

Abstract
Nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1) is required for nuclear nicotinamide adenine mononucleotide (NAD+) biosynthesis in all nucleated cells, and despite its functional ubiquity, mutations in this gene lead to an isolated retinal degeneration. The mechanisms underlying how mutant NMNAT1 causes disease are not well understood, nor is the reason why the pathology is confined to the retina. Using a mouse model of NMNAT1-associated retinal degeneration that harbors the p.Val9Met mutation, we tested the hypothesis that decreased function of mutant NMNAT1 has a greater effect on the levels of NAD+ in the retina than elsewhere in the body. Measurements by liquid chromatography with tandem mass spectrometry showed an early and sustained decrease of NAD+ in mutant retinas that was not observed in other tissues. To understand how consumers of nuclear NAD+ are affected by the reduced availability of NAD+ in mutant retinas, poly(ADP-ribose) polymerase (PARP) and nuclear sirtuin activity were evaluated. PARP activity was elevated during disease progression, as evidenced by overproduction of poly(ADP-ribose) (PAR) in photoreceptors, whereas histone deacetylation activity of nuclear sirtuins was not altered. We hypothesized that PARP could be activated because of elevated levels of oxidative stress; however, we did not observe oxidative DNA damage, lipid peroxidation, or a low glutathione to oxidized glutathione ratio. Terminal deoxynucleotidyl transferase dUTP nick end labeling staining revealed that photoreceptors appear to ultimately die by apoptosis, although the low NAD+ levels and overproduction of PAR suggest that cell death may include aspects of the parthanatos cell death pathway.
Introduction
Nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1)-associated retinal degeneration is an isolated recessive disease that leads to severe and early-onset vision loss (1–4). NMNAT1 is one of three NMNAT enzymes that are required for the biosynthesis of nicotinamide adenine mononucleotide (NAD+) (5). Although the NMNAT isozymes share function, they are encoded by separate genes and localize to different subcellular compartments; NAD+ is regenerated in the nucleus by NMNAT1, in the cytosol and Golgi by NMNAT2, and in mitochondria by NMNAT3 (5). Therefore, NMNAT1 maintains the pool of NAD+ that supports essential nuclear functions, such as DNA repair and the regulation of gene expression, in all tissues in the body (6) except blood (7).
The mechanisms underlying why mutations in NMNAT1 cause disease only in the retina, despite the ubiquitous function of this enzyme, are unclear. Thus far, experiments using fibroblasts from patients with NMNAT1-associated retinal degeneration indicate that pathogenic mutations in NMNAT1 lead to varying degrees of loss-of-function, either by directly interfering with catalysis (1) or by causing protein folding defects under conditions of cellular stress (8). Mutant alleles that cause NMNAT1-associated disease may be either hypomorphic or null, although the observation that a global Nmnat1 knockout in mice is embryonic lethal suggests that some NMNAT1 function must be maintained for survival (9). In humans and in mice with NMNAT1-associated disease (1–4,10), the residual NMNAT1 function is apparently insufficient for retinal cell viability and the deficit cannot be compensated by NMNAT2 or NMNAT3 activity (9).
To further investigate the biology of NMNAT1-associated retinal degeneration, we tested the hypothesis that decreased NAD+ biosynthesis by mutant NMNAT1 has a greater effect on NAD+ pathway-related metabolites in the retina than elsewhere in the body. NAD+ pathway-related metabolites were measured in seven tissue types collected from a mouse model of NMNAT1-associated retinal degeneration (10), before and during retinal degeneration. This model harbors the p.Val9Met-Nmnat1 (Nmnat1V9M) mutation. We have shown previously that purified recombinant human NMNAT1 with the p.Val9Met mutation leads to a 63% reduction in NMNAT1 enzymatic activity (1). Nmnat1V9M/V9M mice have morphologically normal retinas at 3 weeks of age, as observed by in vivo and ex vivo imaging, mild photoreceptor loss at 4 weeks of age, and severe photoreceptor loss by 8 weeks of age (10). Using this model, we then investigated whether mutant NMNAT1 affects the function of NAD+ consumers that are important for nuclear homeostasis and cell survival, specifically poly(ADP-ribose) polymerase (PARP) and nuclear sirtuins (11–13). The PARP1 isoform supports the repair of DNA damage, and when activated, usage of NAD+ by this enzyme can increase to as much as 500 times the basal rate, making it the greatest consumer of nuclear NAD+ (14). Nuclear sirtuins require NAD+ to perform histone deacetylation (HDAC) (15), which contributes to regulation of gene expression by facilitating the condensation of chromatin structure (16). To test the hypothesis that elevated levels of oxidative stress contribute to NMNAT1-associated retinal degeneration, we evaluated markers of oxidative stress. In addition, given that the onset of the phenotype occurs in Nmnat1V9M/V9M mice < 2 weeks after eye opening, we assessed whether reducing light exposure to counter a possible accumulation of photo-oxidative damage could delay disease onset and decrease the severity of the degeneration. The cell death pathway leading to the ultimate demise of the cell was also investigated.
Results
Retina-specific alterations in NAD+ and NAD+ pathway-related metabolite levels
To test whether mutant NMNAT1 affects the levels of NAD+ and NAD+ pathway-related metabolites in the retina more profoundly than in other tissues, we performed liquid chromatography with tandem mass spectrometry (LC–MS/MS) on seven tissue types from Nmnat1V9M/V9M mice before they developed retinal degeneration (3 weeks of age) and during disease progression (6 weeks of age). The 31 metabolites included in the test panel are listed in Table 1. In Nmnat1V9M/V9M mice, the retina showed more alterations in metabolite levels than the heart, lung, brain, kidney, liver, and skin at both ages, based on the results from age-matched wildtype littermates. Levels of NAD+ were 23.0% lower (P < 0.0001), whereas levels of its precursor, nicotinamide mononucleotide (NMN), were 8.6-fold higher (P < 0.0001) in Nmnat1V9M/V9M retinas at 3 weeks of age than in the control retinas. Similarly, levels of NAD+ were 31.2% lower (P = 0.002), and levels of NMN were 3.6-fold higher (P < 0.0001) in the Nmnat1V9M/V9M retinas at 6 weeks of age (Fig. 1). The only other tissue to show a significant difference in NAD+ levels in mutant versus the wildtype mice was the lung. The levels of NAD+ were 41.9% (P = 0.048) lower in the lungs of 3-week-old mutant mice than in the lungs of age-matched wildtype mice, but this difference normalized by 6 weeks, despite the high NMN content in the older Nmnat1V9M/V9M mice. In all other tissues from the 3-week-old mutant mice, NMN levels were only modestly higher than those measured in the same tissue type from age-matched wildtype mice, except for the brain and liver in which NMN levels were 1.3-fold (P = 0.04) and 2.1-fold (P = 0.04) higher, respectively. Significantly higher levels of NMN were observed in the brain (2.4-fold, P = 0.0003), the heart (2.0-fold, P = 0.04), and the lungs (4.4-fold, P = 0.04) of 6-week-old mutant mice than in these tissues from the 6-week-old wildtype mice.
Table 1.
Nicotinamide adenine mononucleotide (NAD+) pathway-related metabolites tested by LC–MS/MS
| Metabolite | HMDB | Metabolite | HMDB |
|---|---|---|---|
| 3-Hydroxybutyric acid | HMDB0000011 | Kynurenine | HMDB0000684 |
| 3-Hydroxykynurenine | HMDB0011631 | N1-Methylnicotinamide | HMDB0000699 |
| 4-Hydroxyproline | HMDB0000725 | NAD+ | HMDB0000902 |
| 6-Hydroxypicolinic acid | HMDB0013188 | NADH | HMDB0001487 |
| Adenosine monophosphate | HMDB0000045 | NADP | HMDB0000217 |
| Adenosine triphosphate | HMDB0000538 | NADPH | HMDB0000221 |
| Carnitine | HMDB0000062 | Nicotinamide | HMDB0001406 |
| Cyclic guanosine monophosphate | HMDB0060465 | Nicotinic acid | HMDB0002730 |
| Choline | HMDB0000097 | Nicotinic acid N-oxide | HMDB0001488 |
| Creatinine | HMDB0000562 | Nicotinamide mononucleotide | HMDB0000229 |
| Flavin adenine dinucleotide | HMDB0001248 | Nicotinamide riboside | HMDB00855 |
| Glutamine | HMDB0000641 | Ophthalmic acid | HMDB0005765 |
| Glutathione | HMDB0000125 | Proline (L-Proline) | HMDB0000162 |
| Oxidized glutathione | HMDB0003337 | Trigonelline | HMDB0000875 |
| Guanosine triphosphate | HMDB0001273 | Tryptophan | HMDB0000929 |
| Hypoxanthine | HMDB0000157 |
Figure 1.
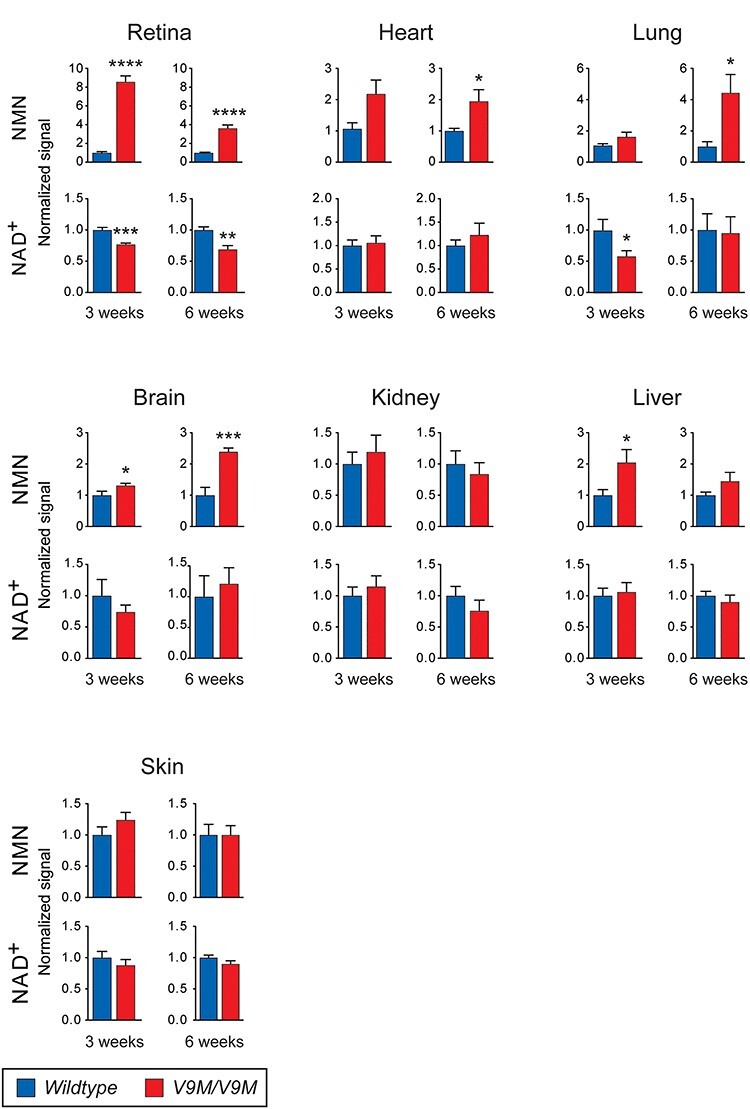
LC–MS/MS measurements of NMN and NAD+ in the retina, heart, lung, brain, kidney, liver, and skin. In comparison to wildtype mice (blue), NMN in Nmnat1V9M/V9M mice (red) is elevated in retina and brain at 3 and 6 weeks of age, in the liver at 3 weeks of age, and in the heart and lung at 6 weeks of age. In Nmnat1V9M/V9M mice, NAD+ is decreased in the retina at both time points and in the lung at 3 weeks of age. Mice per genotype, n = ;7–8, with the exception of brain at 3-weeks (wildtype, n = ;6). Error bars represent SEM. Statistical significance is represented by *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Besides NMN and NAD+, the levels of other metabolites were significantly different in the retinas of mutant versus wildtype mice (Supplementary Material, Fig. S1, Supplementary Material, Table S1). Cyclic guanosine monophosphate (cGMP) in the retina was 21.3% lower (P = 0.02) in 3-week-old mutant mice and 62.7% lower (P < 0.0001) in 6-week-old mutant mice than in age-matched wildtype mice. N1-methylnicotinamide in the retina was 89.0% higher (P = 0.04) in 3-week-old mutant mice and 41.1% higher (P = 0.02) in 6-week-old mutant mice than in age-matched wildtype mice. Higher levels of reduced glutathione (GSH, 36.0%, P = 0.045) and oxidized glutathione (GSSG, 67.5%, P = 0.02) were detected in the retina of 3-week-old mutant mice than in the retinas of the age-matched wildtype mice, whereas levels of guanosine triphosphate (GTP, 39.2%, P = 0.008) were lower in the retinas of 6-week-old mutant mice than the age-matched wildtype mice. Non-retinal tissues had relatively few metabolites that were significantly different in the mutant versus wildtype mice at either age. In some cases, an analyte could not be measured reliably due to low abundance in a given tissue. For example, cGMP could only be measured in the retina, liver and skin.
PARP activity is upregulated in mutant photoreceptors
Based on the observation that NAD+ levels are lower in the retinas of mutant mice than in the retinas of wildtype mice, we hypothesized that the activities of the two major consumers of NAD+ in the nucleus, PARPs and sirtuins (11,15,17), are affected. As PARP is the greatest consumer of nuclear NAD+, we first used an antibody against poly(ADP-ribose) (PAR) to test whether the poly(ADP-ribosyl)ation activity of this enzyme is affected in Nmnat1V9M/V9M mice. In comparison to retinal sections from wildtype mice that showed no evidence of high PARP activity by immunohistochemistry (IHC), puncta of intense labeling in the outer nuclear layer (ONL) were observed in all retinal sections from 4.5 to 6-week-old Nmnat1V9M/V9M mice (Fig. 2A) at a rate of 1.6 ± 0.3 (P = ;0.0001) photoreceptors per row of ONL (Fig. 2B). These puncta are consistent with the pattern reported previously for excessive PAR production in retinal degeneration 1 (rd1) mice (18). Likewise, using the same antibody, quantitative western blot analysis showed a trend for greater PAR content in Nmnat1V9M/V9M retinas than in wildtype retinas at both 3 weeks (20.7%, P = 0.67) and 6 weeks (33.5%, P = 0.48) of age, even with some photoreceptor loss in the Nmnat1V9M/V9M retinas at the later time point (Fig. 2C and D).
Figure 2.
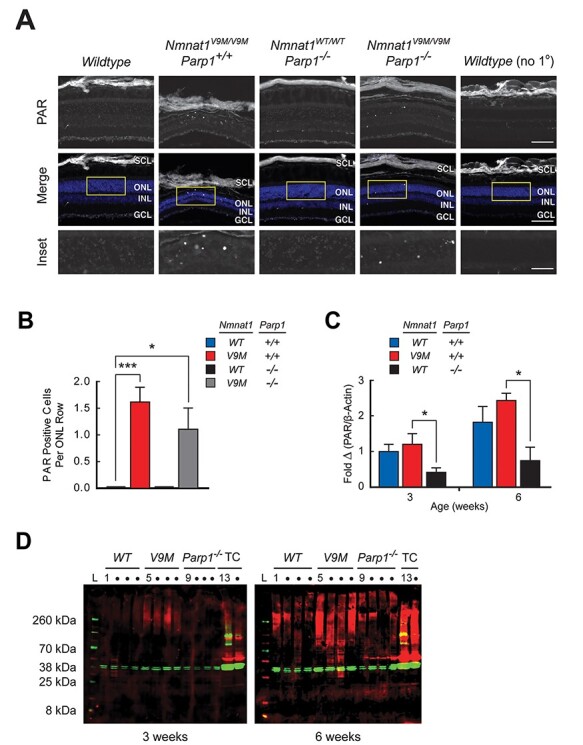
Increased PAR production in photoreceptors of Nmnat1V9M/V9M/Parp+/+ and Nmnat1V9M/V9M/Parp−/− mice. (A) Representative retinal sections from 6-week-old mice are shown, labeled with an antibody against PAR. Low-level PAR production is observed in the wildtype mouse (column 1) and the Nmnat1WT/WT/Parp−/− mouse (column 3), whereas the ONL in the Nmnat1V9M/V9M/Parp+/+ mouse (column 2) shows intense puncta in the ONL, indicating over-production of PAR in photoreceptors. These puncta are not eliminated in the Nmnat1V9M/V9M/Parp−/− mouse (column 4). The low-level PAR signal associated with wildtype mice is not observed in the absence of the primary antibody (column 5). The images of PAR immunoreactivity (top row) are shown merged (middle row) with a DAPI counterstain (blue). The regions outlined in yellow are magnified in the bottom row. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; SCL, sclera. 20x magnification, scale bar = ;100 μm; insets, scale bar = ;40 μm. (B) No PAR-positive cells were observed for wildtype (blue, n = ;6) or Nmnat1WT/WT/Parp1−/− mice (black, n = ;5), whereas ~ 1–2 positive cells per row of ONL were identified for Nmnat1V9M/V9M/Parp1+/+ (red, n = ;8) and Nmnat1WT/WT/Parp1−/− (gray, n = ;4) mice. (C) The quantitation of western blots shows a trend for Nmnat1V9M/V9M/Parp+/+ retinas (red) having increased PAR content in comparison to wildtype retinas (blue) at 3 and 6 weeks. Nmnat1WT/WT/Parp1−/− retinas (black) have reduced PAR content in comparison to the wildtype and Nmnat1V9M/V9M/Parp1−/− retinas at both ages. (D) Western blots using antibodies against PAR (red) and β-actin (green) in neural retinal lysates from wildtype (WT, lanes 1–4), Nmnat1V9M/V9M/Parp1+/+ (V9M, lanes 5–8) and Nmnat1WT/WT/Parp1−/− (Parp1−/−, lanes 9–12) mice at 3 (left blot) and 6 (right blot) weeks of age that were subjected to quantitation. Tissue cultured (TC) ARPE-19 cells treated with H2O2 were used as a positive control (lanes 13–14). Protein ladder (lane L). Statistical significance is represented by *P < 0.05, ***P < 0.001.
Excessive upregulation of PAR production can contribute to neuronal cell death by depleting the nuclear NAD+ pool (19). Therefore, we hypothesized that eliminating PARP1 activity would rescue the retinal degenerative phenotype in the Nmnat1V9M/V9M mice. To test this hypothesis, we crossed the Nmnat1V9M/V9M mice with a Parp1 knockout (Parp1−/−) mouse in which the gene is completely inactivated (20). Knockout of Parp1 alone does not result in an abnormal retinal phenotype (21). IHC showed that PAR production was not completely eliminated in either Nmnat1WT/WT/Parp1−/− or the Nmnat1V9M/V9M/Parp1−/− retinas and that knocking out Parp1 did not protect Nmnat1V9M/V9M/Parp1−/− retinas from degeneration (Fig. 2A). Puncta of intense PAR labeling were observed in the ONL of Nmnat1V9M/V9M/Parp1−/− retinas of 4.5 to 6-week-old mice at a rate of 1.1 ± 0.4 (P = ;0.02) photoreceptors per row (Fig. 2B), as compared with wildtype retinas that, as mentioned above, were clear of this signal. The number of intensely labeled photoreceptors per row of ONL in Nmnat1V9M/V9M/Parp1−/− retinas was not significantly different than that observed in Nmnat1V9M/V9M/Parp1+/+ retinas (P = ;0.43). Like wildtype retinas, no intensely labeled photoreceptors were observed in Nmnat1WT/WT/Parp1−/− retinas. Measurements of retinal thickness from optical coherence tomography (OCT) images collected from the Nmnat1V9M/V9M/Parp1−/− mice were in agreement with the time course that was reported previously for Nmnat1V9M/V9M/Parp1+/+ mice (10), indicating no rescue of the phenotype: 219.3 μm ± 2.5 for Nmnat1V9M/V9M/Parp1−/− versus 225.5 μm ± 3.1 for Nmnat1V9M/V9M/Parp1+/+ (P = 0.19) at 3 weeks of age; 160.0 μm ± 4.8 for Nmnat1V9M/V9M/Parp1−/− versus 165.7 μm for Nmnat1V9M/V9M/Parp1+/+ (value estimated from regression) at 6 weeks of age; and 147.6 μm ± 3.1 for Nmnat1V9M/V9M/Parp1−/− versus 139.3 μm ± 3.5 for Nmnat1V9M/V9M/Parp+/+ (P = 0.09) at 8 weeks of age (Supplementary Material, Fig. S2). Quantitative western blot analysis showed that removal of Parp1 resulted in a trend of reduced PAR production in the Nmnat1WT/WT/Parp1−/− retina at both 3 weeks (58.4%, P = 0.08) and 6 weeks (59.2%, P = 0.14) of age, as compared with that in wildtype littermates. PAR production was significantly lower in the Nmnat1WT/WT/Parp1−/− mice at 3 weeks (65.5%, P = 0.02) and 6 weeks (69.4%, P = 0.02) of age, as compared with Nmnat1V9M/V9M/Parp1+/+ littermates (Fig. 2C and D).
Nuclear sirtuin function is not affected by mutant Nmnat1
In addition to PARPs, sirtuins are the other major consumer of NAD+ in the nucleus (22). We hypothesized that nuclear sirtuin activity would be affected in the retinas of mutant mice due to the lower NAD+ levels that were observed in this tissue. To evaluate sirtuin function, we assessed acetylation patterns using quantitative western blot analyses on retinal tissue from Nmnat1V9M/V9M and wildtype mice at 3 and 6 weeks of age (Fig. 3). We used antibodies against acetylated histone sites: 1) lysine at position 9 of histone 3 (H3K9ac) that is deacetylated by Sirt1 (23) and possibly by Sirt6 (24), a mono (ADP-ribosyl) transferase that may have weak HDAC activity (25); 2) H3K18ac that is deacetylated by Sirt7 (26); and 3) H4K16ac that is deacetylated by Sirt1 (27), Sirt2 (28), which is predominantly cytoplasmic but can be imported to the nucleus (29), and possibly by Sirt6 (27). Antibodies that are reactive to H3 and H4, regardless of the acetylation status, were also used to assess whether expression levels were altered in the mutant mice. There was substantial within-group variability, and none of the histone sites had statistically significant differences in acetylation between the Nmnat1V9M/V9M and wildtype mice at either age tested (Fig. 3A–C). Immunofluorescence imaging of retinal sections from 3-week-old mice that were labeled using the same antibodies corroborated the finding that acetylation levels were unchanged (Supplementary Material, Fig. S3). No statistically significant differences were observed for H3 expression between genotypes at either age (Fig. 3D). However, expression of H4 was 65.1% (P = ;0.02) lower in 3-week-old mutant mice than in aged-matched wildtype mice, although this difference largely resolved by 6 weeks of age (decrease of 25.9%, P = 0.24) (Fig. 3E).
Figure 3.
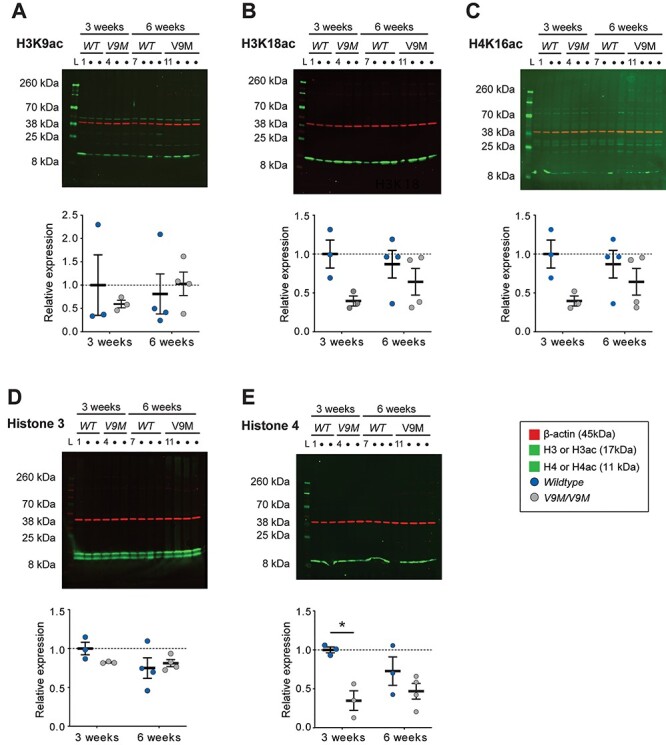
Altered nuclear sirtuin HDAC activity is not detected during Nmnat1-associated disease progression. Quantitative western blot analysis of whole retinal lysate from Nmnat1V9M/V9M mice and wildtype mice at 3 weeks (n = ;3 per genotype) and 6 weeks (n = ;4 per genotype) was performed using antibodies against histone residues that are targets of nuclear sirtuins. Immunoreactivity against (A) acetylated H3K9 (17 kDa, green), (B) acetylated H3K18 (17 kDa, green) and (C) acetylated H4K16 (11 kDa, green) did not significantly differ by genotype at either age, as indicated by the blots (top) or quantitation of those blots (bottom). (D) The expression level of histone 3 (17 kDa, green) did not differ by genotype at either age, whereas (E) histone 4 expression was significantly lower in Nmnat1V9M/V9M mice than in wildtype mice at 3 weeks of age (blot, top; quantitation, bottom). The intensities of the target bands were measured relative to β-actin (45 kDa, red). Statistical significance is represented by *P < 0.05.
Mutant retinas do not have elevated levels of oxidative stress
Given that PAR production was greater in the retinas of mutant mice, and PARP is activated by oxidative DNA damage (30), we hypothesized that there may be elevated oxidative stress in the retinas of mutant mice. To assess levels of oxidative DNA damage, IHC was performed on retinal sections from 4 and 6-week-old wildtype and Nmnat1V9M/V9M mice using an antibody against 8-hydroxydeoxyguanosine (8-OH-dG) (31) (Supplementary Material, Fig. S4A). There were no apparent differences in 8-OH-dG staining between mutant and wildtype retinas at either age tested. To further examine whether there are increases in oxidative stress in mutant retinas, we used LC–MS/MS to measure the levels of GSH, which is an important antioxidant, and GSSG (Supplementary Material, Fig. S4B). A decreased ratio of GSH:GSSG indicates increased oxidative stress (32); however, this effect was not observed in the mutant retinas at either 3 weeks (0.03 ± 0.002 for Nmnat1V9M/V9M versus 0.05 ± 0.02 for wildtype, P = ;0.25) or 6 weeks (0.20 ± 0.05 for Nmnat1V9M/V9M versus 0.37 ± 0.07 for wildtype, P = ;0.10) of age. To assess levels of oxidative damage to lipids, we performed fluorometric measurements of lipid peroxidation in the retinas of 3 and 6-week-old mice, using the malondialdehyde (MDA) assay (Supplementary Material, Fig. S4C). No statistically significant differences in the levels of lipid peroxidation were observed between mutant and wildtype retinas at 3 weeks of age (1.2 ± 0.2 for Nmnat1V9M/V9M versus 2.2 ± 0.8 for wildtype, P = ;0.40) or at 6 weeks of age (1.2 ± 0.2 for Nmnat1V9M/V9M versus 2.4 ± 0.8 for wildtype, P = ;0.40). Consistent with these findings, no differences were observed by IHC with the use of an antibody against 4-hydroxynonenal (4-HNE) (Supplementary Material, Fig. S4D), which is a byproduct of lipid peroxidation (33), or an antibody against nitrotyrosine (Supplementary Material, Fig. S4E), which is a marker of nitrogen free radicals, in retinas of 3, 4.5, and 6-week-old mutant and wildtype mice (34).
Light exposure can result in elevated levels of photo-oxidative stress in photoreceptors (35,36). Given that the retinas of Nmnat1V9M/V9M mice develop normally before beginning to degenerate within two weeks after eye opening (10), we considered that photo-oxidative stress could be a potential disease mechanism. To test this hypothesis, we measured retinal thickness in Nmnat1V9M/V9M and wildtype mice that had been raised and maintained in the dark from birth. As for all experiments described thus far, these mice were pigmented, given their C57Bl/6J genetic background. The degeneration profiles for mutant mice raised in the dark versus the light were the same, based on retinal thickness measurements at 3, 6, and 12 weeks of age (Supplementary Material, Fig. S5A). For example, at 12 weeks of age, retinas of mutant mice raised in the dark were slightly thinner than those of mutant mice raised in the light (dark: 120.1 μm ± 7.4 versus light: 135.9 μm ± 4.5, P < 0.03), and retinas of dark-raised wildtype mice were also slightly thinner than of those raised in the light (dark: 213.3 μm ± 1.6 versus light: 225.5 μm ± 3.1 versus, P < 0.03).
We hypothesized that non-pigmented mice would have greater vulnerability to the effects of mutant Nmnat1 due to the increased amount of light that would reach the retina, relative to their abovementioned pigmented counterparts. Therefore, we outcrossed the Nmnat1V9M/V9M mice with non-pigmented wildtype CD1 mice. The degeneration profiles of the dark-raised and the light-raised non-pigmented mice were indistinguishable (Supplementary Material, Fig. S5B). For example, at 12 weeks of age, retinal thicknesses in the dark-raised and light-raised mutant mice were not significantly different from each other (dark: 126.3 μm ± 9.9 versus light: 131.7 μm ± 1.5, P > 0.99), nor were the dark-raised and the light-raised wildtype mice (dark: 224.6 μm ± 1.1 versus light: 226.5 μm ± 1.8, P > 0.99).
Mutant photoreceptors may undergo apoptotic cell death
Excess PAR production can lead to parthanatos (‘death by PAR’), a programmed cell death pathway that arises from a hyperactivation of PARPs, resulting in depletion of nuclear NAD+ (19) such that neither PARPs nor other consumers of nuclear NAD+ (e.g. sirtuins) function properly (18,19,37,38). As we observed high levels of PAR and low levels of NAD+ in the retinas of mutant mice, aspects of parthanatos may contribute to cell death. However, we also addressed whether apoptosis may play a role in this disease by subjecting retinas from 4.5 and 6-week-old Nmnat1V9M/V9M mice and age-matched wildtype littermates to the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Fig. 4). At 4.5 weeks of age, more TUNEL positive photoreceptors per row of ONL were observed in mutant retinas than in wildtype retinas (1.35 ± 0.36 versus 0.02 ± 0.02, P = 0.02), and a similar trend was observed at 6 weeks of age (0.74 ± 0.26 versus 0.03 ± 0.03, P = ;0.06).
Figure 4.
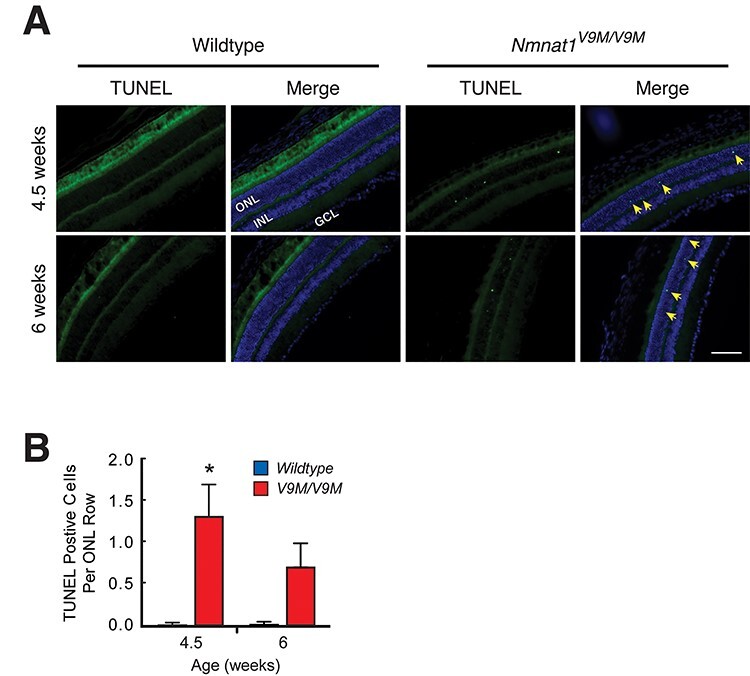
Apoptosis may play a role in photoreceptor cell death in Nmnat1V9M/V9M mice. (A) Representative images at 4.5 (top row) and 6 (bottom row) weeks of age show that wildtype retinas were clear of the TUNEL-positive photoreceptors (first and second columns), whereas TUNEL-positive nuclei (green puncta, yellow arrows) were observed in the ONL of every Nmnat1V9M/V9M mouse tested at both ages (third and fourth columns). TUNEL labeling (first and third columns) is shown merged with a DAPI counterstain (blue, second and fourth columns). GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. 20x magnification, scale bar = ;100 μm. (B) Quantification of TUNEL-positive photoreceptors per row of ONL for wildtype (blue) and Nmnat1V9M/V9M (red) retinas at 4.5 (n = ;3 per genotype) and 6 weeks (n = ;4 per genotype). Statistical significance is represented by *P < 0.05.
Discussion
Using the Nmnat1V9M/V9M mouse model of NMNAT1-associated retinal degeneration, this study identified that NAD+ and NAD+ pathway-related metabolite levels are dysregulated to a greater extent in the retina than in other tissues, both immediately before and during disease progression. Increased NMN in parallel with decreased NAD+ in mutant retinas is consistent with reduced NMNAT1 enzymatic function. We also observed overproduction of PAR, suggesting that activation of PARPs may be driving NAD+ depletion. This phenotype could not be rescued by knocking out Parp1, which suggests that other members of the PARP family of enzymes are involved. Despite the lower levels of NAD+ in mutant retinas, nuclear sirtuin-mediated HDAC function was unaffected. Markers used to assess levels of oxidative stress were no different between Nmnat1V9M/V9M and wildtype mice raised in normal vivarium conditions. In addition, raising the mutant mice in constant darkness to mitigate potential accumulation of photo-oxidative damage did not delay disease onset or lessen the severity of degeneration. We identified that mutant photoreceptors appear to die ultimately by apoptosis, although the changes in NAD+ content and PAR production suggest that this process may be accompanied by aspects of the parthanatos cell death pathway (19).
Identifying whether mutant NMNAT1 affects NAD+ levels, and whether it does so differently across tissues, is fundamental for understanding how NMNAT1-associated disease disrupts cells and why the pathology is isolated to the retina. In the present study, LC–MS/MS measurements in Nmnat1V9M/V9M mice revealed a greater decrease of NAD+ in the retina than in any of the six other tissue types analyzed, both before and during degeneration. The decrease of NAD+, accompanied by a parallel increase of the NAD+ precursor, NMN, in the mutant retina at 3 and 6 weeks of age indicates an early and sustained reduction in NMNAT1 function. The 23% decrease of NAD+ that is observed in the Nmnat1V9M/V9M retina prior to the onset of retinal degeneration is consistent with substantial loss of the nuclear NAD+ pool, given that the nucleus and cytosol hold half of the cellular content of NAD+ (39,40), with some crosstalk between the two compartments (41). Although the levels of retinal NAD+ appeared to decline further by 6 weeks of age, this effect could be due to the loss of some photoreceptors. This interpretation is consistent with the decrease in NMN that was observed at 6 weeks of age, relative to the levels at the 3-week time point. In addition to NAD+ and NMN, other NAD+ pathway-associated metabolites were altered in the retina, cGMP being a notable example. Photoreceptor toxicity due to decreased availability of cGMP is consistent with several photoreceptor diseases (42), including GUCY2D-associated retinal degeneration, which also causes early onset vision loss (43,44). The decrease in the cGMP precursor, GTP, at 6 weeks suggests that the effect of mutant NMNAT1 on the cGMP pool may be indirect. The early and sustained increase in N1-methylnicotinamide could either be an outcome of feedback that alters nicotinamide usage or of a response to disease-related changes in retinal vasculature that are observed in the Nmnat1V9M/V9M model (10,45).
The sustained decrease of NAD+ and the extent of NAD+ pathway-related metabolite disruption in the retina was not observed elsewhere. The only other tissue to show a decrease in NAD+ was the lung at 3 weeks of age, but this organ appears to resolve the deficit over time, possibly by either upregulating NMNAT2 activity or decreasing nuclear NAD+ usage. However, increased NMN content in the lung at 6 weeks indicates continued NMNAT1 dysfunction. NMN was also elevated in the heart, liver, and brain, indicating that the accumulation of NMN is benign or protective; this interpretation is consistent with findings from previous studies (46–48). The lack of an effect on NAD+ in the brain shows that neural cells, as a class, are not broadly sensitive to mutant NMNAT1 and that the retina is unique in this respect. NAD+ levels in the retina may be specifically affected by mutant NMNAT1 because of the high metabolic activity of retinal cells (5,49–52).
Given the decrease of NAD+ in the Nmnat1V9M/V9M retina, we considered whether the functions of major consumers of nuclear NAD+ are altered in this tissue, namely PARP and nuclear sirtuins. PARP activity was increased in the photoreceptors, demonstrated by excessive PAR production, which has the potential to drive cell death by nuclear NAD+ depletion (19). This finding appears to present a paradox in that a decrease in available nuclear NAD+ could be anticipated to limit PAR production. We hypothesize that the decrease in nuclear NAD+ limits PARP from being able to fully rectify cellular events that activate PAR production, thereby causing a sustained upregulation in PARP activity until the NAD+ pool is depleted. However, knocking out Parp1 does not rescue the phenotype in Nmnat1V9M/V9M mice, as it does for rd1 mice that also have upregulated PAR production in photoreceptors (21). Although PARP1 is responsible for 85–90% of PAR production (11), we found that eliminating PARP1 alone in the Nmnat1V9M/V9M retina is not sufficient to prevent excess PAR from accumulating. This result suggests that other PARPs are involved in the observed increase in PAR production, as several of the other 17 members of the PARP/tankyrase superfamily, particularly PARP2, also have poly(ADP-ribosyl)ation activity (11,53). Increased PAR production and low cellular NAD+ content have been associated with reduced sirtuin function (11,12,54). Therefore, we tested whether nuclear sirtuin-mediated HDAC activity was decreased in the Nmnat1V9M/V9M retina. No alterations in acetylation patterns were observed, suggesting that sirtuin activity in the retina is normal.
We hypothesized that elevated levels of oxidative stress may lead to cell death, as oxidative DNA damage has been shown to be a potent activator of PARP. However, measurement of the GSH:GSSG ratio by LC–MS/MS (55), assessment of lipid peroxidation levels using the MDA assay, and IHC analysis of markers for oxidative DNA damage, lipid peroxidation, and nitrogen free radicals suggest that oxidative stress is not elevated in the retinas of Nmnat1V9M/V9M mice. By raising mice in darkness, we also confirmed that photo-oxidative stress does not have a role in the disease progression. Alternatively, the decrease in cGMP levels may erroneously signal that the retina is under constant illumination, leading to metabolic stress-induced photoreceptor damage that cannot be attenuated by shielding the retina from light (56,57). Nonetheless, we found that Nmnat1V9M/V9M photoreceptor degeneration may be ultimately mediated by apoptosis, as indicated by the TUNEL assay. This result is corroborated by a recent study in which Nmnat1 was knocked-down in embryonic mouse retinal explants and apoptotic cells were observed (58). Apoptosis may also be accompanied by aspects of parthanatos (‘death by PAR’), as we observed decreased NAD+ and excess PAR production, which are hallmarks of this cell death pathway.
This study aimed to identify mechanisms underlying NMNAT1-associated retinal degeneration and the isolated nature of this disease. We observed a retina-specific decrease of NAD+ and an increase of PAR within that tissue. The increased PAR production and evidence suggestive of apoptosis were localized to photoreceptor cells during the first few weeks of disease progression. This result is consistent with our previous findings that photoreceptors are the first cell type to degenerate in mutant Nmnat1 mice (10) and that delivery of NMNAT1 to these animals by gene therapy must target photoreceptors to be effective (59). Continuing to clarify the pathophysiology of NMNAT1-associated retinal degeneration should not only inform future therapy development for this disease, but it should also provide valuable insights into NAD+ metabolism and nuclear homeostasis.
Materials and Methods
Animal husbandry
Mice were bred and maintained in the Schepens Eye Research Institute Animal Care Facility where they were fed 4% fat rodent diet and water ad libitum and, except when otherwise stated, housed in a 12-hour light/12-hour dark cycle. This study conformed to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and all procedures were approved by the Animal Care and Use Committee of the Schepens Eye Research Institute.
Mouse lines
The Nmnat1V9M/V9M mouse line was derived from a founder identified during an N-ethyl-N-nitrosourea mutagenesis screen (10). During the present study, this line was maintained on the C57Bl/6J genetic background. To generate a non-pigmented version of the Nmnat1V9M/V9M mouse model, Nmnat1V9M/V9M mice were outcrossed with wildtype CD1 mice (CD-1 IGS mouse, Charles River Laboratories, Wilmington, MA) until no pigmentation was apparent in the fur, skin, or eyes. The non-pigmented Nmnat1WT/V9M mice were then were intercrossed to generate the homozygous mutant and wildtype littermates used for testing. The Nmnat1V9M/V9M mouse line (C57Bl/6J background), the Nmnat1V9M/V9M mouse line (CD-1 background) and the Nmnat1V9M/V9M/Parp1−/− line (C57Bl/6J background) were all maintained separately. Experiments were performed on age-matched littermates generated from breeding mice that were heterozygous for the gene(s) of interest. Parp1−/− mice (129S-Parp1tm1Zqw/J, The Jackson Laboratory, Bar Harbor, ME) were engineered to have an in-frame insertion of a promoter-less neomycin cassette containing a TGA stop codon in exon 2 of Parp1, creating a targeted knockout allele (20).
Genotyping
Genotyping for the p.V9M mutation was performed by Sanger sequencing following DNA amplification by the polymerase chain reaction (PCR), as previously described (10).
Genotyping for the targeted Parp1 knockout allele was determined by gel electrophoresis after DNA was amplified using a multiplex PCR strategy. A common forward primer, 5′-GGGAAAGTCCCACACTGGTA-3′, located in exon 2, was used to amplify both the wildtype and knockout alleles (Supplementary Material, Fig. S6). The reverse primer for the wildtype allele, 5′-GCCGTCTTCTTGACCTTCTG-3′, was also located in exon 2, but in a region downstream of the location of the neomycin cassette insertion in the knockout allele. This primer pair specified a 131 bp amplicon in the wildtype allele, as well as a 926 bp amplicon in knockout allele that the thermocycling conditions did not permit to be amplified. For the knockout allele, the reverse primer, 5′-AGTGACAACGTCGAGCACAG-3′, was located within the neomycin cassette and specified a 275 bp amplicon. The 10 μl PCR reactions had final concentrations of 200 μmol/L for each primer, 200 nmol/L for each of the dNTPs (dATP, dGTP, dTTP and dCTP), 2 mmol/L MgCl2, and 1 unit of Hot FirePol DNA polymerase (Solis BioDyne, Tartu, Estonia). The thermocycling protocol was 95 °C for 14 min; 30 cycles of 95 °C for 45 s, 63 °C for 30 s and 72 °C for 30 s; 72 °C for 10 min.
Steady state metabolomics using LC–MS/MS
Nmnat1V9M/V9M mice and age-matched wildtype littermates were euthanized at 3 and 6 weeks of age by cervical dislocation, which is the preferred method for metabolite analysis (60). Immediately after sacrifice, the neural retinas were collected by the rapid ‘winkling’ procedure that does not require enucleation (61,62), placed in separate 1.5 ml microcentrifuge tubes, and snap-frozen in liquid nitrogen. Samples of brain, heart, liver, lung, kidney, and skin were also dissected, frozen, and stored in the same manner. Frozen brain was powdered using a liquid nitrogen-cooled mortar and pestle set (H37260-0100, SP Bel-Art, Wayne, NJ). To extract the metabolites, tissues were homogenized with 40:40:20 acetonitrile:methanol:water with 0.1 M formic acid at 4 ul/mg, neutralized with NH4HCO3 after 3 min, and then filtered using PVDF syringe filters (17 mm diameter, 0.2 μm pore). Samples (50 μl) were dried and then reconstituted using a 30:70 ratio of mobile phases A and B. Metabolites were then analyzed as previously described (63). Each metabolite was tuned with standards for optimal transitions, and the extracted multiple reaction monitoring peaks were integrated using MultiQuant version 3.0.3 software (AB Sciex, Framingham, MA). Brains were harvested from different mice than the rest of the tissues, which were harvested together, minimizing the dissection time per animal. For multi-tissue mice at 3 weeks (wildtype, n = ;7; Nmnat1V9M/V9M, n = ;8) and 6 weeks (wildtype, n = ;8; Nmnat1V9M/V9M, n = ;8) and for brain at 3 weeks (wildtype, n = ;6; Nmnat1V9M/V9M, n = ;8) and 6 weeks (wildtype, n = ;7; Nmnat1V9M/V9M, n = ;7).
Measurements of retinal thickness
Detailed methods for in vivo retinal imaging by spectral-domain OCT (Bioptogen, Morrisville, NC) are described elsewhere (10). Briefly, the average retinal thickness value was calculated from four evenly spaced measurements that were obtained using virtual calipers within the instrument’s imaging software (InVivoVue). The measurements were made from the nerve fiber layer to the retinal pigment epithelium in an optical section (B-scan) ~200 μm ventral to the center of the optic disc. For measurements presented in Supplementary Material, Figure S2: At 3, 6, and 8 weeks, Nmnat1V9M/V9M/Parp1−/− (n = ;7, 6, 7); at 3 and 8 weeks, wildtype (n = ;9, 10) and Nmnat1V9M/V9M/Parp1+/+ (n = ;10, 6). Data for the wildtype mice and Nmnat1V9M/V9M/Parp1+/+ mice are adapted from Greenwald et al., American Journal of Pathology (2016), with the regression used to calculate the values for the 6-week timepoint.
Quantitative western blotting
Retinas were harvested at 3 and 6 weeks of age, and both retinas from individual mice were pooled in each tube. Samples were homogenized in RIPA lysis buffer (Sigma, R0278) and sonicated. Protein was quantified using the Pierce BCA Protein Assay Kit (23 225, Thermo Scientific, Waltham, MA). In total, 25 μg protein was loaded into a 4–20% Tris-Glycine gel (XP04205BOX, Novex WedgeWell, Invitrogen, Carlsbad, CA) using 4X protein sample loading buffer (928-40 004, LI-COR, Lincoln, NE) and electrophoresed with the XCell SureLock Mini-Cell (El0001, ThermoFisher, Waltham, MA) using Tris-Glycine SDS running buffer (LC2675, Novex, Life Technologies, Carlsbad, CA). The protein was transferred to a nitrocellulose membrane (IPFL00010, Immobilon-FL Membrane, Millipore, Burlington, MA) using the Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad, 1 703 930, Hurcules, CA) and Tris-Glycine transfer buffer diluted in 20% methanol (LC3675, ThermoFisher). Membranes were incubated in blocking buffer (927-60001, Intercept Blocking Buffer, LI-COR, Lincoln, NE) for 1 h at room temperature with subsequent overnight incubation at 4 °C with the primary antibody of interest and a primary antibody against beta-actin (Table 2), each diluted in blocking buffer. After washing 3 times for 5 min each wash, the blots were then incubated in secondary antibody (1:5000, 926-68070, IRDye 680 RD goat anti-mouse, LI-COR Biosciences; 1:5000, 926-32211, IRDye 800 CW goat anti-rabbit, LI-COR Biosciences) at room temperature for 1 h. The blots were again washed 3 times for 5 min each wash and the signal was visualized using the Odyssey CLX imaging system (LI-COR Biosciences). Quantifications were performed using Image Studio software (LI-COR Biosciences). Samples sizes for the PAR western blots were 4 mice per genotype (wildtype, Nmnat1V9M/V9M/Parp1+/+, and Nmnat1WT/WT/Parp1−/−) at 3 and 6 weeks. Samples sizes for histone western blots were 3 mice per genotype (wildtype and Nmnat1V9M/V9M/Parp1+/+) at 3 weeks and 4 mice per genotype at 6 weeks.
Table 2.
Primary antibodies used for immunohistochemistry (IHC) and western blot (WB)
| Antibody | Host | Dilution (IHC) | Dilution (WB) | Company | Catalog # |
|---|---|---|---|---|---|
| 4-HNE | Rabbit | 1:500; 1:1000 | — | Alpha Diagnostic Intl, San Antonio, TX | HNE11-S |
| 8-OH-dG | Mouse | 1:500; 1:1000 | — | StressMarq, Victoria, CAN | SMC-155 |
| β-actin | Rabbit | — | 1:5000 | Abcam, Cambridge, UK | Ab8227 |
| β-actin | Mouse | — | 1:5000 | Abcam, Cambridge, UK | Ab8226 |
| H3 | Rabbit | — | 1:2500 | Abcam, Cambridge, UK | Ab1791 |
| H3-Lys9ac | Rabbit | 1:1000 | 1:1000 | Active Motif, Carlsbad, CA | 39 586 |
| H3-Lys18ac | Rabbit | 1:1000 | 1:1000 | Active Motif, Carlsbad, CA | 39 130 |
| H4 | Rabbit | — | 1:1000 | Abcam, Cambridge, UK | Ab177840 |
| H4-Lys16ac | Rabbit | 1:1000 | 1:1000 | Active Motif, Carlsbad, CA | 39 168 |
| Nitrotyrosine | Rabbit | 1:500; 1:1000 | — | Millipore, Burlington, MA | 06-284 |
| PAR [10H] | Mouse | 1:500 | 1:500 | GeneTex, Irvine, CA | GTX75054 |
To generate positive control samples for anti-PAR western blots, cultured ARPE-19 cells were treated with hydrogen peroxide (H2O2). The ARPE-19 cells (CRL-2302, ATCC, Manassas, VA) were maintained in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F12) media (11320082, ThermoFisher Scientific, Waltham, MA) that was supplemented with 10% fetal bovine serum (SH30071.03, GE Healthcare, Chicago, IL). Cells were cultured in 6-well plates (3516, Corning, Corning, NY) and treated with 300 or 500 uM H2O2 (BDH76090–3, VWR, Radnor, PA) for 20 min, then washed with phosphate-buffered saline (PBS), and finally homogenized in RIPA Lysis Buffer (Sigma, R0278) for western blotting.
Preparation of histological retinal sections
Immediately after euthanasia by CO2 asphyxiation, mice were perfused through the heart with heparinized (2 units/ml) PBS (pH 7.4), followed by 2% paraformaldehyde (PFA) using a Masterflex peristaltic pump (Cole-Parmer, Vernon Hills, IL). Before enucleation, the eyes were marked for orientation by making a mild corneal scar with a small vessel cauterizer (#18000-00, Fine Science Tools, Foster City, CA) slightly anterior to the superior limbus. The enucleated eyes were incubated for 30 min in 2% PFA before removal of the cornea (leaving a small portion for orientation), iris, and lens. The remaining eyecups were incubated in 2% PFA for 30 min and then immersed in 30% sucrose until having sunk to the bottom of the vial. The eyecups were then embedded in freezing medium (O.C.T. Compound, Fisher Scientific, Waltham, MA) and cryofrozen on ethanol-soaked dry ice. Each block contained samples from experimental and control animals to ensure that specimens being compared were treated identically throughout the experiment. Finally, the blocks were sliced into 10 μm sections by cryotomy, and the sections were collected on microscopy slides.
Immunohistochemistry
Cryosectioned specimens were washed in PBS 2 times for 10 min, permeabilized in PBS + 0.5% Triton-X 100 for 10 min, and then washed again in PBS for 5 min. Next, the specimens were incubated for 1 h in blocking buffer (PBS, 5% goat serum, 1% BSA, 0.2% Triton-X 100) before being incubated for 1 h in primary antibody (Table 2) diluted in blocking buffer. The specimens were then washed in PBS 3 times for 5 min each wash, before being incubated for 1 h in the appropriate secondary antibody (goat anti-mouse Alexa Fluor 555, A21422, Invitrogen, Carlsbad, CA; goat anti-rabbit Alexa Fluor 555, A32732, Invitrogen) diluted 1:500 in PBS + 0.3% Triton-X 100. The specimens were washed again in PBS 3 times for 5 min each wash, incubated with DAPI (1:10 000, D21490, Invitrogen) for 3–5 min, washed in PBS twice for 10 min each wash, and coverslipped with mounting media (Fluoromount-G, 00-4958-02, ThermoFisher). All steps were completed at room temperature. A negative control (i.e. the same specimens carried through the staining procedure with no primary antibody) was included for every experiment. Samples sizes for wildtype and Nmnat1V9M/V9M PAR study mice, at 4.5 to 6 weeks of age, were as follows: wildtype, n = ;6; Nmnat1V9M/V9M, n = ;8; Nmnat1WT/WT/Parp1−/−, n = ;5; and Nmnat1V9M/V9M/Parp1−/− mice, n = ;4. For antibody labeling against each acetylated histone (H3K9, H3K18, and H4K16) in wildtype and Nmnat1V9M/V9M mice at 3 weeks of age, 3 mice were tested per genotype. For antibody labeling against each 4-HNE and nitrotyrosine, 3 mice were tested per genotype at 4.5 and 6 weeks of age. For antibody labeling against 8-OH-dG, 4 wildtype and 3 Nmnat1V9M/V9M were tested at 4 weeks of age and > 8 mice were tested per genotype at 6 weeks of age.
TUNEL assay
TUNEL staining for cryopreserved tissue was performed according to the manufacturer’s instructions (#1684795, Roche, Basel, Switzerland). This assay is described by the manufacturer to preferentially label DNA strand breaks that occur during apoptosis, allowing this mode of cell death to be distinguished from strand breaks associated with necrosis. This protocol included a negative control in which the label solution without terminal transferase was used instead of the TUNEL reaction mixture, and it included a positive control in which the specimen was incubated with DNase I to induce DNA strand breaks before labeling. Samples sizes were n = ;3 per genotype at 4.5 weeks of age and n = ;4 per genotype at 6 weeks of age.
Malondialdehyde assay
Retinas were harvested at 3 weeks and 4 weeks of age. Both retinas from each mouse (n = ;3 per genotype at each age, except n = ;4 for 3-week-old Nmnat1V9M/V9M mice) were pooled in a tube and were immediately flash frozen in liquid nitrogen. Samples were thawed on ice, and the assay was performed according to manufacturer’s instructions for fluorometric analysis (ab118970, Abcam, Cambridge, UK).
Microscopy
Retinal sections that had been either immunolabeled or subjected to the TUNEL assay were imaged by fluorescence microscopy (Eclipse Ti, Nikon, Tokyo, Japan). Fiji image processing software (64) was used to make linear brightness/contrast adjustments across images and to merge color channels. Counts of PAR and TUNEL positive photoreceptors were performed in images with widths showing a 450 μm field.
Dark housing of mice
A subset of mice was kept in constant darkness from birth until the single and terminal experimental time point in which the retinas were imaged by OCT. A dedicated dark room was not available in the vivarium, so mice were housed in custom opaque black plastic cages (Lab Products, Houston, TX) with blackout canopies above and below the cages on the rack. The cages were positioned in the bottom two rows of the rack to further minimize the amount of light. All mouse colony maintenance duties were performed by the investigators only after all sources of light that are visible to mice were turned off or blocked. These tasks were completed under long-wavelength illumination using a filtered (Supergel 26, Rosco, Stamford, CT) headlamp which emits light at wavelengths beyond the spectral absorption of murine cone photopigments (65). Light measurements were acquired inside the cage using a digital lux meter (Model LX1330B, Dr. Meter), and were close to zero, within the 4% noise tolerance of the device. Sample sizes at 3, 6, and 12 weeks: dark-raised pigmented mice, wildtype (n = ;5, 9, 6) and Nmnat1V9M/V9M (n = ;5, 9, 3); dark-raised non-pigmented mice, wildtype (n = ;4, 3, 3) and Nmnat1V9M/V9M (n = ;5, 3, 3); light-raised non-pigmented mice, wildtype (n = ;5, 5, 5) and Nmnat1V9M/V9M (n = ;7, 4, 3). Sample sizes for light-raised pigmented mice at 3 and 12 weeks: wildtype (n = ;9, 7) and Nmnat1V9M/V9M (n = ;10, 7). Data for light-raised mice are adapted from Greenwald et al., American Journal of Pathology (2016), with the regression used to calculate the values for the 6-week timepoint.
Statistical analyses
For all statistical analyses, data are reported as the mean ± standard error of mean (SEM). For each metabolite analyzed, the unpaired t-test was used to compare the means of the wildtype and mutant mice for each tissue at each time point. Statistics were not calculated across time points because the samples were tested by LC–MS/MS in separate batches. The values for left and right retinas were averaged for each mouse before statistical analysis. Median absolute deviation was used to identify outliers based on a conservative rejection criterion factor of 4 (66). For each LC–MS/MS assay, data from the Nmnat1V9M/V9M mice were normalized to the wildtype measurements for the purpose of presentation. PAR and TUNEL positive photoreceptors were manually counted in microscopy images by an observer who was masked to the genotypes of the mice. For the counts of PAR positive photoreceptors, the results for all genotypes were compared using the one-way analysis of variance (ANOVA), followed with the Dunnett post hoc test to account for Type 1 errors associated with multiple comparisons. For the TUNEL quantification, the unpaired t-test was used to compare wildtype with Nmnat1V9M/V9M retinas within each age, and the Holm–Sidak method was used to correct for multiple comparisons. For quantitative western blot analyses of PAR, a one-way ANOVA was used, followed with the Tukey post hoc test. Retinal thickness measurements were evaluated using either the one-way (Supplementary Material, Fig. S2) or two-way (Supplementary Material, Fig. S5) ANOVA, followed with the Dunnett or Tukey post hoc test, respectively. For histone-related measurements and the MDA assay, statistical significance was determined using the unpaired t-test, corrected for multiple comparisons via the Holm–Sidak method. PAR and histone western blot data were normalized to the respective wildtype result at 3 weeks of age.
Conflict of Interest Statement. None.
Supplementary Material
Funding
This work was supported by the National Eye Institute (EY012910 to E.A.P, EY026030 to J.D.).
References
- 1. Falk, M.J., Zhang, Q., Nakamaru-Ogiso, E., Kannabiran, C., Fonseca-Kelly, Z., Chakarova, C., Audo, I., Mackay, D.S., Zeitz, C., Borman, A.D.et al. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet., 44, 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koenekoop, R.K., Wang, H., Majewski, J., Wang, X., Lopez, I., Ren, H., Chen, Y., Li, Y., Fishman, G.A., Genead, M.et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet., 44, 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perrault, I., Hanein, S., Zanlonghi, X., Serre, V., Nicouleau, M., Defoort-Delhemmes, S., Delphin, N., Fares-Taie, L., Gerber, S., Xerri, O.et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet., 44, 975–977. [DOI] [PubMed] [Google Scholar]
- 4. Chiang, P.W., Wang, J., Chen, Y., Fu, Q., Zhong, J., Chen, Y., Yi, X., Wu, R., Gan, H., Shi, Y.et al. (2012) Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet., 44, 972–974. [DOI] [PubMed] [Google Scholar]
- 5. Chiarugi, A., Dolle, C., Felici, R. and Ziegler, M. (2012) The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer, 12, 741–752. [DOI] [PubMed] [Google Scholar]
- 6. Lau, C., Niere, M. and Ziegler, M. (2009) The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci (Landmark Ed), 14, 410–431. [DOI] [PubMed] [Google Scholar]
- 7. Mori, V., Amici, A., Mazzola, F., Di Stefano, M., Conforti, L., Magni, G., Ruggieri, S., Raffaelli, N. and Orsomando, G. (2014) Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One, 9, e113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sasaki, Y., Margolin, Z., Borgo, B., Havranek, J.J. and Milbrandt, J. (2015) Characterization of Leber's congenital Amaurosis-associated NMNAT1 mutants. J. Biol. Chem., 290, 17228–17238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conforti, L., Janeckova, L., Wagner, D., Mazzola, F., Cialabrini, L., Di Stefano, M., Orsomando, G., Magni, G., Bendotti, C., Smyth, N.et al. (2011) Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J., 278, 2666–2679. [DOI] [PubMed] [Google Scholar]
- 10. Greenwald, S.H., Charette, J.R., Staniszewska, M., Shi, L.Y., Brown, S.D., Stone, L., Liu, Q., Hicks, W.L., Collin, G.B., Bowl, M.R.et al. (2016) Mouse models of NMNAT1-Leber congenital Amaurosis (LCA9) recapitulate key features of the human disease. Am. J. Pathol., 186, 1925–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cantó, C., Sauve, A.A. and Bai, P. (2013) Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med., 34, 1168–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim, M.Y., Zhang, T. and Kraus, W.L. (2005) Poly(ADP-ribosyl) ation by PARP-1: 'PAR-laying' NAD+ into a nuclear signal. Genes Dev., 19, 1951–1967. [DOI] [PubMed] [Google Scholar]
- 13. Zhang, T. and Kraus, W.L. (2010) SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta, 1804, 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferro, A.M. and Olivera, B.M. (1982) Poly(ADP-ribosylation) in vitro. Reaction parameters and enzyme mechanism. J. Biol. Chem., 257, 7808–7813. [PubMed] [Google Scholar]
- 15. Ying, W. (2013) Roles of NAD (+) , PARP-1, and sirtuins in cell death, ischemic brain injury, and synchrotron radiation X-ray-induced tissue injury. Scientifica (Cairo), 2013, 691251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKinsey, T.A., Zhang, C.L. and Olson, E.N. (2001) Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev., 11, 497–504. [DOI] [PubMed] [Google Scholar]
- 17. Bai, P. and Canto, C. (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab., 16, 290–295. [DOI] [PubMed] [Google Scholar]
- 18. Paquet-Durand, F., Silva, J., Talukdar, T., Johnson, L.E., Azadi, S., vanVeen, T., Ueffing, M., Hauck, S.M. and Ekstrom, P.A. (2007) Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci., 27, 10311–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alano, C.C., Garnier, P., Ying, W., Higashi, Y., Kauppinen, T.M. and Swanson, R.A. (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci., 30, 2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang, Z.Q., Auer, B., Stingl, L., Berghammer, H., Haidacher, D., Schweiger, M. and Wagner, E.F. (1995) Mice lacking ADPRT and poly(ADP-ribosyl) ation develop normally but are susceptible to skin disease. Genes Dev., 9, 509–520. [DOI] [PubMed] [Google Scholar]
- 21. Sahaboglu, A., Tanimoto, N., Kaur, J., Sancho-Pelluz, J., Huber, G., Fahl, E., Arango-Gonzalez, B., Zrenner, E., Ekstrom, P., Lowenheim, H.et al. (2010) PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS One, 5, e15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koch-Nolte, F., Fischer, S., Haag, F. and Ziegler, M. (2011) Compartmentation of NAD+-dependent signalling. FEBS Lett., 585, 1651–1656. [DOI] [PubMed] [Google Scholar]
- 23. Adamkova, K., Yi, Y.J., Petr, J., Zalmanova, T., Hoskova, K., Jelinkova, P., Moravec, J., Kralickova, M., Sutovsky, M., Sutovsky, P.et al. (2017) SIRT1-dependent modulation of methylation and acetylation of histone H3 on lysine 9 (H3K9) in the zygotic pronuclei improves porcine embryo development. Journal of animal science and biotechnology, 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Michishita, E., McCord, R.A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., Cheung, P., Kusumoto, R., Kawahara, T.L., Barrett, J.C.et al. (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature, 452, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liszt, G., Ford, E., Kurtev, M. and Guarente, L. (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem., 280, 21313–21320. [DOI] [PubMed] [Google Scholar]
- 26. Barber, M.F., Michishita-Kioi, E., Xi, Y., Tasselli, L., Kioi, M., Moqtaderi, Z., Tennen, R.I., Paredes, S., Young, N.L., Chen, K.et al. (2012) SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature, 487, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. D'Onofrio, N., Servillo, L. and Balestrieri, M.L. (2018) SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid. Redox Signal., 28, 711–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Serrano, L., Martínez-Redondo, P., Marazuela-Duque, A., Vazquez, B.N., Dooley, S.J., Voigt, P., Beck, D.B., Kane-Goldsmith, N., Tong, Q., Rabanal, R.M.et al. (2013) The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev., 27, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eldridge, M.J.G., Pereira, J.M., Impens, F. and Hamon, M.A. (2020) Active nuclear import of the deacetylase Sirtuin-2 is controlled by its C-terminus and importins. Sci. Rep., 10, 2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lan, L., Nakajima, S., Oohata, Y., Takao, M., Okano, S., Masutani, M., Wilson, S.H. and Yasui, A. (2004) In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. U. S. A., 101, 13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sova, H., Jukkola-Vuorinen, A., Puistola, U., Kauppila, S. and Karihtala, P. (2010) 8-Hydroxydeoxyguanosine: a new potential independent prognostic factor in breast cancer. Br. J. Cancer, 102, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Halprin, K.M. and Ohkawara, A. (1967) The measurement of glutathione in human epidermis using glutathione reductase. J. Invest. Dermatol., 48, 149–152. [DOI] [PubMed] [Google Scholar]
- 33. Esterbauer, H. and Cheeseman, K.H. (1990) Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol., 186, 407–421. [DOI] [PubMed] [Google Scholar]
- 34. Ahsan, H. (2013) 3-Nitrotyrosine: a biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol., 74, 1392–1399. [DOI] [PubMed] [Google Scholar]
- 35. Krishnamoorthy, R.R., Crawford, M.J., Chaturvedi, M.M., Jain, S.K., Aggarwal, B.B., Al-Ubaidi, M.R. and Agarwal, N. (1999) Photo-oxidative stress down-modulates the activity of nuclear factor-kappaB via involvement of caspase-1, leading to apoptosis of photoreceptor cells. J. Biol. Chem., 274, 3734–3743. [DOI] [PubMed] [Google Scholar]
- 36. Natoli, R., Jiao, H., Barnett, N.L., Fernando, N., Valter, K., Provis, J.M. and Rutar, M. (2016) A model of progressive photo-oxidative degeneration and inflammation in the pigmented C57BL/6J mouse retina. Exp. Eye Res., 147, 114–127. [DOI] [PubMed] [Google Scholar]
- 37. David, K.K., Andrabi, S.A., Dawson, T.M. and Dawson, V.L. (2009) Parthanatos, a messenger of death. Front Biosci. (Landmark Ed), 14, 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Andrabi, S.A., Kim, N.S., Yu, S.W., Wang, H., Koh, D.W., Sasaki, M., Klaus, J.A., Otsuka, T., Zhang, Z., Koehler, R.C.et al. (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U. S. A., 103, 18308–18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alano, C.C., Tran, A., Tao, R., Ying, W., Karliner, J.S. and Swanson, R.A. (2007) Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res., 85, 3378–3385. [DOI] [PubMed] [Google Scholar]
- 40. Wang, S., Yang, X., Lin, Y., Qiu, X., Li, H., Zhao, X., Cao, L., Liu, X., Pang, Y., Wang, X.et al. (2013) Cellular NAD depletion and decline of SIRT1 activity play critical roles in PARP-1-mediated acute epileptic neuronal death in vitro. Brain Res., 1535, 14–23. [DOI] [PubMed] [Google Scholar]
- 41. Cambronne, X.A., Stewart, M.L., Kim, D., Jones-Brunette, A.M., Morgan, R.K., Farrens, D.L., Cohen, M.S. and Goodman, R.H. (2016) Biosensor reveals multiple sources for mitochondrial NAD(+). Science, 352, 1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Power, M., Das, S., Schutze, K., Marigo, V., Ekstrom, P. and Paquet-Durand, F. (2020) Cellular mechanisms of hereditary photoreceptor degeneration - focus on cGMP. Prog. Retin. Eye Res., 74, 100772. [DOI] [PubMed] [Google Scholar]
- 43. Williams, M.L., Coleman, J.E., Haire, S.E., Aleman, T.S., Cideciyan, A.V., Sokal, I., Palczewski, K., Jacobson, S.G. and Semple-Rowland, S.L. (2006) Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med., 3, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perrault, I., Rozet, J.M., Calvas, P., Gerber, S., Camuzat, A., Dollfus, H., Chatelin, S., Souied, E., Ghazi, I., Leowski, C.et al. (1996) Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet., 14, 461–464. [DOI] [PubMed] [Google Scholar]
- 45. Nejabati, H.R., Mihanfar, A., Pezeshkian, M., Fattahi, A., Latifi, Z., Safaie, N., Valiloo, M., Jodati, A.R. and Nouri, M. (2018) N1-methylnicotinamide (MNAM) as a guardian of cardiovascular system. J. Cell. Physiol., 233, 6386–6394. [DOI] [PubMed] [Google Scholar]
- 46. Tarantini, S., Valcarcel-Ares, M.N., Toth, P., Yabluchanskiy, A., Tucsek, Z., Kiss, T., Hertelendy, P., Kinter, M., Ballabh, P., Süle, Z.et al. (2019) Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol., 24, 101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kiss, T., Nyúl-Tóth, Á., Balasubramanian, P., Tarantini, S., Ahire, C., Yabluchanskiy, A., Csipo, T., Farkas, E., Wren, J.D., Garman, L.et al. (2020) Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. GeroScience, 42, 527–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yoshino, J., Baur, J.A. and Imai, S.I. (2018) NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab., 27, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fu, Y. and Yau, K.W. (2007) Phototransduction in mouse rods and cones. Pflugers Arch., 454, 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yau, K.W. and Hardie, R.C. (2009) Phototransduction motifs and variations. Cell, 139, 246–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rozanowska, M. and Sarna, T. (2005) Light-induced damage to the retina: role of rhodopsin chromophore revisited. Photochem. Photobiol., 81, 1305–1330. [DOI] [PubMed] [Google Scholar]
- 52. Ames, A., 3rd, Li, Y.Y., Heher, E.C. and Kimble, C.R. (1992) Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J. Neurosci., 12, 840–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dawson, V.L. and Dawson, T.M. (2004) Deadly conversations: nuclear-mitochondrial cross-talk. J. Bioenerg. Biomembr., 36, 287–294. [DOI] [PubMed] [Google Scholar]
- 54. Houtkooper, R.H., Cantó, C., Wanders, R.J. and Auwerx, J. (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev., 31, 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Owen, J.B. and Butterfield, D.A. (2010) Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol., 648, 269–277. [DOI] [PubMed] [Google Scholar]
- 56. Fain, G.L. and Lisman, J.E. (1993) Photoreceptor degeneration in vitamin a deprivation and retinitis pigmentosa: the equivalent light hypothesis. Exp. Eye Res., 57, 335–340. [DOI] [PubMed] [Google Scholar]
- 57. Fain, G.L. and Lisman, J.E. (1999) Light, Ca2+, and photoreceptor death: new evidence for the equivalent-light hypothesis from arrestin knockout mice. Invest. Ophthalmol. Vis. Sci., 40, 2770–2772. [PubMed] [Google Scholar]
- 58. Kuribayashi, H., Baba, Y., Iwagawa, T., Arai, E., Murakami, A. and Watanabe, S. (2018) Roles of Nmnat1 in the survival of retinal progenitors through the regulation of pro-apoptotic gene expression via histone acetylation. Cell Death Dis., 9, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Greenwald, S.H., Brown, E.E., Scandura, M.J., Hennessey, E., Farmer, R., Pawlyk, B.S., Xiao, R., Vandenberghe, L.H. and Pierce, E.A. (2020) Gene therapy preserves retinal structure and function in a mouse model of NMNAT1-associated retinal degeneration. Mol. Ther. Meth. Clin. Dev., 18, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhu, S., Yam, M., Wang, Y., Linton, J.D., Grenell, A., Hurley, J.B. and Du, J. (2018) Impact of euthanasia, dissection and postmortem delay on metabolic profile in mouse retina and RPE/choroid. Exp. Eye Res., 174, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Winkler, B.S. (1972) The electroretinogram of the isolated rat retina. Vis. Res., 12, 1183–1198. [DOI] [PubMed] [Google Scholar]
- 62. Anderson, R.E., Maude, M.B., McClellan, M., Matthes, M.T., Yasumura, D. and LaVail, M.M. (2002) Low docosahexaenoic acid levels in rod outer segments of rats with P23H and S334ter rhodopsin mutations. Mol. Vis., 8, 351–358. [PubMed] [Google Scholar]
- 63. Grenell, A., Wang, Y., Yam, M., Swarup, A., Dilan, T.L., Hauer, A., Linton, J.D., Philp, N.J., Gregor, E., Zhu, S.et al. (2019) Loss of MPC1 reprograms retinal metabolism to impair visual function. Proc. Natl. Acad. Sci. U. S. A., 116, 3530–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jacobs, G.H., Fenwick, J.C., Calderone, J.B. and Deeb, S.S. (1999) Human cone pigment expressed in transgenic mice yields altered vision. J. Neurosci., 19, 3258–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leys C., L.C., Klein O., Bernard P., Licata, L. (2013) Detecting outliers: do not use standard deviation around the mean, use absolute deviation around the median. J. Exp. Soc. Psychol., 49, 764–766. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.