, responsible for the commercialization of ceramide analogs to induce mitophagy.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- Baazim H, Schweiger M, Moschinger M, Xu H, Scherer T, Popa A, Gallage S, Ali A, Khamina K, Kosack L, et al. (2019). CD8+ T cells induce cachexia during chronic viral infection. Nat. Immunol 20, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektas M, Jolly PS, Müller C, Eberle J, Spiegel S, and Geilen CC (2005). Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: role of Bcl-2 expression. Oncogene 24, 178–187. [DOI] [PubMed] [Google Scholar]
- Bektas A, Schurman SH, Gonzalez-Freire M, Dunn CA, Singh AK, Macian F, Cuervo AM, Sen R, and Ferrucci L (2019). Age-associated changes in human CD4+ T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging (Albany NY) 11, 9234–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho VA, Galvani S, Engelbrecht E, Liu C, Swendeman SL, Kono M, Proia RL, Steinman L, Han MH, and Hla T (2015). HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 523, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneuil N (2007). Ageing laws for the human frontal cortex. Ann. Hum. Biol 34, 484–492. [DOI] [PubMed] [Google Scholar]
- Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, Hippenstiel S, Dingeldey M, Kruse B, Fauchere F, et al. (2020). SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 587, 270–274. [DOI] [PubMed] [Google Scholar]
- Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, Brisendine A, Anderton K, Cusack SL, Maines LW, et al. (2017). A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res 23, 4642–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronietzki AW, Schuster M, and Schmitz I (2015). Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol 93, 25–34. [DOI] [PubMed] [Google Scholar]
- Brown-Borg HM, and Anderson RM (2017). Metabolic adventures in aging research. Mol. Cell. Endocrinol 455, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, and Verdin E (2019). From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri I, Lin MT, Zheng K, Keller-McGandy CE, Betensky RA, Johns DR, Beal MF, Standaert DG, and Simon DK (2005). Somatic mitochondrial DNA mutations in single neurons and glia. Neurobiol. Aging 26, 1343–1355. [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Vaena SG, Thyagarajan K, Chatterjee S, Al-Khami A, Selvam SP, Nguyen H, Kang I, Wyatt MW, and Baliga U (2019). Pro-survival lipid sphingosine-1-phosphate metabolically programs T cells to limit anti-tumor activity. Cell Rep. 28, 1879–1893.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne DP, Hatle KM, Fortner KA, D’Alessandro A, Thornton TM, Yang R, Torralba D, Tomás-Cortázar J, Jun YW, Ahn KH, et al. (2016). Fine-tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity 44, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Thyagarajan K, Kesarwani P, Song JH, Soloshchenko M, Fu J, Bailey SR, Vasu C, Kraft AS, Paulos CM, et al. (2014). Reducing CD73 expression by IL1β-Programmed Th17 cells improves immunotherapeutic control of tumors. Cancer Res. 74, 6048–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, Fu J, Zhang J, Nguyen H, and Kang I (2018). CD38-NAD+ axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. 27, 85–100.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Kim JA, Kim TH, Li HY, Shin KO, Lee YM, Oh S, Pewzner-Jung Y, Futerman AH, and Suh SH (2015). Altering sphingolipid composition with aging induces contractile dysfunction of gastric smooth muscle via K(Ca) 1.1 upregulation. Aging Cell 14, 982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, and Janssen EM (2015). Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J. Immunol 195, 2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, and Mattson MP (2001). Sphingomyelin and ceramide as regulators of development and lifespan. Mech. Ageing Dev 122, 895–908. [DOI] [PubMed] [Google Scholar]
- Cutler RG, Thompson KW, Camandola S, Mack KT, and Mattson MP (2014). Sphingolipid metabolism regulates development and lifespan in Caenorhabditis elegans. Mech. Ageing Dev 143–144, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dany M, and Ogretmen B (2015). Ceramide induced mitophagy and tumor suppression. Biochim. Biophys. Acta 1853, 2834–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dany M, Gencer S, Nganga R, Thomas RJ, Oleinik N, Baron KD, Szulc ZM, Ruvolo P, Kornblau S, Andreeff M, and Ogretmen B (2016). Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128, 1944–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, Blanco EM, Alfranca A, Cussó L, Desco M, et al. (2020). T cells with dysfunctional mitochondria induce multimorbidity and pre-mature senescence. Science 368, 1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodig S, Čepelak I, and Pavić I (2019). Hallmarks of senescence and aging. Biochem. Med. (Zagreb) 29, 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia GG, and Miller RA (2002). Age-dependent defects in TCR-triggered cytoskeletal rearrangement in CD4+ T cells. J. Immunol 169, 5021–5027. [DOI] [PubMed] [Google Scholar]
- Gencer S, Oleinik N, Kim J, Panneer Selvam S, De Palma R, Dany M, Nganga R, Thomas RJ, Senkal CE, Howe PH, and Ogretmen B (2017). TGF-βreceptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci. Signal 10, eaam7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, Spite M, and Nagy L (2019). Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat. Immunol 20, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, and Spiegel S (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt P, Ostkotte D, Nolte H, Gerl MJ, Jais A, Brunner HL, Sprenger H-G, Awazawa M, Nicholls HT, and Turpin-Nolan SM (2019). CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell 177, 1536–1552.e23. [DOI] [PubMed] [Google Scholar]
- Hannun YA, and Obeid LM (2018). Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol 19, 175–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski SM, and Conzelmann A (2002). LAG1 puts the focus on ceramide signaling. Int. J. Biochem. Cell Biol 34, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH 3rd, Dang VM, Appleton J, O’Connell MP, Cheng P, et al. (2016). sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-R, Hong MS, Dan JM, and Kang I (2006). Altered IL-7Ralpha expression with aging and the potential implications of IL-7 therapy on CD8+ T-cell immune responses. Blood 107, 2855–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk CJ, and Miller RA (1999). Age-sensitive and -insensitive pathways leading to JNK activation in mouse CD4(+) T-cells. Cell. Immunol 197, 83–90. [DOI] [PubMed] [Google Scholar]
- Kishton RJ, Sukumar M, and Restifo NP (2017). Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 26, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Geltink RI, Edwards-Hicks J, Apostolova P, O’Sullivan D, Sanin DE, Patterson AE, Puleston DJ, Ligthart NAM, Buescher JM, Grzes KM, et al. (2020). Metabolic conditioning of CD8+ effector T cells for adoptive cell therapy. Nat. Metab 2, 703–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page A, Dupuis G, Larbi A, Witkowski JM, and Fülöp T (2018). Signal transduction changes in CD4+ and CD8+ T cell subpopulations with aging. Exp. Gerontol 105, 128–139. [DOI] [PubMed] [Google Scholar]
- Li J, Wang J, Kang AS, and Sacitharan PK (2020). Mapping the T cell response to COVID-19. Signal Transduct. Target. Ther 5, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Huang X, Withers BR, Blalock E, Liu K, and Dickson RC (2013). Reducing sphingolipid synthesis orchestrates global changes to extend yeast lifespan. Aging Cell 12, 833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Jimenez CP, Eling N, Chen H-C, Vallejos CA, Kolodziejczyk AA, Connor F, Stojic L, Rayner TF, Stubbington MJT, Teichmann SA, et al. (2017). Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinova EA (1998). Influence of sphingolipids on T lymphocyte activation. Biochemistry (Mosc.) 63, 102–110. [PubMed] [Google Scholar]
- Mattoo H, Faulkner M, Kandpal U, Das R, Lewis V, George A, Rath S, Durdik JM, and Bal V (2009). Naive CD4 T cells from aged mice show enhanced death upon primary activation. Int. Immunol 21, 1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellouk A, and Bobé P (2019). CD8+, but not CD4+ effector/memory T cells, express the CD44highCD45RBhigh phenotype with aging, which displays reduced expression levels of P2X7 receptor and ATP-induced cellular responses. FASEB J. 33, 3225–3236. [DOI] [PubMed] [Google Scholar]
- Mendelsohn BA, Bennett NK, Darch MA, Yu K, Nguyen MK, Pucciarelli D, Nelson M, Horlbeck MA, Gilbert LA, Hyun W, et al. (2018). A high-throughput screen of real-time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol. 16, e2004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas PS, Liu L, Moon PK, Joshi AU, Dove C, Mhatre S, Contrepois K, Wang Q, Lee BA, Coronado M, et al. (2019). Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol 20, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoliu I, Scherer M, Beguelin F, DaSilva L, Mari D, Salvioli S, Martin F-PJ, Capri M, Bucci L, Ostan R, et al. (2014). Serum profiling of healthy aging identifies phosphor- and sphingolipid species as markers of human longevity. Aging (Albany NY) 6, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov YN, Naumova EN, Yassai MB, and Gorski J (2011). Selective T cell expansion during aging of CD8 memory repertoires to influenza revealed by modeling. J. Immunol 186, 6617–6624. [DOI] [PubMed] [Google Scholar]
- Nganga R, Oleinik N, Kim J, Selvam SP, De Palma R, Johnson KA, Parikh RY, Gangaraju V, Peterson Y, Dany M, et al. (2019). Receptor-interacting Ser/Thr kinase 1 (RIPK1) and myosin IIA-dependent ceramidosomes form membrane pores that mediate blebbing and necroptosis. J. Biol. Chem 294, 502–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG (2004). Mitochondrial membrane potential and aging. Aging Cell 3, 35–40. [DOI] [PubMed] [Google Scholar]
- Novgorodov SA, Szulc ZM, Luberto C, Jones JA, Bielawski J, Bielawska A, Hannun YA, and Obeid LM (2005). Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J. Biol. Chem 280, 16096–16105. [DOI] [PubMed] [Google Scholar]
- O’Sullivan D, van der Windt GJ, Huang SC-C, Curtis JD, Chang C-H, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, et al. (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleinik N, Kim J, Roth BM, Selvam SP, Gooz M, Johnson RH, Lemasters JJ, and Ogretmen B (2019). Mitochondrial protein import is regulated by p17/PERMIT to mediate lipid metabolism and cellular stress. Sci. Adv 5, eaax1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Yang Z, Zhang R, Arnaboldi P, Lu G, Li Q, Wang W, Zhang B, Cui M, Zhang H, et al. (2011). Potentiation of Th17 cytokines in aging process contributes to the development of colitis. Cell. Immunol 266, 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneer Selvam S, De Palma RM, Oaks JJ, Oleinik N, Peterson YK, Stahelin RV, Skordalakes E, Ponnusamy S, Garrett-Mayer E, Smith CD, and Ogretmen B (2015). Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal 8, ra58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneer Selvam S, Roth BM, Nganga R, Kim J, Cooley MA, Helke K, Smith CD, and Ogretmen B (2018). Balance between senescence and apoptosis is regulated by telomere damage-induced association between p16 and caspase-3. J. Biol. Chem 293, 9784–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-W, Park W-J, and Futerman AH (2014). Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta 1841, 671–681. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache I, Kamocki K, Poirier C, Pewzner-Jung Y, Laviad EL, Schweitzer KS, Van Demark M, Justice MJ, Hubbard WC, and Futerman AH (2013). Ceramide synthases expression and role of ceramide synthase-2 in the lung: insight from human lung cells and mouse models. PLoS ONE 8, e62968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewzner-Jung Y, Ben-Dor S, and Futerman AH (2006). When do Lasses (longevity assurance genes) become CerS (ceramide synthases)? Insights into the regulation of ceramide synthesis. J. Biol. Chem 281, 25001–25005. [DOI] [PubMed] [Google Scholar]
- Pulko V, Davies JS, Martinez C, Lanteri MC, Busch MP, Diamond MS, Knox K, Bush EC, Sims PA, Sinari S, et al. (2016). Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat. Immunol 17, 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, Loh L, Olshanksy M, Naeem H, Tsyganov K, et al. (2018). Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 23, 3512–3524. [DOI] [PubMed] [Google Scholar]
- Rana A, Oliveira MP, Khamoui AV, Aparicio R, Rera M, Rossiter HB, and Walker DW (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun 8, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revuelta M, and Matheu A (2017). Autophagy in stem cell aging. Aging Cell 16, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riebeling C, Allegood JC, Wang E, Merrill AH, and Futerman AH (2003). Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acylCoA donors. J. Biol. Chem 278, 43452–43459. [DOI] [PubMed] [Google Scholar]
- Ron-Harel N, Sharpe AH, and Haigis MC (2015). Mitochondrial metabolism in T cell activation and senescence: a mini-review. Gerontology 61, 131–138. [DOI] [PubMed] [Google Scholar]
- Roy AK, Oh T, Rivera O, Mubiru J, Song CS, and Chatterjee B (2002). Impacts of transcriptional regulation on aging and senescence. Ageing Res. Rev 1, 367–380. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, and Kauppinen A (2018). The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res. Rev 48, 1–10. [DOI] [PubMed] [Google Scholar]
- Scheffel MJ, Helke K, Lu P, Bowers JS, Ogretmen B, Garrett-Mayer E, Paulos CM, and Voelkel-Johnson C (2017). Adoptive transfer of ceramide synthase 6 deficient splenocytes reduces the development of colitis. Sci. Rep 7, 15552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenten D, Nish SA, Yu S, Yan X, Lee HK, Brodsky I, Pasman L, Yordy B, Wunderlich FT, Brüning JC, et al. (2014). Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity 40, 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sckisel GD, Bouchlaka MN, Monjazeb AM, Crittenden M, Curti BD, Wilkins DE, Alderson KA, Sungur CM, Ames E, Mirsoian A, et al. (2015). Out-of-sequence signal 3 paralyzes primary CD4(+) T-cell-dependent immunity. Immunity 43, 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvam SP, and Ogretmen B (2013). Sphingosine kinase/sphingosine 1-phosphate signaling in cancer therapeutics and drug resistance. Handb. Exp. Pharmacol 216, 3–27. [DOI] [PubMed] [Google Scholar]
- Sentelle RD, Senkal CE, Jiang W, Ponnusamy S, Gencer S, Selvam SP, Ramshesh VK, Peterson YK, Lemasters JJ, Szulc ZM, et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol 8, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, and Wright WE (2007). Hallmarks of telomeres in ageing research. J. Pathol 211, 114–123. [DOI] [PubMed] [Google Scholar]
- Simula L, Pacella I, Colamatteo A, Procaccini C, Cancila V, Bordi M, Tregnago C, Corrado M, Pigazzi M, and Barnaba V (2018). Drp1 controls effective T cell immune-surveillance by regulating T cell migration, proliferation, and cMyc-dependent metabolic reprogramming. Cell Rep. 25, 3059–3073.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, and Spiegel S (2020). Sphingosine-1-phosphate signaling: a novel target for simultaneous adjuvant treatment of triple negative breast cancer and chemotherapy-induced neuropathic pain. Adv. Biol. Regul 75, 100670. [DOI] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sofi MH, Heinrichs J, Dany M, Nguyen H, Dai M, Bastian D, Schutt S, Wu Y, Daenthanasanmak A, Gencer S, et al. (2017). Ceramide synthesis regulates T cell activity and GVHD development. JCI Insight 2, e91701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopel N, Kölle J, Dumendiak S, Koch S, Reichel M, Rhein C, Kornhuber J, and Finotto S (2019). Immunoregulatory role of acid sphingomyelinase in allergic asthma. Immunology 156, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner MW, Thapa D, Zhang M, Gibson GA, Calderon MJ, St Croix CM, and Scott I (2016). α-Lipoic acid promotes α-tubulin hyperacetylation and blocks the turnover of mitochondria through mitophagy. Biochem. J 473, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulc ZM, Bielawski J, Gracz H, Gustilo M, Mayroo N, Hannun YA, Obeid LM, and Bielawska A (2006). Tailoring structure-function and targeting properties of ceramides by site-specific cationization. Bioorg. Med. Chem 14, 7083–7104. [DOI] [PubMed] [Google Scholar]
- Thomas RJ, Oleinik N, Panneer Selvam S, Vaena SG, Dany M, Nganga RN, Depalma R, Baron KD, Kim J, Szulc ZM, and Ogretmen B (2017). HPV/E7 induces chemotherapy-mediated tumor suppression by ceramide-dependent mitophagy. EMBO Mol. Med 9, 1030–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayssac M, Hannun YA, and Obeid LM (2018). Role of sphingolipids in senescence: implication in aging and age-related diseases. J. Clin. Invest 128, 2702–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K, and Futerman AH (2000). Ceramide as a second messenger: sticky solutions to sticky problems. Trends Cell Biol. 10, 408–412. [DOI] [PubMed] [Google Scholar]
- Wang D, Malo D, and Hekimi S (2010). Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1α in long-lived Mclk1+/− mouse mutants. J. Immunol 184, 582–590. [DOI] [PubMed] [Google Scholar]
- Wang H, Abajobir AA, Abate KH, Abbafati C, Abbas KM, Abd-Allah F, Abera SF, Abraha HN, Abu-Raddad LJ, and Abu-Rmeileh NM; GBD 2016 Mortality Collaborators (2017). Global, regional, and national under-5 mortality, adult mortality, age-specific mortality, and life expectancy, 1970–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1084–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson KR, Berg KM, Kapitonov D, Payne SG, Miner AS, Bittman R, Milstien S, Ratz PH, and Spiegel S (2007). Sphingosine-1-phosphate and the immunosuppressant, FTY720-phosphate, regulate detrusor muscle tone. FASEB J. 21, 2818–2828. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, and Chandel NS (2014). Futility sustains memory T cells. Immunity 41, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten-Blanks LG, Song P, Senkal CE, and Ogretmen B (2007). Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J. 21, 3386–3397. [DOI] [PubMed] [Google Scholar]
- Yang CC, Chen D, Lee SS, and Walter L (2011). The dynamin-related protein DRP-1 and the insulin signaling pathway cooperate to modulate Caenorhabditis elegans longevity. Aging Cell 10, 724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JK, Xu R, Jeong E, Mileva I, Truman J-P, Lin CL, Wang K, Snider J, Wen S, Obeid LM, et al. (2016). Aging-related elevation of sphingoid bases shortens yeast chronological life span by compromising mitochondrial function. Oncotarget 7, 21124–21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelnik ID, Ventura AE, Kim JL, Silva LC, and Futerman AH (2020). The role of ceramide in regulating endoplasmic reticulum function. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1865, 158489. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Liu X, Le W, Xie L, Li H, Wen W, Wang S, Ma S, Huang Z, Ye J, et al. (2020). A human circulating immune cell landscape in aging and COVID-19. Protein Cell 11, 740–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated during this study are available at Gene Expression Omnibus (GEO) with the accession number GSE148498.