

Abstract
Hypertrophic cardiomyopathy (HCM) is a genetic disease of the myocardium characterized by a hypertrophic left ventricle with a preserved or increased ejection fraction. Cardiac hypertrophy is often asymmetric, which is associated with left ventricular outflow tract obstruction. Myocyte hypertrophy, disarray and myocardial fibrosis constitute the histological features of HCM. HCM is a relatively benign disease but an important cause of sudden cardiac death in the young and heart failure in the elderly.
Pathogenic variants (PVs) in genes encoding protein constituents of the sarcomeres are the main causes of HCM. PVs exhibit a gradient of effect sizes, as reflected in their penetrance and variable phenotypic expression of HCM. MYH7 and MYBPC3, encoding β-myosin heavy chain and myosin binding protein C, respectively, are the two most common causal genes and responsible for ~40% of all HCM cases but a higher percentage of HCM in large families. PVs in genes encoding protein components of the thin filaments are responsible for ~5% of the HCM cases. Whereas pathogenicity of the genetic variants in large families has been firmly established, ascertainment causality of the PVs in small families and sporadic cases is challenging. In the latter category PVs are best considered as probabilistic determinants of HCM.
Deciphering the genetic basis of HCM has enabled routine genetic testing and has partially elucidated the underpinning mechanism of HCM as increased number of the myosin molecules that are strongly bound to actin. The discoveries have led to the development of mavacamten that targets binding of the myosin molecule to actin filaments and imparts beneficial clinical effects. In the coming years the yield of the genetic testing is expected to be improved and the so-called “missing causal gene” be identified. The advances are also expected to enable development of additional specific therapies and editing of the mutations in HCM.
Subject Terms: Genetics, Heart Failure, Hypertrophy
Keywords: Hypertrophy, Genetics, Mutation, Sudden Cardiac Death, Heart Failure, Myosin heavy chain, Sarcomere, diastolic heart failure
INTRODUCTION
The main objective is to discuss the genetic basis of hypertrophic cardiomyopathy (HCM) from a prospective of deterministic and probabilistic approaches in discerning the pathogenicity of the genetic variants (GVs). The second objective is to highlight impact of the genetic discoveries in providing fundamental insights into the pathogenesis and hence, enabling specific treatment of patients with HCM. Other aspects of HCM are briefly discussed in order to provide a context for the genetic discoveries. For a conventional review, the readers are referred to a recent review co-authored by Dr. Braunwald, whose seminal characterization of the clinical and hemodynamic aspects of HCM has formed the essence of our current understanding of this intriguing disease.1
Henri Liouville, a French pathologist has been credited for describing the first case of HCM, a case likely with left ventricular outflow tract obstruction (LVOTO) in 1869 (described in2). In the same year, Frederick Miescher isolated DNA, which he described as “nuclein”. (described in3) In the next few decades, HCM was merely a pathological entity, detected at autopsy. In 1949, William Evans recognized the familial occurrence of HCM and referred to the disease as “familial cardiomegaly”4. A few years later, Donald Teare described the familial nature of asymmetric septal hypertrophy, which he described as muscular hamartoma5. An autosomal dominant mode of inheritance of HCM was illustrated by J.A.P. Paré and colleagues in a large French-Canadian family in 1961.6 About 30 years later, Christine and Jonathan Seidman identified the first mutation in the gene encoding the myosin heavy chain 7 (MYH7) protein in the family that was described by Paré.7. The seminal discovery paved the way for partial delineation of the molecular genetic basis of HCM. The advances ushered in the era of molecular genetic testing and laid the foundation for design of specific therapies for HCM.8
DEFINITION
HCM is defined based on its phenotypic features, which are cardiac hypertrophy, a non-dilated left ventricle, and a preserved or an enhanced left ventricular ejection fraction (LVEF). Cardiac hypertrophy in an adult is defined as a maximum left ventricular end diastolic wall thickness of >13 mm on an echocardiogram or other imaging techniques. In children, a Z-score of greater 2 of maximum wall thickness, matched for sex and age, is used to detect cardiac hypertrophy. A maximum wall thickness of ≥ 15 mm in adults is used to define HCM, which offers a greater specificity at the cost of under-detection.
Expression of cardiac hypertrophy is age-dependent, evolving gradually during the second to fourth decades of life.9, 10 Cardiac hypertrophy may be regional, involving only a segment of the left ventricle, rendering it difficult to detect on imaging modalities. The presence of confounding factors, such as concomitant hypertension, valvular diseases, athletes’ heart, and the phenocopy conditions further compound the accurate diagnosis of HCM. Phenotypic features, such as the asymmetric nature of hypertrophy with a predominant involvement of the interventricular septum, a small left ventricular cavity size, and LVOTO could enhance an accurate diagnosis. In a subset of patients, hypertrophy is predominantly or exclusively restricted to left ventricular apex, which is referred to as apical HCM. It has the characteristic electrocardiographic findings notable for a deep T wave inversion in the precordial leads.
EPIDEMIOLOGY
The estimated prevalence of HCM in the general adult population, diagnosed based on the presence of cardiac hypertrophy, is between 1:300 to 1:600.11, 12 Incorporation of more sensitive diagnostic tools, family information, and the genetic data increases the population prevalence of HCM to ~1:250 individuals in the general population13, 14. HCM, as an autosomal disease, does not show a predilection toward a specific ethnic backgrounds, sex, or geographic locations, albeit sex and ethnic backgrounds influence its phenotypic expression.15, 16,17, 18
MOLECULAR GENETIC BASIS OF HCM
Classically, HCM is a quintessential single gene disorder with an almost exclusive autosomal dominant mode of inheritance. Genetic linkage and co-segregation analysis in large families, exhibiting an autosomal dominant model of inheritance, have led to partial elucidation of the molecular genetic basis of HCM. Christine and Jonathan Seidman identified the p.Arg403Glu (p.R403Q) mutation in the MYH7 gene, encoding sarcomere protein MYH7, as the first mutation for familial HCM.7 Subsequently, over a dozen causal genes were identified, which led to a near complete elucidation of the molecular genetic basis of HCM in large families.19–21 In about half of the cases, either the family size is too small or the disease is sporadic. The molecular genetic etiology of HCM in the sporadic cases and small families is less well-defined, as unequivocal ascertainment of causality in such cases is exceedingly challenging. To set the stage and gain some insights into the challenges encountered in full delineation of the genetic basis of HCM, a brief discussion on the genome-wide spectrum, population frequencies, and functions of the GVs is presented. An expanded discussion of these topic is provided in a recent review.22
A primer on the spectrum of the GVs in the human genome
Each human genome, comprised of ~ 3.2 × 109 base pairs, differs in approximately 4 to 5 million nucleotides from the reference genome.23–25 The variation is mainly the consequence of the error rate of the DNA replication machinery that is estimated to be ~ 1.3 ×10−8 per nucleotide.26–28 Consequently, each genome introduces approximately 60 de novo variants (absent in the parents’ genomes) to the population pool.26–29 Common variants are typically shared across the populations; however, the vast majority of the rare variants are restricted to specific populations.25,30
GVs exhibit a continuum of size variation, ranging from a single base to the whole-chromosome (Table 1). Variants affecting a single nucleotide, referred to as single nucleotide variants (SNVs) or single nucleotide polymorphism (SNP), are the most common variants, totally about 4 million in each genome. Small insertion/deletion (indels) variants are the second most common variants in the human genome.
TABLE 1.
Spectrum of Genetic Variants in the Human Genome
| Category | Description | Relevance to HCM |
|---|---|---|
| Nonsynonymous SNVs (nsSNVs) |
|
|
| Insertion/deletion variants (Indels) |
|
|
| Splice junction variants |
|
|
| Synonymous SNVs (silent variants) (synSNVs) |
|
|
| Regulatory variants |
|
|
| Intronic and intergenic variants |
|
|
| Structural variants (SVs) |
|
|
Abbreviations:
SNVs: Single nucleotide polymorphism; HCM: Hypertrophic cardiomyopathy; PV: Pathogenic variant s; MYBPC3: Myosin binding protein C3; Indel: insertion/deletion
Functional characteristics of the GVs
The wide spectrum of the variants in the genomes of apparently healthy individuals demonstrates the challenging nature of ascertaining their functional significance and clinical pathogenicity.23, 24 In silico prediction of pathogenicity is based on the anticipated effects of the GVs on the structure and function of the genome and the involved proteins, such as the effects on polarity, charge, and hydropathy of the involved amino acid, secondary and tertiary structures of the mRNAs and proteins, as well as the catalytic activities of the involved proteins. (reviewed in31) Likewise, evolutionary conservation of the involved codon and the population frequency of the variant denote provisional functionality, requiring validation based on the clinical and experimental evidence. However, experimental data, while valuable, are subject to the experimental conditions, which often are not reflective of the in vivo biology in human, as functional and clinical significance of the GVs is often influenced by the milieu in which they occur. Likewise, population-level functional data, although valuable, often are not fully applicable at the individual level, necessitating the focus on the individual in assessing function and pathogenicity of the GVs.
At the genetic level, the loss-of-function (LoF) variants are predicted to impair protein-coding function of the involved genes. Accordingly, non-sense, gain-of-stop codon, canonical splice site, and frame-shift indel variants, which lead to expression of unstable premature truncated proteins, are predicted to be LoF variants. A LoF variant might also delete an entire exon or the gene itself. Throughout the text LoF is used to denote the predicted function, unless the LoF variant is experimentally documented. A gain-of-function (GoF) variant is defined biologically, as it affords the protein a new function, such as interaction with a new protein, or enhances its enzymatic activity.
Categorization of the GVs for clinical applications
Clinical significance of the GVs to some extent is reflective of their functional significance, albeit not all functional variants impart clinically discernible effects. This is amply evident in the personal genome sequencing data showing the presence of several hundred predicted (p)LoF variants in each genome in the absence of a discernible related clinical phenotype.23, 24,25 On the other hand, all clinically significant variants are expected to be functional, even though their functions might not be evidence, when the clinical phenotype is detected.
Clinical impacts of the functional variants are also determined by the tolerance of the involved gene to the LoF variants, which varies across the genes. The intolerance reflects a gradual elimination of the most deleterious variants from the genome by natural selection. Accordingly, a LoF variants in a gene that exhibits a strong natural selection against LoF variants, i.e., the gene that is intolerant to the LoF variants, is expected to impart larger clinical effects than a LoF variant in a gene that is tolerant to LoF variants. Consequently, LoF variants in genes that are intolerant to LoF variants are rare in the general population and when present, they are typically pathogenic. In contrast, LoF in genes that are tolerant to the LoF variants, are often found in the general population with no discernible phenotype. Thus, information on the pathogenicity of LoF is partially embedded in the population frequency of a LoF variant.
Clinically, the GVs are categorized into pathogenic variants (PVs), likely (L)PVs, variants of unknown significance, likely benign variants and benign variants (Table 2). In addition, a subset of GVs affects function or level of the involved protein without a clear clinical significance. The focus in clinical genetics is on the PVs, defined as variants for which there is strong evidence for involvement in the pathogenesis of the disease, and to a lesser extent on LPVs. At the broader genetic parlance, any change in the nucleotide sequence is considered a mutation. At the clinical genetic level, however, a mutation is defined as a variant with a population frequency of ≤1%. Given the plethora of the rare variants in each genome and to imply a clinical significance, the term mutation should be reserved only for a subset of the PVs for which there is a well-established robust and unequivocal evidence of causality in the phenotype.
TABLE2.
Categorization of the Genetic Variant for Clinical Applications in HCM
| Category | Definition | Characteristics |
|---|---|---|
| PVs | Variants for which there is a strong genetic evidence for their involvement in the pathogenesis of HCM |
|
| LPVs | As for PVs, except that genetic evidence for their involvement in the pathogenesis of HCM is less robust |
|
| Functional variants with undefined clinical significance | Variants affect gene expression and/or protein function but are not associated with a clinical phenotype |
|
| Variants with unknown significance | Undefined functional or clinical significance |
|
| Likely benign variants | These variants are not expected to cause discernible phenotypic effects. They might have modest effects on gene expression but are not related to HCM |
|
| Benign variants | Variants with no discernible functional, biological, or clinical effects |
|
Abbreviations:
PVs: Pathogenic variants; LPVs: Likely pathogenic variants; LoF: Predicted loss of function; CADD: Combined Annotated Dependent Depletion, which is a score calculated upon integration of more than 60 genomic features of single nucleotide variants and indels.
PVs and the clinical phenotype
Expression of the clinical phenotype of HCM is the consequence of non-linear and stochastic interactions among multiple genetic and non-genetic factors. PVs are the key determinants of the phenotype but their penetrance varies according to their effect sizes and the genetic backgrounds of the individuals. Penetrance of the PVs is partially determined by the impact of the PVs on the structure and function of the genes and the encoded proteins with those totally disrupting the protein function exhibiting the highest penetrance. In addition, the penetrance is expected to be higher for PVs that are located in the proteins that are proximal to the phenotype, i.e., directly related to the pathogenesis of the phenotype, as opposed to PVs in proteins that are distant or indirectly related to the phenotype. Moreover, functional variants in other genes and proteins related to the phenotype are expected to influence penetrance of the main PVs and modify phenotypic expression of the phenotype. Hence, the latter group of the functional variants are referred to as the modifier variants (MVs).32–34 The MVs are neither sufficient nor necessary to cause HCM but in aggregate impart a large influence on its phenotypic expression. Consequently, in the classic single gene HCM, the phenotype is also a polygenic trait, influenced by a large number of functional variants. The collective effect sizes of the MVs and non-genetic factors increase inversely with the effect size of the main PV. Accordingly, when the effect size of the main PV is large, the contribution of the MVs and non-genetic factors is relatively modest and vice versa.
Deterministic vs. Probabilistic approach to understanding the genetic basis of HCM
The conventional deterministic approach to genetic studies of HCM, as a quintessential single gene disease, posits that the presence of the single PV or mutation is necessary and sufficient to cause HCM, albeit with a variable penetrance and expressivity. The contributions of the deterministic approach to elucidation of the genetic basis of HCM in large families, exhibiting an autosomal dominant mode of inheritance, are irrefutable. The approach has led to identification of the most common causal genes in familial HCM. The deterministic approach to some extent, however, is oblivious to the gradient of effect sizes of the PVs and the continuity of the phenotypic expression at the molecular and clinical levels. Consequently, the deterministic approach cannot be successfully applied to decipher the genetic basis of HCM in small families wherein co-segregation and linkage cannot be established; and to sporadic cases, wherein the causality of a given PV cannot be ascertained unambiguously. Consequently, the genetic basis of HCM has remained obscure in about half of the cases.
The probabilistic approach surmises that PVs in genes encoding proteins directly or indirectly involved in the biological networks pertinent to the phenotype contribute to the pathogenesis of the HCM, albeit the extent of contributions varies according to the effect size of the variants. It also conjectures that there is a genetic hierarchy in the pathogenesis of the phenotype. Accordingly, PVs in genes whose encoded proteins are involved in biological pathways directly relevant to the pathogenesis of HCM have a higher probability of exerting larger effect sizes than those residing in genes that are indirectly related to the phenotype. Similarly, PVs in genes that are expressed at high levels in cardiac myocytes, the primary cell type involved in HCM, are expected to impart larger effects on the phenotype than those that are expressed at low levels. Moreover, PVs in genes that are highly intolerant to the LoF or missense variants are expected to make larger contributions to the phenotype than those in genes that are somewhat tolerant to LoF or missense variants.
The probabilistic approach considers the phenotype in HCM as a polygenic trait secondary to contributions, large and small, of multiple PVs (Figure 1). Consequently, it shifts the focus from identification of a single PV to multiple PVs in inducing the phenotype in HCM, particularly in small families and the sporadic cases. The approach is predominantly pertinent to the PVs that do not exhibit a high penetrance and therefore, do not show robust evidence of co-segregation and genetic linkage. Given the complexity of the clinical phenotype, a probabilistic approach is preferable in discerning determinants of the phenotype in HCM. According to this notion, PVs in genes responsible for the classic single gene HCM, such as those in MYH7 and MYBPC3, are expected to be enriched in the sporadic cases with HCM. Further extension of this notion posits that such PVs are also expected to be enriched in individuals who exhibit an exaggerated hypertrophic response to external stimuli, such as pressure overload (aortic stenosis or systemic arterial hypertension) or physical exercise (professional athletes). Finally, the probabilistic stipulates that the PVs involving pathways that are relevant to the phenotype, directly or indirectly, account for the so-called “missing causal genes”, as discussed later. The clinical impact of the approach is amply evident in clinical genetic testing, which thus-far has had a relatively low yield by focusing in identifying the PVs in genes already known to cause HCM.35 The main caveat with the probabilistic approach is to the risk of over-interpretation and improper clinical conclusions. The proper utilization of the approach requires a clear understanding of the molecular genetics, biology and pathogenesis of HCM.
Figure 1. Gradient of effect sizes of the pathogenic variants (PVs) and their population frequencies in the context of familial and sporadic HCM.
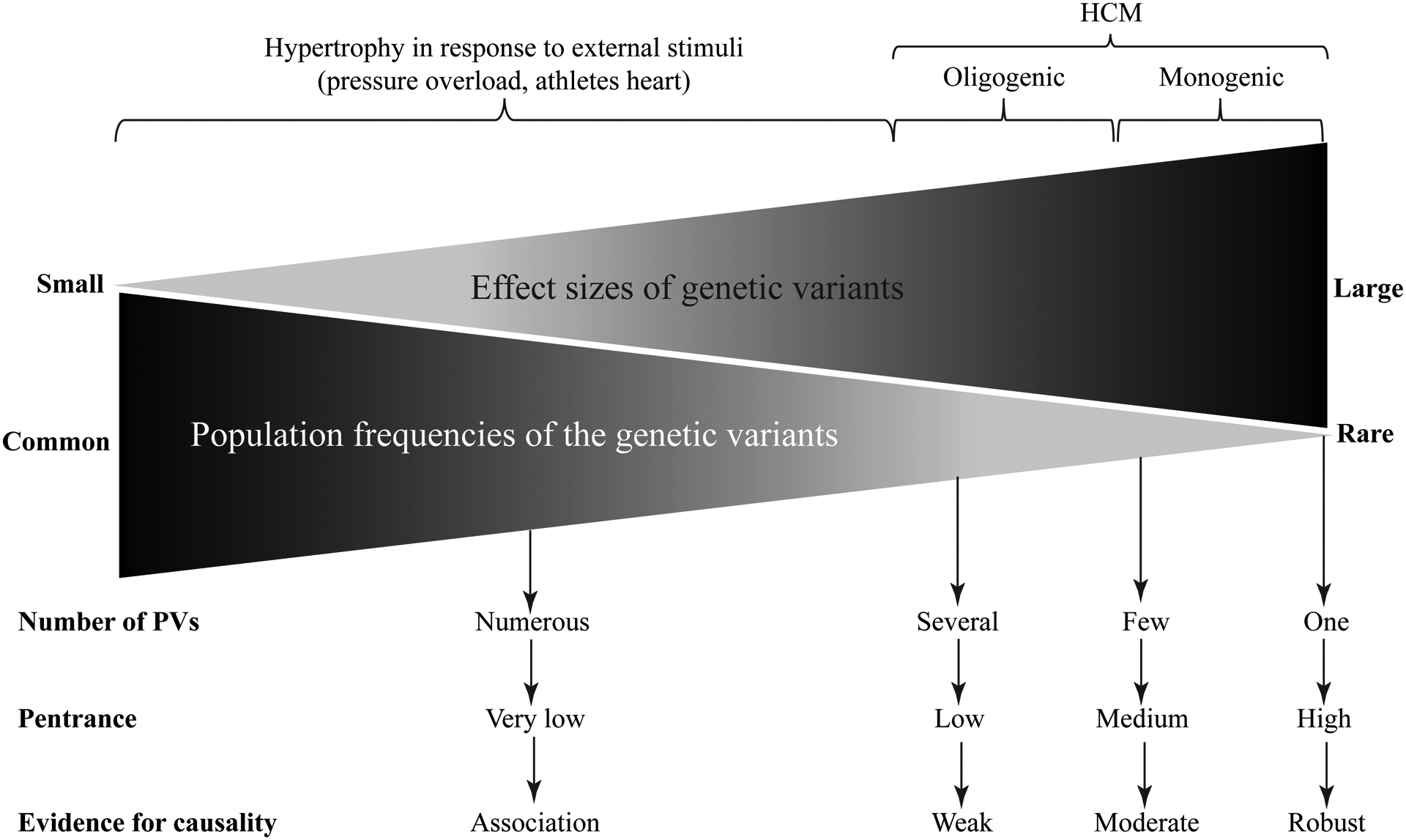
A schematic presentation of the effect sizes of the PVs that vary from small to large. The effect size typically inversely correlates with the population frequency of the variant. Rare variants with large effect sizes are more common in familial HCM and those with moderate effect sizes are often found in the sporadic cases and in small families with HCM.
Specific genes implicated as causes of HCM
In keeping with the complexity of the clinical phenotype, a large number of genes are involved in the pathogenesis of HCM. Genetic data, from the very early on, identified HCM mainly as a disease of the sarcomeres. Sarcomeres are orderly structures in the straited muscles, which are comprised of thick and thin filaments, whose highly regulated ATP-dependents interaction generate the force of muscle contraction (Figure 2). Several specific genes, according to the biological functions of the encoded proteins, are briefly discussed.
Figure 2. Schematic illustration of sarcomere proteins involved in HCM.
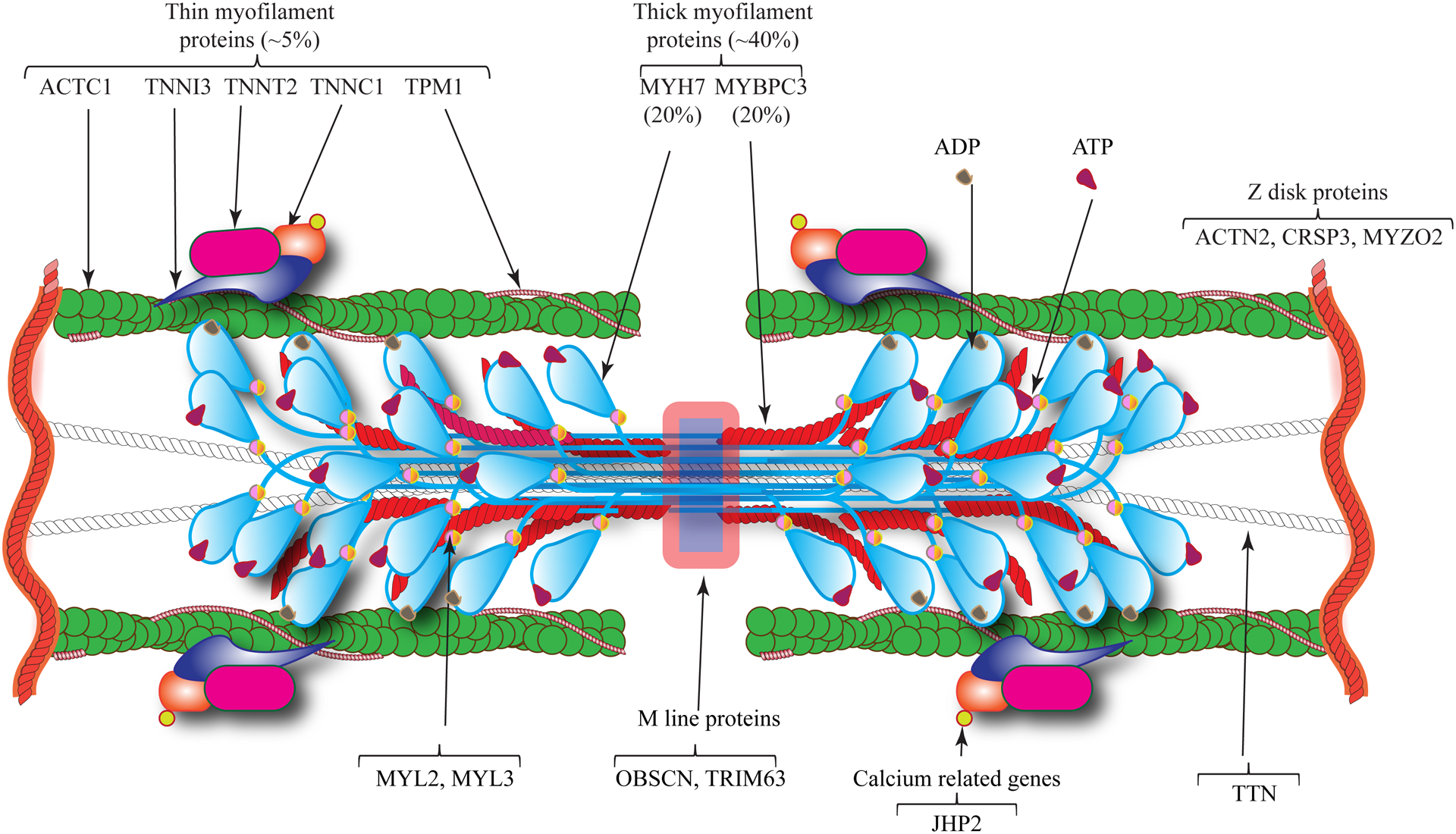
HCM is mainly a disease of sarcomere proteins, which are comprised of thick and thin filaments. Thick filaments are mainly composed of myosin heavy chain 7 (MYH7) and myosin binding protein C3 (MYBPC3), which are the most commonly affected proteins by mutations causing HCM. Thin filaments are comprised of cardiac α actin 1 (ACTC1), troponin complex (TNNT2, TNNI3, and TNNC1) and α tropomyosin (TPM1), which are affected by mutations in about 5% of the HCM cases. Examples of the Z and M line proteins implicated in HCM are also illustrated, which are rare causes of HCM.
Genes encoding thick filament proteins of the sarcomere:
The main constituents of the thick filaments involved in HCM are MYH7 and MYBPC3, which are responsible for force generation.
MYH7:
The gene codes for MYH7 or β-MYH, which comprises about 1/3 of the sarcomere proteins. It is predominantly expressed in cardiac myocytes and to a lesser extent in slow fibers of the skeletal muscles (https://www.proteinatlas.org/). The protein has 1,935 amino acid and is ~ 220 kDa in molecular weight. Structurally, it has distinct domains including a globular head, which is the sites of ATP binding and hydrolysis as well as binding to the actin molecule in the thin filament. It contains a lever arm or a hinge region, which is followed by the α-helical coil-coil rod domain. The latter comprises the majority of the amino acids in the protein. MYH7 is the main muscle motor molecule and essential for muscle contraction. It forms a hexameric complex composed of two MYH7, 2 myosin light chain 2 (MYL2), and 2 myosin light chain 3 (MYL3). The hexameric complex interacts with actin through the actin-binding domains, and hydrolyzes ATP to ADP and inorganic phosphate to generate the force of contraction.
Several hundred PVs and LPVs in the MYH7 gene, primarily missense variants and a few nonsense and indels variants, have been identified in patients with HCM (https://www.ncbi.nlm.nih.gov/clinvar/). The population frequency of each mutation is low, as each is found in less than 1% of the HCM cases. The founder mutations, which are uncommon in the MYH7 gene, have a higher population frequency in specific populations. Overall, PVs in the MYH7 gene are found in approximately 20% of patients with HCM cases but aa higher number of large families with HCM. MYH7 shows selection against LoF variants, reflected in the loss-of-function observed/expected upper bound fraction (LOEUF) index of 0.57 in the gnomAd database (a low LOEUF score indicates strong selection toward LoF variants). Given its key biological functions in cardiac myocytes and its well-established role in HCM, PVs in the MYH7 gene rank among the highest in the hierarchy of the genetic causes of HCM.
MYBPC3:
The gene codes for myosin binding protein C3, which is associated with the thick and thin filaments of the sarcomere in cardiac myocytes (Figure 2). The protein has 1,274 amino acids and a molecular weight of ~ 140 kDa. MYBPC3 binds to MYH7 and cardiac actin and regulates acto-myosin interactions. It undergoes extensive post-translational modifications (PTMs), including phosphorylation by PKA, PKC, GSK3β, and other kinases at multiple sites, which collectively modulates its interactions with other sarcomere proteins and hence, muscle contraction.36
Heterozygous mutations in the MYBPC3 gene are found in about 20% of the HCM cases and in a higher number of large HCM families.37,38, 39 A notable feature of mutations in the MYBPC3 gene is over-representation of the truncating mutations, such as indels, splicing, and nonsense mutations, as opposed to other well-established causal genes, whereby the missense mutations prevail.38, 39 The truncating mutations typically activate the nonsense-mediated decay (NMD) and the ubiquitin-proteasome system (UPS), resulting in haplo-insufficiency of the MYBPC3 protein.40
The population frequencies of specific MYBPC3 mutations are rare with the exception of the founder mutations, which are responsible for a significant number of HCM cases in the European countries and in Japan.41–43 A common 25-bp deletion in intron 32 of MYBPC3 gene has been associated with cardiomyopathies, including HCM in the South Asian population.44 This 25-bp variant is in linkage disequilibrium with an intronic variant, which introduces a cryptic splice acceptor site in intron 13, resulting in inclusion of 50 intronic nucleotide in the transcript, and subsequent premature termination of the MYBPC3 protein.45 The MYBPC3 gene is tolerant to missense and LoF variants (LOUEF:0.96). Despite its well-established role in HCM, PVs in the MYBPC3 have to be analyzed further to implicate their causal role in HCM.
MYL2 and MYL3:
Genes encoding MYL2 and MYL3 were amongst the first set of genes identified as causes of HCM.46 The proteins are part of the myosin hexamer and interact with the hinge region of the MYH7 protein. Upon phosphorylation by myosin light chain kinase (MLCK), they regulate cardiac contractility. Both proteins have well-established roles in muscle contraction and are highly expressed in cardiac myocytes. MYL2 and MYL3 are uncommon causes of HCM, together being responsible for <1% of the HCM cases. The MYL3 and MYL2 genes are largely tolerant to missense and LoF variants. Therefore, pathogenicity of the variants identified in the MYL2 and MYL3 genes has to be assessed for the presence of co-segregation, whenever feasible, and in the context of the population frequency of the variants as well as the functional data. Overall, given their relatively high levels of expression in cardiac myocytes and the existing genetic data, PVs in these genes are plausible candidates to cause HCM.
TTN:
The gene encodes the giant sarcomere protein titin (TTN), which expands from M to Z lines, i.e., half of the sarcomere length. It is almost exclusively expressed in cardiac myocytes and skeletal muscles. The gene is intolerant to LoF (LOEUF: 0.35) but tolerant to missense variants. Truncating variants in the TTN gene are major causes of familial and sporadic dilated cardiomyopathy (DCM) but are rare in HCM.47 Overall, PVs in the TTN gene rank relatively low as causes of HCM.
MYH6:
Myosin heavy chain 6 protein, encoded by the MYH6 gene, comprises less than 10% of the total MYH protein, with MYH7 being the main isoform in the human cardiac ventricular myocytes. Despite being the minor myosin isoform, its transcripts are abundant in the human cardiac myocytes. The gene is mostly tolerant to missense and LoF variants (LOEUF:0.86). Variants in the MYH6 have been reported in HCM cases.48, 49 However, robust genetic evidence to implicate PVs in the MYH6 gene as causes of HCM are lacking. Given that MYH6 comprises a small fraction of the total MYH pool in cardiac myocytes, heterozygous PVs in the MYH6 gene, by affecting only a very small fraction of the sarcomere MYH protein (likely <5%), are not expected to cause HCM.
Genes encoding thin filament proteins of the sarcomere:
The thin filaments in cardiac myocytes are comprised of a polymer of ACTC1 and the troponin/tropomyosin complex. The troponin/tropomyosin complex is formed by cardiac troponin C (TNNC1), which is the calcium sensing unit of the complex, cardiac troponin I (TNNI3), which is the inhibitory component, and cardiac troponin T (TNNT2), which anchor the troponin complex to tropomyosin (TPM1). The latter places the troponin/tropomyosin complex on ACTC1, forming the thin filaments (Figure 2).
TNNT2:
The gene codes for TNNT2, which constitute about 5% of the sarcomere proteins. The TNNT2 gene was among the first genes whose causality in HCM was established through genetic linkage.21 The finding along with identification of mutations in other major sarcomere proteins established HCM mainly as a disease of sarcomere.21 A large number of PVs and LPVs in the TNNT2 gene, mostly missense variants, have been identified in patients with HCM. The TNNT2 gene is tolerant to LoF variants (LOEUF:0.97). In view of its key role in cardiac myocyte function, and robust evidence for the causal role of this gene in HCM, PVs in the TNNT2 gene should be assessed further to imply a causal role in the pathogenesis of HCM.
TNNI3, TNNC1, and TPM1:
The genes encode thin filament proteins TNNI3, TNNC1, and TPM1, respectively. Mutations in the TNNI3, TNNC1, and TPM1 genes are uncommon causes of HCM (<5% of the HCM cases). The three genes are partially intolerant to missense and LoF variants and in view of their functions and well-established role in HCM, PVs in these genes are plausible candidates to cause HCM.
ACTC1:
The gene encodes ACTC1, which is among the least tolerant genes to missense and LoF variants. ACTC1 is a major component of the thin filaments and together with troponin/tropomyosin complex interact with the MYH7 protein to generate force of muscle contraction. Mutations in the ACTC1 gene are rare causes of HCM.50 Given the highly conserved nature of ACTC1 and its major role in cardiac myocyte contraction, PVs in the ACTC1 gene benefit from a high probability of exerting large effect sizes and causing HCM (LOEUF:0.48).
Genes encoding the protein constituents of the Z lines (or Z discs or Z bands):
The Z lines anchor the end of the thin filaments, providing mechanical stability for contraction and relaxation of the filaments and transmission of the force of contraction to neighboring units (Figure 2). The Z lines in cardiac muscle are comprised of a large number of proteins, including α actinin 2 (ACTN2), myozenin 2 (MYOZ2), cysteine and glycine-rich protein 3 (CSRP3), titin-cap (TCAP), and Four and half LIM domain 1 (FHL1), to name a few.
MYOZ2:
The encode protein MYOZ2 is exclusively expressed in cardiac myocytes and skeletal muscles, where it interacts with ACTN2, and others and presumably regulates myofibrillogenesis and calcium-dependent signaling pathways. The gene is tolerant to LoF and missense variants. PV and LPVs in the MYOZ2 have been implicated in sporadic as well as familial HCM.51, 52 Overall, PVs in MYOZ2 are expected to exert moderate effects sizes on the phenotypic expression of HCM, as the causal or the modifier variants.
ACTN2:
The gene is predominantly expressed in cardiac and skeletal muscle, where it binds to the thin filament protein actin and regulates sarcomere function. The gene is very intolerant to LoF variants (LOEUF:0.24). PVs in the ACTN2 gene have been associated with skeletal myopathy as well as various forms of cardiomyopathies, including sporadic HCM.53, 54
CSRP3:
The encoded protein is a small multi-functional protein referred to as CSRP3, which was formerly known as muscle lim protein (MLP). The protein is highly expressed in cardiac and skeletal myocytes and interacts with a number of sarcomere and cytoskeletal proteins as well as cardiac transcription factors. Mutations in the CSRP3 gene are implicated in HCM with the strongest evidence emerging from linkage analysis in a large family.55 The gene is, however, tolerant to missense and LoF variants. Therefore, PVs in this gene alone are not expected to cause HCM, but act in concert with PVs in other genes relevant to the pathogenesis of HCM.
TCAP:
Rare variants in the TCAP gene, which encodes titin-cap, have been reported in patients with HCM.56 The protein regulates assembly of the TTN protein in cardiac and skeletal muscle. The gene is tolerant to missense but tolerant to LoF variants (LOEUF:0.95). This gene, like the TTN gene, does not appear to be a major cause of HCM.
FHL1:
The FHL1 gene is located on the X chromosome and encodes the FHL1 protein, which is expressed predominantly in skeletal muscle, cardiac myocytes, smooth muscle cells, and to a lesser extent in other cell types. The gene is highly intolerant to the LoF (LOEUF:0.28) and to a lesser degree to missense variants. PVs in the FHL1 gene have been predominantly associated with skeletal myopathy but also have been reported in sporadic cases with HCM.57 In addition, FHL1 is considered a modifier gene for cardiac hypertrophy in HCM.58, 59 Overall, PVs in FHL1 gene are expected to exert modest effect sizes on the expression of HCM and are unlikely to cause HCM in the absence of skeletal myopathy.
Genes coding for proteins located at the M line (M band):
Whereas the Z lines largely anchor the thin filaments, the M lines, located in the middle of the sarcomere, are considered anchors for the thick filaments. In addition to the structural proteins, several other proteins with enzymatic functions are localized to the M lines. Notable examples of proteins that are localized to the M lines are obscurin (OBSCN), myomesin 2 (MYOM2), and tripartite motif containing 63 (TRIM63).
OBSCN:
The gene codes for obscurin, which is a large protein predominantly expressed in the skeletal muscle and to a lesser extent in cardiac and tongue muscles. It interacts with TTN and MYBPC3 and also has signaling functions and kinase activity. The gene is somewhat tolerant to LoF (LOEUF:0.87) and tolerant to missense variants. A few PVs in the OBSCN gene have been associated with various forms of cardiomyopathies, including HCM.60
TRIM63:
The gene encodes TRIM63 or muscle ring finger 1 (MuRF1) protein, which is predominantly expressed in cardiac and skeletal myocytes. The protein is an E3 ubiquitin ligase, which tags the thick filament proteins for degradation by the UPS. The gene is tolerant to LoF and missense variants. LoF variants in the TRIM63 gene have been associated with HCM in small families and sporadic cases.61 The mechanisms involve impaired ubiquitination of MYH7 and MYBPC3 and activation of the MTOR-S6K pathways.61 Likewise, homozygous LoF in the TRIM63 gene have been associated with an autosomal -recessive form of familial HCM.62 Overall, the data points to the modest to moderate effect sizes of the PVs in the TRIM63 gene in HCM.
MYOM2:
The encoded protein cross links TTN and MYH7 at the M line and is implicated in sarcomerogenesis. The gene is predominantly expressed in cardiac and skeletal muscle. Variants in the MYOM2 gene have been associated with HCM.63
Gene regulating calcium homeostasis in cardiac muscle:
Intracellular Ca2+ concentration regulates acto-myosin interaction and generation of the force of muscle generation.
JPH2:
The encoded protein junctophilin 2 is a major component of the junctional membrane complex involved in calcium homeostasis and excitation-contraction coupling. Expression of this gene is enriched in cardiac, skeletal, and smooth muscles, the latter in various organs, including colon and urinary bladder. The gene is intolerant to LoF (LOEUF:0.14) and missense variants. PVs are enriched in patients with sporadic HCM, as compared to the general population.64 PVs in the JPH2 gene are expected to exert moderate effect sizes on the phenotypic expression of HCM.
PLN:
The gene encode phospholamban, which is predominantly expressed in cardiac myocytes where it functions as a regulator of Ca2+ homeostasis through inhibiting ATP2A2 (SERCA2a) activity. Consequently, it is a negative regulator of cardiac contraction. PVs in the PLN gene are best known to cause DCM. A rare truncating variant has been reported in HCM.65, 66
Genes encoding sarcomere-associated proteins:
Several other genes with indirect role in regulating sarcomere structure and function are also implicated as causes of HCM.
FLNC:
FLNC is one of the latest genes implicated in HCM but most prominently in DCM.67, 68 The gene is highly intolerant to LoF and to a lesser extent to missense variants. Its expression is enriched in cardiac myocytes and more so in the skeletal muscle. The protein interacts and cross-links with actin and is presumed to regulate organization of the cytoskeletal protein in response to stress. FLNC gene is intolerant to LoF variants (LOEUF:0.25). FLNC variants have been identified in sporadic cases and in small families with HCM.67, 68
ALPK3:
The gene, which codes for α kinase 3, is most abundantly expressed in cardiac and skeletal muscle and implicated in myocyte differentiation. The gene is tolerant to missense and LoF variants. PVs variants in this gene, including homozygous truncating variants, have been reported in patients with HCM alone or in conjunction with other PVs.69, 70
CAV3:
The gene codes for caveolin 3, which is a component of caveolae of the cytoplasmic membrane. The gene is expressed in cardiac and skeletal muscle, where the encoded protein interacts with G-protein alpha subunits, the dystrophin complex, and the T-tubule system. CAV3 is implicated in regulating myogenic differentiation, endocytosis, mechanosensing, signaling, and mitochondrial homeostasis, among others. The gene is tolerant to missense and LoF variants. PVs in the CAV3 gene are associated with skeletal myopathy and long QT syndrome 9 and various forms of cardiomyopathies, including HCM.71
CRYAB:
The gene encodes crystalline αB, which is a member of small heat shock proteins, with chaperon-like activity. The gene is expressed in multiple cell types but is enriched in cardiac and skeletal muscles. The gene is tolerant to LoF and missense variants. PVs in the CRYAB gene have been reported mostly in patients with DCM and in a few patients with sporadic HCM. Overall, PVs and LPVs are not expected to emerge as major contributors to the phenotypic expression of HCM.
Common and founder mutations in HCM
With the exception of a few founder and population-specific mutations, the population frequency of each specific mutation in HCM is less than 1%.35 The p.Arg502Trp and p.Val762Asp in the MYBPC3, the latter in a Japanese population, have been reported at a relatively high frequencies in the HCM populations, ranging from 1 to 4%35, 72, 73. Likewise, the c.927-2A>G founder mutation in the MYBPC3 gene is found in about 50% of the patients with HCM in Iceland.41 Several other population-specific founder mutations, mostly in the genes coding for sarcomere proteins have been reported.74–76
Digenic and oligogenic HCM
HCM in large families exhibits an autosomal dominant pattern of inheritance and is an archetypical single gene disorders, whereby the presence of a single PV, typically with a large effect size, is sufficient to cause the disease. The severity of the phenotype, however, is determined not only by the main PV but also by additional PVs with smaller effect sizes, i.e., the MVs. On the other end of the spectrum is the sporadic form of HCM, which often is the consequence of a de novo PVs and on occasion aggregation of multiple low penetrance PVs, each exerting modest to moderate effect sizes and collectively leading to the clinical phenotype of HCM. Approximately, 5% of the HCM cases are estimated to be digenic or oligemic in etiology.35, 70, 77, 78 The presence of multiple PVs is typically associated with more pronounced phenotype, such as severe cardiac hypertrophy or LVOTO35, 70, 77, 78. The prevalence of the digenic and oligogenic HCM and the impact of multiple PVs on the clinical manifestations of HCM remain to be fully understood.
Missing causal genes
The PVs responsible for HCM have been identified in less than half of the cases and small families with HCM but in almost all large families. The difficulty in ascertaining the remaining causal genes pertains to the modest to moderate effect sizes of the responsible PVs and consequently, incomplete penetrance, or the de novo nature of the variants. In the latter situations, unambiguous ascertainment of causality is exceedingly challenging, resulting in the so-called “missing causal gene” in HCM.79
To identify the “missing causal genes” it is necessary to shift from a deterministic, which assumes PVs with large effect sizes are the causes of HCM, to a probabilistic approach, which posits multiple PVs with moderate and modest effect sizes collectively are responsible for HCM in the sporadic cases and in small families. In the probabilistic approach, all PVs in genes encoding proteins involved in the networks pertaining to the HCM phenotype are expected to contribute to expression of the phenotype in HCM. However, detection of the PVs in the pathways that pertain to the HCM phenotype alone, is insufficient to conclude a causal role, as the effects are often context-dependent. Therefore, the findings should be analyzed in the context of HCM phenotype, its pathogenesis, and myocardial biology, including gene expression. Identification of the so-called “missing causal genes” in HCM would require genome-wide analysis to detect the candidates and more importantly, to gain a clear understanding of the biological functions of the encoded proteins.
GENETIC TESTING
Genetic testing, typically by whole exome sequencing (WES), is routinely performed in patients with HCM.35 The objective is to find the PV and LPVs, which would provide insights into the pathogenesis of HCM, and ultimately, to prevention of the evolving phenotype, and attenuation or reversal of the established disease. The most informative yield of genetic testing is in the familial setting, whereby co-segregation of the variant of interest with the phenotype could be ascertained to deduce causality. Cascade screening of the family members at risk could lead to identification of those who carry the PVs. Such individuals undergo frequent clinical evaluations to detect the disease early, and to intervene, as clinically indicated.80, 81 Moreover, identification of the family members who have not inherited the culprit PV(s) would abrogate unnecessary frequent clinical evaluations. Moreover, genetic testing could lead to identification of the phenocopy conditions, including storage diseases and the athletes’ hearts. Approximately 3% of the individuals who have undergone genetic testing for HCM are found to have PVs in genes known to cause phenocopy conditions.35 The distinction is clinically important as the treatment of HCM from the phenocopy conditions differ, particularly with the availability of specific therapies.
Despite its informativity, the current yield of genetic testing in HCM, by WES is rather disappointing, as only in ~ 30% to 50% of probands with HCM, a candidate PV is identified.35 There are a number of reasons, in addition the current deterministic approach, that lead to a relatively low yield of genetic testing in HCM. (reviewed in22) Another shortcoming is the current approach to variant detection, including by WES, which is based on the Willie Sutton idiom, as the existing data show that the majority of the discovered mutations reside in the coding region. Nevertheless, the approach excludes the vast majority of the variants in the genome. The whole genome sequencing (WGS) approach, on the other hand, requires extensive computational analysis of a complex set of data, which is challenging. In addition, the current techniques are based on short-read sequencing, which results in imperfect variants detection and do not provide a robust platform for detection of large indels and structural variations. A number of alternative approaches, such as long-read sequencing and single molecule sequencing, have been developed. However, none is ready for clinical genetic testing, partly because of a relatively high error rate.82
Genetic testing has modest utility in predicting the phenotypic outcomes, partly because the outcomes are the consequence of stochastic and non-linear interactions among multiple determinants. The effect size of a specific culprit PV in HCM diminishes with the remoteness of the phenotype to the genotype, because of the contributions of other competing determinants. Accordingly, a PV is expected to exert a greater effect on the transcript level of the involved gene than on the risk of heart failure or sudden cardiac death (SCD), as the latter are also affected by a much larger number of genetic and non-genetic factors. The “mutation load”, however, seems to be an important determinant of severity of the phenotype in HCM, as the presence of multiple PVs in genes with key roles in cardiac myocyte biology.35, 70, 77, 78
The main challenge in genetic testing is in interpretation of the genetic findings in terms of their clinical significance. This is particularly the case in small families and sporadic cases, as each genome contains a large number of PVs and LPVs. To extract robust and accurate information from the genetic data, it is important to garner knowledge not only about how to discern the PVs, but also in understanding the role of the involved proteins in cardiac muscle biology and function as well as the pathogenesis of the HCM, while keeping in mind the complexities of the clinical phenotype. Finally, genetic counseling and guidance on exercise and physical activities are routinely incorporated in the care of patients with HCM.
GENETIC-BASED INSIGHTS INTO THE PATHIOGENESIS OF HCM
Familial HCM is mainly an autosomal dominant disease, and hence, only one copy of the gene carries the causal variant. By definition the primary defect in HCM is the causal mutation or the PVs. For simplicity, the focus is on a single PV in providing the impetus for the initiation of a cascade of events that leads to the pathological phenotypes, such as myocyte hypertrophy, disarray, and fibrosis, and the clinical phenotypes, such as heart failure with preserved ejection fraction (HFpEF), arrhythmias, and SCD. Nonetheless, the mechanisms discussed below are also applicable when multiple PVs are operant, whereby, each provides an impetus, often overlapping with another, for the instigation of the phenotype.
The mutation operates mainly by changing the amino acid sequence or reducing expression level of the encoded protein. The former mechanism is referred to as a dominant-negative (or poison-peptide) effect and the latter to haplo-insufficiency (Figure 3). A dominant-negative mechanism is more common in HCM in keeping with the majority of the known mutations being missense mutation. A haplo-insufficiency mechanism is more common in HCM caused by mutations in the MYBPC3 gene, as the majority of mutations in this gene are frameshift and stop codon mutations.38, 39, 83, 84.
Figure 3. Pathogenesis of HCM.
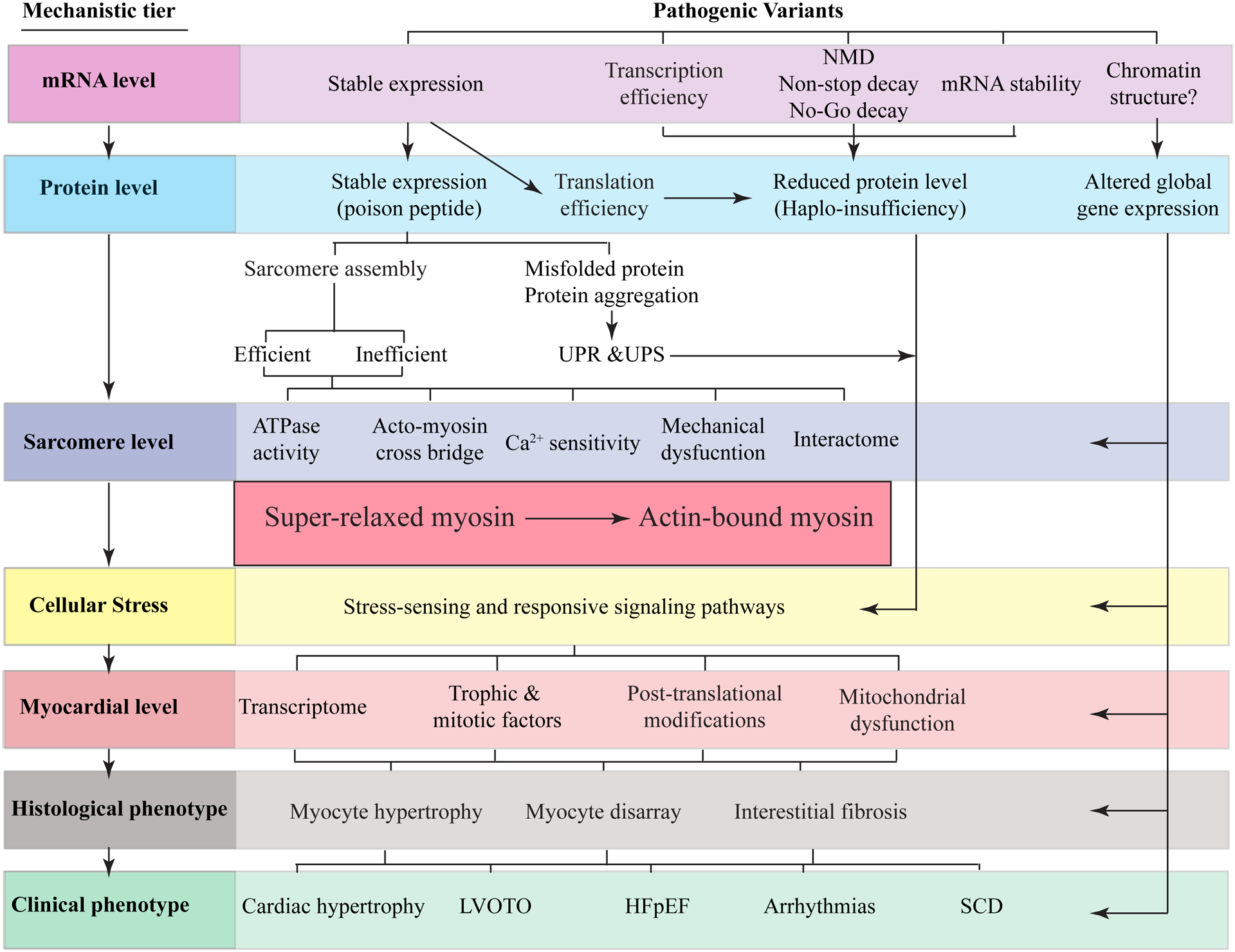
The primary defect in HCM is the mutation in the sarcomere proteins, composed of thick and thin filaments, M line and the Z disks (or lines). The effects of the mutations on mRNA, protein, sarcomere, myocyte, myocardium (molecular level), histology, and clinical phenotypes are depicted as sequential layers. A change in the amino acid sequence in the sarcomere protein (dominant-negative effect) or the deficiency of a sarcomere protein (haplo-insufficiency) instigates a series of proximal functional defects in cardiac sarcomeres, such as altered calcium sensitivity and ATPase activity. A consequence of these initial defects is a shift in the number of myosin molecules in the super-relaxed state with a low ATPase activity toward the myosin molecules that are in strong bound state with the actin molecule and have a high ATPase activity. The changes activate expression of a stress-sensing (mechanical, biochemical, and energetics) intermediary molecular phenotypes, such as altered transcriptomics and expression of trophic and mitotic factors. The latter set of the molecular changes induce histological and morphological changes in the myocardium, such as myocyte hypertrophy and fibrosis, which collectively lead to the clinical phenotypes, such as cardiac arrhythmias and heart failure.
Effects of the mutations on protein expression:
The missense mutations are typically incorporated into the expressed full-length protein. However, the mutation could affect mRNA transcription rate, stability, and its translation efficiency, leading to a modest reduction in the level of the mutant protein.85, 86 Transcripts containing a premature termination codon are detected by the surveillance machinery and degraded by the NMD pathway.87 Similarly, the elongated transcripts are surveyed by the non-stop decay pathway, are released from the ribosomes to prevent translation, and are degraded by the exosomes.88 Moreover, the No-Go mRNA decay pathway stops the ribosomal translation of the mRNA followed by its degradation by endonucleolytic cleavage near the stalled site.89 The net effect of such quality surveillance mechanisms is prevention or reduction in the expression levels of the truncated proteins. Those transcripts escaping the NMD are translated as truncated or elongated proteins, which are typically degraded by the protein quality control mechanisms.90 There is, however, partial allelic compensation with the wild type allele compensating for the fraction of the lost transcripts. Despite, these surveillance mechanisms, if and whenever a truncated or an elongated protein is stably expressed, it functions through a dominant -negative mechanism.
The mutation could also affect protein folding and its tertiary structure, which typically result in degradation of the misfolded protein by the UPS. (reviewed in91) However, accumulation of the misfolded protein could lead to endoplasmic reticulum stress and activation of the unfolded protein response, and aggregation myopathy.
Effects of the mutations on protein and sarcomere structure and function:
The mutant protein, whenever stably expressed, exhibits impaired biological functions, such as ATPase activity, its interactions with its binding partners (interactome), and/or incorporation into sarcomeres. Filaments incorporating the mutant sarcomere protein often exhibit impaired calcium-dependent acto-myosin cross-bridge cycling, altered Ca2+ sensitivity of the troponin complex, and impaired force generation (Figure 3).
Impaired acto-myosin cross-bridge cycling has emerged as the key mechanisms in the pathogenesis of HCM. (reviewed in92) In brief, binding of the ATP molecule to the globular head of MYH7 leads to dissociation of MYH7 from the ACTC1 molecule, whereas subsequent hydrolysis of ATP to ADP and inorganic phosphate by the MYH7 results in association of the two molecules (Figure 4). The release of the inorganic phosphate induces conformation changes in the MYH7 protein, flexion of the myosin lever arm, and the consequent displacement of the actin filament by about 10 nm, which generates the force of muscle contraction. (reviewed in92) It is estimated that under physiological conditions during diastole, about 10% of the myosin molecules are bound to the actin filaments, which increase to about 30% during systole.92 The actin-bound myosin molecules have a high ATP turnover rate and produce the force of muscle contraction.93(Figure 4) Approximately 50 to 60% of the myosin molecules are unbound to actin, have a very low ATP turnover rate, and are considered to be in a super-relaxed state.94 The remaining 30 to 40% of myosin molecules are weakly associated with the actin molecule and exhibit an intermediary ATP turnover rate. A prevailing hypothesis in the pathogenesis of HCM posits that the causal mutations shift the number of the myosin molecules that are in the super-relaxed state toward the actin-bound state.95, 96 The shift is regulated by the intra-cellular Ca2+ concentration, cytosolic ADP/ATP content, and phosphorylation states of the sarcomere proteins, such as MYL2, MYL3, and MYBPC3. The ensuing increase in the number of actin-bound myosin molecules in HCM results in increased myocyte contractility and ATP utilization, which instigates a cascade of the secondary molecular events that induce the histological, morphological, and the clinical phenotype of HCM.
Figure 4. Impaired acto-myosin interaction in the pathogenesis of HCM.
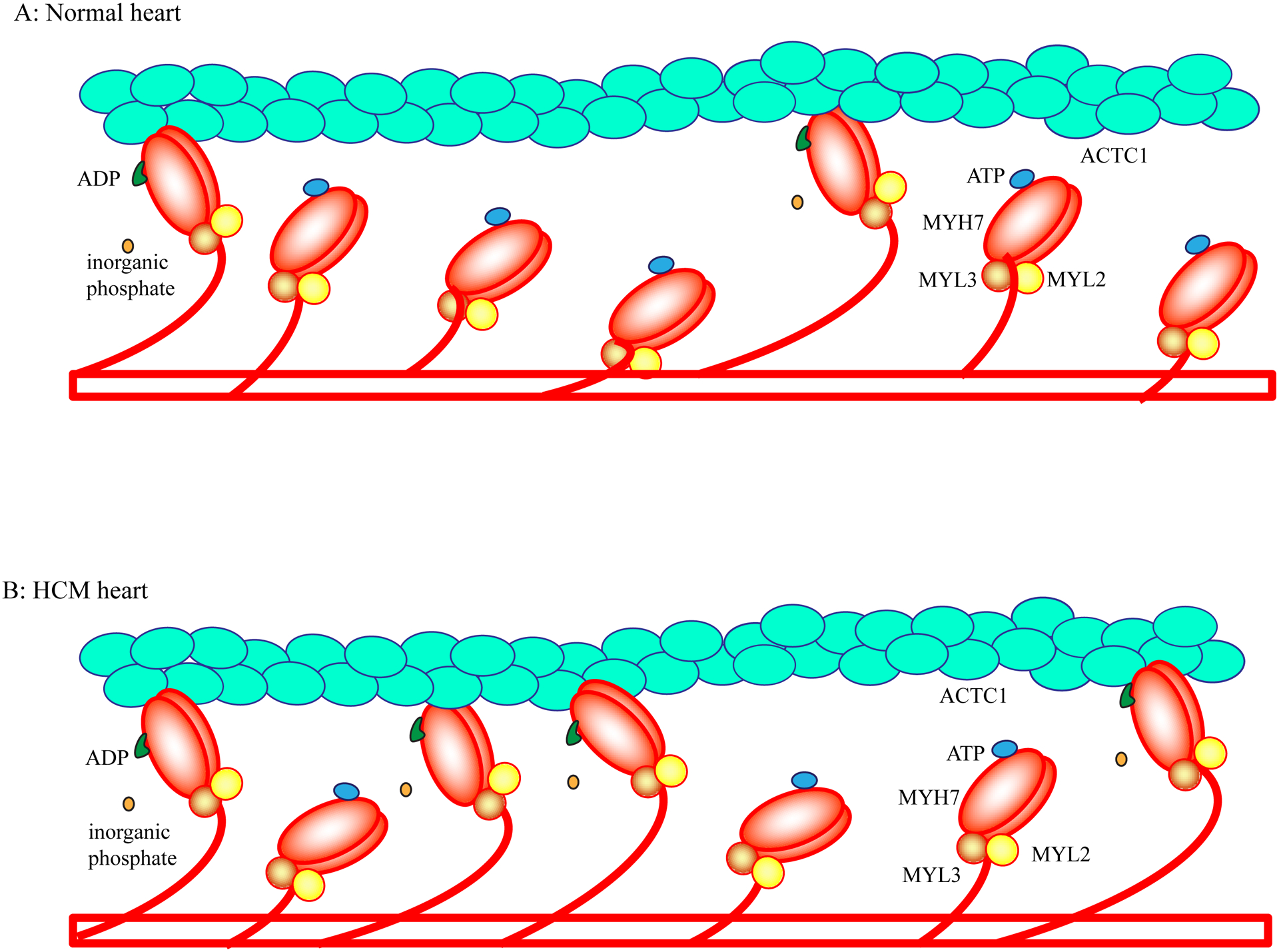
Binding of ATP to the globular head of myosin results in its dissociation from the thin filament actin, whereas hydrolysis of ATP to ADP and inorganic phosphate leads to association of the MYH7 and ACTC1 molecules. Release of the inorganic phosphate induces conformational changes in the MYH7 and flexion of the myosin globular head at lever arm over ACTC1 in the thin filament. The flexion displaces the thin filament by about 10 nm.
In the normal heart, at any given moment during diastole, only a small fraction of the MYH7 molecules (estimated to be about 10%) is bound to the actin (ACTC1) molecule. Approximately 50–60% of the myosin molecules are totally dissociated from the actin molecule and are in the super-relaxed state. The remaining myosin molecules are weakly associated with the actin filaments (Panel A). In HCM caused by mutations in the MYH7 and MYBPC3 genes, there is a shift from myosin molecules in the super-relax state to the myosin molecules in the strong bound state with the actin molecule (Panel B). The increased number of acto-myosin complex (strong bound state) is responsible for increased contractility and impaired relaxation.
In accord with the diversity of the mutations and genes involved, a diverse array of molecular and cellular functional defects is implicated in the pathogenesis of HCM.97–102 Likewise, there are differences in the functional phenotypes induced by the PVs in different genes. For example, mutations in the MHY7 gene exhibit more pronounced effects on the myofibrillar ATPase activity and a higher energy cost for generation of muscle tension those located in the MYBPC3 gene.97, 98 The increased cost of tension generation in the presence of mutant sarcomere protein results in a reduced ratio of cardiac phosphocreatine to ATP in the heart and myocardial bioenergetic deficiency in patients with HCM.103, 104
Differential Ca2+ sensitivity of myofibril function is also implicated in the pathogenesis of HCM caused by mutations in genes encoding thick and thick filament proteins. Specifically, mutations in the thin filament proteins, in contrast to those in the thick filament proteins, increase Ca2+ sensitivity of myofibrillar ATPase activity, and enhance maximum force generation100, 102, 105–107. Moreover, expression levels of the mutant transcripts and Ca2+ sensitivity of maximal force generation also vary at the cellular level.86 Finally, there is considerable molecular heterogeneity, including differences in gene expression, among myocardial regions, which could influence the effects of the mutant protein on regional phenotypic expression in HCM.108, 109 Collectively, the data suggest the presence of mosaic molecular and cellular functional defects among different myocytes and myocardial regions in HCM
Functional defects observed at the molecular and cellular levels seem to occur early and precede the development of cardiac hypertrophy.98, 110,101. Development and progression of cardiac hypertrophy, is also associated with secondary modifications of the sarcomere proteins, including phosphorylation of TNNI3 and MYBPC3, which are known to affect sarcomere functions106, 111. Functional changes collectively induce biochemical, bioenergetics, or mechanical stress and lead to expression of stress-responsive genes and activation of stress-sensing signaling pathways, which mediate the subsequent downstream phenotypes (Figure 3).
Pathogenesis of the histological phenotype in HCM:
Three main histological features of HCM include myocyte hypertrophy, myocyte disarray, and myocardial fibrosis. The prevailing hypothesis posits that all three main histological phenotypes are largely the consequences of activation of the intermediary molecular pathways, ranging from stress-sensing signaling pathways to expression of trophic and mitotic factors followed by secondary gene expression, partially similar to those activated in other forms of myocardial stress and injury.112 The extent of the histological changes often reflects the severity of the disease and is a marker for poor clinical outcomes, including risk of heart failure, cardiac arrhythmias, and SCD113, 114.
Cardiac myocyte hypertrophy is the consequence of increased expression of trophic factors and activation of the signaling pathways involved in cardiac hypertrophy, such as the MAPK and calcineurin pathways, among others.52, 61, 115–118 Likewise, myocardial fibrosis, which is common in HCM, results from increased expression and secretion of the mitotic factors, such as TGFβ1 from cardiac myocytes and activation of the corresponding downstream pro-fibrotic cascades in cardiac myocytes as well as fibroblasts.116, 119 Myocyte disarray, the pathological hallmark of HCM, is conventionally thought to result from altered myocardial architecture. Recently, however, cell-to-cell heterogeneity in gene expression, including in the expression levels of the mutant protein, and consequently, cell-to-cell functional differences among cardiac myocytes, have been advocated as a mechanism for disorganized architecture and orientation in the myocardium.86, 109
Pathogenesis of the clinical phenotype in HCM:
The clinical phenotype of HCM is the consequence of cardiac myocyte hypertrophy and myocardial fibrosis in the background of abnormal gene expression, including those affecting Ca2+ homeostasis and myocyte relaxation. Notable clinical features of HCM are cardiac hypertrophy, asymmetric septal hypertrophy, enhanced LVEF, LVOTO, HFpEF, cardiac arrhythmias, and SCD. These phenotypes are affected not only by the primary defect but also by the secondary changes that occur in the myocardium, such as PTMs of the sarcomere and the related proteins, as well as by non-genetic and external factors.
Cardiac hypertrophy is primary due to myocyte hypertrophy, as discussed earlier. Predilection of cardiac hypertrophy toward the interventricular septum, apical hypertrophy, and other forms of localized hypertrophy, a characteristic feature of HCM, likely reflects differential gene expression among myocardial regions, which is partly a consequence of differential regional wall stress, although direct evidence in support of such mechanisms in HCM is scant.108, 109
A notable physiological feature of HCM is an enhanced left ventricular global systolic function, as measured by the LVEF, or a hyperdynamic left ventricle, despite reduced regional myocardial strain and tissue Doppler velocities.80, 81, 120–122. The phenotype likely reflects the increased number of myosin molecules that are in bound state with the thin filament actin during cardiac cycle, as discussed earlier (Figure 4). Increased Ca2+ sensitivity of the myofilament filaments is also a major determinant of cardiac systolic function. A number of other secondary changes, such as phosphorylation of the sarcomere proteins or other PTMs are also known to affect cardiac function. These changes are extensive and involve almost all sarcomere proteins and are beyond the scope of the present review. Myocardial relaxation is commonly impaired in HCM and left ventricular compliance is reduced, indicating diastolic dysfunction, which is particularly accentuated during exercise, providing an explanation for exercise intolerance and exertional dyspnea. Impaired relaxation is a major determinant of HFpEF, which manifests in a subset of patients with HCM, particularly in those with a long-standing history of severe LVOTO. Impaired myocardial relaxation and HFpEF in part reflect the increased number of aacto-myosin complex, decreased sensitivity of the acto-myosin dissociation to cytosolic ATP, and the secondary molecular changes in the myocardium, such as impaired Ca2+ uptake during diastole as well as PTMs of the sarcomere proteins. Likewise, histological changes in the myocardium, such as fibrosis, also contribute to diastolic dysfunction and HFpEF.
An intriguing physiological phenotype of HCM is the LVOTO, which is present at rest in approximately a third of the patients and could be provoked in another third with maneuvers, such as Valsalva, or infusion of inotropic agents, such as dobutamine. LVOTO is considered a consequence of encroachment of the hypertrophied septum toward the outflow tract, which along with the hyperdynamic ejection resulting in systolic anterior motion of the mitral valve leaflets, causing an obstruction to blood flow through the LVOT. Consequently, the presumed mechanisms for the expression of LVOTO in a subset of patients with HCM are regional differences in gene expression, wall stress due to hemodynamic forces, and likely the developmental anatomical variations of the interventricular septum and mitral leaflets.
Cardiac arrhythmias are common in HCM and on occasional lead to syncope and SCD. Non-sustained ventricular tachycardia (NSVT) is present in about a quarter of patients with HCM but sustained ventricular tachycardia and ventricular fibrillation are uncommon.123, 124 Supraventricular arrhythmias, including atrial fibrillation, are also relatively common and present in about 20% of patients with HCM, particularly in those with LVOT obstruction.125–127. Cardiac arrhythmias in HCM are likely the consequences of a combination of changes in the biophysical characteristics of ion channels, secondary to PTMs in cardiac myocytes harboring the mutant sarcomere protein, altered myocardial architecture; such as fibrosis, and dysregulated gene expression. (reviewed in128)
Genotype-Phenotype correlation in HCM:
A large number of genotype-phenotype correlation studies have been described, albeit mostly in small data sets, which are prone to spurious results. As a general principle, PVs with large effect sizes would be expected to influence the clinical expression of HCM more than those with small effect sizes. Besides this principle and as discussed earlier, the effect of the PV on the distant phenotypes, such as the clinical phenotype of HFpEF and SCD is relatively modest. This modest effect is in part because of the multiplicity of the factors that are involved in influencing the clinical phenotype, which through competition diluting the influence of the PVs.
The majority of patients with HCM, particularly those without LVOTO, experience a normal life span free of significant limitations or symptoms, consistent with the modest effect of the PVs on survival.129, 130 Approximately, 5 to10% of patients with HCM, particularly those with LVOTO develop cardiac systolic dysfunction and heart failure with reduced ejection fraction (HFrEF).131 The role of the underlying PVs in susceptibility of patients to HFrEF is unclear.
There are sex-dependent differences in the phenotypic expression of HCM, as women exhibit HCM at an older age but are at a higher risk of heart failure than men.15, 16,17, 18 However, sex-dependent differences in the clinical expression of HCM is not related to the underlying genetic causes, as HCM is typically not an X-liked disorder. The differences are likely secondary to effects of the sex hormones on the phenotype.
The most dreadful complication of HCM is SCD, which occurs in 0.5% to 2% per year in adults with HCM.124, 132, 133 The major risk factors for SCD include a family history of SCD, unexplained syncope, ventricular tachycardiac, including frequent NSVT, severe cardiac hypertrophy and myocardial fibrosis.124 Although a number of initial studies suggested genotype-dependent differences in the risk of SCD in HCM, there is no consistent evidence to suggest the underlying PVs are major determinants of the risk of SCD in HCM.
GENETIC-BASED INSIGHTS INTO CLINICAL MANAGEMENT OF PATIENTS WITH HCM
The important components in the management of patients with HCM are evaluation for the risk of SCD, HFpEF, LVOTO, atrial fibrillation, and thromboembolic complications. Despite the extensive genetic heterogeneity of HCM, a similar pharmacological and surgical approaches are used in treatment of patients with HCM. Beta-adrenergic receptor blockers are the mainstay of pharmacological therapy of symptomatic patients with HCM. The L-type calcium channel blockers, such as verapamil and diltiazem are also used in symptomatic patients, including in those with LVOTO. Disopyramide, a negative inotropic agent, is used, in conjunction with beta blockers, to reduced LVOTO,134 Diuretics are used carefully, when there is evidence of increased left ventricular end diastolic pressure, pulmonary congestion, and elevated B-type natriuretic peptide levels. Patients with atrial fibrillation require cardioversion or rate control and anticoagulation, the latter, using a direct oral anticoagulant. Those at high risk for SCD typically undergo implantation of a cardioverter/defibrillator (ICD), which is highly effective in aborting malignant rhythm and hence, by inference, SCD.135 Surgical myectomy (marrow’s procedure) and alcohol septal ablation are highly effective in alleviating LVOTO, improving symptoms, and alleviating HFpEF, regardless of the genotype.136
One of the fundamental contributions of genetic discovery has been partial elucidation of the molecular pathogenesis of HCM, which has led to the development of specific molecules that target the underpinning mechanisms. Mavacamten, which targets the acto-myosin interaction, imparts a dose-dependent reduction in the MYH7 ATPase activity and hence, left ventricular contractility.8 The drug has been advanced to the phase III clinical trials and the initial results have been favorable.137, 138 Treatment with mavacamten improved NYHA functional class and peak oxygen consumption and reduced outflow tract gradient in patients with HCM and LVOTO.137 Likewise, it has beneficial effects on plasma levels of cardiac biomarkers in patients with non-obstructive form of HCM.138 The effects of this drug on hard clinical endpoints such as mortality, SCD, or hospitalization because of heart failure remain to be determined. The main concern with the use of mavacamten is its negative inotropic effect, which is its main mechanism of action, and hence, the risk of inducing HFrEF. When used judiciously, mavacamten reduces LVOTO without a significant drop in LVEF.137 The clinical use of this drug, once approved, would require individualized dose titration and close monitoring to avoid HFrEF.
There have been a number of other experimental therapies for HCM, including angiotensin II receptor blockade, 3-hydroxy-3-methyglutaryl-coenzyme A reductase inhibitors (statins), mineralocorticoid receptor blockers, and a N-acetylcysteine, none, however, has imparted notable beneficial effects in HCM patients, despite the evidence for their beneficial effects in model organisms.116–119, 139–146 Likewise, gene therapies approaches are only at the experimental phase and are not expected to advance to clinical use in the foreseeable future.
PERSPECTIVE
Advances in the sequencing technologies, such as the development of more accurate long-read WGS approaches are expected to shift the genetic testing approach from WES to WGS and hence, enhance detection of the PVs in HCM. The shift along with interpretation of the PVs as probabilistic determinants of the phenotype in HCM are expected to elucidate a comprehensive landscape of the genetic basis of HCM and resolve the current enigma of the so-called “missing causal genes”. The frequencies of the newly discovered genes and PVs in the HCM populations are expected to be low, indicating the presence of extensive genetic diversity of HCM. Likewise, the majority of the newly discovered PVs and genes are expected to exert modest to moderate effect sizes. Consequently, the phenotype in HCM will be recognized as a continuum, in one end being an archetypical single gene disorder caused by a major PV with a large effect size and on the opposite end of the spectrum an oligogenic or even polygenic trait caused by the cumulative effects of multiple PVs, each exerting a modest effect size and modifying the phenotype.
Clinical applications of genetic discoveries are expected to remain largely limited to cascade screening and the distinction of HCM from the phenocopy conditions, such as storage diseases. Genotype-phenotype correlation studies are not expected to yield consistent and robust associations, because of the numerosity of the determinants and remoteness of the clinical phenotypes to the genotype.
The model organisms, including iPSC-CMs, despite their shortcomings, would be expected to continue providing insights in to the molecular pathogenesis of HCM. Given the diversity of the mechanisms involved, one might surmise that a single therapeutic approach will not applicable to all HCM cases. Consequently, the focus is expected to shift to gene- and base-editing approaches, which are currently are at the early stages of development, inefficient, and have considerable off target effects. The success in the clinical applications of gene and base-editing will depend on fundamental discoveries to enhance the efficiency of the approach and abrogate the off-target effects. The progress, which is unavoidable, will enable the clinician-scientists to gain deep insights into molecular genetics, pathogenesis, and clinical aspects of HCM and practice Sir William Osler conviction that “the good physician treats the disease; the great physician treats the patient who has the disease”
FUNDING SUPPORT
The author was supported in part by NIH, National Heart, Lung and Blood Institute (NHLBI: 1R01HL132401 and R01HL151737), NIH: S10 OD018135, Leducq Foundation (14 CVD 03), The Ewing Halsell Foundation, George and Mary Josephine Hamman Foundation, and the TexGen Fund from Greater Houston Community Foundation.
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- ACTC1
Cardiac α actin 1
- ACTN2
α actinin 2
- ALPK3
α kinase 3
- CAV3
Caveolin 3
- CNV
Copy number variant
- CRYAB
crystalline αB
- CSRP3
Cysteine and glycine-rich protein 3
- DCM
Dilated cardiomyopathy
- EF
Ejection fraction
- FHL1
Four and a half LIM domain 1
- FLNC
Filamin C
- GoF
Gain-of-function
- GV
Genetic variant
- HCM
Hypertrophic cardiomyopathy
- HFpEF
Heart failure with preserved ejection fraction
- HFrEF
Heart failure with reduced ejection fraction
- INDEL
Insertion/deletion (typically involving < 50 nucleotides)
- JPH2
Junctophilin 2
- LOEUF
Loss-of-function observed/expected upper bound fraction
- LoF
Loss-of-function
- LPV
Likely pathogenic variant
- LVEF
Left ventricular ejection fraction
- LVOTO
left ventricular outflow tract obstruction
- MV
Modifier variant
- MYBPC3
Myosin binding protein C3
- MYH7
Myosin heavy chain 7
- MYL2
Myosin light chain 2
- MYL3
Myosin light chain 3
- MYOM2
Myomesin 2
- MYOZ2
Myozenin 2
- NMD
Non-sense mediated decay
- nsSNV
Non-synonymous SNV
- OBSCN
Obscurin
- PLN
Phospholamban
- PTM
Post-translational modification
- PV
Pathogenic variant
- SCD
Sudden cardiac death
- SNV/SNP
Single nucleotide variant/single nucleotide polymorphism
- SV
Structural variant
- SynSNV
Synonymous SNV
- TCAP
Titin capping protein
- TNNC1
Cardiac troponin C
- TNNI3
Cardiac troponin I
- TNNT2
Cardiac troponin T
- TPM1
α tropomyosin
- TRIM63
Tripartite motif containing 63
- TTN
Titin
- UPS
Ubiquitin-proteasome system
- WES
Whole exome sequencing
- WGS
Whole genome sequencing
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
None
REFERENCES
The author apologizes to the authors whose valuable work was not cited, simply because of the vast number of such articles in the literature. For genetic variants the readers are referred to https://www.ncbi.nlm.nih.gov/clinvar/, for expression data in all genes discussed in the manuscript to https://www.proteinatlas.org/) and for data on gene tolerance to predicted loss-of-function variants to https://gnomad.broadinstitute.org/.
- 1.Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121:749–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braunwald E Hypertrophic cardiomyopathy: The first century 1869–1969. Glob Cardiol Sci Pract. 2012;2012:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahm R Friedrich miescher and the discovery of DNA. Dev Biol. 2005;278:274–288 [DOI] [PubMed] [Google Scholar]
- 4.Evans W Familial cardiomegaly. Br Heart J. 1949;11:68–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teare D Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;20:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pare JA, Fraser RG, Pirozynski WJ, Shanks JA, Stubington D. Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy. Am J Med. 1961;31:37–62 [DOI] [PubMed] [Google Scholar]
- 7.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, Seidman JG. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62:999–1006 [DOI] [PubMed] [Google Scholar]
- 8.Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351:617–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charron P, Dubourg O, Desnos M, et al. Diagnostic value of electrocardiography and echocardiography for familial hypertrophic cardiomyopathy in a genotyped adult population. Circulation. 1997;96:214–219 [DOI] [PubMed] [Google Scholar]
- 10.Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med. 1986;315:610–614 [DOI] [PubMed] [Google Scholar]
- 11.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–789 [DOI] [PubMed] [Google Scholar]
- 12.Zou Y, Song L, Wang Z, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in china: A population-based echocardiographic analysis of 8080 adults. Am J Med. 2004;116:14–18 [DOI] [PubMed] [Google Scholar]
- 13.Bick AG, Flannick J, Ito K, et al. Burden of rare sarcomere gene variants in the framingham and jackson heart study cohorts. Am J Hum Genet. 2012;91:513–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65:1249–1254 [DOI] [PubMed] [Google Scholar]
- 15.Nijenkamp L, Bollen IAE, Niessen HWM, Dos Remedios CG, Michels M, Poggesi C, Ho CY, Kuster DWD, van der Velden J. Sex-specific cardiac remodeling in early and advanced stages of hypertrophic cardiomyopathy. PLoS One. 2020;15:e0232427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberly LA, Day SM, Ashley EA, et al. Association of race with disease expression and clinical outcomes among patients with hypertrophic cardiomyopathy. JAMA Cardiol. 2020;5:83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rowin EJ, Maron MS, Wells S, Patel PP, Koethe BC, Maron BJ. Impact of sex on clinical course and survival in the contemporary treatment era for hypertrophic cardiomyopathy. J Am Heart Assoc. 2019;8:e012041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lakdawala NK, Olivotto I, Day SM, et al. Associations between female sex, sarcomere variants and clinical outcomes in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020 [DOI] [PubMed] [Google Scholar]
- 19.Watkins H, McKenna WJ, Thierfelder L, et al. Mutations in the genes for cardiac troponin t and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. 1995;332:1058–1064 [DOI] [PubMed] [Google Scholar]
- 20.Niimura H, Bachinski LL, Sangwatanaroj S, et al. Mutations in the gene for cardiac myosin-binding protein c and late-onset familial hypertrophic cardiomyopathy. N Engl J Med. 1998;338:1248–1257 [DOI] [PubMed] [Google Scholar]
- 21.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. Alpha-tropomyosin and cardiac troponin t mutations cause familial hypertrophic cardiomyopathy: A disease of the sarcomere. Cell. 1994;77:701–712 [DOI] [PubMed] [Google Scholar]
- 22.Marian AJ. Clinical interpretation and management of genetic variants. JACC Basic Transl Sci. 2020;5:1029–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levy S, Sutton G, Ng PC, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876 [DOI] [PubMed] [Google Scholar]
- 25.Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palamara PF, Francioli LC, Wilton PR, et al. Leveraging distant relatedness to quantify human mutation and gene-conversion rates. Am J Hum Genet. 2015;97:775–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Besenbacher S, Liu S, Izarzugaza JM, et al. Novel variation and de novo mutation rates in population-wide de novo assembled danish trios. Nat Commun. 2015;6:5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campbell CD, Chong JX, Malig M, et al. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet. 2012;44:1277–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang H, Thomas PD. Tools for predicting the functional impact of nonsynonymous genetic variation. Genetics. 2016;203:635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marian AJ. Modifier genes for hypertrophic cardiomyopathy. Curr Opin Cardiol. 2002;17:242–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daw EW, Lu Y, Marian AJ, Shete S. Identifying modifier loci in existing genome scan data. Ann Hum Genet. 2008;72:670–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daw EW, Chen SN, Czernuszewicz G, Lombardi R, Lu Y, Ma J, Roberts R, Shete S, Marian AJ. Genome-wide mapping of modifier chromosomal loci for human hypertrophic cardiomyopathy. Hum Mol Genet. 2007;16:2463–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: Expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–888 [DOI] [PubMed] [Google Scholar]
- 36.Gupta MK, Robbins J. Post-translational control of cardiac hemodynamics through myosin binding protein c. Pflugers Arch. 2014;466:231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232 [DOI] [PubMed] [Google Scholar]
- 38.Millat G, Bouvagnet P, Chevalier P, et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J Med Genet. 2010;53:261–267 [DOI] [PubMed] [Google Scholar]
- 39.Helms AS, Thompson AD, Glazier AA, et al. Spatial and functional distribution of mybpc3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020;13:396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vignier N, Schlossarek S, Fraysse B, et al. Nonsense-mediated mrna decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein c mutant levels in cardiomyopathic mice. Circ Res. 2009;105:239–248 [DOI] [PubMed] [Google Scholar]
- 41.Adalsteinsdottir B, Teekakirikul P, Maron BJ, et al. Nationwide study on hypertrophic cardiomyopathy in iceland: Evidence of a mybpc3 founder mutation. Circulation. 2014;130:1158–1167 [DOI] [PubMed] [Google Scholar]
- 42.Calore C, De Bortoli M, Romualdi C, Lorenzon A, Angelini A, Basso C, Thiene G, Iliceto S, Rampazzo A, Melacini P. A founder mybpc3 mutation results in hcm with a high risk of sudden death after the fourth decade of life. J Med Genet. 2015;52:338–347 [DOI] [PubMed] [Google Scholar]
- 43.Kubo T, Kitaoka H, Okawa M, et al. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac myosin-binding protein c gene among japanese. J Am Coll Cardiol. 2005;46:1737–1743 [DOI] [PubMed] [Google Scholar]
- 44.Dhandapany PS, Sadayappan S, Xue Y, et al. A common mybpc3 (cardiac myosin binding protein c) variant associated with cardiomyopathies in south asia. Nat Genet. 2009;41:187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harper AR, Bowman M, Hayesmoore JBG, et al. Reevaluation of the south asian mybpc3(delta25bp) intronic deletion in hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020;13:e002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC, Rayment I, Sellers JR, Fananapazir L, Epstein ND. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet. 1996;13:63–69 [DOI] [PubMed] [Google Scholar]
- 47.Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: Identification of a novel disease gene. Biochem Biophys Res Commun. 1999;262:411–417 [DOI] [PubMed] [Google Scholar]
- 48.Carniel E, Taylor MR, Sinagra G, et al. Alpha-myosin heavy chain: A sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation. 2005;112:54–59 [DOI] [PubMed] [Google Scholar]
- 49.Rubattu S, Bozzao C, Pennacchini E, et al. A next-generation sequencing approach to identify gene mutations in early- and late-onset hypertrophic cardiomyopathy patients of an italian cohort. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mogensen J, Klausen IC, Pedersen AK, Egeblad H, Bross P, Kruse TA, Gregersen N, Hansen PS, Baandrup U, Borglum AD. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest. 1999;103:R39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Osio A, Tan L, Chen SN, Lombardi R, Nagueh SF, Shete S, Roberts R, Willerson JT, Marian AJ. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2007;100:766–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruggiero A, Chen SN, Lombardi R, Rodriguez G, Marian AJ. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc Res. 2013;97:44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chiu C, Bagnall RD, Ingles J, Yeates L, Kennerson M, Donald JA, Jormakka M, Lind JM, Semsarian C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J Am Coll Cardiol. 2010;55:1127–1135 [DOI] [PubMed] [Google Scholar]
- 54.Prondzynski M, Lemoine MD, Zech AT, et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol Med. 2019;11:e11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geier C, Gehmlich K, Ehler E, et al. Beyond the sarcomere: Csrp3 mutations cause hypertrophic cardiomyopathy. Hum Mol Genet. 2008;17:2753–2765 [DOI] [PubMed] [Google Scholar]
- 56.Hayashi T, Arimura T, Itoh-Satoh M, et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol. 2004;44:2192–2201 [DOI] [PubMed] [Google Scholar]
- 57.Friedrich FW, Wilding BR, Reischmann S, et al. Evidence for fhl1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet. 2012;21:3237–3254 [DOI] [PubMed] [Google Scholar]
- 58.Christodoulou DC, Wakimoto H, Onoue K, et al. 5’rna-seq identifies fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest. 2014;124:1364–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim DS, Roberts R, Marian AJ. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: Insight into the pathogenesis of phenotypes. J Am Coll Cardiol. 2001;38:1175–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arimura T, Matsumoto Y, Okazaki O, Hayashi T, Takahashi M, Inagaki N, Hinohara K, Ashizawa N, Yano K, Kimura A. Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2007;362:281–287 [DOI] [PubMed] [Google Scholar]
- 61.Chen SN, Czernuszewicz G, Tan Y, Lombardi R, Jin J, Willerson JT, Marian AJ. Human molecular genetic and functional studies identify trim63, encoding muscle ring finger protein 1, as a novel gene for human hypertrophic cardiomyopathy. Circ Res. 2012;111:907–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salazar-Mendiguchia J, Ochoa JP, Palomino-Doza J, et al. Mutations in trim63 cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart. 2020;106:1342–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Auxerre-Plantie E, Nielsen T, Grunert M, et al. Identification of myom2 as a candidate gene in hypertrophic cardiomyopathy and tetralogy of fallot, and its functional evaluation in the drosophila heart. Dis Model Mech. 2020;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsushita Y, Furukawa T, Kasanuki H, Nishibatake M, Kurihara Y, Ikeda A, Kamatani N, Takeshima H, Matsuoka R. Mutation of junctophilin type 2 associated with hypertrophic cardiomyopathy. J Hum Genet. 2007;52:543–548 [DOI] [PubMed] [Google Scholar]
- 65.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. Pln-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: Summary of the literature and implications for genetic testing. Am Heart J. 2011;161:165–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2007;43:337–343 [DOI] [PubMed] [Google Scholar]
- 67.Valdes-Mas R, Gutierrez-Fernandez A, Gomez J, et al. Mutations in filamin c cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326. [DOI] [PubMed] [Google Scholar]
- 68.Gomez J, Lorca R, Reguero JR, et al. Screening of the filamin c gene in a large cohort of hypertrophic cardiomyopathy patients. Circ Cardiovasc Genet. 2017;10. [DOI] [PubMed] [Google Scholar]
- 69.Almomani R, Verhagen JM, Herkert JC, et al. Biallelic truncating mutations in alpk3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67:515–525 [DOI] [PubMed] [Google Scholar]
- 70.Li L, Bainbridge MN, Tan Y, Willerson JT, Marian AJ. A potential oligogenic etiology of hypertrophic cardiomyopathy: A classic single-gene disorder. Circ Res. 2017;120:1084–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hayashi T, Arimura T, Ueda K, et al. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2004;313:178–184 [DOI] [PubMed] [Google Scholar]
- 72.Saltzman AJ, Mancini-DiNardo D, Li C, et al. Short communication: The cardiac myosin binding protein c arg502trp mutation: A common cause of hypertrophic cardiomyopathy. Circ Res. 2010;106:1549–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Page SP, Kounas S, Syrris P, Christiansen M, Frank-Hansen R, Andersen PS, Elliott PM, McKenna WJ. Cardiac myosin binding protein-c mutations in families with hypertrophic cardiomyopathy: Disease expression in relation to age, gender, and long term outcome. Circ Cardiovasc Genet. 2012;5:156–166 [DOI] [PubMed] [Google Scholar]
- 74.Jaaskelainen P, Helio T, Aalto-Setala K, et al. A new common mutation in the cardiac beta-myosin heavy chain gene in finnish patients with hypertrophic cardiomyopathy. Ann Med. 2014;46:424–429 [DOI] [PubMed] [Google Scholar]
- 75.Jaaskelainen P, Helio T, Aalto-Setala K, et al. Two founder mutations in the alpha-tropomyosin and the cardiac myosin-binding protein c genes are common causes of hypertrophic cardiomyopathy in the finnish population. Ann Med. 2013;45:85–90 [DOI] [PubMed] [Google Scholar]
- 76.Alders M, Jongbloed R, Deelen W, et al. The 2373insg mutation in the mybpc3 gene is a founder mutation, which accounts for nearly one-fourth of the HCM cases in the netherlands. Eur Heart J. 2003;24:1848–1853 [DOI] [PubMed] [Google Scholar]
- 77.Blair E, Price SJ, Baty CJ, Ostman-Smith I, Watkins H. Mutations in cis can confound genotype-phenotype correlations in hypertrophic cardiomyopathy. J Med Genet. 2001;38:385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Girolami F, Ho CY, Semsarian C, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010;55:1444–1453 [DOI] [PubMed] [Google Scholar]
- 79.Marian AJ. The case of “missing causal genes” and the practice of medicine: A sherlock holmes approach of deductive reasoning. Circ Res. 2016;119:21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagueh SF, McFalls J, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue doppler imaging predicts the development of hypertrophic cardiomyopathy in subjects with subclinical disease. Circulation. 2003;108:395–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ. Tissue doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001;104:128–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ameur A, Kloosterman WP, Hestand MS. Single-molecule sequencing: Towards clinical applications. Trends Biotechnol. 2019;37:72–85 [DOI] [PubMed] [Google Scholar]
- 83.Rottbauer W, Gautel M, Zehelein J, et al. Novel splice donor site mutation in the cardiac myosin-binding protein-c gene in familial hypertrophic cardiomyopathy. Characterization of cardiac transcript and protein. J Clin Invest. 1997;100:475–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that mybpc3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222 [DOI] [PubMed] [Google Scholar]
- 85.Tripathi S, Schultz I, Becker E, et al. Unequal allelic expression of wild-type and mutated beta-myosin in familial hypertrophic cardiomyopathy. Basic research in cardiology. 2011;106:1041–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kraft T, Montag J, Radocaj A, Brenner B. Hypertrophic cardiomyopathy: Cell-to-cell imbalance in gene expression and contraction force as trigger for disease phenotype development. Circ Res. 2016;119:992–995 [DOI] [PubMed] [Google Scholar]
- 87.Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mrna decay for genome engineering and personalized medicine. Cell. 2016;165:1319–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mrna surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261 [DOI] [PubMed] [Google Scholar]
- 89.Siwaszek A, Ukleja M, Dziembowski A. Proteins involved in the degradation of cytoplasmic mrna in the major eukaryotic model systems. RNA Biol. 2014;11:1122–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, Eschenhagen T, Zolk O. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein c mutants. Cardiovasc Res. 2005;66:33–44 [DOI] [PubMed] [Google Scholar]
- 91.Schlossarek S, Frey N, Carrier L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J Mol Cell Cardiol. 2014;71:25–31 [DOI] [PubMed] [Google Scholar]
- 92.Spudich JA. Hypertrophic and dilated cardiomyopathy: Four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nag S, Trivedi DV. To lie or not to lie: Super-relaxing with myosins. Elife. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stewart MA, Franks-Skiba K, Chen S, Cooke R. Myosin atp turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci U S A. 2010;107:430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anderson RL, Trivedi DV, Sarkar SS, et al. Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci U S A. 2018;115:E8143–E8152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McNamara JW, Li A, Smith NJ, Lal S, Graham RM, Kooiker KB, van Dijk SJ, Remedios CGD, Harris SP, Cooke R. Ablation of cardiac myosin binding protein-c disrupts the super-relaxed state of myosin in murine cardiomyocytes. J Mol Cell Cardiol. 2016;94:65–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Witjas-Paalberends ER, Piroddi N, Stam K, et al. Mutations in myh7 reduce the force generating capacity of sarcomeres in human familial hypertrophic cardiomyopathy. Cardiovasc Res. 2013;99:432–441 [DOI] [PubMed] [Google Scholar]
- 98.Witjas-Paalberends ER, Guclu A, Germans T, et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovasc Res. 2014;103:248–257 [DOI] [PubMed] [Google Scholar]
- 99.Bloemink M, Deacon J, Langer S, Vera C, Combs A, Leinwand L, Geeves MA. The hypertrophic cardiomyopathy myosin mutation r453c alters atp binding and hydrolysis of human cardiac beta-myosin. J Biol Chem. 2014;289:5158–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, Marian AJ. Differential interactions of thin filament proteins in two cardiac troponin t mouse models of hypertrophic and dilated cardiomyopathies. Cardiovasc Res. 2008;79:109–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagueh SF, Chen S, Patel R, Tsybouleva N, Lutucuta S, Kopelen HA, Zoghbi WA, Quinones MA, Roberts R, Marian AJ. Evolution of expression of cardiac phenotypes over a 4-year period in the beta-myosin heavy chain-q403 transgenic rabbit model of human hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2004;36:663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Solaro RJ, Varghese J, Marian AJ, Chandra M. Molecular mechanisms of cardiac myofilament activation: Modulation by ph and a troponin t mutant r92q. Basic research in cardiology. 2002;97 Suppl 1:I102–110 [DOI] [PubMed] [Google Scholar]
- 103.Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J Am Coll Cardiol. 2003;41:1776–1782 [DOI] [PubMed] [Google Scholar]
- 104.Roberts R, Marian AJ. Can an energy-deficient heart grow bigger and stronger? J Am Coll Cardiol. 2003;41:1783–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Harada K, Potter JD. Familial hypertrophic cardiomyopathy mutations from different functional regions of troponin t result in different effects on the ph and ca2+ sensitivity of cardiac muscle contraction. J Biol Chem. 2004;279:14488–14495 [DOI] [PubMed] [Google Scholar]
- 106.Kraft T, Witjas-Paalberends ER, Boontje NM, et al. Familial hypertrophic cardiomyopathy: Functional effects of myosin mutation r723g in cardiomyocytes. J Mol Cell Cardiol. 2013;57:13–22 [DOI] [PubMed] [Google Scholar]
- 107.Gupte TM, Haque F, Gangadharan B, et al. Mechanistic heterogeneity in contractile properties of alpha-tropomyosin (tpm1) mutants associated with inherited cardiomyopathies. J Biol Chem. 2015;290:7003–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cordero-Reyes AM, Youker K, Hamilton DJ, Torre-Amione G, Marian AJ, Nagueh SF. Molecular, cellular, and functional characterization of myocardial regions in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2012;5:419–422 [DOI] [PubMed] [Google Scholar]
- 109.Litvinukova M, Talavera-Lopez C, Maatz H, et al. Cells of the adult human heart. Nature. 2020;588:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fraysse B, Weinberger F, Bardswell SC, et al. Increased myofilament ca2+ sensitivity and diastolic dysfunction as early consequences of mybpc3 mutation in heterozygous knock-in mice. J Mol Cell Cardiol. 2012;52:1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sequeira V, Wijnker PJ, Nijenkamp LL, et al. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res. 2013;112:1491–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58–60 [DOI] [PubMed] [Google Scholar]
- 113.O’Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:867–874 [DOI] [PubMed] [Google Scholar]
- 114.Schelbert EB, Piehler KM, Zareba KM, et al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, Valdivia HH, Day SM. Genotype-dependent and -independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation. 2016;134:1738–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Teekakirikul P, Eminaga S, Toka O, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires tgf-beta. J Clin Invest. 2010;120:3520–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Senthil V, Chen SN, Tsybouleva N, Halder T, Nagueh SF, Willerson JT, Roberts R, Marian AJ. Prevention of cardiac hypertrophy by atorvastatin in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res. 2005;97:285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, Marian AJ. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lim DS, Lutucuta S, Bachireddy P, Youker K, Evans A, Entman M, Roberts R, Marian AJ. Angiotensin ii blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nagueh SF, Kopelen HA, Lim DS, Zoghbi WA, Quinones MA, Roberts R, Marian AJ. Tissue doppler imaging consistently detects myocardial contraction and relaxation abnormalities, irrespective of cardiac hypertrophy, in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2000;102:1346–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kramer CM, Reichek N, Ferrari VA, Theobald T, Dawson J, Axel L. Regional heterogeneity of function in hypertrophic cardiomyopathy. Circulation. 1994;90:186–194 [DOI] [PubMed] [Google Scholar]
- 122.Urbano-Moral JA, Rowin EJ, Maron MS, Crean A, Pandian NG. Investigation of global and regional myocardial mechanics with 3-dimensional speckle tracking echocardiography and relations to hypertrophy and fibrosis in hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2014;7:11–19 [DOI] [PubMed] [Google Scholar]
- 123.Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: An independent marker of sudden death risk in young patients. J Am Coll Cardiol. 2003;42:873–879 [DOI] [PubMed] [Google Scholar]
- 124.O’Mahony C, Jichi F, Pavlou M, et al. Hypertrophic Cardiomyopathy Outcomes I. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (hcm risk-scd). Eur Heart J. 2014;35:2010–2020 [DOI] [PubMed] [Google Scholar]
- 125.Olivotto I, Cecchi F, Casey SA, Dolara A, Traverse JH, Maron BJ. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517–2524 [DOI] [PubMed] [Google Scholar]
- 126.Siontis KC, Geske JB, Ong K, Nishimura RA, Ommen SR, Gersh BJ. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical correlations, and mortality in a large high-risk population. J Am Heart Assoc. 2014;3:e001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Debonnaire P, Joyce E, Hiemstra Y, Mertens BJ, Atsma DE, Schalij MJ, Bax JJ, Delgado V, Marsan NA. Left atrial size and function in hypertrophic cardiomyopathy patients and risk of new-onset atrial fibrillation. Circ Arrhythm Electrophysiol. 2017;10. [DOI] [PubMed] [Google Scholar]
- 128.Marian AJ, Asatryan B, Wehrens XHT. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovasc Res. 2020;116:1600–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, Rakowski H. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39:638–645 [DOI] [PubMed] [Google Scholar]
- 130.Klarich KW, Attenhofer Jost CH, Binder J, Connolly HM, Scott CG, Freeman WK, Ackerman MJ, Nishimura RA, Tajik AJ, Ommen SR. Risk of death in long-term follow-up of patients with apical hypertrophic cardiomyopathy. Am J Cardiol. 2013;111:1784–1791 [DOI] [PubMed] [Google Scholar]
- 131.Kubo T, Ochi Y, Baba Y, Sugiura K, Takahashi A, Hirota T, Yamanaka S, Yamasaki N, Doi YL, Kitaoka H. Elevation of high-sensitivity cardiac troponin t and left ventricular remodelling in hypertrophic cardiomyopathy. ESC Heart Fail. 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374:2441–2452 [DOI] [PubMed] [Google Scholar]
- 133.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the united states, 1980–2006. Circulation. 2009;119:1085–1092 [DOI] [PubMed] [Google Scholar]
- 134.Sherrid MV, Barac I, McKenna WJ, Elliott PM, Dickie S, Chojnowska L, Casey S, Maron BJ. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45:1251–1258 [DOI] [PubMed] [Google Scholar]
- 135.Maron BJ, Spirito P, Ackerman MJ, et al. Prevention of sudden cardiac death with implantable cardioverter-defibrillators in children and adolescents with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2013;61:1527–1535 [DOI] [PubMed] [Google Scholar]
- 136.Seggewiss HN R; Schaff H Hypertrophic obstructive cardiomyopathy: Surgical myectomy vs septal ablation. Circulation Research. 2017 [DOI] [PubMed] [Google Scholar]
- 137.Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (explorer-hcm): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759–769 [DOI] [PubMed] [Google Scholar]
- 138.Ho CY, Mealiffe ME, Bach RG, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–2660 [DOI] [PubMed] [Google Scholar]
- 139.Tsybouleva N, Zhang L, Chen S, Patel R, Lutucuta S, Nemoto S, DeFreitas G, Entman M, Carabello BA, Roberts R, Marian AJ. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109:1284–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Marian AJ, Senthil V, Chen SN, Lombardi R. Antifibrotic effects of antioxidant n-acetylcysteine in a mouse model of human hypertrophic cardiomyopathy mutation. J Am Coll Cardiol. 2006;47:827–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shimada YJ, Passeri JJ, Baggish AL, et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013;1:480–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nagueh SF, Lombardi R, Tan Y, Wang J, Willerson JT, Marian AJ. Atorvastatin and cardiac hypertrophy and function in hypertrophic cardiomyopathy: A pilot study. Eur J Clin Invest. 2010;40:976–983 [DOI] [PubMed] [Google Scholar]
- 143.Maron MS, Chan RH, Kapur NK, Jaffe IZ, McGraw AP, Kerur R, Maron BJ, Udelson JE. Effect of spironolactone on myocardial fibrosis and other clinical variables in patients with hypertrophic cardiomyopathy. Am J Med. 2018;131:837–841 [DOI] [PubMed] [Google Scholar]
- 144.Marian AJ, Tan Y, Li L, et al. Hypertrophy regression with n-acetylcysteine in hypertrophic cardiomyopathy (halt-hcm): A randomized, placebo-controlled, double-blind pilot study. Circ Res. 2018;122:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Olivotto I, Camici PG, Merlini PA, et al. Efficacy of ranolazine in patients with symptomatic hypertrophic cardiomyopathy: The restyle-hcm randomized, double-blind, placebo-controlled study. Circ Heart Fail. 2018;11:e004124. [DOI] [PubMed] [Google Scholar]
- 146.Ho CY, Lakdawala NK, Cirino AL, et al. Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: A pilot randomized trial to modify disease expression. JACC Heart Fail. 2015;3:180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]