

Abstract
Mutations in CHCHD10, coding for a mitochondrial intermembrane space protein, are a rare cause of autosomal dominant amyotrophic lateral sclerosis. Mutation-specific toxic gain of function or haploinsufficiency models have been proposed to explain pathogenicity. To decipher the metabolic dysfunction associated with the haploinsufficient p.R15L variant, we integrated transcriptomic, metabolomic and proteomic data sets in patient cells subjected to an energetic stress that forces the cells to rely on oxidative phosphorylation for ATP production. Patient cells had a complex I deficiency that resulted in an increased NADH/NAD+ ratio, diminished TCA cycle activity, a reorganization of one carbon metabolism and an increased AMP/ATP ratio leading to phosphorylation of AMPK and inhibition of mTORC1. These metabolic changes activated the unfolded protein response (UPR) in the ER through the IRE1/XBP1 pathway, upregulating downstream targets including ATF3, ATF4, CHOP and EGLN3, and two cytokine markers of mitochondrial disease, GDF15 and FGF21. Activation of the mitochondrial UPR was mediated through an upregulation of the transcription factors ATF4 and ATF5, leading to increased expression of mitochondrial proteases and heat shock proteins. There was a striking transcriptional up regulation of at least seven dual specific phosphatases, associated with an almost complete dephosphorylation of JNK isoforms, suggesting a concerted deactivation of MAP kinase pathways. This study demonstrates that loss of CHCHD10 function elicits an energy deficit that activates unique responses to nutrient stress in both the mitochondria and ER, which may contribute to the selective vulnerability of motor neurons.
Introduction
Autosomal dominant mutations in coiled-helix coiled-helix domain containing protein 10 (CHCHD10) were recently identified as rare genetic causes of amyotrophic lateral sclerosis (ALS) (1–13). Most predicted pathogenic variants are present in the N-terminal half of the protein and are rarely found in the defining CHCH-domain in the C-terminus. The mutation c.44C > A (predicting p. R15L) has been reported in sporadic and familial ALS, and motor neuron disease in four studies (1,4,6,14). CHCHD10 is a soluble 14 kDa mitochondrial protein that is upregulated in stress conditions and localizes to the mitochondrial intermembrane space where it forms a complex with its paralogue CHCHD2 (15,16). However, the precise function of CHCHD10 remains unknown.
Mitochondrial dysfunction has long been suggested to contribute to ALS disease pathology, but prior to the identification of mutations in CHCHD10, these deficiencies were thought to be pleiotropic effects. Although all reported pathogenic mutations in CHCHD10 are autosomal dominant, the molecular basis of pathogenicity of CHCHD10 is variable. For instance, the p.S59L variant reported in patients with ALS-FTD (frontotemporal dementia) is associated with a fragmented mitochondrial network, protein aggregates in mitochondria, and the activation of the integrated stress response (ISR), ascribed to a toxic gain of function (2,17–19). On the other hand, the p.R15L and p.G66V variants, in which reduced levels of CHCHD10 protein are associated with mitochondrial respiratory chain dysfunction, appear to be haploinsufficient (15,20,21).
Emerging lines of evidence suggest a link between the endoplasmic reticulum (ER) unfolded protein response and neurodegenerative diseases like ALS (22). This unfolded protein stress response is designed to maintain and recover ER proteostasis through three distinct signalling pathways: PRKR-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring kinase 1 (IRE1) (23). In particular, the IRE1 signalling cascade has been suggested as potential target for treatment as downstream targets such as XBP1 and CHOP have been shown to play a role in several ALS disease models, and knockdown of XBP1 provides significant protection against neuronal death and delays onset of disease (24–27).
To investigate the cellular and metabolic remodelling caused by the CHCHD10 variant p.R15L, we collected metabolomic, transcriptomic and proteomic datasets on patient fibroblasts. We cultured patient and rescued cells (expressing wild-type CHCHD10) in medium containing either glucose or galactose as a nutrient source. Glucose-free galactose medium imposes an energetic stress, as it forces cells to rely solely on oxidative phosphorylation (OXPHOS) for ATP production. Our data demonstrate a global remodelling of mitochondrial and cellular metabolic pathways, and the activation of an unfolded protein (UPR) stress response mediated by the IRE1/XBP1 pathway in the ER, and by ATF4 and ATF5 in mitochondria, in patient cells under energetic stress conditions.
Results
To uncover the mechanisms of pathogenesis in patient fibroblasts carrying the CHCHD10 p.R15L variant (hereafter referred to as ‘patient’), we set out to compare them to patient cells expressing a wild-type CHCHD10 cDNA (hereafter called ‘rescue’). Defects in mitochondrial metabolism are often exacerbated by energetic stress, and we previously showed that about 20% of patient cells died over 24 hours after transfer to galactose medium, while the remaining cells remained attached to the plate, but did not re-enter the cell cycle (15). This phenotype could be rescued by retroviral expression of a wild-type CHCHD10 cDNA that restored CHCHD10 to near control levels. To investigate potential metabolic re-wiring and cellular stress responses in patient cells, the transcriptome, proteome and metabolome of patient and rescue cells were determined by RNA sequencing, tandem mass tag (TMT) mass spectrometry and targeted metabolomic analyses in response to the galactose challenge. To profile the initial response, we compared the patient to the rescued cells grown in glucose medium (‘Glucose’) or in galactose medium for 48 hours (‘Galactose’) (Fig. 1A).
Figure 1 .
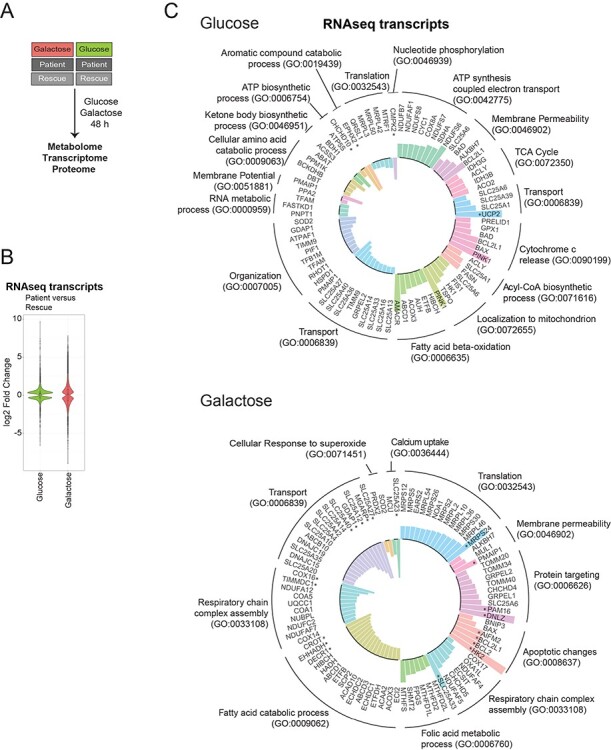
Altered biological processes revealed by RNAseq analysis. (A) Schematic representation of the omics experiments. Patient and rescue cells were grown in glucose or galactose media for 48 h and subsequently, the metabolome, transcriptome and proteome were analysed. ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (B) Violin plot representing the distribution of log2 fold change of expression for transcripts (patient vs rescue) in the two conditions (‘Glucose’, green and ‘Galactose’, red) with an adjusted P-value < 0.05 (FDR < 0.05). The effect size was larger for the ‘Galactose’ condition. (C) Circular bar plot depicting mitochondrial transcripts with a minimum log2 fold change (patient vs rescue) of ±0.3 in ‘glucose’ and ‘galactose’, grouped based on the GO term: biological processes. Bars facing upwards from the inner circle represent the upregulated transcripts, and bars facing inwards represent the downregulated transcripts. Asterisk ‘*’ indicates transcripts with a log2 fold change >1.
RNAseq analysis reveals reorganization of one-carbon metabolism
To investigate transcriptional alterations in patient cells and the response to galactose stress, we first determined steady-state mRNA levels by RNA sequencing. Differentially expressed transcripts in ‘Glucose’ and ‘Galactose’ media showed a substantially different distribution; more transcripts had a larger fold change in ‘Galactose’ compared to ‘Glucose’ (Figs 1B, Supplementary Material, Fig. S1A and B, Supplementary Material, Table S1). We first focused on transcripts for proteins found in MitoCarta2.0 (28) to ascertain which mitochondrial processes were most affected in the patient cells.
In ‘Glucose’, enrichment analysis of differentially expressed transcripts for the gene ontology (GO) term for biological processes (BP), identified an upregulation of pathways involving ATP synthesis, the respiratory chain and the tricarboxylic acid cycle (TCA) cycle (Fig. 1C, ‘Glucose’). Interestingly, transcripts of the acyl-CoA biosynthetic process, for example ATP citrate synthase (ACLY), fatty acid synthetase (FASN) and the citrate transporter (SLC25A1) were upregulated in glucose even though the biosynthesis of fatty acids would seem to be counterproductive in a state of compromised energy supply. Beta-oxidation of fatty acids, a catabolic pathway, was upregulated as well. The most upregulated transcript was uncoupling protein 2 (UCP2), a protein involved in protection against oxidative stress and regulation of reactive oxygen species (ROS). Downregulated transcripts encompassed several mitochondrial transporters including SLC25A27 (UCP4), SLC25A14 (UCP5), SLC25A33, SLC25A36 (pyrimidines) and SLC25A13 (aspartate/malate). Transcripts coding for enzymes involved in ketone body metabolism, 3-hydroxybutyrate dehydrogenase 1 (BDH1) and acyl-CoA synthetase short chain 3 (ACSS3), were downregulated. Lastly, the mitochondrial UMP-CMP kinase (CMPK2), responsible for the phosphorylation of dUMP and dCMP, was downregulated, potentially effecting the local supply of deoxynucleotides as essential precursors for mitochondrial DNA replication and transcription.
Mitochondrial processes downregulated in patient cells in ‘Galactose’ included the catabolism of fatty acids, transcripts of respiratory chain complex assembly, in particular the assembly of complexes I and IV, and a number of mitochondrial transporters such as SLC25A27 (UCP4) and SLC25A14 (UCP5) (Fig. 1C, ‘Galactose’). Both have the highest level of expression in the brain, where they have been suggested to uncouple oxidative phosphorylation, exerting a protective role in cells exposed to mitochondrial defects (29,30). The aspartate/glutamate transporter SLC25A12, which catalyses the calcium-dependent exchange of cytoplasmic glutamate with mitochondrial aspartate, was also downregulated in patient cells in galactose (31).
Upregulated transcripts in patient in ‘Galactose’ included those involved in mitochondrial translation, protein targeting, apoptosis and the assembly of the respiratory chain complexes. However, the transcripts with a log2 fold change >1 mainly involved apoptotic pathways (Fig. 1C, ‘Galactose’). We also observed an upregulation of transcripts of the mitochondrial folate cycle, linked to cellular one-carbon metabolism, which is in part responsible for nucleic acid production, and therefore cellular growth. A more detailed analysis of gene transcripts involved in one-carbon metabolism revealed several changes in this pathway in the patient (Fig. 2A, B). Transcripts for mitochondrial proteins involved in folate synthesis, MTHFD2L, MTHFD2, MTHFD1L and SHMT2 were all upregulated, whereas transcripts for the cytosolic arm of the one-carbon cycle, DHFR, SHMT1, TYMS and MTHFD1 were downregulated in galactose. Mitochondrial folate metabolism supplies the cytosol with formate for purine synthesis, and for the synthesis of NADPH in mitochondria (32,33). The same tendency was observed comparing patient cells in ‘Glucose’ versus patient cells in ‘Galactose’, suggesting a marked shift in one carbon metabolism in patient cells in ‘Galactose’ in particular (Supplementary Material, Fig. S1C, D). Lastly, transcripts of serine metabolism were downregulated in both ‘Glucose’ and ‘Galactose’ conditions, suggesting that serine is not synthesized de novo from glucose but rather directly obtained from the culture media or through the conversion of glycine to serine (Fig. 2A and B).
Figure 2 .
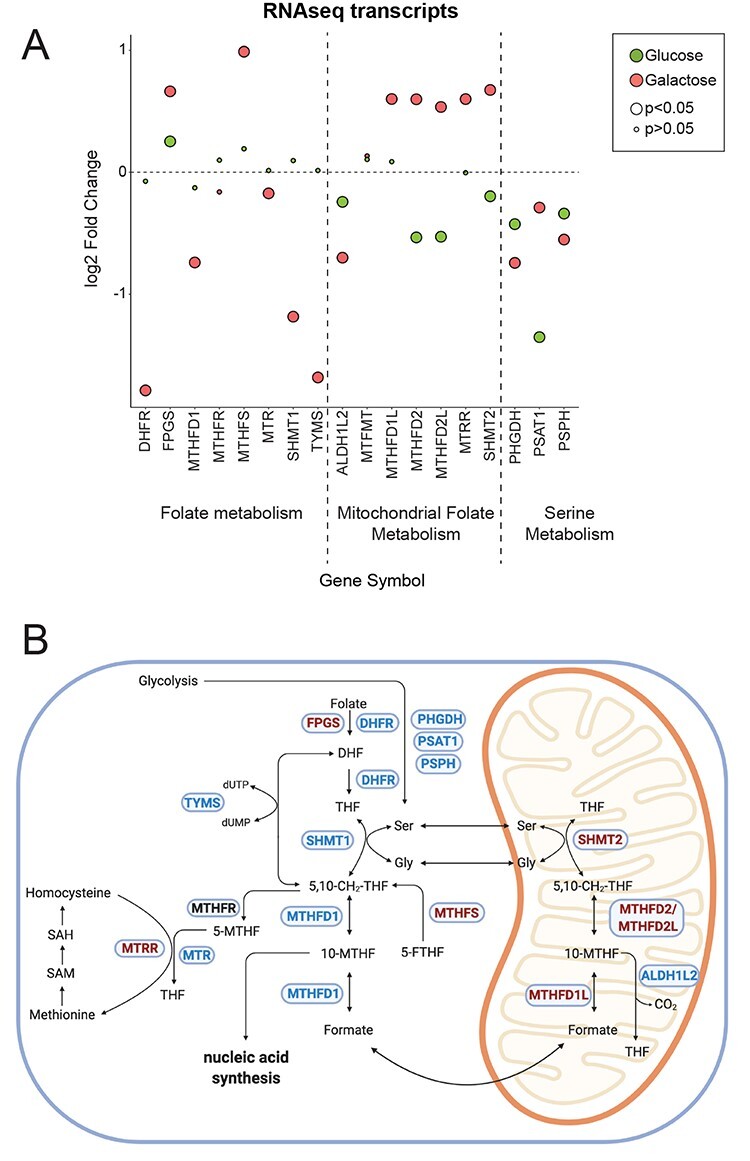
RNAseq analysis reveals changes in one-carbon metabolism. (A) Dot plot representing the fold change of the transcripts of the folate and serine metabolism. Fold change is represented in log2 scale. Large circles represent significant values (FDR < 0.05), small circles show non-significant values (FDR > 0.05). ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (B) Graphical representation of enzymes and metabolites of the one-carbon metabolism. Selected enzymes are shown in blue boxes for cytosolic and mitochondrial folate metabolism as well as serine and methionine metabolism. Upregulated transcripts in patient cells in galactose are shown in red and downregulated transcripts in blue. This scheme was created with BioRender.
In summary, in ‘Glucose’ patient cells upregulated transcripts of the mitochondrial respiratory chain, the TCA cycle and fatty acid beta-oxidation presumably to compensate for the lack of mitochondrial energy production. In ‘Galactose’ on the other hand, transcripts of mitochondrial translation and mitochondrial protein import were upregulated in response to the energy deficit. Upregulation of transcripts of one-carbon metabolism in mitochondria would act to increase the supply of formate from mitochondria to the cytosol for the production of purines, and to supply mitochondria with more NADPH. Interestingly, the mitochondrial aspartate/glutamate carrier was downregulated, which could lead to a deficiency of energy equivalents in mitochondria and a lack of mitochondrial aspartate in the cytosol.
Metabolomic analysis reveals a high NADH/NAD+ ratio that stalls the TCA cycle in patient cells
Metabolomic analyses were performed in the four groups described above on whole cells. Using a targeted assay for 116 metabolites involved in bioenergetics, oxidative stress and mitochondrial function, the levels of 85 metabolites were successfully determined across all samples. Multi-variant data analysis using orthogonal transformation through principal component analysis (PCA) or hierarchical clustering with heatmap analysis showed tight clustering of the replicates and clear differences among the four conditions analysed, demonstrating the quality of the data (Figs 3A and Supplementary Material, Fig. S2A). The first component PC1 showed that the treatment effect of glucose medium versus galactose medium was clearly separated, particularly for the patient cells, explaining 55% of the variance in the dataset. Moreover, we were able to separate the effect of the genotype within both the galactose or glucose condition, through the second component analysis PC2, explaining 18% of the variance. The genotype difference was larger in ‘Galactose’ than in ‘Glucose’ (Fig. 3A). The fold-change distribution of differentially detected metabolites in the two conditions ‘Glucose’ and ‘Galactose’ was larger in the ‘Galactose’ condition showing log2 fold changes ranging from +3 to −2.5 in galactose versus +1 to −2.5 in glucose (Fig. 3B, Supplementary Material, Table S2).
Figure 3 .
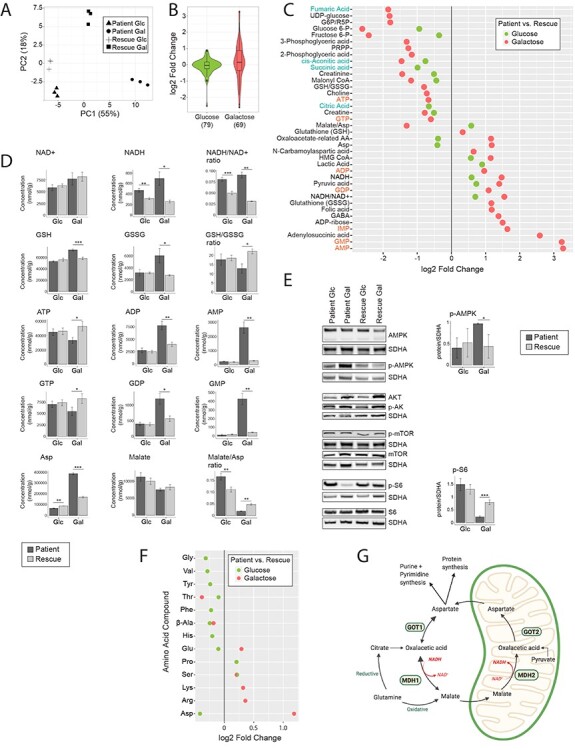
Metabolomic analysis reveals a high NADH/NAD+ ratio, which stalls the TCA cycle in patient cells. (A) Principal component analysis of the overall metabolomic profile. Symbols convey information about the four experimental groups and their three replicates. (B) Violin plot representing the distribution of the log2 fold change (patient/rescue) for all measured metabolites. Box plots indicate the median and the first and third quartile. The number of metabolites in each group is indicated in parentheses below the graph. ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (C) Dot plot representing the fold change of the most changed metabolites for each condition. Fold change is represented in log2 scale and the data show only metabolites with significant differences with a P-value <0.05. The P-value is computed by Welch’s test. Metabolites of nucleotides are highlighted in orange font and TCA cycle intermediates in blue font. ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (D) Bar plots representing the most significantly changed metabolites. Concentrations are depicted in nmol/g and ratios are pure ratios. Values for the patient cells are shown in dark grey and values for the rescue cells are shown in light grey. P-values were computed by Welch’s test, *P < 0.05, **P < 0.01, ***P < 0.001. (E) Immunoblot analysis of proteins involved in the mTOR pathway. Whole cell extracts from patient and rescue fibroblasts grown in glucose and galactose for 2 days were separated by SDS-PAGE and probed with antibodies against indicated proteins. SDHA was used as a loading control. Bar plot indicates the quantification of three and four immunoblot analyses for p-AMPK and p-S6, respectively, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001. (F) Dot plot representing the fold change of the most changed amino acid levels for each condition. Fold change is represented in log2 scale and data show only metabolites with significant differences with a P-value <0.05. The P-value is computed by Welch’s test. ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (G) Graphical representation of enzymes and metabolites of the malate aspartate shuttle. Selected enzymes are shown in green boxes for cytosolic and mitochondrial processes. This scheme was created with BioRender.
Redox imbalance caused by high levels of NADH in the patient
The metabolomic analysis revealed that the NADH/NAD+ ratio was significantly increased in both ‘Glucose’ and ‘Galactose’ in patient cells. Normally, when control cells are put in galactose the NADH/NAD+ ratio decreases (34), which was evident in the rescued cell line (Fig. 3C and D). The complex I deficiency in the patient cells likely accounted for the increase in the NADH/NAD+ ratio in galactose.
Additionally, the glutathione antioxidant defence pathway was activated in patient cells in galactose. Total levels of glutathione were increased (GSH + GSSG), and the GSH:GSSG ratio was decreased (Fig. 3C and D), potentially to deal with increased oxidative stress in galactose.
The TCA cycle is stalled in the patient
Analysis of the common upregulated and downregulated metabolites in glucose and galactose revealed decreased levels of TCA-cycle intermediates, including cis-aconitic acid, succinate, fumarate and citrate (Fig. 3C, Supplementary Material, Table S2), suggesting a stalling of the TCA cycle. However, the level of pyruvate, a substrate entering the TCA cycle was increased (Fig. 3C). The TCA cycle is regulated by ATP and NADH levels (35), which were decreased and increased, respectively (Fig. 3C). The conversion of pyruvate into acetyl-CoA, acetyl-CoA into citrate and ketoglutarate to succinate is NADH-dependent and likely inhibited in the patient, as NADH levels were high. ADP activates the conversion from pyruvate into acetyl-CoA, acetyl-CoA into citrate and isocitrate into ketoglutarate, and its levels were increased, producing counteracting signals (Fig. 3C and D). Overall, however, the data suggested that flux through the TCA cycle was decreased in both ‘Glucose’ and ‘Galactose’.
High levels of aspartate do not rescue the stalled TCA cycle
With the exception of aspartate, other detected amino acids were not significantly altered in the patient cells in either ‘Glucose’ or ‘Galactose’ (Fig. 3F). Aspartate plays a key role in mitochondrial metabolism and is involved in the shuttling of electrons across the inner mitochondrial membrane through the malate–aspartate shuttle (Fig. 3G) (35). The malate/aspartate ratio thus serves as an indirect indicator of the NADH/NAD+ ratio and the energy state of the cell. A dysregulation of the malate/aspartate ratio was observed in both glucose and galactose, but, in opposite directions (Fig. 3D and F). The shuttle is responsible for producing NADH in mitochondria where it is reoxidized to NAD+ by respiratory chain complex I. The mitochondrial asparate/glutamate transporter SLC25A12 was downregulated in patient cells in galactose at the transcript level (Fig. 1C), which could lead to diminished transport of aspartate outside of mitochondria and a reduction of NADH inside of mitochondria. In the cytosol, aspartate is converted to malate by GOT1 and MDH1 (36). Interestingly, the enzyme GOT1 was increased at the transcript level in the patient in galactose. GOT1 normally metabolizes aspartate to oxalacetic acid, which is then converted by MDH1 into malate. This cycle allows the supply of electrons for the mitochondrial respiratory chain. However, when the electron transport chain is inhibited, GOT1 produces aspartate, which serves as a substrate to synthesize purines and pyrimidines, compensating for the lack of mitochondrial aspartate synthesis (36,37) (Fig. 3G). The increased levels of aspartate and GOT1 therefore suggest that aspartate is produced by GOT1 in the cytosol to support the proliferation of patient cells. At the same time, the mitochondrial pyrimidine carrier SLC25A33 is markedly upregulated in patient cells in ‘Galactose’ (Fig. 1C), suggesting an increased pyrimidine import into mitochondria.
Reduced energy equivalents in the patient in galactose
The metabolomic analysis revealed that nucleotide mono- and diphosphates (IMP, AMP/ADP and GMP/GDP) were the most highly increased metabolites in patient cells in ‘Galactose’, resulting in a marked increase in the AMP/ATP and GMP/GTP ratios (Fig. 3C and D), and reflecting impaired mitochondrial ATP production due to the complex I deficiency. Concomitantly, the metabolite adenylosuccinic acid required for the conversion of IMP to AMP was also markedly increased as well as ADP-ribose and folate, which are involved in the biosynthesis of purines and pyrimidines (Fig. 3C and D). Formyl folate is the precursor for inosine monophosphate, which itself is the precursor of GMP and AMP. In addition, the level of S-adenosylmethionine (SAM) a metabolite of the methionine cycle, which is linked to the folate cycle, was increased (Supplementary Material, Fig. S2B). The level of its counterpart S-adenosylhomocysteine (SAH) was below the detection limit (Supplementary Material, Fig. S2B). These data indicate that increased folate levels lead to an increased generation of nucleotides IMP, AMP and GMP, as major enzymes of the folate cycle in the cytosol were transcriptionally downregulated (Fig. 2A and B).
The energy balance is tightly monitored in patient cells in galactose
Concomitant with the markedly disturbed AMP/ATP ratio, we detected an increase in Thr172-phosphorylated AMP-activated protein kinase (pAMPK), signalling low cellular energy levels (38) (Fig. 3E). Another sensor of cellular energy balance is the mechanistic/mammalian target of rapamycin (mTOR) pathway. mTOR1 is a serine/threonine kinase that responds to a number of physiological inputs including nutrients (amino acids), oxygen level, hormones and energy status (AMP/ATP ratio) (39). Generally, mTORC1 is activated when the energy supply is sufficient and is negatively regulated by pAMPK when energy levels are low. The reduced phosphorylation of ribosomal protein S6 in patient cells in ‘Galactose’ suggests that the mTORC1 pathway was inhibited in this condition (Fig. 3E). The levels of AKT (serine/threonine kinase), phosphorylated AKT, mTOR and phosphorylated mTOR were unchanged (Fig. 3E).
In summary, NADH/NAD+ ratios were high in both ‘Glucose’ and ‘Galactose’, which translated into a marked cellular energy deficit in ‘Galactose’. Glycolysis rescued the inefficient energy supply from mitochondria in ‘Glucose’ as evidenced by the increased production of lactate that we previously reported (15); however, in ‘Galactose’ glycolysis was undetectable as expected. High levels of NADH presumably stalled the TCA cycle as the amounts of NADH produced cannot be used by the respiratory chain to generate ATP due to the complex I deficiency. Subsequently, entry level substrates of the TCA cycle, like pyruvate and aspartate accumulated. Aspartate was additionally generated by GOT1 in the cytosol, possibly in an attempt to rescue the proliferation defect of the patient in galactose.
Integration of proteomics and metabolomics confirms reduction in TCA cycle activity at the protein level
Changes in the proteome of isolated crude mitochondria from patient and rescue cells were determined by quantitative proteomic analysis using TMT technology. We identified 1901 unique proteins in all conditions, of which 414 proteins were mitochondrial based on MitoCarta2.0 (28), accounting for 21.8% of the total proteins in the data set (Fig. 4A). We elected to investigate crude mitochondrial pellets, rather than whole cell lysates, for this analysis to increase the sensitivity of detection of mitochondrial proteins, but we realize that these preparations will contain variable amounts of contaminants, especially from other heavy membranes in the cell. Thus, comparisons of the non-mitochondrial proteins between samples could potentially be skewed by the degree of contamination, which may not the same between samples. With this caveat, and applying a significance level of 5% (false discovery rate, FDR), we identified differentially expressed proteins for each condition (Figs 4B and Supplementary Material, Fig. S3, Supplementary Material, Table S3). In ‘Glucose’, several complex I subunits were downregulated (NDUFV1, NDUFS8, NDUFA7, NDUFB8) confirming the results from our first study (15) (Supplementary Material, Fig. S3). Among the most significantly upregulated proteins were four proteins in the glycolytic pathway, glyceraldehyde-3-phosphate dehydrogenase (GAPDHS), fructose-bisphosphate aldolase A (ALDOA), phosphopyruvate hydratase (ENO1) and phosphoglycerate kinase 1 (PGK1) supporting the increased activity of the glycolytic pathway in patient cells in ‘Glucose’. Phosphoserine aminotransferase 1 (PSAT1), responsible for the conversion of the glycolytic intermediate 3-phosphoglycerate, an intermediate of glycolysis, to serine (Fig. 2B), was also upregulated in ‘Glucose’ (Supplementary Material, Fig. S3). Serine is used through the folate cycle to produce purines. Lastly, glutathione S-transferase P (GSTP1) and glutathione S-transferase omega 1 (GSTO1), two detoxification enzymes that catalyze the conjugation of glutathione to chemical mutagens and protect against products of oxidative stress, were upregulated (Supplementary Material, Fig. S3).
Figure 4 .
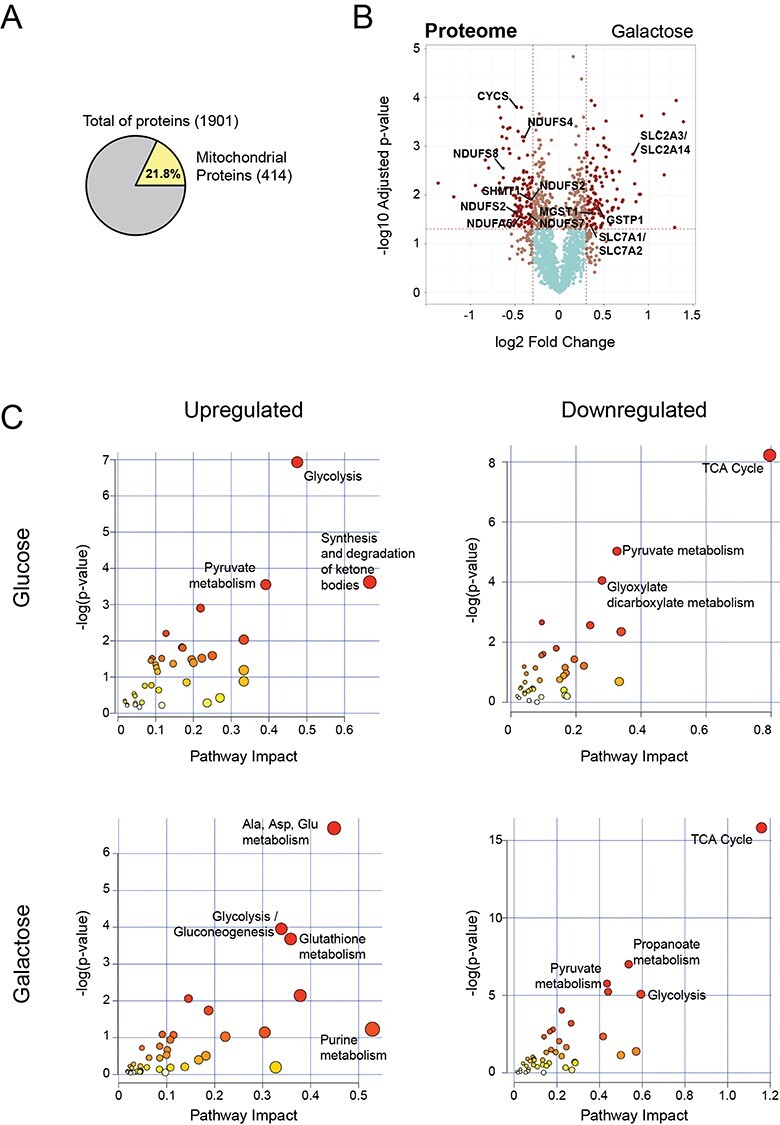
Integration of proteomics and metabolomics confirms stalled TCA cycle at the protein level. (A) Pie chart representing the total number of proteins identified in all conditions by TMT quantitative proteomics analysis. Number of nuclear encoded mitochondrial proteins (MitoCarta2.0) are indicated in yellow, as well as the percentage relative to the total. (B) Volcano plot representing the results of differential protein expression analysis between patient and rescue cells in galactose. x-axis values represent the log2 fold change and y-axis values represent the -log10 of the adjusted P-value. Selected mitochondrial proteins are labelled. (C) Joint pathway enrichment analysis performed with the MetaboAnalyst tool of significant up- or downregulated metabolites and proteins in glucose and galactose condition. Scatterplots represent P-values from integrated enrichment analysis and impact values from pathway topology analysis using KEGG metabolic pathways. The node colour represents the P-values and the node radius is based on the pathway impact values calculated with degree centrality.
In ‘Galactose’, we observed the downregulation of several complex I subunits (NDUFS8, NDUFS7, NDUFB10, NDUFA5, NDUFS5, NDUFA10, NDUFS4, NDUFS2) as well as cytochrome c (CYCS) (Fig. 4B). Interestingly, many lysosomal proteins were downregulated (ASAH1, HEXB, CTSZ, CLTB, GNS, LAMP1, CTSL, NPC2, CTSK, PSAP, PPT1, DNASE2, CTSD, LGMN, CTSB, ATP6V1G1), in particular cathepsins with cysteine-type peptidase activity (Supplementary Material, Table S3). The cytosolic serine hydroxymethyltransferase (SHMT1), which is responsible for converting serine to glycine and vice-versa, providing one-carbon units for the synthesis of methionine, thymidylate and purines, was downregulated in ‘Galactose’ at the protein and transcript levels (Figs 2A, B and 4B).
The most prominent proteins upregulated in ‘Galactose’ are implicated in ER stress (Supplementary Material, Table S3). Furthermore, several transporters responsible for providing glycolysis and the TCA cycle with substrates, SLC2A1/A3 and SLC2A14 (glucose carrier) and SLC7A1/A2 (arginine, lysine and ornithine transporter) were upregulated. Lastly, we observed an increase in proteins of glutathione metabolism, GSTP1, microsomal glutathione S-transferase (MGST1) and thioredoxin domain containing 12 (TXNDC12) (Fig. 3B, Supplementary Material, Table S3).
To improve the depth of the proteomic study, we used the ‘MetaboAnalyst’ tool for the integration of proteomic and metabolomic data, which allows one to perform pathway enrichment analysis based on differentially detected metabolites and expressed proteins (Fig. 4C). We used the degree of the node (number of nodal connections) to define our enrichment analysis. Nodes with more connections, so-called hubs, have a higher impact in the pathway and, when affected, influence the enrichment more than a less connected node. Glycolysis (ALDOA, LDHA, ENO1, GAPDHS, PGK1, lactic acid, pyruvic acid), the synthesis and degradation of ketone bodies (ACAT2, 3-hydroxy-3-methylglutaryl-CoA), and pyruvate metabolism (LDHA, ACAT2, lactic acid, pyruvic acid) were the most highly induced pathways in ‘Glucose’ (Figs 4C and Supplementary Material, Fig. S3). The increase of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) (Fig. 3C) indicates a stalled mevalonate pathway, responsible for the synthesis of cholesterol and other isoprenoids. The rate limiting step of the mevalonate pathway is in the conversion of HMG-CoA to mevalonate by HMG reductase, which itself is inactivated by high levels of AMP. Therefore, in addition to probable TCA cycle stalling due to high levels of NADH, high levels of AMP predict a reduction of anabolic cellular metabolism. The integration of proteins with metabolites suggested a downregulation of the TCA cycle (DLD, OGDH, MDH2, cis-aconitic acid, succinic acid, citric acid), pyruvate metabolism (PKLR, MDH2, ALDH3A2, DLD, malonyl-CoA), and glyoxylate and dicarboxylate metabolism (MDH2, CAT, citric acid, cis-aconitic acid, glycine).
In ‘Galactose’ on the other hand, we detected an up-regulation of alanine, aspartate and glutamate metabolism (ADSL, aspartic acid, adenylsuccinic acid, argininosuccinic acid, pyruvic acid, ureidosuccinic acid, gamma-aminobutyric acid), glutathione metabolism (ANPEP, TXNDC12, GSTP1, MGST1, oxidized glutathione, NADP) as well as glycolysis and gluconeogenesis (LDHAL6B, LDHA, ENO2, ENO3, PGAM1, PGAM4, TPI1, pyruvate) However, the absence of cellular lactate and intermediates of glycolysis at the metabolite level in ‘Galactose’ (Fig. 3C, Supplementary Material, Table S2) demonstrates that glycolysis does not occur in patient cells in these conditions. As in ‘Glucose’, the TCA cycle was also downregulated in ‘Galactose’ a(DLD, SUCLA2, SUCLG1, IDH3A, MDH2, PDHA1, PDHB, FH, fumaric acid, cis-aconitic acid). The TCA cycle and mitochondrial activity are normally upregulated in galactose in control cells (40–42).
In summary, the energy deficit in patient cells could be compensated by high glycolytic activity in patient in ‘Glucose’, but not in ‘Galactose. Furthermore, high levels of AMP would lead to a stalled mevalonate pathway, which is responsible for the synthesis of sterols, heme A and ubiquinone. The upregulation of glutathione-related proteins in both conditions indicates an increased need to deal with mitochondrial oxidative stress.
Metabolic dysfunction leads to canonical ER UPR in patient cells in galactose
The upregulation of proteins involved in ER stress (Supplementary Material, Table S3), and the fact that mitochondrial and ER functions are tightly coupled, led us to analyse the unfolded protein (UPR) and integrated stress response (ISR) in both organelles. A coordinated activation expression of the mitochondrial unfolded protein response and the integrated stress response of the ER has been proposed previously (43). In response to unfolded proteins in the ER, but also other cellular stress signals, three different arms of the UPR response can be activated through PERK, IRE1 or ATF6. In all cases, the folding chaperone binding immunoglobulin protein (BiP/GRP78/HSPA5) dissociates from PERK, IRE1 or ATF6, respectively, and leads to their oligomerization. The oligomerization of PERK as well as the activation of other kinases through other stress signals such as amino acid stress (GCN2), hypoxia (HRI) and double-stranded RNA through viral infection (PKR) initiates the ISR through the phosphorylation of eukaryotic translation factor 2 alpha (eIF2alpha) (44). This results in the global attenuation of protein synthesis, and the selective translation of stress response proteins. Although transcript levels of PKR (EIF2AK2), and PERK (EIF2AK3) were upregulated, as were the levels of the PERK and GCN2 proteins in patient cells in ‘Galactose’ (Figs 5A, B and Supplementary Material, Fig. S4B), we did not observe increased phosphorylation of PERK, GCN2 or eIF2alpha (Figs 5B and Supplementary Material, Fig. S4B, C). The IRE1 pathway on the other hand is independent of eIF2alpha phosphorylation. Autophosphorylation of the kinase domains of IRE1 lead to stimulation of its endoribonuclease function, resulting in splicing of the mRNA of X-Box binding protein 1 (XBP1), removing a premature stop codon, and converting it into a transcriptional activator (45,46). Our data show that the ER unfolded protein response in patient cells in ‘Galactose’ was activated through IRE1 and XBP1, with no evidence of activation of the ISR through the phosphorylation of eIF2alpha, the level of which was in fact significantly reduced in patient cells in ‘Galactose’ (Fig. 5A and B). Protein phosphatase GADD34 (PPP1R15A), which recruits the protein phosphatase PP1 to dephosphorylate eIF2alpha and attenuate the ISR, was upregulated at the transcript level, possibly indicating a transcriptional response for prolonged UPR signalling (Fig. 5A and B). Consistent with the reduction in eIF2alpha phosphorylation, we observed an upregulation of transcripts involved in both cytoplasmic and mitochondrial translation (Figs 1C and 5C). Persistent activation of IRE1 can result in the phosphorylation of JNK, which promotes apoptosis (47); however, strikingly the phosphorylation of both JNK isoforms p54 and p46 was nearly completely abolished in patient cells in ‘Galactose’ (Fig. 5B), and significantly, the transcripts of at least seven dual specific phosphatases (DUSPs) responsible for JNK, ERK and p38 dephosphorylation were upregulated (Fig. 5D). The activation of IRE1 was associated with the increased translation of several ER quality control genes including transcription factors ATF3 and ATF4, proteases, CHOP/DDIT3 and the cellular oxygen sensor EGLN3 (Fig. 5A, B, D).
Figure 5 .
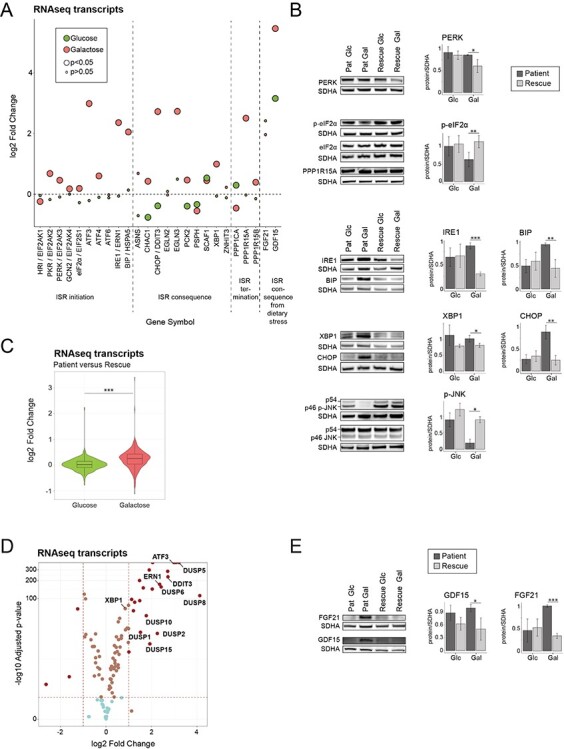
Metabolic dysfunction leads to a canonical ER stress response in patient cells in galactose. (A) Dot plot representing the fold change of the transcripts of ISR in the ER. Fold change is represented in log2 scale. Large circles represent significant values (FDR < 0.05), small circles show non-significant values (FDR > 0.05). ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (B) Immunoblot analysis of the proteins involved in the integrated stress response of the ER. Whole cell extracts from patient and rescue fibroblasts grown in glucose or galactose for 2 days were separated by SDS-PAGE and probed with antibodies against indicated proteins. SDHA was used as a loading control. Bar plot indicates the quantification of four (PERK), 7 (P-eIF2alpha), and three (IRE1, BIP, XBP1, CHOP, p-JNK) immunoblot analyses, normalized to SDHA, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001. (C) Violin plot representing the distribution of the log2 fold change (patient/rescue) for transcripts involved in translation for the glucose and galactose condition. Box plots indicate the median and the first and third quartile. Wilcoxon statistical test, ***P < 0.001. (D) Volcano plot representing the results of differential gene expression analysis between patient and rescue cells in galactose for proteins involved in the ISR in the ER. x-axis values represent the log2 fold change and y-axis values represent the -log10 of the adjusted P-value. Selected genes of interest are labelled. (E) Immunoblot analysis of GDF15 and FGF21. Whole cell extracts from patient and rescue fibroblasts grown in glucose or galactose for 2 days were separated by SDS-PAGE and probed with antibodies against GDF15 and FGF21. SDHA was used as a loading control. Bar plot indicates the quantification of 4 (FGF21) and 3 (GDF15) immunoblot analyses, normalized to SDHA, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001.
Two metabolic cytokines, fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15), associated with mitochondrial disease and proposed as potential biomarkers, are often activated concomitantly with stress responses (48,49). Protein levels of both FGF21 and GDF15 as well as transcript levels of GDF15 were upregulated (Fig. 5A and E). GDF15 transcript levels were upregulated even in ‘Glucose’ (Fig. 5A); however, the upregulation of all the components of the UPR was markedly enhanced in ‘Galactose’ by comparison (Figs 5A, Supplementary Material, Fig. S4A).
Metabolic dysfunction leads to an activation of the mitochondrial UPR
The upregulation of the UPR in the ER as well as the increase of FGF21 and GDF15 led us to investigate the mitochondrial UPR in more detail (Fig. 6A). SIRT3, a mitochondrial NAD+-dependent protein deacetylase that affects gene expression in the nucleus (50), interacts with FOXO3 to activate anti-oxidant genes like superoxide dismutase 2 (SOD2) and catalase (CAT) (51). Although transcript levels of CAT and SOD2 were reduced in the patient (Fig. 6A), the level of CAT protein was increased (Fig. 6B). NAD+-dependent protein deacetylase SIRT7, which increases nuclear respiratory factor 1 (NRF1) activity, and alleviates mitochondrial stress by reducing the expression of mitochondrial translation proteins (52), was increased in our model. ATF4 and ATF5 are transcription factors that activate the mitochondrial UPR (53), and both were increased in patient cells in ‘Galactose’ (Fig. 6A). We also saw a marked increase in the transcript level for PPARGC1A, the transcription coactivator peroxisome proliferation-activated receptor gamma, coactivator 1 alpha (also known as PGC-1α), which enhances oxidative phosphorylation, mitochondrial biogenesis, antioxidant enzyme expression and mitochondrial fatty acid oxidation (Fig. 6A) (54,55), although components of fatty acid catabolic pathway were transcriptionally downregulated (Fig. 1C). Downstream targets of the mitochondrial UPR, including proteases CLPP, HTRA2 and LONP1 and the heat shock proteins HSP70 (HSPA9), HSP60 (HSPD1) and HSP10 (HSPE1) were upregulated at the transcript and/or the protein level, here too, the changes were greater in ‘Galactose’ (Supplementary Material, Fig. S5). In order to clear the cells of malfunctioning organelles, autophagy is generally upregulated during the cellular stress response. In our model, we could identify an upregulation of autophagy markers p62 (SQSTM1) and LC3 (MAP1LC3A/B) at the transcript and the protein level for the patient in ‘Galactose’ (Fig. 6C). This type of increase, particularly in the levels of LC3B is indicative of a higher autophagic flux, which potentially clears damaged organelles in the patient cells (56). Lastly, we identified and increase in apoptotic cleavage of PARP and caspase 3 in patient cells in ‘Galactose’ (Fig. 6D).
Figure 6 .
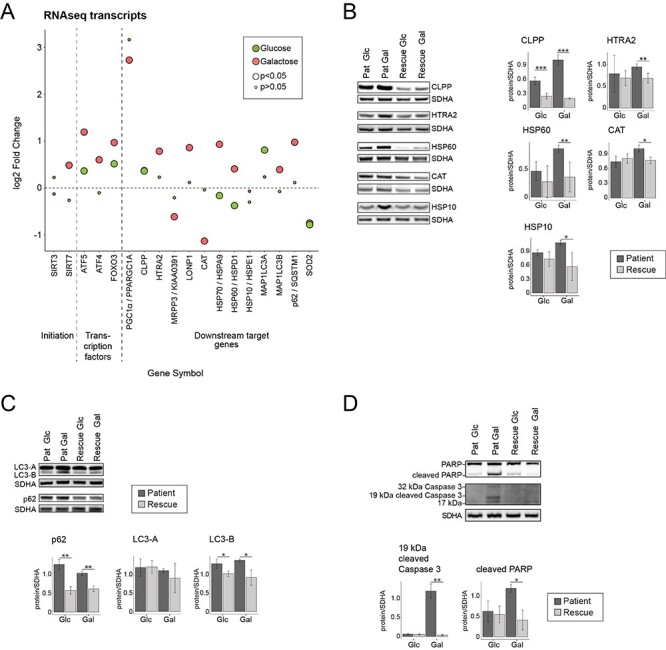
Metabolic dysfunction leads to an activation of the mitochondrial unfolded protein response (mtUPR) and increased autophagy. (A) Dot plot representing the fold change of the transcripts of mitochondrial integrated protein response. Fold change is represented in log2 scale. Large circles represent significant values (FDR < 0.05), small circles show non-significant values (FDR > 0.05). ‘Glucose’ (green) represents the comparison of patient versus rescue cells in glucose. ‘Galactose’ (red) represents the comparison of patient versus rescue cells in galactose. (B) Immunoblot analysis of the proteins involved in mitochondrial integrated stress response. Whole cell extracts from patient and rescue fibroblasts grown in glucose or galactose for 2 days were separated by SDS-PAGE and probed with antibodies against indicated proteins. SDHA was used as a loading control. Bar plot indicates the quantification of three (CLPP, HSP10, CAT), 4 (HTRA2, HSP60) immunoblot analyses, normalized to SDHA, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001. (C) Immunoblot analysis of the proteins involved in autophagy. Whole cell extracts from patient and rescue fibroblasts grown in glucose or galactose for 2 days were separated by SDS-PAGE and probed with antibodies against indicated proteins. SDHA was used as a loading control. Bar plot indicates the quantification of three (LC3-A, LC3-B, p62) immunoblot analyses, normalized to SDHA, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001. (D) Immunoblot analysis of the proteins involved in apoptosis. Whole cell extracts from patient and rescue fibroblasts grown in glucose or galactose for 2 days were separated by SDS-PAGE and probed with antibodies against indicated proteins. SDHA was used as a loading control. Bar plot indicates the quantification of three (cleaved PARP) and four (cleaved CC3) immunoblot analyses, normalized to SDHA, where patient is depicted in dark grey and rescue in light grey, *P < 0.05, **P < 0.01, ***P < 0.001.
In summary, we identified the activation of the ER UPR through the IRE1/XBP1 axis, but not the ISR that is mediated through phosphorylation of eIF2alpha in patient cells in ‘Galactose’. We identified an upregulation at the transcript and protein level of specific downstream targets including CHOP, CLPP, HTRA2, HSP60, HSP10, FGF21 and GDF15 acting to rescue the patient cells in galactose, and lastly, increased autophagic and apoptotic flux possibly induced by CHOP.
Discussion
This study illustrates the metabolic consequences of energetic stress in fibroblast cells harbouring the CHCHD10 p.R15L pathogenic variant. This variant causes a fundamental metabolic reorganization and an activation of the UPR in both the ER and mitochondria in response to a nutrient stress. The UPR response in the ER is activated through IRE1/XBP1 pathway and the mitochondrial UPR through upregulation of the transcription factors ATF4 and ATF5, both presumably acting as pro-survival mechanisms. In addition, the MAP kinase JNK was almost completely unphosphorylated, likely the result of the upregulation of several dual specific protein phosphatases whose substrates include ERK, JNK and p38 (57), obviating pro-apoptotic or inflammatory pathways. A previous study of cybrid cells harbouring mutations in the mitochondrially coded ND1 gene showed that activation of an IRE1-mediated stress response resulted in activation of JNK and p38, and that this was associated with cell death in galactose medium (58). Pharmacologic suppression of MAP kinase activation rescued the cell death phenotype, supporting the notion that a concerted mechanism to dephosphorylate MAP kinases in our model may indeed be a pro-survival strategy.
While the existence of a so-called classical mitochondrial UPR has been well-described in C. elegans, its nature, and indeed its existence in mammals, has been the subject of considerable debate (59) since the first description of the pathway in response to expression of a misfolded mitochondrial protein in a cell culture system (60). It has recently been argued that the mammalian mitochondrial UPR is activated downstream of the ISR (59). The ISR is an evolutionarily conserved signalling network that integrates inputs from a variety of cell stresses including unfolded proteins, viral infection, nutrient deprivation and hypoxia culminating in the phosphorylation but eIF2alpha by one of four specific kinases (61). This prioritizes selective translation of a set of stress response genes with upstream open reading frames (uORFS), such as the transcription factors ATF4 and ATF5. Induction of an ISR has been observed in the context of mitochondrial dysfunction in many cell and animal models (14,62–65). A comprehensive study of mitochondrial dysfunction in Hela cells demonstrated that ATF4 was the master regulator, responsible for transcriptional activation of most target stress genes (62). The triggers for induction of the ISR have been shown to vary, depending on the nature of the mitochondrial defect and the metabolic state of the cell. For instance, in myoblasts treated with OXPHOS inhibitors the ISR is induced by aspartate depletion and activation of the eIF2alpha kinase GCN2, while in myotubes, it is induced in response to hyperpolarization of the inner mitochondrial membrane (63).
Although the nutrient stress in CHCHD10 p.R15L patient cells elicited an upregulation of stress proteins associated with the UPR in both the ER and mitochondria characteristic of an ISR, unexpectedly this was associated with a small but significant decrease in phosphorylated eIF2alpha in patient cells grown in galactose. Consistent with this, we also observed transcriptional upregulation of the regulatory subunit (GADD34) of the protein phosphatase (PPI) that regulates eIF2alpha dephosphorylation. While we have not evaluated the impact of these responses on global cellular translation, it appears that phosphorylation of eIF2alpha, or at least an increase of eIF2alpha phosphorylation, is not a sine qua non for selective translation of the proteins involved in the stress responses related to mitochondrial dysfunction in the cells we studied. The IRE1 pathway, which is one of three different pathways that transduce the ER unfolded protein response (45) appears to be the main sensor of the nutrient-induced stress in the CHCHD10 patient cells we studied. The activation of IRE1 leads to the splicing of XBP1, which renders the protein transcriptionally active and initiates the upregulation of stress target proteins required for protein folding, maturation and degradation, therefore controlling a subset of UPR target genes (66). Moreover, IRE1 promotes a process called ‘regulated IRE1-dependent decay (RIDD), which degrades mRNAs of ER-targeted proteins to reduce the load of incoming proteins into the ER (67). Activation of the IRE1/XBP1 axis does not appear to be a general response to mitochondrial energetic dysfunction as it was not observed in other cellular models in which energetic stress was induced by the application of a mitochondrial uncoupler, mitochondrial translation inhibition, or by application of specific inhibitors of the OXPHOS complexes (62,63).
Recently, two groups reported the activation of the ISR for both the CHCHD10 variant p.S59L (19) and the double knock-out of CHCHD10 and CHCHD2 (68) in mouse models. In the knock-in model, CHCHD10 p.S59L accumulated in protein aggregates and induced activation of mTORC1, an increase in transcription factors specific for the ISR, the secretion of metabolic cytokines FGF21 and GDF15, the upregulation of the serine and one-carbon metabolism, and the downregulation of the respiratory chain complexes in the heart where these effects were examined (19). Although the p.S59L CHCHD10 variant was proposed to act by a dominant gain of function, and the double-knock-out through a complete loss of function, both models showed activation of the ISR. Our results on the haploinsufficient p.R15L CHCHD10 variant were similar in that we observed secretion of the two metabolic cytokines, upregulation of serine and one carbon metabolism, induction of the ATF transcription factors, and expression of characteristic stress survival proteins, all of which have been observed previously in models of mitochondrial dysfunction. However, we did not identify an activation of mTORC1, but rather an inhibition through the phosphorylation of AMPK. Interestingly, AMPK was previously shown to be activated in TDP-43 mouse spinal cords, and reducing AMPK activity led to an increase in survival of motor neurons (69).
We observed increased autophagic and apoptotic flux, possibly mediated by increased levels of CHOP, but not phosphorylated JNK. Phosphorylated JNK is involved in apoptosis through the phosphorylation of BcL2 family of proteins (70), and JNK1 and JNK2 double knock-out cells are resistant to apoptosis. CHCHD10 mutant cells have also been found to be less sensitive to apoptotic cell death stimuli (71,72). The nearly complete dephosphorylation of JNK was an unexpected finding in patient cells grown in galactose, as was the upregulation of several dual specific phosphatases, whose substrate is JNK (73). To our knowledge, this has not been previously reported in the context of mitochondrial dysfunction, and the regulation of this presumably pro-survival strategy deserves further exploration.
Despite numerous functional studies on pathogenic variants, the normal physiological role of CHCHD10 remains unknown. The deletion of the yeast ortholog Mix17 in S. cerevisiae results in the reduction of mitochondrial oxygen consumption (74), implicating it in some aspect of oxidative metabolism and our findings implicate an essential role of CHCHD10 in mitochondrial energy metabolism, directly affecting the mitochondrial respiratory chain. A link between energy metabolism and ALS has long been established, and reduced oxidative phosphorylation has been described previously for other genetic models of ALS (75–78). In human cells, siRNA mediated knockdown of CHCHD10 does not result in a destabilization of OXPHOS complexes (79); however, two CHCHD10 variants have been reported to alter the OXPHOS complex assembly/stability. The CHCHD10 p.S59L variant was shown to destabilize complex V and lead to its disassembly (2), and we previously showed that the complex I defect in p.R15L patient fibroblasts was phenocopied by the CRISPR-Cas9 mediated knockout of CHCHD10 (15). Our proteomics data confirmed the downregulation of complex I subunits in patient cells in both ‘Glucose’ and ‘Galactose’ (Figs 4B and Supplementary Material, Fig. S3) and transcripts for several factors involved in the complex I and IV assembly in patient cells in ‘Galactose’ (Fig. 1C); however, the precise molecular function of CHCHD10 in the assembly of the OXPHOS complexes remains unknown and will require further detailed study.
The ratio between the oxidized and reduced form (NADH) influences many cellular and mitochondrial processes (80). Balanced levels of NAD+ protect and sustain basal metabolic function and health in neurons. In our model, levels of NADH were upregulated in the patient, whereas NAD+ levels were unchanged, resulting in an increased NADH/NAD+ ratio, a universal feature of respiratory chain dysfunction. A recent study showed that the accumulation of NADH due to ongoing production through the enzymatic reactions of MTHFD2/MTHFD2L in mitochondria was toxic to cells with an impaired electron transport chain (81). We observed an increase in transcripts of enzymes of one-carbon metabolism and an increase in total NADH, suggesting that normalization of NADH could be a potential target for rescuing the phenotype of CHCHD10 related disorders. Expression of a cytoplasmic or mitochondrially targeted NADH oxidase was shown to suppress the ISR in cells targeted with complex I or IV inhibitors (63); however, in our hands, expression of a mitochondrially-targeted NADH oxidase (82) failed to rescue either the cell death or the proliferation phenotype in patient cells in ‘Galactose’.
In summary, our study describes how the CHCHD10 p.R15L variant affects mitochondrial metabolism, especially under nutrient stress conditions (Fig. 7). The complex I deficiency leads to an increased NADH/NAD+ ratio, which is exacerbated in ‘Galactose’, a condition that forces the cell to rely solely on mitochondria for energy production. The cell responds to the ensuing proliferation defect by synthesising aspartate from malate through GOT1, and the upregulation of the mitochondrial folate metabolism to supply formate to the cytosol. A high AMP/ATP ratio, results in increased phosphorylation of AMPK, activating catabolic processes on one hand and deactivating mTORC1 on the other. These metabolic changes induce the UPR in the ER through the IRE1/XBP1 axis and the UPR in mitochondria through ATF4 and ATF5. Finally, the patient cells either survive the existing nutrient stress by adapting their metabolism over time or undergo apoptosis, through a JNK-independent mechanism. We speculate, that motor neurons, the cells that are specifically targeted in ALS, are more susceptible to mitochondrial dysfunction and potentially suffer damage much earlier in limited nutrient conditions than do fibroblasts. It is known that ageing results in decreased neuronal uptake of glucose, therefore rendering motor neurons harbouring CHCHD10 p.R15L variant more susceptible to cell death in the ageing individual (83). We therefore propose the careful consideration of targeting mitochondrial metabolism and the UPR response for potential treatments in ALS models of CHCHD10.
Figure 7 .
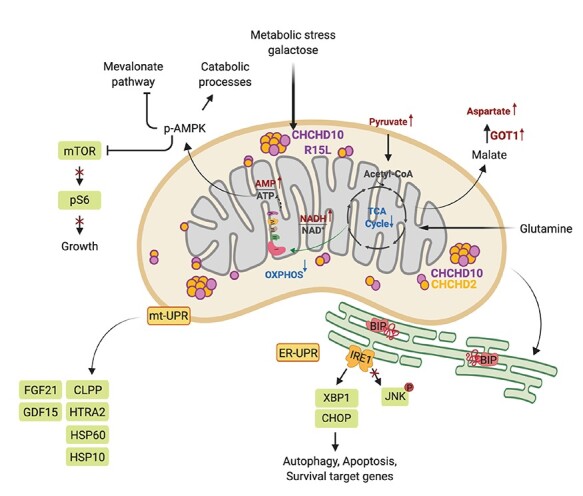
Summary of metabolic responses to the energetic challenge in patient fibroblasts. Mitochondrial metabolism and energy production plays a key role in cellular homeostasis. The CHCHD10 p.R15L variant leads to a complex I deficiency, resulting in an increase in the NADH/NAD+ ratio and a downregulation of the TCA cycle. Under nutrient stress, the energy deficit in leads to an increase in the AMP/ATP ratio, increased phosphorylation of AMPK and the subsequent activation of catabolic pathways and the downregulation of the growth targeted mTOR pathway. This metabolic stress results in the activation of the UPR of the ER through IRE1/XBP1. The upregulation of different proteases and heat shock proteins, and the metabolic cytokines FGF21 and GDF15, demonstrates the simultaneous activation of the mtUPR. Finally, patient cells show increased cell death through apoptotic processes independent of JNK. This scheme was created with BioRender.
Materials and Methods
Human studies
The investigation of cell lines was approved by the institutional review board of the Montreal Neurological Institute, McGill University.
Cell lines and media
A primary fibroblast culture was previously established from a heterozygous carrier of c.44G > T (p.R15L) (patient DNA #8807) diagnosed with sporadic ALS at age 54 (involving his upper limb) and died at the age of 69 (6,15). The variant CHCHD10 p.R15L, has been associated with ALS in several independent studies (1,4,6). Fibroblasts stably overexpressing CHCHD10 in patient cells (rescue) were engineered using retroviral vectors as described previously (15,84). Cells were cultivated in 4.5 g/l glucose DMEM supplemented with 10% foetal bovine serum (FBS) and 5% pen/strep, at 37°C in an atmosphere of 5% CO2. For carbon source-dependent experiments, fibroblasts were cultivated for two days in DMEM with 10% dialyzed FBS supplemented with either 4.5 g/l glucose or 4.5 g/l galactose before harvesting for ‘omics’ experiments.
Mitochondrial isolation
Fibroblasts were washed twice with phosphate buffered saline (PBS), resuspended in ice-cold buffer (250 mM sucrose, 10 mM Tris–HCl, pH 7.4), and homogenized by nitrogen cavitation under 500 psi pressure for 5 min. A post-nuclear supernatant was obtained by centrifugation of the samples twice for 10 min at 600 × g. Mitochondria were pelleted by centrifugation for 10 min at 10 000 × g and washed once in the same buffer. Protein concentration was determined using the Bradford assay.
Antibodies
Antibodies directed against the following proteins were used in this study: SDHA (abcam, ab14715), AMPK (Cell Signaling, 2532), p-AMPK (Thr172) (Cell Signaling, 2535), AKT (Cell Signaling, 9272S), p-AKT (Ser473) (Cell Signaling, 4060S), mTOR (Cell Signaling, 2983P), p-mTOR (Ser2448) (Cell Signaling, 5536P), S6 (Cell Signaling, 2217), p-S6 (Ser235/236) (Cell Signaling, 4858), PERK (Cell Signaling, 5683), p-PERK (Thr980) (Cell Signaling, 3191), GCN2 (Cell Signaling, 3302), p-GCN2 (Thr899) (Abcam, ab75836), eIF2α (Cell Signaling, 2103), p-eIF2α (Ser 51) (SIGMA, SAB4504388), GADD34/PPP1R15A (Abclonal, A17117), IRE1/ERN1 (Cell Signaling, 3294), BIP (abcam, ab21685), XBP1 (Santa Cruz, sc-7160), CHOP/DDIT3 (Cell Signaling, 5554), JNK (Cell Signaling, 9252), p-JNK/SAPK (T183/Y185) (Cell Signaling, 9251), FGF21 (abcam, ab171941), GDF15 (abcam, ab106006), CLPP (Proteintech, 15 698–1-1AP), HTRA2 (Proteintech, 15 775–1-AP), HSP60 (Santa Cruz, sc-136 291), CAT (Abclonal, A11777), HSP10 (Santa Cruz, SC-376313), LC3 (Cell Signaling, D3U4C), SQSTM1/p62 (Cell Signaling, 8025), PARP (Cell Signaling, 9542), cleaved PARP (Cell Signaling, 9541), cleaved caspase 3 (Cell Signaling, 9661S).
SDS-PAGE and immunoblot analysis
Cells were pelleted and lysed with 1.5% n-dodecyl-D-maltoside (DDM) in PBS with cOmplete™ protease inhibitor (Roche) for 15 min on ice. Cells were centrifuged at 20 000 × g for 20 min at 4°C. Protein concentration was determined using Bradford assay. 2× Laemmli buffer was added to 20–30 μg of protein and denatured at 55°C for 15 min. The mixture was run on denaturing 8–12.5% polyacrylamide gels. Separated proteins were transferred onto a nitrocellulose membrane and immunoblot analysis was performed with the indicated antibodies in 5% milk in Tris-buffered saline and Tween 20 (TBST).
RNA extraction and RNA sequencing
To capture the transcriptome, total RNA from patient fibroblasts was extracted and purified by using the RNeasy Mini kit (QIAGEN). Three independent biological replicates were collected of the four chosen conditions, patient in glucose and galactose, rescue in glucose and galactose. RNA quality was tested on a 1% agarose gel and subsequently mRNA sequencing was performed via polyA selection on an Illumina HiSeq 4000 instrument by GENEWIZ, LLC. (South Plainfield, NJ, USA). RNA samples received were quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA), and RNA integrity was checked with 4200 TapeStation (Agilent Technologies, Palo Alto, CA, USA). RNA sequencing library preparation used the NEBNext Ultra RNA Library Prep Kit for Illumina followed by manufacturer’s instructions (NEB, Ipswich, MA, USA). Briefly, mRNA was first enriched with Oligod(T) beads. Enriched mRNAs were fragmented for 15 minutes at 94°C. First strand and second strand cDNA were subsequently synthesized. cDNA fragments were end repaired and adenylated at 3’ends, and universal adapters were ligated to cDNA fragments, followed by index addition and library enrichment by PCR with limited cycles. The sequencing library was validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA) and quantified by using Qubit 2.0 Fluorometer as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, USA). The sequencing libraries were clustered on a single lane of a flow cell. After clustering, the flow cell was loaded on the Illumina HiSeq instrument according to manufacturer’s instructions. The samples were sequenced using a 2×150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS). Raw sequence data (.bcl files) generated from Illumina HiSeq was converted into fastq files and de-multiplexed using Illumina’s bcl2fastq 2.17 software. One mismatch was allowed for index sequence identification.
Transcriptomic analysis
Raw data files from RNA sequencing were analyzed through Genpipes, a Python-based multi-step computational pipeline (85). Paired-ends sequencing reads were clipped for adapter sequence, trimmed for minimum quality (Q30) in 3′ and filtered for minimum length of 32 bp using Trimmomatic (86). Surviving read pairs were aligned to the reference genome, Homo sapiens (GRCh37, Ensembl) genome assembly, using the two-pass method of STAR (87). A gene-level count-based gene quantification against Ensembl annotations was performed using HT-seq count in the intersection-nonempty mode (88). Transcript-level assembly, quantification and differential expression analysis was performed using Cufflinks (89) and Cuffdiff (90). Exploratory analysis was conducted using various functions and packages from R and Bioconductor, differential expression with DEseq (91). Groups were defined as follows: (1) ‘Glucose’: patient in glucose versus rescue in glucose, (2) ‘Galactose’: patient in galactose versus rescue in glucose, (3) ‘Patient’: patient in galactose versus patient in glucose and (4) ‘Rescue’: rescue in galactose versus rescue in glucose. The Wald test was used to generate P-values and log2 fold changes. Genes with an adjusted P-value <0.05 and absolute log2 fold change >1 were considered as differentially expressed genes. RNA sequencing data was deposited to NCBI Gene Expression Omnibus (GEO), GSE144725.
Gene ontology analysis
Gene ontology (GO) analysis of significant differentially expressed transcripts was performed using the annotation tool ENRICHR (https://amp.pharm.mssm.edu/Enrichr/, GO_BP 2018), focusing on the GO term of biological processes (92,93). As an input, gene symbols of genes passing the cut off of log2 fold change of ≷ ± 0.3 were used. A maximum P-value of 0.05 was chosen to select only significant enrichment.
Metabolite extraction and analysis
Metabolite extraction was performed according to the protocol of Human Metabolome Technologies, Inc (HMT). Briefly, 5 million cells were seeded in four replicates for each condition, one for counting and three for the collection of intracellular metabolites in three independent biological replicates. The culture medium was aspirated from each culture plate and cells were washed twice with washing buffer (5% w/w mannitol solution). The washing buffer was aspirated and methanol (LC/MS grade) together with an internal standard solution (H3304–1002, Human Metabolome Technologies, Inc., Tsuruoka, Japan) was added to extract metabolites. Cell extracts were centrifuged for 5 min at 2300 × g at 4°C. The supernatant was transferred into ultracentrifuge filter cups (5 kDa) and centrifuged at 9000 × at 4°C for 3 hours. The tube was set up in a centrifugal evaporator for 6 hours to evaporate the supernatant and obtain the metabolites as precipitate. The dried pellet was re-suspended in 50 μL of Milli_Q water for ce-MS analysis by HMT and processed. Cationic compounds were measured in the positive mode of ce-TOFMS and anionic compounds were measured in the positive and negative modes of ce-MS/MS according to the methods developed by Soga et al. (94–96). Peaks detected by ce-TOFMS and ce-MS/MS were extracted using automatic integration software (MasterHands, Keio University, Tsuruoka, Japan (97) and MassHunter Quantitative Analysis B.04.00, Agilent Technologies, Santa Clara, CA, USA, respectively) in order to obtain peak information including m/z, migration time (MT) and peak area. The peaks were annotated with putative metabolites from the HMT metabolite database based on their MTs in ce and m/z values determined by TOFMS. The tolerance range for the peak annotation was configured at ±0.5 min for MT and ± 10 ppm for m/z. In addition, concentrations of metabolites were calculated by normalizing the peak area of each metabolite with respect to the area of the internal standard and by using standard curves, which were obtained by three-point calibrations.
Metabolic pathway analysis was performed using the integrative tool for joint pathway analysis in the MetaboAnalyst 4.0 platform (http://www.metaboanalyst.ca; (98)). Metabolites and proteins were selected with a cut-off of log2 fold change of ≷ ± 0.2 and a P-value <0.05. Selected parameters were: hypergeometric test for enrichment analysis, degree centrality for topology analysis, metabolic pathways for pathway database and ‘combine query’ as the integration method.
Proteome extraction and analysis
Fibroblasts were grown to 90% confluency in 15 cm dishes to lead to a minimum of 2 × 106 cells per condition. Cells were rinsed 2–3 times with 1× PBS to remove cell culture media and pelleted at 600 × g. Mitochondria were isolated as described above and lysed with lysis buffer (50 mM HEPES, 1% SDS, 150 mM NaCl). The lysate was centrifuged at 16 000 × g for 10 minutes at 4°C. Protein concentration was determined in the supernatant by BCA Protein Assay Kit (Thermo Fisher Scientific, #23227). About, 100 μg of protein was transferred into a new tube and the final volume was adjusted to 100 μL with 100 mM triethylammonium bicarbonate (TEAB). About, 5 μL of 200 mM Tris(2-carboxyethyl)phosphine) (TCEP) was added and samples were incubated at 55°C for 1 hour. About, 5 μL of 375 mM iodoacetamide was added to the mix and let stand for 30 minutes protected from light at room temperature. About, 600 μL of pre-chilled acetone (−20°C) was added and the mix was put at −20°C over-night to precipitate. Samples were centrifuged at 8000 × g for 10 minutes at 4°C. The tubes were carefully inverted to decant the acetone and let dry for 30 min at room temperature. Samples were digested, labelled and analysed by mass spectrometry on an Orbitrap Fusion instrument (Thermo Scientific), which was operated in data-dependent acquisition (DDA) and multi-notch MS3 mode at the Institute de Recherches Cliniques (IRCM) de Montreal. The isobaric labelling of the peptides was performed using TMT 10-plex labelling reagents (Thermo Fisher Scientific). The patient cells in glucose and the rescue cells in galactose were measured with three independent biological replicates, the patient cells in galactose and the rescue cells in glucose were measured in two independent biological replicates. Peptide identification and quantification was done with Proteome Discoverer version 2.1.0.81. The protein database used for the analysis was downloaded from Uniprot on 4 April 2015. Differentially expressed proteins were identified by performing Welch’s t-statistic test, comparing patient cells with rescue cells in glucose and galactose. Values were normalized by subtraction of the mean from the log2 values of the comparison ratios. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018806 and 10.6019/PXD018806.
Bioinformatic and statistical analysis
Pathways, transcripts and proteins selected in each condition were filtered after Benjamini-Hochberg correction at an adjusted P-value <0.05 (FDR 5%). All data analysis and plots were generated using R and RStudio and modified using Adobe Illustrator CC. For the rest of the analysis, data were expressed as mean ± SEM, and P-values were calculated using two-tailed Welch’s t-test for pairwise comparisons of metabolites and proteins and the Wald as well as the Wilcoxon test for transcripts. Statistical tests were performed using RStudio and GraphPad Prism 6.0 (*P < 0.05; **P < 0.01; ***P < 0.001; n ≥ 3).
Supplementary Material
Acknowledgements
We thank Hana Antonicka, Aurel Besse-Patin and Mari Aaltonen for fruitful discussions and critique of the manuscript and Logan Walsh for the help with the analysis of the RNAseq data. We also wish to thank Denis Faubert and his team at the IRCM for the collaboration with the mass spectrometry analysis.
Conflict of Interest statement. The authors report no conflict of interest.
Funding
This work was supported by grants from the Canadian Institutes of Health Research, Parkinson Canada and the Muscular Dystrophy Association to E.A.S. I.S. was financially supported by fellowships from Parkinson Canada and Healthy Brain for Healthy Lifes (HBHL, McGill University).
Author contribution
I.R. planned and executed all the experiments and wrote the manuscript. W.W. executed several immunoblot experiments. E.A.S. supervised the project and wrote the manuscript.
References
- 1. Muller, K., Andersen, P.M., Hubers, A., Marroquin, N., Volk, A.E., Danzer, K.M., Meitinger, T., Ludolph, A.C., Strom, T.M. and Weishaupt, J.H. (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain, 137, e309. [DOI] [PubMed] [Google Scholar]
- 2. Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E.C., Lacas-Gervais, S., Fragaki, K., Berg-Alonso, L., Kageyama, Y., Serre, V., Moore, D.G.et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain, 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chaussenot, A., Le Ber, I., Ait-El-Mkadem, S., Camuzat, A., deSeptenville, A., Bannwarth, S., Genin, E.C., Serre, V., Auge, G., French research network on, F.T.D et al. (2014) Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging, 35, 2884.e1-2884.e4. [DOI] [PubMed] [Google Scholar]
- 4. Johnson, J.O., Glynn, S.M., Gibbs, J.R., Nalls, M.A., Sabatelli, M., Restagno, G., Drory, V.E., Chio, A., Rogaeva, E. and Traynor, B.J. (2014) Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain, 137, e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ronchi, D., Riboldi, G., Del Bo, R., Ticozzi, N., Scarlato, M., Galimberti, D., Corti, S., Silani, V., Bresolin, N. and Comi, G.P. (2015) CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain, 138, e372. [DOI] [PubMed] [Google Scholar]
- 6. Zhang, M., Xi, Z., Zinman, L., Bruni, A.C., Maletta, R.G., Curcio, S.A., Rainero, I., Rubino, E., Pinessi, L., Nacmias, B.et al. (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain, 138, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dols-Icardo, O., Nebot, I., Gorostidi, A., Ortega-Cubero, S., Hernandez, I., Rojas-Garcia, R., Garcia-Redondo, A., Povedano, M., Llado, A., Alvarez, V.et al. (2015) Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain, 138, e400. [DOI] [PubMed] [Google Scholar]
- 8. Zhou, Q., Chen, Y., Wei, Q., Cao, B., Wu, Y., Zhao, B., Ou, R., Yang, J., Chen, X., Hadano, S.et al. (2017) Mutation screening of the CHCHD10 gene in Chinese patients with amyotrophic lateral sclerosis. Mol. Neurobiol., 54, 3189–3194. [DOI] [PubMed] [Google Scholar]
- 9. Chio, A., Mora, G., Sabatelli, M., Caponnetto, C., Traynor, B.J., Johnson, J.O., Nalls, M.A., Calvo, A., Moglia, C., Borghero, G.et al. (2015) CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging, 36, 1767.e3-1767.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen, S., He, J., Tang, L., Zhang, N. and Fan, D. (2017) CHCHD10 mutations in patients with amyotrophic lateral sclerosis in mainland China. Neurobiol. Aging, 54, 214.e7-214.e10. [DOI] [PubMed] [Google Scholar]
- 11. Ryan, M., Zaldivar Vaillant, T., McLaughlin, R.L., Doherty, M.A., Rooney, J., Heverin, M., Gutierrez, J., Lara-Fernandez, G.E., Pita Rodriguez, M., Hackembruch, J.et al. (2019) Comparison of the clinical and genetic features of amyotrophic lateral sclerosis across Cuban, Uruguayan and Irish clinic-based populations. J. Neurol. Neurosurg. Psychiatry, 90, 659–665. [DOI] [PubMed] [Google Scholar]
- 12. Lehmer, C., Schludi, M.H., Ransom, L., Greiling, J., Junghanel, M., Exner, N., Riemenschneider, H., van derZee, J., Van Broeckhoven, C., Weydt, P.et al. (2018) A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med., 10, e8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kurzwelly, D., Kruger, S., Biskup, S. and Heneka, M.T. (2015) A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain, 138, e376. [DOI] [PubMed] [Google Scholar]
- 14. Khan, N.A., Nikkanen, J., Yatsuga, S., Jackson, C., Wang, L., Pradhan, S., Kivela, R., Pessia, A., Velagapudi, V. and Suomalainen, A. (2017) mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab., 26, 419–428 e415. [DOI] [PubMed] [Google Scholar]
- 15. Straub, I.R., Janer, A., Weraarpachai, W., Zinman, L., Robertson, J., Rogaeva, E. and Shoubridge, E.A. (2018) Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet., 27, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang, X., Wu, B.P., Nguyen, D., Liu, Y.T., Marani, M., Hench, J., Benit, P., Kozjak-Pavlovic, V., Rustin, P., Frank, S.et al. (2018) CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet., 27, 3881–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Genin, E.C., Bannwarth, S., Lespinasse, F., Ortega-Vila, B., Fragaki, K., Itoh, K., Villa, E., Lacas-Gervais, S., Jokela, M., Auranen, M.et al. (2018) Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis., 119, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Genin, E.C., Madji Hounoum, B., Bannwarth, S., Fragaki, K., Lacas-Gervais, S., Mauri-Crouzet, A., Lespinasse, F., Neveu, J., Ropert, B., Auge, G.et al. (2019) Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10(S59L/+) mouse. Acta Neuropathol., 138, 123–145. [DOI] [PubMed] [Google Scholar]
- 19. Anderson, C.J., Bredvik, K., Burstein, S.R., Davis, C., Meadows, S.M., Dash, J., Case, L., Milner, T.A., Kawamata, H., Zuberi, A.et al. (2019) ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol., 138, 103–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brockmann, S.J., Freischmidt, A., Oeckl, P., Muller, K., Ponna, S.K., Helferich, A.M., Paone, C., Reinders, J., Kojer, K., Orth, M.et al. (2018) CHCHD10 mutations p.R15L and p.G66V cause motoneuron disease by haploinsufficiency. Hum. Mol. Genet., 27, 706–715. [DOI] [PubMed] [Google Scholar]
- 21. Penttila, S., Jokela, M., Bouquin, H., Saukkonen, A.M., Toivanen, J. and Udd, B. (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol., 77, 163–172. [DOI] [PubMed] [Google Scholar]
- 22. Hetz, C. and Mollereau, B. (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci., 15, 233–249. [DOI] [PubMed] [Google Scholar]
- 23. Hughes, D. and Mallucci, G.R. (2019) The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J., 286, 342–355. [DOI] [PubMed] [Google Scholar]
- 24. Jaronen, M., Goldsteins, G. and Koistinaho, J. (2014) ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front. Cell. Neurosci., 8, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ito, Y., Yamada, M., Tanaka, H., Aida, K., Tsuruma, K., Shimazawa, M., Hozumi, I., Inuzuka, T., Takahashi, H. and Hara, H. (2009) Involvement of CHOP, an ER-stress apoptotic mediator, in both human sporadic ALS and ALS model mice. Neurobiol. Dis., 36, 470–476. [DOI] [PubMed] [Google Scholar]
- 26. Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., Martinez, G., Cuervo, A.M., Brown, R.H. and Glimcher, L.H. (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev., 23, 2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Medinas, D.B., Gonzalez, J.V., Falcon, P. and Hetz, C. (2017) Fine-tuning ER stress signal transducers to treat amyotrophic lateral sclerosis. Front. Mol. Neurosci., 10, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Calvo, S.E., Clauser, K.R. and Mootha, V.K. (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res., 44, D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramsden, D.B., Ho, P.W., Ho, J.W., Liu, H.F., So, D.H., Tse, H.M., Chan, K.H. and Ho, S.L. (2012) Human neuronal uncoupling proteins 4 and 5 (UCP4 and UCP5): structural properties, regulation, and physiological role in protection against oxidative stress and mitochondrial dysfunction. Brain Behav., 2, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoang, T., Smith, M.D. and Jelokhani-Niaraki, M. (2012) Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4 and UCP5. Biochemistry, 51, 4004–4014. [DOI] [PubMed] [Google Scholar]
- 31. Thangaratnarajah, C., Ruprecht, J.J. and Kunji, E.R. (2014) Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat. Commun., 5, 5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinez-Reyes, I. and Chandel, N.S. (2014) Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov., 4, 1371–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng, Y., Lin, T.Y., Lee, G., Paddock, M.N., Momb, J., Cheng, Z., Li, Q., Fei, D.L., Stein, B.D., Ramsamooj, S.et al. (2018) Mitochondrial one-carbon pathway supports cytosolic folate integrity in cancer cells. Cell, 175, e1517, 1546–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryall, J.G., Dell'Orso, S., Derfoul, A., Juan, A., Zare, H., Feng, X., Clermont, D., Koulnis, M., Gutierrez-Cruz, G., Fulco, M.et al. (2015) The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell, 16, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berg, J.M. and Stryer, L. (2002) Biochemistry, 5th edn. W H Freeman, New York. [Google Scholar]
- 36. Birsoy, K., Wang, T., Chen, W.W., Freinkman, E., Abu-Remaileh, M. and Sabatini, D.M. (2015) An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell, 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lane, A.N. and Fan, T.W. (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res., 43, 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herzig, S. and Shaw, R.J. (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol., 19, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bond, P. (2016) Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr., 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lane, R.S., Fu, Y., Matsuzaki, S., Kinter, M., Humphries, K.M. and Griffin, T.M. (2015) Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res. Ther., 17, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aguer, C., Gambarotta, D., Mailloux, R.J., Moffat, C., Dent, R., McPherson, R. and Harper, M.E. (2011) Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE, 6, e28536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ostergaard, S., Olsson, L. and Nielsen, J. (2001) In vivo dynamics of galactose metabolism in Saccharomyces cerevisiae: metabolic fluxes and metabolite levels. Biotechnol. Bioeng., 73, 412–425. [DOI] [PubMed] [Google Scholar]
- 43. Melber, A. and Haynes, C.M. (2018) UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res., 28, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pakos-Zebrucka, K., Koryga, I., Mnich, K., Ljujic, M., Samali, A. and Gorman, A.M. (2016) The integrated stress response. EMBO Rep., 17, 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Adams, C.J., Kopp, M.C., Larburu, N., Nowak, P.R. and Ali, M.M.U. (2019) Structure and molecular mechanism of ER stress Signaling by the unfolded protein response signal activator IRE1. Front. Mol. Biosci., 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang, S., Xing, Y. and Liu, Y. (2019) Emerging roles for the ER stress sensor IRE1alpha in metabolic regulation and disease. J. Biol. Chem., 294, 18726–18741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Almanza, A., Carlesso, A., Chintha, C., Creedican, S., Doultsinos, D., Leuzzi, B., Luis, A., McCarthy, N., Montibeller, L., More, S.et al. (2019) Endoplasmic reticulum stress signalling - from basic mechanisms to clinical applications. FEBS J., 286, 241–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lehtonen, J.M., Forsstrom, S., Bottani, E., Viscomi, C., Baris, O.R., Isoniemi, H., Hockerstedt, K., Osterlund, P., Hurme, M., Jylhava, J.et al. (2016) FGF21 is a biomarker for mitochondrial translation and mtDNA maintenance disorders. Neurology, 87, 2290–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scholle, L.M., Lehmann, D., Deschauer, M., Kraya, T. and Zierz, S. (2018) FGF-21 as a potential biomarker for mitochondrial diseases. Curr. Med. Chem., 25, 2070–2081. [DOI] [PubMed] [Google Scholar]
- 50. Scher, M.B., Vaquero, A. and Reinberg, D. (2007) SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev., 21, 920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tseng, A.H., Shieh, S.S. and Wang, D.L. (2013) SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med., 63, 222–234. [DOI] [PubMed] [Google Scholar]
- 52. Mohrin, M., Shin, J., Liu, Y., Brown, K., Luo, H., Xi, Y., Haynes, C.M. and Chen, D. (2015) Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science, 347, 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Teske, B.F., Fusakio, M.E., Zhou, D., Shan, J., McClintick, J.N., Kilberg, M.S. and Wek, R.C. (2013) CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell, 24, 2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Austin, S. and St-Pierre, J. (2012) PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci., 125, 4963–4971. [DOI] [PubMed] [Google Scholar]
- 55. Cheng, C.F., Ku, H.C. and Lin, H. (2018) PGC-1alpha as a pivotal factor in lipid and metabolic regulation. Int. J. Mol. Sci., 19, 3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Glick, D., Barth, S. and Macleod, K.F. (2010) Autophagy: cellular and molecular mechanisms. J. Pathol., 221, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Patterson, K.I., Brummer, T., O'Brien, P.M. and Daly, R.J. (2009) Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J., 418, 475–489. [DOI] [PubMed] [Google Scholar]
- 58. Soustek, M.S., Balsa, E., Barrow, J.J., Jedrychowski, M., Vogel, R., Jan, S., Gygi, S.P. and Puigserver, P. (2018) Inhibition of the ER stress IRE1alpha inflammatory pathway protects against cell death in mitochondrial complex I mutant cells. Cell Death Dis., 9, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anderson, N.S. and Haynes, C.M. (2020) Folding the mitochondrial UPR into the integrated stress response. Trends Cell Biol., 30, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhao, Q., Wang, J., Levichkin, I.V., Stasinopoulos, S., Ryan, M.T. and Hoogenraad, N.J. (2002) A mitochondrial specific stress response in mammalian cells. EMBO J., 21, 4411–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Costa-Mattioli, M. and Walter, P. (2020) The integrated stress response: from mechanism to disease. Science, 368, eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quiros, P.M., Prado, M.A., Zamboni, N., D'Amico, D., Williams, R.W., Finley, D., Gygi, S.P. and Auwerx, J. (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol., 216, 2027–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mick, E., Titov, D.V., Skinner, O.S., Sharma, R., Jourdain, A.A. and Mootha, V.K. (2020) Distinct mitochondrial defects trigger the integrated stress response depending on the metabolic state of the cell. elife, 9, e49178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bao, X.R., Ong, S.E., Goldberger, O., Peng, J., Sharma, R., Thompson, D.A., Vafai, S.B., Cox, A.G., Marutani, E., Ichinose, F.et al. (2016) Mitochondrial dysfunction remodels one-carbon metabolism in human cells. elife, 5, e10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Restelli, L.M., Oettinghaus, B., Halliday, M., Agca, C., Licci, M., Sironi, L., Savoia, C., Hench, J., Tolnay, M., Neutzner, A.et al. (2018) Neuronal mitochondrial dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep., 24, 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee, A.H., Iwakoshi, N.N. and Glimcher, L.H. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol., 23, 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hollien, J. and Weissman, J.S. (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science, 313, 104–107. [DOI] [PubMed] [Google Scholar]
- 68. Liu, Y.T., Huang, X., Nguyen, D., Shammas, M.K., Wu, B.P., Dombi, E., Springer, D.A., Poulton, J., Sekine, S. and Narendra, D.P. (2020) Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum. Mol. Genet., 29, 1547–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lim, M.A., Selak, M.A., Xiang, Z., Krainc, D., Neve, R.L., Kraemer, B.C., Watts, J.L. and Kalb, R.G. (2012) Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J. Neurosci., 32, 1123–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tsuruta, F., Sunayama, J., Mori, Y., Hattori, S., Shimizu, S., Tsujimoto, Y., Yoshioka, K., Masuyama, N. and Gotoh, Y. (2004) JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J., 23, 1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tournier, C., Hess, P., Yang, D.D., Xu, J., Turner, T.K., Nimnual, A., Bar-Sagi, D., Jones, S.N., Flavell, R.A. and Davis, R.J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science, 288, 870–874. [DOI] [PubMed] [Google Scholar]
- 72. Genin, E.C., Plutino, M., Bannwarth, S., Villa, E., Cisneros-Barroso, E., Roy, M., Ortega-Vila, B., Fragaki, K., Lespinasse, F., Pinero-Martos, E.et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med., 8, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ha, J., Kang, E., Seo, J. and Cho, S. (2019) Phosphorylation dynamics of JNK Signaling: effects of dual-specificity phosphatases (DUSPs) on the JNK pathway. Int. J. Mol. Sci., 20, 6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Longen, S., Bien, M., Bihlmaier, K., Kloeppel, C., Kauff, F., Hammermeister, M., Westermann, B., Herrmann, J.M. and Riemer, J. (2009) Systematic analysis of the twin cx(9)c protein family. J. Mol. Biol., 393, 356–368. [DOI] [PubMed] [Google Scholar]
- 75. Jung, C., Higgins, C.M. and Xu, Z. (2002) Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem., 83, 535–545. [DOI] [PubMed] [Google Scholar]
- 76. Mattiazzi, M., D'Aurelio, M., Gajewski, C.D., Martushova, K., Kiaei, M., Beal, M.F. and Manfredi, G. (2002) Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem., 277, 29626–29633. [DOI] [PubMed] [Google Scholar]
- 77. Menzies, F.M., Cookson, M.R., Taylor, R.W., Turnbull, D.M., Chrzanowska-Lightowlers, Z.M., Dong, L., Figlewicz, D.A. and Shaw, P.J. (2002) Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain, 125, 1522–1533. [DOI] [PubMed] [Google Scholar]
- 78. Wiedemann, F.R., Manfredi, G., Mawrin, C., Beal, M.F. and Schon, E.A. (2002) Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem., 80, 616–625. [DOI] [PubMed] [Google Scholar]
- 79. Burstein, S.R., Valsecchi, F., Kawamata, H., Bourens, M., Zeng, R., Zuberi, A., Milner, T.A., Cloonan, S.M., Lutz, C., Barrientos, A.et al. (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet., 27, 160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stein, L.R. and Imai, S. (2012) The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab., 23, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang, L., Garcia Canaveras, J.C., Chen, Z., Wang, L., Liang, L., Jang, C., Mayr, J.A., Zhang, Z., Ghergurovich, J.M., Zhan, L.et al. (2020) Serine catabolism feeds NADH when respiration is impaired. Cell Metab., 31, 809–821 e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Titov, D.V., Cracan, V., Goodman, R.P., Peng, J., Grabarek, Z. and Mootha, V.K. (2016) Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science, 352, 231–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yin, F., Sancheti, H., Patil, I. and Cadenas, E. (2016) Energy metabolism and inflammation in brain aging and Alzheimer's disease. Free Radic. Biol. Med., 100, 108–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lochmuller, H., Johns, T. and Shoubridge, E.A. (1999) Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res., 248, 186–193. [DOI] [PubMed] [Google Scholar]
- 85. Bourgey, M., Dali, R., Eveleigh, R., Chen, K.C., Letourneau, L., Fillon, J., Michaud, M., Caron, M., Sandoval, J., Lefebvre, F.et al. (2019) GenPipes: an open-source framework for distributed and scalable genomic analyses. Gigascience, 8, giz037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bolger, A.M., Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T.R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Anders, S., Pyl, P.T. and Huber, W. (2015) HTSeq--a python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Trapnell, C., Williams, B.A., Pertea, G., Mortazavi, A., Kwan, G., vanBaren, M.J., Salzberg, S.L., Wold, B.J. and Pachter, L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Trapnell, C., Hendrickson, D.G., Sauvageau, M., Goff, L., Rinn, J.L. and Pachter, L. (2013) Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol., 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen, E.Y., Tan, C.M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G.V., Clark, N.R. and Ma'ayan, A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kuleshov, M.V., Jones, M.R., Rouillard, A.D., Fernandez, N.F., Duan, Q., Wang, Z., Koplev, S., Jenkins, S.L., Jagodnik, K.M., Lachmann, A.et al. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res., 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Soga, T. and Heiger, D.N. (2000) Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal. Chem., 72, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 95. Soga, T., Ueno, Y., Naraoka, H., Ohashi, Y., Tomita, M. and Nishioka, T. (2002) Simultaneous determination of anionic intermediates for Bacillus subtilis metabolic pathways by capillary electrophoresis electrospray ionization mass spectrometry. Anal. Chem., 74, 2233–2239. [DOI] [PubMed] [Google Scholar]
- 96. Soga, T., Ohashi, Y., Ueno, Y., Naraoka, H., Tomita, M. and Nishioka, T. (2003) Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res., 2, 488–494. [DOI] [PubMed] [Google Scholar]
- 97. Sugimoto, M., Wong, D.T., Hirayama, A., Soga, T. and Tomita, M. (2010) Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics, 6, 78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chong, J., Soufan, O., Li, C., Caraus, I., Li, S., Bourque, G., Wishart, D.S. and Xia, J. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res., 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.