

Abstract
The sigma receptor system has been classified into two distinct subtypes, sigma 1 (σ1R) and sigma 2 (σ2R). Sigma 1 receptors (σ1Rs) are involved in many neurodegenerative diseases and different central nervous system disorders such as Alzheimer's disease, Parkinson's disease, schizophrenia, and drug addiction, and pain. This makes them attractive targets for developing radioligands as tools to gain a better understanding of disease pathophysiology and clinical diagnosis. Over the years, several σ1R radioligands have been developed to image the changes in σ1R distribution and density providing insights into their role in disease development. Moreover, the involvement of both σ1Rs and σ2Rs with cancer make these ligands, especially those that are σ2R selective, great tools for imaging different types of tumors. This review will discuss the principles of molecular imaging using PET and SPECT, known σ1R radioligands and their applications for labelling σ1Rs under different disease conditions. Furthermore, this review will highlight σ1R radioligands that have demonstrated considerable potential as biomarkers, and an opportunity to fulfill the ultimate goal of better healthcare outcomes and improving human health.
Molecular imaging studies have paved the road for the development of successful σ1R ligands currently in clinical trials.
Introduction
The concept of what sigma 1 receptors (σ1Rs) are has evolved significantly over the past 40 years. Currently, σ1Rs are known to be a unique class of chaperone proteins that regulate protein folding, oxidative stress, and cell homeostasis, and are involved in many pharmacological events that make them an attractive, validated therapeutic target. σ1Rs gained a lot of interest in the past 25 years with a total of 1102 articles published from 1992–2017 demonstrating intensive efforts employed in the area of medicinal chemistry to develop selective ligands to probe the associated, putative pharmacologies.1 σRs are classified into 2 subtypes: sigma 1 (σ1Rs) and sigma 2 (σ2Rs). They differ in protein size, tissue expression, and pharmacological and drug selectivity profiles.2–4 σRs are widely distributed in the central nervous system (CNS) in areas involved in pain modulation, memory, emotions, and motor functions, and the periphery where they are expressed mainly in the heart, liver, spleen, lungs, kidneys, adrenal glands, and gastrointestinal tract.2,5–11 Previously, they were misclassified as opioid receptors due to their high affinity to (+)-benzomorphans.2–4 Subsequently, they were further incorrectly thought to be the phencyclidine (PCP) binding site at the glutamate NMDA receptors because SKF-10047 can bind to the PCP site and PCP can bind to σRs.12 Later, it was confirmed that σRs are orphan receptors and are now recognized as a unique class of chaperone proteins.5,13–15
The σ1Rs comprise 223 amino acids. The amino acid sequence shares more than 90% identity across species with no similarity to any other mammalian proteins and less than 30% homology with the fungal enzyme C8–C7 sterol isomerase, although it lacks C8–C7 isomerase activity.5,16 The sequence of the ligand-binding domain of σ1Rs is highly conserved across species, while the transmembrane helices are poorly conserved.17 The σ2Rs have a molecular weight between 18–22 kDa. Previously, it has been claimed that the σ2R binding sites are located in the progesterone receptor membrane component 1 (PGRMC1) protein complex,18 but recent studies emphasized that both are different proteins.19 In 2017, the σ2Rs were cloned by Alon et al.20 who identified the σ2Rs as the endoplasmic reticulum (ER)-resistant TMEM97 transmembrane protein, which is involved in cholesterol trafficking, homeostasis, and cell growth regulation. The crystal structure of the σ2Rs has not been resolved due to the lack of selective ligands.
σ1Rs are chaperone proteins located at the mitochondria associated membrane (MAM) of the endoplasmic reticulum (ER). At the MAM, the ER supplies Ca2+ directly to the mitochondria through inositol 1,4,5-trisphosphate receptors (IP3Rs). σ1Rs are Ca2+ sensitive chaperones that form a complex with another chaperone protein, immunoglobulin heavy chain-binding protein (BiP). Upon ER stress and depletion of Ca2+, σ1Rs dissociate from BiP, sustain the proper conformation of the IP3Rs, and regulate Ca2+ signalling into the mitochondria. The location of σ1Rs at the MAM and the fine-tuning mechanism they exert on mitochondrial Ca2+ signaling supports many of their reported functions such as: regulation of protein folding/degradation, and involvement in cell survival and cellular stress responses.13,21,22 Also, several studies have reported that σ1Rs translocate to the plasma membrane and the nuclear membrane where they can associate with different protein targets to regulate their action through protein–protein interactions, such as ion channels (potassium, calcium, sodium) and G protein-coupled receptors, mainly glutamate, NMDA, and μ opioid.21,23–25
σ1Rs were first cloned in 1996 from guinea pigs, followed with subsequent cloning from human placental choriocarcinoma, and mouse and rat tissues.5,16,26,27 Since the cloning of σ1Rs and the generation of σ1R knockout mice in 2003,28 the research on σ1Rs has made enormous progress and has provided a greater understanding of the physiological and pathological roles of σ1Rs.1 Human σ1Rs were crystalized with two ligands in 2016. The crystal structure of the σ1Rs showed a triangular trimer with a single transmembrane domain in each protomer.17 Interestingly, σ1Rs exist in different dynamic oligomerization states that change based on the bound ligands. Fluorescence resonance energy transfer (FRET) studies revealed that antagonists stabilize higher oligomeric states, while agonists favour dissociation of these complexes.29,30 Although many efforts have been employed to find the endogenous ligand, no small molecule endogenous ligand has been concretely identified yet for σ1Rs. Interestingly, some endogenous molecules show high/moderate binding affinities to σ1Rs; nevertheless, no consensus has been reached on a single ligand. Some of the proposed σ1R endogenous ligands are neurosteroids, such as progesterone (Kiσ1 = 270 nM). Also, N-alkyl amines, sphingosine and its derivatives such as l-threo-sphingosine (Kiσ1 = 20 nM) and d-erythro-sphingosine (Kiσ1 = 140 nM) were presented as endogenous ligands. Most recently, N,N-dimethyltryptamine (DMT) (Kiσ1 = 14 750 nM) was suggested as the endogenous ligand for σ1Rs.4,31–35 Over the past five decades, ligands with diverse structures and flexibility that bind to σ1Rs with high to moderate affinity and low selectivity were reported. Some of these ligands are marketed prescription drugs such as haloperidol (antipsychotic, dopamine antagonist), fluoxetine (antidepressant, selective serotonin reuptake inhibitor), donepezil (Alzheimer's disease, cholinesterase inhibitor), and pentazocine (analgesic, opioid agonist). Although these compounds were very useful in aiding researchers to identify the role of σ1Rs in different diseases, some of these compounds are not selective enough for σ1Rs to draw definitive conclusions. Some of these drugs displayed higher or equal affinity at sigma receptors compared to their approved therapeutic target. For example, haloperidol was reported to bind with equal affinity to both sigma receptors and D2 receptors in rat brain (Ki = 2.8 nm).36 However, another study reported haloperidol to have lower affinity at D2Rs in rat (total) striatum (Ki = 10 nM).37
Many inconsistent results or off target activities have been reported, which complicate the interpretation of the actual contribution of σ1Rs. Recently, with the help of ligand design strategies and imaging techniques, high affinity, selective ligands have been discovered to probe the receptors and explore their diverse biological contributions. However, no feasible in vitro functional assays for σ1Rs have been accepted to determine downstream signalling pathways that discriminate between agonists and antagonists. Identifying the functional activity of σ1R ligands remains challenging and really needs further investigation.22,38
Identification of σ1R ligand functional activity (how to differentiate/discriminate between agonists and antagonists in the absence of functional assays)
Generally, σ1R ligands are characterized by radioligand binding assays and some predictive approaches have been used to identify the agonist/antagonist profile which include:
Behavioral pharmacological assays
Pain related behaviors (allodynia and hyperalgesia) were evaluated against sigma ligands. It has been well established that σ1R agonists (e.g. pentazocine) diminish opioid analgesic activity, while σ1R antagonists (e.g. haloperidol) have been demonstrated to potentiate opioid analgesia in both CD-1 mice and Sprague-Dawley rats, as well as being endowed with antiallodynic effects in different pain models.39–41 Ligands that induce the same phenotype as pentazocine are commonly accepted as σ1R agonists, whereas compounds that show the same effect as haloperidol are considered as antagonists. Responses have been measured by using different animal models of pain such as formalin or capsaicin induced pain models, chronic constriction injury (CCI) assays, tail flick assays, and Von Frey assays.42
On the other hand, animal behavioral studies using a cocaine-induced convulsion model have been helpful to discriminate between σR agonists and antagonists. Pretreatment of mice with σR antagonists before the administration of a convulsive dose of cocaine were reported to have protective effects and attenuate cocaine induced behavioral toxicity, lethality, and locomotor stimulatory effects. Meanwhile, σR agonists worsened the behavioral toxicity of cocaine and exacerbated the convulsive effects of cocaine.43–45
Furthermore, agonist or antagonist profiles of novel compounds could also be determined by their effects on 1,3-di(2-tolyl)guanidine (DTG)-induced acute dystonic reactions in rats, an established functional assay for σR activity. Compounds are microinjected into the rat red nucleus where they are considered to be agonists if they elicit dystonia. Antagonists were reported to attenuate σR agonist-induced dystonic head postures.46,47
Genetic (knockout mice)
Knockout mice help to understand the role and the function of genes that have been inactivated. The difference between the normal behavior or physiology of knockout mice and that of wild type mice infers the possible gene function. For example, in σ1R knockout mice, attenuation of pain behaviors in different pain models and enhancement of morphine mechanical antinociception were observed, which is consistent with the observation that σ1R antagonists showed antinociception in pain models.28,48–50 Knockout mice are a helpful tool that may predict the functional activity of σ1R ligands. There is always the caveat of compensatory mechanisms, however.
Molecular biology (antisense oligonucleotides)
Antisense oligonucleotides have been utilized in knockdown expression methods in which downregulation of the targeted receptor occurs to study gene functions.51 With σ1R antisense oligodeoxynucleotides, enhanced analgesia to morphine as well as blockade of cocaine acquisition, attenuation of cocaine induced convulsions, and reduction in cocaine induced locomotor stimulatory effects were observed.43,52 These effects are all consistent with σ1R antagonist activity.
Competition binding assay with phenytoin (DPH)
DPH was proposed as an allosteric modulator of σ1Rs that modify the binding affinity of σ1R ligands.53 DPH increases the affinity of σ1R agonists to the active state and does not increase the affinity of the antagonists. Thus, these results suggest that DPH can be used as a predictive tool to differentiate between σ1R agonists and antagonists.
Fluorescence resonance energy transfer (FRET) biosensor assay
Biosensor assays have the ability to detect ligand-mediated conformational changes of σ1Rs induced by agonist or antagonist binding. This technique is based on the use of cyan and yellow fluorescent proteins (CFP and YFP, respectively), which upon ligand binding based on their agonist or antagonist profile will lead to real-time fluorescence resonance energy transfer (FRET) changes in living cells. The agonist binding will lead to a decrease in the FRET signal, while the antagonist will increase the FRET signal. Thus, σ1R ligand agonist/antagonist profiles can be predicted.54
The alteration of the σ1R oligomerization state upon binding of agonists and antagonists
Agonist binding resulted in dissociation of the multimers into monomers and dimers and induced an outward facing conformation of dopamine transporter (DAT), thus enhancing cocaine binding and behavioral responses. However, the antagonist stabilized the higher order of oligomerization without changing the DAT conformation.29,55
σ1R ligands in clinical trials
Both agonists and antagonists are of great interest as potential therapeutic candidates against σ1R related diseases. Selective and high affinity σ1R ligands (10 compounds) have been developed previously and advanced to clinical trials for Alzheimer's disease, depression, neuropsychiatric disorders, schizophrenia, major depressive disorder, and anxiety. Unfortunately, these compounds failed and were discontinued in clinical development.56
To date, three σ1R agonists are in clinical trials (Fig. 1); the first one is ANAVEX®2-73 (blarcamesine), a mixed muscarinic receptor/σ1R ligand (Kiσ1 = 850 nM; Kiσ2 = inactive).57 ANAVEX®2-73 is currently in a phase III clinical trial for Alzheimer's disease (ClinicalTrials.gov identifier: NCT03790709), as well as a phase II clinical trial for treatment of cognitive impairment in Parkinson's disease patients with dementia (ClinicalTrials.gov identifier: NCT03774459), an observational study for event-related potential (ERP) biomarkers in subjects with schizophrenia and healthy volunteer subjects (ClinicalTrials.gov identifier: NCT04025502), and a phase II clinical trial in Rett syndrome patients. Recently, Anavex Life Sciences announced that the FDA granted the Fast Track designation for the clinical development program for the treatment of Rett syndrome (ClinicalTrials.gov identifier: NCT03758924).58 The second ligand is SA4503 (cutamesine), a selective σ1R agonist (Kiσ1 = 4.6 nM; Kiσ2 = 63 nM), which has completed a phase II clinical trial for acute ischemic stroke (ClinicalTrials.gov identifier: NCT00639249), and a phase II clinical trial for major depressive disorder (ClinicalTrials.gov identifier: NCT00551109).59 The third agonist is pridopidine (ACR16 or Huntexil). Initially, it was classified as a dopamine stabilizer; however, recently, it was found to be a selective σ1R agonist (Kiσ1 = 70 nM) at the lower end of the active dose known to produce neurochemical and behavioral effects in rats. At this dose, it displayed 100 fold selectivity over the dopamine D2 receptor (Ki = 7520 nM).60,61 Pridopidine is currently in a phase II clinical trial to evaluate its safety and efficacy for treating levodopa induced dyskinesia in patients with Parkinson's disease (ClinicalTrials.gov identifier: NT03922711).62 It also completed a phase III clinical trial for the treatment of motor symptoms of Huntington's disease (ClinicalTrials.gov identifier: NCT00665223). Recently, it was selected for inclusion in a novel platform trial for amyotrophic lateral sclerosis (ALS) by the Sean M. Healey & AMG Center for ALS at Massachusetts General Hospital.
Fig. 1. Successful σ1R ligands in clinical trials.

On the other hand, σ1R antagonists in clinical trials (Fig. 1) are led by E-52862 (S1RA), the first-in-class potential σ1R antagonist (Kiσ1 = 17 nM; Kiσ2 = 6300 nM), currently in a phase II clinical trial in Europe (EudraCT number: 2012-000398-21) for pain management as a monotherapy for neuropathic pain of different etiology and as an adjuvant therapy for opioids.63 In addition, [18F]FTC-146 is the most highly selective σ1R antagonist reported to date (Kiσ1 = 0.0025 nM; Kiσ2 = 364 nM), and is currently in a phase I clinical trial as a PET/MRI diagnostic agent to pinpoint sites of nerve damage, identify the source of pain generation and monitor treatment responses in complex regional pain syndrome (CRPS) and sciatica patients, and chronic neuropathic and/or nociceptive pain to investigate changes in σ1R expression for chronic pain. [18F]FTC-146 is a tool to help identify the correlation between nerve injury, σ1R expression, and pain generation (ClinicalTrials.gov identifier: NCT02753101).64–66
Role of σ1R activation or inhibition in chronic neurological diseases
σ1Rs are involved in many pharmacological events and functions throughout the CNS, such as signal transduction,67 memory, recognition, emotion, and modulation of the neurotransmitters dopamine,68 acetylcholine,69 serotonin,70 and glutamate.71
Neurodegenerative diseases
The pathophysiology of neurodegenerative diseases is complex; however, there is a common factor that involves dysfunction at the mitochondrial, endoplasmic reticulum, and synapse axes.72 Therefore, the location of σ1Rs at the MAM makes them attractive targets for studying neurodegenerative diseases and developing diagnostic biomarkers to monitor disease progression and develop potential therapeutics. Several studies have shown the involvement of σ1Rs in neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and juvenile amyotrophic lateral sclerosis (ALS).73,74 Remarkably, the expression levels of σ1Rs were found to be altered in the brain of patients who suffer from different neurodegenerative diseases.75,76
Furthermore, the activation of σ1Rs attenuates reactive oxygen species (ROS) at the ER, suppresses oxidative stress, and are involved in cellular defense against neurodegenerative disorders.73,76–79 Of additional interest is the resultant increase in the expression of protective genes such as the antiapoptotic protein bcl-2, after activation of σ1Rs. Thus, σ1Rs may also contribute to neuroprotection.57,80,81 Therefore, determination of the expression level of σ1Rs in the CNS could be useful in the diagnosis of AD, PD, and ALS.
Alzheimer's disease (AD)
AD is a progressive brain disorder characterized by slow destruction of memory and thinking skills and considered as the most common cause of dementia. Both σRs are involved in many cellular pathways that affect brain plasticity, learning and memory processes, and AD progression. For recent reviews on the role of sigma receptors in AD, see these selected references (ref. 78, 82–84). Additionally, postmortem and PET neuroimaging studies revealed that AD patients had experienced an idiopathically low density of σ1Rs in the hippocampus compared to healthy individuals.84–86 So, activation of σ1Rs was examined for the treatment of AD, whereas σ1R agonists were reported to attenuate memory deficit and showed neuroprotective and anti-amnesic properties. This is thought to be due to the σ1Rs' modulatory role on Ca2+ mobilization, regulation of oxidative stress, antiapoptotic effect, regulation of glutamate release and increases in acetylcholine secretion.85–89 The σ1R agonist, ANAVEX®2-73, is in a phase III clinical trial for AD, which activates σ1Rs, and has demonstrated the ability to reduce crucial pathophysiological signs of AD such as beta amyloid, hyperphosphorylated tau, and increased inflammation. ANAVEX®2-73 has also shown a dose dependent improvement in cognitive functions. Moreover, the σ2R allosteric antagonist Elayta (CT1812), is currently in phase I/II clinical trials for mild to moderate AD treatment (ClinicalTrials.gov identifier: NCT02907567). Elayta displaces the toxic beta amyloid oligomers and prevents their binding to neurons, which in turn prevents downstream synaptotoxicity and protects against memory loss.90
Both σ1R agonists and σ2R antagonists showed a neuroprotective effect, anti-amnesic activity, and improvement in patients' cognitive functions. Radioligands for σRs could be used as potential diagnostic biomarkers through brain imaging to afford insights about AD pathophysiology and monitor therapeutic efficacy.
Amyotrophic lateral sclerosis (ALS)
ALS is a neurodegenerative disease that is characterized by the loss of motor neurons in the brain and spinal cord leading to paralysis and early death. Complex pathophysiological mechanisms contribute to ALS such as neuronal injury from excitotoxicity, mitochondrial dysfunction, increased reactive oxygen species, and endoplasmic reticulum stress responses (which initiate protein degradation). Also, motor neuron damage can lead to activation of microglia and astrocytes, which further contributes to neurodegeneration.91 Since σ1Rs are located at the MAM, regulate Ca2+ homeostasis, modulate neuronal excitability, and are highly expressed on the motor neurons in the spinal cord, it is believed that σ1Rs could be involved with ALS progression and serve as a potential target for ALS pharmacotherapy. Moreover, several lines of evidence suggest that σ1R alterations or mutations lead to motor neuron degeneration and progression of ALS. It is of potential importance that low levels of σ1Rs were observed in ALS patients.92–95 σ1R activation by agonists, such as PRE-084, pridopidine, and SA4503, showed neuroprotection and reduced microglial and astroglial reactivity in the transgenic superoxide dismutase 1 (SOD1) mouse model.96,97 It also prevented the loss of neuromuscular connections, motor axons, and motor neuron cell bodies in the spinal cord and increased animal survival.96–99
Parkinson's disease (PD)
PD is a neurodegenerative disease, which affects motor functions, characterized by the gradual loss of dopaminergic neurons in the substantia nigra. Several studies have suggested that σ1Rs are linked to PD because they are expressed in the substantia nigra and are known to modulate dopamine release via different mechanisms.100,101 Moreover, in two different human clinical trials, low σ1R density has been observed in early PD patients compared to healthy volunteers when the PET radioligand [11C]SA4503 was utilized.102,103 In patients with PD, the binding potential of [11C]SA4503 to σ1Rs in the anterior putamen (10.8 ± 4.2) was lower compared to that in normal individuals (12.2 ± 5.0). In addition, the binding potential of [11C]SA4503 was significantly lower on the more affected (9.1 ± 4.4) than the less affected (12.4 ± 4.3) side of the anterior putamen of PD patients. Meanwhile, σ1R knockout mice showed increases in α-synuclein aggregation and phosphorylation, a major constituent of Lewy bodies that are believed to play a critical role in the pathogenesis of PD. Also, the loss of dopaminergic neurons in the substantia nigra was observed in σ1R knockout mice.101 Consequently, σ1R activation has been reported to restore synaptic connectivity and protect nigrostriatal dopamine neurons against degeneration.104,105 In a unilateral 6-hydroxydopamine (6-OHDA) lesion model of parkinsonism in mice, the σ1R agonist, pridopidine, demonstrated a neuroprotective effect, and showed an increase in dopaminergic fiber density in the striatum, restorative plasticity, and upregulation of neurotrophic factors (BDNF). However, 6-OHDA-lesioned mice with σ1R knockout did not show the beneficial effects of pridopidine.104 It was reported that daily administration of PRE-084, a σ1R agonist, utilizing a murine model with induced nigrostriatal degeneration has shown significant motor recovery and an increase in striatal dopaminergic fiber density suggesting the therapeutic potential of σ1R agonists in PD.105
Huntington's disease (HD)
HD is a hereditary neurodegenerative disease associated with the production of mutant huntingtin protein (mHtt) and characterized by the gradual, progressive loss of neurons in the brain, which leads to motor and cognitive impairments.106 The loss in function of normal proteins and the production of mutant proteins result in the disruption of multiple intracellular pathways, apoptosis, mitochondrial dysfunction, oxidative stress, ER stress, and autophagy.107 Since σ1Rs are activated under ER-stress, they may be implicated in the ER-related degradation of the mHtt. Several studies have shown that activation of σ1Rs provides a neuroprotective role in HD; for example, the σ1R agonist PRE084 demonstrated a neuroprotective effect by decreasing ROS levels, exerting antioxidant effects and increasing antiapoptotic effects by affecting NF-kB signaling.108 Administration of the σ1R agonist, (+)-3-PPP, resulted in a neuroprotective effect and an increase in the density of the neuronal cultures in mice.109 Meanwhile, pridopidine improved motor performance and survival in the R6/2 and Yac128 HD mouse models.110 It was also suggested that the agonistic activity of pridopidine at σ1Rs resulted in modulation of ER stress, especially the PKR-like ER-localized eIF2a kinase (PERK) pathway.111
Neuropsychiatric disorders
The first pharmacological activity reported for σRs upon binding of the prototypic ligand, (±)-SKF10047 (Kiσ1 = 44.8 nM, σ2/σ1 = 95.1), was inducing psychotomimetic effects.34 Later, σ1Rs were reported to be involved in neuronal plasticity. Neuronal plasticity is the ability of the nervous system to form new neuronal connections and compensate for injury. Basically, changes in the structure, function and organization of neurons occur in response to new experiences and injuries. Disruption in neuronal plasticity and reduction in dendritic spine density have been reported to be implicated in the pathophysiology of neuropsychiatric disorders such as depression and schizophrenia.112 These findings suggested the contribution of σRs to neuropsychiatric disorders. Moreover, σ1Rs provide a defense mechanism against oxidative and ER stress that might be triggered under psychological stress or neuropathological conditions.113 Thus, the antipsychotic potential of σ1R ligands has been explored with great interest.114–116
Depression
σ1Rs are involved in the pathophysiology of depression because of depressive-like behaviors that develop in σ1R knockout mice.117 Additionally, σ1R agonists have been reported to have antidepressant activity.118,119 It has been suggested that σ1Rs' antidepressant activity is due to modulation of serotonin, noradrenaline, and glutamate neurotransmission.70,71,120,121 Remarkably, clinically used antidepressants (e.g. imipramine) bind with high to moderate affinity to σ1Rs. It was suggested that antidepressants enhance the brain-derived neurotrophic factor (BDNF) signaling, which induce glutamate release through activation of the PLC-γ/IP3/Ca2+ pathway due to their binding to σ1Rs.71
Schizophrenia
σ1Rs are also involved in the pathophysiology of schizophrenia as a result of their modulation of dopaminergic neurotransmitters. The symptoms of schizophrenia include positive and negative symptoms, cognitive impairment, and social isolation. E-5842, a σ1R ligand, was reported to increase dopamine release in the striatum and its neurochemical profile is similar to that of atypical antipsychotics.122–124 It was reported that σ1R agonists (e.g. pregnenolone and dehydroepiandrosterone) were effective against negative and cognitive symptoms of schizophrenia and demonstrated antipsychotic activity without producing extrapyramidal side effects.116,125–129 The antidepressant drug, fluvoxamine, is a selective serotonin reuptake inhibitor (SSRI) as well as a σ1R agonist with high affinity (Ki = 36 nM). Fluvoxamine at therapeutic doses binds to σ1Rs, which was confirmed using PET imaging studies on human brain.130 Its efficacy in treating cognitive impairments and negative symptoms in some schizophrenic patients is suggested to be through σ1R activation.131–133 Furthermore, the σ1R density was reported to be low in the postmortem brain, predominantly in the temporal cerebral cortex, of schizophrenic patients compared to age-matched, normal postmortem controls.134,135 Previously, five σ1R antagonists [panamesine (EMD57445), eliprodil (SL82.0715), rimcazole (BW234U), BMY14802 (BMS181100), and DuP734] progressed to clinical trials for the treatment of positive symptoms of schizophrenia. However, they were not effective against positive symptoms. Interestingly, eliprodil and rimcazole were effective against the negative symptoms of schizophrenia.77,116,136
Pain and analgesia
Under normal physiological (non-sensitizing) conditions, σ1Rs do not modify normal sensory mechanical or thermal perception. However, they are activated and effective under pathological or sensitizing conditions such as nerve injury, chronic pain, inflammation, allodynia, and hyperalgesia, and they are associated with neurophysiopathological changes.56 σ1Rs are considered an endogenous anti-opioid system since σ1R agonists reduce opioid analgesia, while σ1R antagonists potentiate opioid analgesia and restore normal nociceptive thresholds. It was well established that σ1Rs have a significant role in pain modulation and have been associated with nerve injury and neuroinflammation.137,138 Moreover, σ1R antagonists demonstrated antiallodynic effects in neuropathic and neurogenic pain.38,49,138–140 σ1R antagonists are considered as potential biomarkers to locate nerve injury and neuroinflammation.64 CM304, a σ1R antagonist, has shown promising anti-allodynic activity in different animal models of neuropathic pain and nociceptive pain.141 Also, σ1Rs are known to be upregulated at the site of partial sciatic nerve ligation.64,142 Interestingly, two σ1R antagonists are now in clinical trials: E5286 (phase II) for pain management/neuropathic pain, and [18F]FTC-146 (phase I) as a diagnostic agent to pinpoint nerve damage in sciatica and complex regional pain syndrome (CRPS). These findings together with the large body of the literature about the role of σ1Rs in pain modulation suggest that σ1Rs are a promising class of pharmacotherapeutics for pain in the future.
The proposed mechanism of antinociception of σ1R antagonists involves inhibition of glutamate release, regulation of the activity of different targets involved in pain pathways such as ion channels (Na+, K+, Ca+) and G-protein coupled receptors (cannabinoid CB1 receptors, serotonin 5-HT1A and 5-HT2A receptors, glutamate NMDA and mu opioid receptor), and activation of descending inhibitory systems.56
Addiction
σ1R activation is associated with the addictive, neurotoxic, and reinforcing effects of many abused drugs (cocaine, methamphetamine, and alcohol). Preclinical studies on male rodents suggested that σ1R antagonists inhibit behaviors related to alcohol use disorders (AUDs) and reduce alcohol consumption and alcohol-seeking behavior.143 So, σ1Rs might be a promising target for treating AUDs and superior to the current Food and Drug Administration (FDA)-approved drugs for AUDs that have limited efficacy such as disulfiram, naltrexone, and acamprosate.143 Further human studies are needed to confirm the efficacy in humans.143 Additionally, σ1R upregulation was found after chronic self-administration of methamphetamine to rats. σ1Rs are a promising therapeutic target as well for the treatment of methamphetamine addiction.124,144 Moreover, antagonism of σ1R led to the attenuation of psychostimulant-induced effects, such as cocaine induced seizures, hyper-locomotion, sensitization, and changed the gene and protein expression that was upregulated by cocaine administration.145,146
Role of σ1Rs in cancer
σ1Rs are highly expressed in different types of cancer, such as brain, breast, prostate, and colorectal cancer and chronic myeloid leukemia cell lines. Their upregulation in several cancers has attracted much research focused around tumor imaging. Hence, imaging of σ1Rs with radioligands might contribute to a better understanding of the tumor physiology and the pathophysiological function of σ1Rs, and aid in the development of novel antineoplastic drugs. For a comprehensive review about σ1R radioligands developed for cancer imaging, readers are referred to the following reference (ref. 147). Besides, σ1R antagonists show a strong ability to inhibit cancer cell proliferation in vitro and in vivo.148–151 For example, the σ1R antagonist, 1-(4-iodophenyl)-3-(2-adamantyl)guanidine (IPAG), resulted in an unfolded protein response (UPR) followed by autophagy and apoptosis in cancer cells.151 Additionally, σ1R ligands were reported to regulate cancer cell electrical plasticity.149,152 Although the exact mechanism is still inconclusive, σ1Rs are reported to inhibit cancer cell proliferation via upregulation of anti-apoptotic pathways, involvement in protein homeostasis, a pathway known to be involved in cell death and cancers, and regulation of membrane electrical activities. It is noteworthy that both σRs are overexpressed in different tumors,153 but σ2Rs show higher expressions and overwhelming evidence of being useful biomarkers for tumor proliferation. σ2R ligands have been demonstrated to be useful tools in imaging solid tumors, and as potential therapeutics for cancer treatment.154–156
Role of σ1Rs in cardiac dysfunction
Although both σRs were found to be expressed in the heart by ligand binding studies in the early 1990s,157 the physiological function of cardiac σRs remains unknown and limited studies have been done to explore their role in the heart. Recently, Chowdhury et al. reported that σ1Rs regulate normal mitochondrial organization and size in the heart of mice. Additionally, σ1R knockout mice demonstrated cardiac dysfunction associated with accumulations of irregularly shaped mitochondria and defects in their respiratory function.158 These findings suggested that σ1Rs exert a cytoprotective effect, regulate cardiac hemodynamics and are needed to maintain normal cardiac contractility. More clinical research is required to define the physiological function of σ1Rs in the heart and evaluate the potential therapeutic role of σ1Rs in cardiovascular diseases.
Shedding light on σ1Rs and molecular imaging
σ1Rs are now recognized as potential therapeutic targets that have a putative role in many diseases. It has been reported that σ1Rs can be involved in reducing the symptoms of some neurodegenerative disorders, but can lead to the establishment of other diseases.73,159 Consequently, σ1Rs have been considered as an attractive target and have gained more attention in the drug discovery field for their potential therapeutic value. These observations confirmed the importance of studying σ1Rs in many neurodegenerative diseases, CNS disorders, tumor progression, pain, addiction, and cardiac dysfunction to assess their possibility as a promising target for therapeutic development.77,160,161
Over the past decades, σ1Rs have been studied using different imaging techniques. These techniques made and continue to have a significant impact in the recognition of σ1Rs as “a Pluripotent Modulator in Living Systems”.21 Atomic force microscopy imaging and confocal imaging techniques have been used to identify the interaction between σ1Rs and other proteins (receptors and ion channels), which is not the scope of this review. This in turn helped with understanding the pathophysiology and the functional crosstalk between σ1Rs and other proteins.21 Moreover, developing radioligands for imaging σ1Rs in vitro and in vivo helped to confirm that the σ1R is a unique protein and does not belong to the opioid or NMDA receptor families.162 This also provided insights about the available σR subtypes and their anatomical distribution,9 and identified high affinity σ1R ligands.
In addition, σ1R radioligands had a critical role in understanding the receptor pharmacology and its contributions in many diseases. Bibliometric analysis of the scientific publications focused on σ1Rs in the last 25 years indicated that research efforts were previously focused more on neuroimaging, addiction, and psychiatric disorders; however, neurodegenerative diseases, neuroprotection, and pain are currently attracting the most attention.1 Interestingly, the top two keywords were “Positron Emission Tomography (PET)” and “Neuroprotection,” respectively.1 This indicates the importance of imaging in the discovery of σ1Rs' roles in neurodegenerative and neuropsychiatric diseases through preclinical and clinical studies. The following sections discuss the role of imaging in drug discovery, frequently used techniques and radioligands, and the application of σ1R radioligands in molecular imaging.
Imaging in drug discovery
Nuclear medicine functional imaging (molecular imaging) is a type of medical imaging that noninvasively creates a visual representation of the internal aspects of the body and determines the biological/molecular processes in normal and diseased states to identify abnormalities. The principles of in vivo molecular imaging depend on detecting the energetic particles (radiation) emitted from a radioactive material (radioisotope) upon decaying by gamma scintigraphy, single photon emission computed tomography (SPECT), or positron emission tomography (PET). Imaging data obtained are processed by computers to produce 2D and 3D images. These images can be used for diagnosis and detection of functional processes in living systems quantitatively.
Functional imaging nowadays has a great impact on drug discovery and development and valuable contributions in the pharmaceutical industry. It is considered as a major tool in preclinical development, translational research, clinical diagnosis, clinical trials, and life sciences. It facilitates the visualization of the biological activities in animals, without the need to use invasive techniques, which require the sacrifice of the animals. For a review on the advantages and limitations of ex vivo autoradiography versus molecular imaging, the reader is directed to this reference (ref. 163). Molecular imaging has been utilized by different fields such as oncology, cardiology and mostly neuroscience due to the inaccessibility of the human brain.
The advancement in nuclear medicine technology and the use of powerful non-invasive instrumentation allow for the use of radioligands as diagnostic biomarkers. Radioligands have become important tools in improving the drug discovery process through the quantitative assessment of radioligand distribution, determination of target expression levels in different tissues, characterization and validation of many targets, and confirmation of target engagement under many pathological conditions in living systems.164 The assessment of target distribution and expression levels became easier, which in turn helped with the design of safer and more efficacious treatments. Moreover, the use of these imaging techniques in animals and humans has helped to delineate normal physiological and pathological conditions resulting in improvements in understanding disease pathophysiology, monitoring disease progression, and earlier diagnosis as well as follow-up treatments.
Radioligands used in clinical diagnosis
Classification of radioligands
Radioligands used for PET or SPECT can be classified depending on how the radiolabel is introduced into the imaging agent.165 In the first class, the imaging agent is the radionuclide itself (e.g. [18F]sodium fluoride is a PET imaging agent for osteosarcoma). The second class has the radiolabels attached (atomic substitute) to or pendent from the target molecule (e.g. [18F]fluorodeoxyglucose (18F-FDG) is a marker for tissue glucose uptake and monitoring tumor metabolism). Meanwhile, in class three, the radionuclides are incorporated within a molecule as an isotopic modification. The selected molecule usually is a ligand that binds specifically to the target of interest; for detailed information, the reader is encouraged to read ref. 165.
Criteria for an ideal radioligand
The ideal imaging agent should demonstrate high affinity (at the nanomolar or picomolar range), with a high selectivity profile over other targets, high in vivo stability, and high uptake at the target tissue. Also, high specific binding with minimal nonspecific binding is important to ensure more detailed results and avoid incorrect interpretation of the imaging data. Mintun et al. reported a mathematical model that can provide quantitative characterization of drug binding sites for in vivo PET imaging.166 This can be achieved by calculating the binding potential (BP), which is equivalent to the product of the maximum drug specific binding concentration (Bmax) and the reciprocal of the radioligand binding affinity (KD), BP = BmaxKD−1. Thus, BP reflects the potential of a given tissue for ligand-binding site interaction and provides accurate characterization of drug–receptor kinetics in living subjects. A suitable tissue kinetic profile is desirable as well; good radioligands should demonstrate fast and reversible binding kinetics, a considerable washout period and adequate clearance because slow pharmacokinetics will limit the clinical utility of the radioligand. Moreover, the radiation risk should be within an acceptable range with low potential toxicity and a relatively low total radiation dose to the patient per unit of initial activity after administration.167 In the case of brain imaging, high brain to blood ratios and good blood–brain barrier (BBB) permeabilities are essential. Moreover, for radioligands with short half-lives, a rapid uptake into the brain is essential to ensure a pseudo-equilibrium has been reached before the decay of the radionuclide. In addition to the aforementioned, radiometabolites generated via peripheral metabolism should not be able to cross the BBB.
Challenges of imaging agents in humans
One of the challenges of imaging agents is their stability in the human body. For example, a [18F] PET radioligand can be metabolized by defluorination and result in non-specific accumulation of [18F]fluoride radioactivity in bone, which will affect the quality of imaging, the quantitation of PET signals and the utility of the radioligand. So, with fluorine radioligands, little to no accumulation in bone is desirable during the scan time to ensure reliable results and quantification of the PET signals.168 Also, the same concept is applied to [123I] as it can undergo deiodination.169 Moreover, the presence of radiometabolites would limit the usefulness of the radioligand and affect the kinetic analysis. In particular, if the metabolite is active and binds to the target with a different affinity, the quantification of the signals will be complicated. Meanwhile, if the radiometabolite is inactive, nonspecific binding that will affect the signal to noise ratio may increase.163 Therefore, radioligands are preferred to have good in vivo metabolic stability.
Another challenge is that the incorporation of the radioisotope into a molecule may change its chemical and physical properties which may affect its binding affinity, pharmacokinetic properties or biological activity.163,169 One of the factors to be considered when choosing the type of the radiolabelled nuclei is the radiosynthesis step, the time of the introduction of the radiolabelled atom. This is one of the challenges that face [18F]fluoride PET tracers synthesis. For example, if the radiosynthesis of the [18F]fluoride is the last step to obtain the final PET tracer, the radiochemical yield will be high. But, if more steps are required after the introduction of the radiolabelled [18F], reduction of the radiochemical yield will occur due to the longer production time and its relatively short half-life (109.8 min).170
Non-invasive imaging techniques (PET and SPECT) in drug discovery
The use of SPECT and PET imaging in drug discovery is common.164,171 Different factors should be considered before choosing the imaging technique such as the resolution, sensitivity, cost, availability of scanners and equipment, availability of the radiolabelled tracer and ease of synthesis, and the clinical use (e.g.: repeated dosing).165
Single-photon emission computed tomography (SPECT) imaging
Principle: a nuclear medicine 3D tomographic imaging technique that directly detects the gamma rays emitted from a radioactive isotope upon decaying, using gamma cameras that surround the body. The cameras acquire many 2-D images from multiple angles, and then a tomographic reconstruction algorithm is applied to generate a 2D or 3D data set. The total time of a scan is around 15–20 minutes.
Generally, the patient is injected with a diagnostic radiolabelled probe that has affinity for a specific target, where it will accumulate. When the radioisotope decays, gamma radiation is emitted, and captured. The resultant computationally generated images show the distribution of the radiolabelled probe within the patients' body that can be interpreted and used for diagnosis. The isotopes suitable for SPECT are thallium201Tl, technetium 99mTc, gallium 67Ga, iodine 123I, iodine 125I, and iodine 131I. The half-lives for their gamma emission are 73 h, 6 h, 78.26 h, 13.2 h, 59.49 days, and 8 days, respectively. The most widely used SPECT radiolabels for biomolecule labelling are the radiometal 99mTc and radioiodine 123I. The [99mTc] Tc isotope has the advantages of a moderate half-life (6 h), which is suitable for clinical use, besides its convenient production and the availability of an in-house generator (molybdenum-99). Meanwhile, 123I has been used clinically as a radionuclide for SPECT because of its longer half-life (13.2 h) and easy synthesis, and its gamma emissions are ideal for sodium-iodide-based SPECT detectors.165
The radioisotope 131I is used for therapeutic applications, while the radioisotope 125I has been used in nuclear medicine imaging mainly for in vitro or ex vivo assays due to its radioactive emission of a total of 21 low-energy (∼20–500 eV) Auger electrons compared to 11 Auger electrons emitted from 123I. These Auger electrons have been found to do little cellular damage and their radiotoxicity depends strongly on their distribution within the cell. Thus, in order to reduce the exposure risk of Auger electrons, the subcellular distribution should be considered. For example, the diagnostic use of these radiopharmaceuticals should localize the Auger electrons in the cytoplasm of cells, while therapeutic use in cancer should direct the radiochemical to the tumor cell nucleus.172 The radioactive emission has limited the utility of 125I as an in vivo diagnostic agent. However, it has been reported to be used for in vivo SPECT, or SPECT/CT studies mainly for tumor imaging of small animals.173–177 The long half-life of the 125I isotope has enabled ex vivo biodistribution studies to verify the in vivo data. Moreover, 125I is the radionuclide of choice for radioimmunoassays. SPECT imaging is the most common in clinical imaging because of its advantages over PET that make it widely available. Some of its advantages are that the radioisotopes are more easily obtained, less expensive and have long half-lives that allow for the observation of biological processes up to several hours after the administration of the radioisotope.164 The gamma scanning equipment is less expensive and a cyclotron is not needed for preparation of the radioisotopes on site, which add to the reduced cost.
Positron emission tomography (PET) imaging
Principle: a radioactive nuclide (PET tracers) emits a positron and neutrino upon conversion of a proton to a neutron using a cyclotron. When the positron collides with an electron (antiparticle), an annihilation process occurs; two gamma photons are generated in opposite directions. The resulting signals are recorded when a PET scanner detects these emissions concurrently. Thus, the origin of the irradiation can be identified. In this case, PET scanners detect gamma rays emitted indirectly from a positron-emitting radioligand.170,178
PET scans produce higher spatial resolution images and exhibit higher sensitivity compared to SPECT and other imaging methods such as computed tomography (CT) or standard magnetic resonance imaging (MRI). The improved resolution and sensitivity permit better detection of detailed brain areas and early dementia where there are no clinical signs or little structural changes have occurred that are hard to detect to identify pathological conditions by CT or MRI. Together with the quantitative nature of PET scans, PET imaging is a useful tool in diagnosis of brain diseases and neurodegenerative diseases, neuroimaging, and cancer biology.171,179 The main drawback of PET scanning is the short half-lives of the radionuclides. The PET radioligands decay rapidly, so they have to be synthesized prior to imaging studies. This requires the synthesis and the use of the tracers to be within the half-lives of the radiolabelled molecules. Consequently, limited time is allowed for clinical use and detection in the body is dedicated for short tasks. Other drawbacks of PET scanning to be considered are the availability of an onsite cyclotron to prepare the radioligands and the high financial cost.
The most commonly used non-metallic positron-emitting radionuclides are 11C, 13N, 15O, and 18F and less commonly 76Br, and 124I, with their half-lives being 20.4 min, 9.96 min, 2.03 min, 109.8 min, 16.1 h, and 4.18 days, respectively. Early PET tracers utilized 11C isotopes due to their synthetic feasibility. However, if a potent ligand containing a fluorine atom is available, the 18F isotope is a superior PET tracer due to the longest decay half-life that enables enough time for radiosynthesis and detection. Because of this, 18F ligands do not require a cyclotron close to the bedside and they can be synthesized offsite and shipped to imaging clinics.165 Also, the lower positron energy (0.64 MeV) of the 18F isotope compared to that of the 11C isotope (0.96 MeV) results in the production of images with higher resolution.180
Dual modalities
Hybrid biomedical imaging modalities combine CT or MRI with SPECT or PET such as SPECT/CT or PET/CT and more recently, PET/MRI scanners. They allow the correlation of the functional imaging information with the anatomical information, which resulted in tremendous advancements in the imaging field that produce more precise 3D localization of the tissues that expressed high radioactivity. These multimodality diagnostic imaging techniques have become important tools in clinical diagnosis, treatment planning, and therapy monitoring. Remarkably, PET/MRI has a great advantage of combining the high sensitivity and molecular imaging properties of PET with the ability of MRI to penetrate tissues and provide superior soft tissue contrast, and detect anatomical details with high spatial resolution and low noise.181,182 The complementary role of PET/MRI has opened new opportunities in non-invasive imaging to visualize both biochemical and anatomical changes and provide more accurate measurements of radioligand uptake.64,183 The clinical use of PET/MRI and the [18F]FTC146 PET tracer was reported for imaging peripheral nerve injury and the origin of chronic pain in humans successfully, which was not accessible using only CT or MRI.64,65
Radiotheranostics
Radiotheranostics is a term used in nuclear medicine that describes the use of radiolabelled probes that have both diagnostic imaging and targeted therapeutic components. Currently, this is a highly active area of research mainly in the field of oncology. Radiotheranostics contributes to the concept of personalized precision medicine, and represents a tool for improving patient outcomes, enhancement of therapy efficacy, and predicting adverse effects.184–186
σ1Rs are highly expressed in different types of tumors and several peer-reviewed studies show the therapeutic and diagnostic potential of σ1R ligands in cancer.154,187–189 Therefore, σ1R targeted radionuclide therapies are considered to be radiotheranostics.186 The imaging, or diagnostic component, identifies the extent of sigma receptor expression in the tumor. This information is used as a diagnostic biomarker that can determine the efficacy of the σ1R probe as a therapy and measure tumor shrinking.186 Ogawa, K. introduced some σ1R radiolabelled probes as “a companion diagnostic test of therapeutic agents,”184 and reported the use of a radiolabelled σR ligand for receptor radionuclide therapy for the first time.190 The iodinated vesamicol derivative (+)-2-[4-(4-iodophenyl)piperidino]cyclohexanol [(+)-pIV] is a σ1R ligand that showed high affinity (Ki = 1.30 nM) at σ1Rs over VAChT (Ki = 1260 nM).191 The analogous radioiodine labelled derivative (+)-[125I]pIV showed high accumulation in DU-145 tumor-bearing mice, where DU-145 is a human prostate cancer cell line overexpressing the σ1Rs. Accordingly, Ogawa, K et al. supposed that the use of the therapeutic radioiodine 131I, which emits beta particles, instead of 125I to label the sigma ligand (+)-pIV would create a radiotheranostic agent.190 (+)-[131I]pIV was prepared and showed significant tumor growth inhibition in DU-145-bearing cancer mice compared to the control group upon single administration, Fig 2.190 This finding suggested that (+)-[131I]pIV could be a potential radionuclide therapy and further studies are required to reduce the nonspecific radioactivity reported at the liver and kidneys due to its high lipophilicity.
Fig. 2. Structure of (+)-pIV, (+)-BrV–OH, and modified aza-vesamicol derivative 1.

Moreover, further studies for development of radiohalogen labelled σ1R ligands were reported. Different (+)-pIV analogs having the α-particle emitting radionuclide halogen, astatine-211 (211At), were also reported as a radionuclide therapy, which gained much consideration as a candidate for clinical use in the future. However, its properties have not yet been fully characterized.192
In addition, σ1R radiobrominated analogs of pIV were synthesised. (+)-pBrV (Kiσ1 = 2.4 nM) exhibited high tumor uptake in mice. However, the radioactivity was retained in the liver and kidneys after blocking studies which was expected due to its high lipophilicity. Modified analogs with an extra hydroxyl group were developed that exhibited lower lipophilicity. (+)-4-[1-(2-Hydroxycyclohexyl)piperidine-4-yl]-2-bromophenol, (+)-BrV–OH(Kiσ1 = 60.3 nM), was selected for distribution and blocking studies. Initially, the 77Br isotope was developed because of its long half-life of 57.0 h, and it is an Auger electron emitter and can be used for radiotherapy. It displayed lower lipophilicity than the parent compound and high tumor uptake at early time points but faster clearance even from the brain and the tumor which may be due to its lower affinity. Moreover, the PET tracer, (+)-[76Br]BrV–OH, showed high uptake in tumors via σ1Rs. This PET tracer might be a promising imaging agent, but its affinity is not sufficient and further modification is warranted to increase the affinity without increasing the lipophilicity to improve its biodistribution.193
Recently, a series of aza-vesamicol derivatives, with varying alkyl chain lengths between a piperazine ring and a benzene ring was developed to improve the radioiodine labeled probes for σ1R imaging. The binding affinity at σ1Rs increased depending on the length of the alkyl chain and the highest affinity derivative 2-(4-(3-phenylpropyl)piperazin-1-yl)cyclohexan-1-ol (Ki = 5.8 nM) is compound 1, Fig. 2. Its radioiodine labeled probe [125I] showed high accumulation in σ1R expressing DU-145 cells both in vitro and in vivo, which was confirmed by blocking studies using haloperidol. Compared to the parent compound, [125I]1 showed better biodistribution as a σ1R imaging probe at 24 h post-injection.184
Radiotheranostics could be also applied in pain management and [18F]FTC-146 could be considered as a potential agent. The diagnostic agent [18F]FTC-146 was able to accumulate at injured sciatic nerves created in a rat model and accurately detected the peripheral nerve injury and neuroinflammatory areas, which correlated to pain sensitivity, using PET/MR imaging and ex vivo autoradiography. Also, this study indicated that σ1Rs are upregulated in areas of nerve damage at the site of partial sciatic nerve ligation in the spared nerve injury (SNI) rat model.64 In a human clinical trial using [18F]FTC-146, a successful treatment course was realized after the source of chronic knee pain was localized. This led to the surgical removal of an intraarticular synovial lipoma that showed high [18F]FTC-146 uptake using PET/MRI, which resulted in complete reversal of the chronic knee pain.183 Interestingly, the analogous cold ligand, CM304, showed antiallodynic activity in mouse neuropathic pain models: chronic constriction injury assay and cisplatin-induced neuropathy assay.141 Moreover, CM304 displayed antinociceptive activity in induced chemical and inflammatory pain.141 In addition, ultrasound-guided direct injection of CM304 into the neuroma of SNI rats resulted in reduction of the mechanical allodynia in animals experiencing neuropathic pain.64 Thus, [18F]FTC-146 could be considered as a radiotheranostic agent that has the potential to precisely identify the location of σ1Rs and their expression level to diagnose peripheral nerve injury, and enable image-guided treatment and at the same time provide pain relief. However, the short plasma half-life of CM304 (t1/2 = 2.3 h) in Sprague Dawley rats has hindered its development as a therapeutic/analgesic.194
Applications of σ1R radioligands in molecular imaging
The development of σ1R radioligands has been under investigation for a number of years. These radioligands helped with understanding σ1R pathophysiology and linking the apparent pharmacological events to σ1R binding. Therefore, radioligand imaging probes are powerful tools in studying the complex role of σ1Rs under physiological and pathological conditions, quantifying the down- or upregulation, and monitoring disease progression and therapeutic outcomes. In theory, imaging studies could also allow for improved diagnosis and the development of new therapeutic approaches.
σR radioligand tools for preclinical imaging studies
In vitro radioligand binding studies are important for probing new receptors and confirming their existence in certain tissues as well as identifying high affinity and selective ligands that can be selected for further evaluation. These assays continue to play a central role in drug discovery and preclinical studies.
However, most of the earlier studies used to visualize both σRs were carried out with nonselective compounds that did not completely discriminate between both subtypes. In addition, some previous compounds that are reported to bind σRs were not highly selective over other drug targets or proteins. To add further ambiguity, as reported by Leitner ML et al., usually both sigma receptor subtypes are co-localized, but exist in different ratios.195
Some of the pharmacological tools used as σ1R agonists are PRE-084, (+)-pentazocine, DTG, and (+)-SKF-10047, which could induce some action or change in receptor function or location. Antagonists that have been studied such as BD-1047, BD-1063, and NE-100 may be more suited to understand localization of receptors. These agonists and antagonists were the most used blocking agents in radiolabelled binding studies. Some of them ([3H] NE-100, [3H] pentazocine, [3H] DTG, [3H] SKF-10047) were used as the radioligand in binding assays. Even with their shortcomings, these hallmark ligands played a critical role in assessing the involvement of σ1Rs in different pharmacological activities.
[3H](+)-Pentazocine, a benzomorphan derivative, is the prototype σ1R agonist (Kiσ1 = 3.1 nM; Kiσ2 = 1542 nM; σ1/σ2 = 500) and is used as the gold standard radioligand in binding assays. It was developed into an enantiomerically pure radioligand by De Costa et al., Fig 3.196 However, pentazocine has significant limitations: it is difficult to synthesize and degrades over time, resulting in increased background levels. Several ligands were synthesized to label σ1Rs and develop better and selective radioprobes and proposed as a replacement of [3H](+)-pentazocine but few candidates displayed real selectivity to σ1Rs over other targets and none have been widely accepted as a replacement. Two of the best candidates, [3H]-BHDP and [3H]-SN56, have not seemed to gain traction as replacements, although they are much more selective. Table 1 summarizes the radioligand affinity (Kd) and the density of available receptors (Bmax) of the proposed replacements compared to those of pentazocine.
Fig. 3. Radiolabelled σR selective ligands for preclinical studies. * indicates the radiolabelled proton site.

Equilibrium dissociation constant (Kd) and maximal density of binding site (Bmax) values of the most selective σ1R radioligands in rat brain.
[3H]-BHDP is a potent and selective σ1R ligand that displayed high affinity in rat liver mitochondria and rat brain membranes with similar Kd values (Kd = 2–3 nM), Fig. 3. It demonstrated 100 fold selectivity over σ2 and low affinity (μM range) for most of the 32 receptors examined.197 The receptor profile of [3H]-BHDP suggests that it could be a potent and selective σ1R ligand in binding experiments.
It is noteworthy that SN56 has been reported as a highly selective σ1R ligand (Kiσ1 = 0.56 nM; Kiσ2 = nM; σ1/σ2 > 1000) and demonstrated a high selectivity profile over 16 targets, Fig 3.198 Its tritiated derivative [3H]-SN56 was examined for its application as a tritium radioligand in competition binding assays. [3H]-SN56 displayed several advantages over pentazocine: high affinity (70-fold higher than that of pentazocine) and selectivity for σ1Rs with specific, saturable, and reversible binding to the σ1Rs, facile synthesis in high yields, and chemical stability. These results suggested [3H]-SN56 to be a favorable alternative for [3H](+)-pentazocine in radioligand binding assays to study σ1Rs.199
[3H]DTG, a tritiated radiolabelled analog of DTG, is a non-selective sigma receptor agonist that has high affinity for both σRs, Fig. 3. However, it is still used for in vitro binding assays to determine the binding affinities of new compounds at σ2Rs in the presence of (+)-pentazocine (to block binding to σ1R sites). This is because no selective σ2R ligand has been accepted and used in binding assays up to this time.6,201 Therefore, further investigations to develop selective σ2R probes are warranted to explore their pharmacological/physiological role in different diseases.
Some of the compounds used previously for studying σRs have affinity to other therapeutic targets such as, haloperidol. Haloperidol is a dopamine D2 antagonist and marketed as an antipsychotic drug. It has high affinity at both σRs (Kiσ1 = 3.0 nM, Kiσ2 = 54.0 nM) and demonstrates a nonselective σR antagonist activity, Fig 3.202 Haloperidol is the most frequently used σR antagonist for in vitro and in vivo biodistribution blocking studies as a blocking agent to confirm the uptake, distribution, blood brain barrier penetration, specific binding and selective labelling of σRs by the tested radioligands.
σ1R radioligands investigated in human clinical trials
Selective σ1R radiolabelled compounds have been developed for studying in vitro and in vivo biological activities to elucidate their role in different diseases. Accordingly, the development of imaging probes for σ1Rs in the human body, especially the brain, has become of great interest to many research groups. Despite many PET and SPECT radioligands being developed, few compounds have been evaluated in humans to visualize σ1Rs and investigate their density in human brain: [11C]SA4503, [18F]FPS, [11C]nemonapride, [123I]TPCNE, (S)-[18F]fluspidine, and [18F]FTC-146, Fig. 4. Interestingly, the [18F]haloperidol PET tracer has been used to study brain uptake and distribution in healthy volunteers and schizophrenic patients, but it could not be used for selective labelling of σRs due to its high affinity to the D2 receptor and low σR selectivity profile.203
Fig. 4. Radiolabelled σ1R ligands tested in humans.

[11C]SA4503
In 2000, the first selective σ1R PET radioligand, [11C]SA4503, was developed by Kawamura et al.204 and evaluated in human brain in 2001.205 SA4503 showed high σ1R affinity (Kiσ1 = 4.4 nM, Kiσ2 = 242), moderate affinity for the vesicular acetylcholine transporters (VAChT, Ki = 50.2 nM) and emopamil binding protein (EBP), and low affinity over other 29 targets.206–208 However, [11C]SA4503 did not bind to VAChT in rat brain.207 Preclinical evaluation of [11C]SA4503 using PET studies suggested that it is a potential radioligand for mapping σ1Rs in human brain.209–211 PET imaging studies in Alzheimer's and Parkinson's patients showed successful visualization of σ1Rs where reduced σ1R densities in their brains were reported.86,102 Tumor uptake studies have been conducted using [11C]SA4503 that supports the role of σ1Rs in cancer.212–214
Currently, SA4503 (cutamesine), is being investigated in multiple clinical trials for the treatment of many σ1R involved diseases. However, there are some limitations of [11C]SA4503 such as the short half-life of the 11C isotope, which limits its use as a diagnostic agent, the requirement of an onsite cyclotron, and the relatively slow kinetics due to its high affinity and low rate of dissociation.215
[18F]FPS and derivatives
The second PET radioligand evaluated in healthy human volunteers for brain imaging was [18F]FPS (Kiσ1 = 4.3 nM). However, it displayed high affinity to σ1Rs (Kd = 0.5 nM), which resulted in slow clearance with no significant washout from the brain and did not reach transient equilibrium by 4 h after administration.216 [18F]FPS is not a suitable candidate for neuroimaging, so different analogs were developed to improve its pharmacokinetic parameters and synthesize tracers with lower affinity such as [18F]SFE, the fluoroethyl derivative of [18F]FPS. It exhibited lower affinity for σ1Rs (Kd = 5 nM) and faster clearance,217,218 but no human clinical data have been reported for this compound. Recently, the synthesis of fluorinated ligands related to [18F]FPS has been reported. The authors claimed that these compounds might have potential as σR ligands (binding data not available).219
[11C]Nemonapride
[11C]Nemonapride binds with high affinity to dopamine D2 receptors in the striatum and sigma receptors in the cerebral cortex and cerebellum where there are no D2 receptors. It was used in PET imaging studies to image σRs in the cerebellum of PD patients who are suffering from levodopa-induced dyskinesia (LID). PET studies indicated an increase in the σR cerebellar binding in dyskinetic patients with PD, which was reduced after pallidal surgery. This reduction in σR binding and the improvement from dyskinesia suggested the association of σRs in the pathogenesis of PD.220
[123I]TPCNE
[123I]TPCNE (1(trans-iodopropen-2-yl)-4-[(4-cyanophenoxy)methyl]piperidine) is a σ1R ligand (Kiσ1 = 0.67 nM, Kiσ2 = 38.8 nM) that showed a low selectivity profile over σ2Rs (σ1/σ2 = 50). It was employed in human trials utilizing SPECT imaging and demonstrated high brain uptake. A blocking study using haloperidol suggested that binding was specific to σ1Rs. However, binding in the posterior cingulate area was not affected by haloperidol pretreatment, which could not be accounted for.
The high affinity resulted in an irreversible binding profile and the radioligand did not clear over 30 h. Thus, no further studies have been reported for [123I]TPCNE.221
[18F]Fluspidine
[18F]Fluspidine is a spirocyclic piperidine derivative that exhibited high affinity and selectivity toward σ1Rs, and high metabolic stability in vitro and in vivo.222 It has two enantiomers that show different affinities toward σ1Rs, the R isomer (Kiσ1 = 0.57 nM, σ2/σ1 = 1330) and the S isomer (Kiσ1 = 2.3 nM).222,223 Both isomers have been used to image σ1Rs in mice and piglets to investigate their respective in vivo kinetics and suitability for σ1R imaging in humans.224,225 Both enantiomers were also investigated in several tumor cell lines, and PET/CT imaging of brain tumors in mice was conducted. High tumor uptake supports the use of both tracers as potential PET imaging agents for brain tumor.188 Also, (S)-(−)-[18F]fluspidine exhibited fast and reversible kinetics in the brain and was selected for a first-in-human PET/CT study to investigate σ1Rs in the brain (German Clinical Trials Register ID: DRKS00008321).226 The results indicate that (S)-(−)-[18F]fluspidine is a potential PET imaging agent for clinical investigation of σ1Rs. Hence, the utility of (S)-(−)-[18F]fluspidine for quantifying pathological changes (via determining σ1R expression) in major depressive disorder was evaluated.226 Recently, metabolic stability studies have been conducted in vitro and in humans for (S)-(−)-[18F]fluspidine. Human plasma metabolic stability studies for (S)-(−)-[18F]fluspidine showed that 91% of the drug remained unchanged at 30 min post injection. This information indicates that (S)-(−)-[18F]fluspidine is a suitable candidate for PET imaging of σ1Rs.227 However, no more information is currently available about its imaging performance in humans.
[18F]FTC-146
[18F]FTC-146 is a selective σ1R antagonist (>1000 fold over σ2Rs) that showed a picomolar affinity at σ1Rs (Kiσ1 = 0.00025 nM) which might be responsible for its slow pharmacokinetics in humans. Preclinical studies showed high brain uptake and favourable pharmacokinetics in rodents (mouse, rat) and non-human primates (monkey).168,228 It has been used as a PET/MRI diagnostic agent currently in phase 1 clinical trials for identifying the source of pain generation in complex regional pain syndrome (CRPS), sciatica patients, and chronic neuropathic and/or nociceptive pain.65
Successful σ1R radioligands in animal studies
In the past two decades, different classes of compounds have been evaluated for imaging both σRs by PET and SPECT. Previous comprehensive overviews discuss the development of PET and/or SPECT radioligands for both σRs.77,170,229–232 Therefore, we will introduce the recent successful radioligands used for imaging σ1Rs in animals.
To date, there is no potential 99mTc-labeled σ1R SPECT imaging agent that has advanced to human clinical trials. Previous agents developed for preclinical tumor imaging either did not report in vitro affinity or have micromolar affinities. In addition, there are several challenges that hinder the development of 99mTc-based CNS receptor imaging agents. First, there is a need for a chelating agent to form a complex with the transition metal (99mTc). Then, there must be integration of the metal complex into the σ1R ligand. This change to the parent molecule might affect the size and configuration of the final tracer, ultimately affecting brain uptake and target engagement. In 2014, Wang, X. et al., reported a series of cyclopentadienyl tricarbonyl 99mTc complexes as potent σ1R SPECT radioligands.233 This study used 99mTc-labeled σ1R-targeting radioligands which contained a [(Cp-R)99mTc(CO)3] core that allowed for integration of the σ1R ligand to the metal complex via linkers. Initially, rhenium (Re) analogs were synthesized to determine if these complexes could retain the binding affinity at σ1Rs, and then 99mTc labelled radioligands were synthesized. The [99mTc]5 radioligand, Fig. 5, has the advantages of a σ1R nanomolar affinity (Kiσ1 = 2.11, σ2/σ1 = 14.5), high initial brain uptake (2 min post-injection), and specific binding to σ1Rs in normal brain confirmed by the reduction in radioligand uptake upon pre-treatment with haloperidol.
Fig. 5. Structure of the σ1R SPECT imaging agent [99mTc]5.
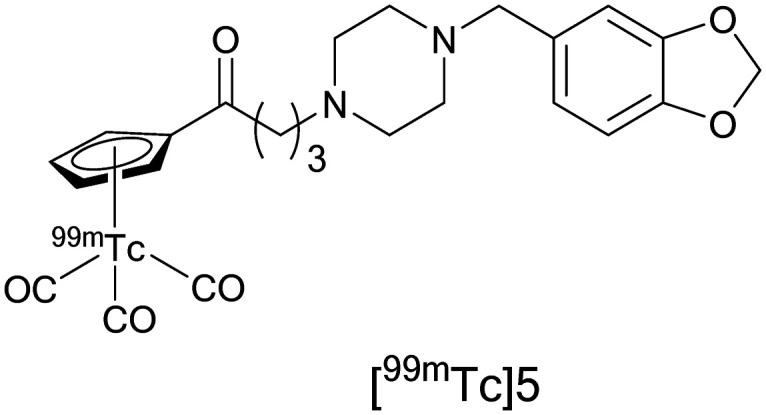
Compound [99mTc]5 demonstrated high metabolic stability in mouse brain, where 94% of radioactive species present in the mouse brain corresponded to the parent compound at 15 min post-injection and radiometabolites detected in the plasma did not enter the brain. The radioligand uptake in the C6 glioma and DU145 cell lines was significantly reduced in a time and dose dependent manner when haloperidol, DTG, and SA4503 were utilized as pre-blocking agents. Further evaluation of [99mTc]5 as a potential in vivo SPECT radioligand for imaging σ1Rs in solid tumors was conducted in C6 glioma-bearing mice, and high specific binding of [99mTc]5 to σ1Rs was observed in the tumor. These results represent a nice advancement in the development of 99mTc-labelled radioligands and further investigations are warranted.233
Efforts are ongoing in the search for an optimal 18F-labeled benzylpiperazine derivative for PET imaging. Among them are a new series of benzylpiperazine derivatives, which were reported as selective σ1R ligands with high affinity (Kiσ1 = 0.31–4.19 nM) and high subtype selectivity (Kiσ2/σ1 = 50–2448).234 Three of the fluoroethoxy analogs also exhibited high selectivity toward the vesicular acetylcholine transporter, VAChT (Ki = 99–18 252), and were chosen for radiolabeling. Radioligands [18F]2, [18F]3, and [18F]4 displayed high initial brain uptake in mice (8.37–11.48% ID per g at 2 min), Fig. 6. In addition to the high selectivity for σ1Rs, these ligands are not substrates for permeability-glycoprotein (P-gp) and had limited defluorination in vivo. [18F]2 and [18F]3 display fast kinetics in the mouse brain and low brain-to-blood ratios, while [18F]4 displayed high brain-to-blood ratios and high in vivo metabolic stability. However, [18F]4 displayed slow kinetics in the mouse brain that limited its application for human neuroimaging. [18F]4 can serve as a lead compound for further structural modifications to explore new potential radioligands for σ1Rs with suitable kinetics for imaging σ1Rs in the brain.
Fig. 6. Structure of 18F-labeled benzylpiperazine derivatives [18F]2, [18F]3, and [18F]4.

Generally, studies that monitor long-term brain recovery post-stroke are limited; however, it is important to study the changes that occur in the brain after stroke to allow for better treatment. Hence, Henderson et al.235 used a multi-modal imaging approach in rats to image the biological recovery process after stroke, neuroinflammation, and neurodegeneration. This approach combines MRI, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and PET imaging. MRI was used for visualizing the infarct 48 h after stroke, while PET and MALDI-MS were used for depiction of biological mechanisms occurring in the long-term recovery (3 months post-stroke).
MALDI-MS imaging has the advantage of providing high spatial resolution imaging for more than one compound, either exogenous or endogenous, in the same experiment. Accordingly, the translocator protein 18 kDa (TSPO) tracer [18F]DPA-714 was used as a biomarker of brain injury and inflammation, whereas σ1R radioligand N-(2-benzofuranylmethyl)-N′-[4-(2-fluoroethoxy) benzyl]piperazine, [18F]IAM6067 (Kiσ1 = 2.6 nM, σ2/σ1 = 187),236 was used as a biomarker for neurodegeneration, Fig. 7. Since stroke can cause a disruption of calcium signaling that may lead to neuronal cell death,237 σ1Rs can serve as a potential neuronal biomarker. [18F]IAM6067 PET scans showed no decrease of the σ1R tracer in the infarct area compared with the rest of the brain, and [18F]DPA-714 PET scans showed no inflammation or TSPO over-expression. These results suggested that the brain has stabilized post-stroke and remodeling of the brain structure had occurred. However, ex vivo MALDI-MS imaging was carried out to investigate lipid biomarker changes that cannot be detected by PET scans in stroke recovery. MALDI-MS imaging showed differences in the lipid profile (e.g. phosphatidylcholine and sphingomyelin) between the scar region and the rest of the brain. This finding indicates that lipid metabolism remains altered in the brain 3 months after the ischemic attack, suggesting that recovery processes are still in play. Clearly, further investigations into the exact role of the lipid biomarkers are needed.235
Fig. 7. Selected successful σ1R radioligands tested in animal studies.

Most PET radioligands used for σ1R imaging are labeled either with carbon-11 or fluorine-18, which have short half-lives (20.4 min and 109.8 min, respectively). Gangangari et al. claimed that tumor imaging requires a radioligand with a long half-life.238 The long half-life radioligand will compensate for the slow possible binding kinetics of σ1R ligands. It will also help to achieve equilibrium, and allow for monitoring the kinetics and receptor occupancy not only for hours, but also for days post administration. This, in turn, will permit the visualization of drug induced apoptosis in cancer cells and assess the efficacy of σ1R antagonists in animal models and clinical trials. The iodine radioisotopes, iodine-123 (t1/2 = 13.22 h), iodine-124 (t1/2 = 4.2 days), and iodine-125 (t1/2 = 59.49 days), offer this advantage of long half-lives. These radioiodinated ligands will provide the longer time necessary for assessment of drug efficacy in cancer treatment and have a potential application in tumor imaging.
For example, σ1R antagonists bearing a benzamide scaffold labelled with [123I] and [125I] were reported for SPECT imaging of σ1Rs in melanoma.239,240 These compounds were able to image the tumor and the results were confirmed by the ex vivo biodistribution studies. Unfortunately, the non-selectivity of the benzamide derivatives makes it difficult to assess σ1R expression due to simultaneous binding to melanin receptors. Another example is the iodine radiolabeled analog, [124I]IPAG, Fig. 7. IPAG [1-(4-iodophenyl)-3-(2-adamantyl)guanidine] is a high affinity selective σ1R antagonist (Ki = 2.8 nM) with a slow clearance from background tissues (around 24 h).241 [124I]IPAG was used to image the upregulation of σ1Rs in vitro in the MCF-7 breast cancer cell line and in vivo in two separate PET imaging studies of MCF-7 tumor-bearing mice and mice bearing LNCaP (prostate) tumors, where the tumors were established in athymic nude mice by injecting 107 cells subcutaneously. PET images at 24 h post administration in MCF7 tumor-bearing mice showed that [124I]IPAG accumulated in the tumor and was clearly visualized. Interestingly, the tumor could not be delineated after 4 h because of the slow clearance from tissues. The high background activity associated with non-target binding clears over time and allows for better detection at 24 h.238 The exponential one phase decay curve of [124I]IPAG showed preferential clearance of the radioligand from the blood, liver, spleen, and muscles while being retained in the tumor for 72 h. The long half-life of the 124I isotope allows PET imaging for an extended time post [124I]IPAG administration (144 h, ∼0.17% ID per g) that facilitates preferential retention of the radioligand activity only in σ1R expressing tissues (tumor, liver, and salivary glands) that retain the radioactivity and improve tumor delineation.238 PET studies using LNCaP tumor bearing mice revealed comparable results with studies done using MCF7 tumor-bearing mice. The biodistribution of [124I]IPAG in mice bearing MCF-7 tumors at 4 h post injection was 0.28 ± 0.01% ID per g in the brain.238 Previously, in a different study, the distribution of [125I]IPAG in mouse brain was reported (specific binding in the cerebellum, 0.64% injected dose; striatum, 0.58%; thalamus, 0.54%; cortex, 0.53%; and hippocampus, 0.46%).242 About 0.6% of the injected dose per g of [125I]IPAG penetrated the blood brain barrier in the mouse brain. Thereby, [124I]IPAG could be potentially used as an imaging agent for brain tumors as hypothesized by the authors, which will require further investigations. The availability of high affinity σ1R PET or SPECT radioligands that display long half-lives could serve as useful tools to image the upregulation of σ1Rs in cancer. This in turn, helps with monitoring the efficacy of cancer therapy in a noninvasive manner.
Compound [18F]6 is a promising PET tracer as a biomarker for early diagnosis in AD animal models, Fig. 7. [18F]6, 1-(4-fluorobenzyl)-4-[(tetrahydrofuran-2-yl)methyl]piperazine, has nanomolar affinity at σ1Rs and moderate selectivity for σ2Rs (Kiσ1 = 3.2, Kiσ2 = 168, σ2/σ1 = 52) with more than 2000-fold selectivity over VAChT and negligible affinity at 10 other CNS targets. PET/MRI studies demonstrated high specific binding at σ1Rs in rat brain and high brain uptake with high brain-to-blood ratios. In addition, [18F]6 was highly stable in vivo as it represented 95% of the total radioactivity in the mouse brain at 60 min post-injection. No signs of peripheral radiometabolites were observed as being able to enter the brain. The compound also has low lipophilicity (log D = 0.76) with suitable kinetics, as the maximum concentration in the brain was reached within 2 min, and was then washed out steadily with time. Moreover, [18F]6 was used to investigate the changes in σ1R expression in the pre-AD stage using SAMP8 mice, a model of AD that displays age-related cognitive decline close to that in humans. In ex vivo autoradiography, a significant reduction in [18F]6 uptake was found in the cortex, striatum, hippocampus, and cerebellum of SAMP8 compared to control mice, indicating the ability of [18F]6 to predict changes in σ1Rs in AD animal models before the emergence of Aβ deposition. Thus, σ1Rs might be a useful biomarker for early diagnosis of AD in preclinical stages and more investigations are warranted for greater understanding of the role of σ1Rs in AD progression.167
PB212 is a selective σ1R antagonist that exhibits high affinity (Ki = 0.030 nM) and 596 fold selectivity over σ2Rs (Ki = 17.9 nM), Fig 7.243In vitro autoradiography experiments using [11C]PB212 revealed high binding in the brain of both wild type and σ1R knockout mice. This was indicative of nonspecific binding and unsuitability for brain imaging of sigma receptors. Meanwhile, high and specific binding of [11C]PB212 was observed in the spleen tissues of both CD1 mice and Wistar rats. This result was confirmed by in vivo PET imaging studies in Wistar rats and blocking studies using haloperidol and fluspidine. Therefore, [11C]PB212 could be used to image σ1R expression in the periphery and further studies are required.244
Recently, σ1Rs were reported to be involved in maintaining normal cardiac contractility. σ1R knockout mice showed cardiac contractile dysfunction, while σ1R inhibition (in wildtype mice) resulted in atrial fibrosis and atrial electrical remodelling.158,245 Moreover, cutamesine (SA4503) is a σ1R agonist in phase II clinical trials for ischemic stroke.246 However, the physiological role of cardiac σ1Rs remains unknown and further investigations (clinical research is encouraged) targeting cardiac diseases should be done to evaluate its therapeutic potential in cardiovascular diseases. Utilization of [11C]donepezil for PET imaging opens the gates for revisiting or reassessing an overlooked σ1R ligand as a target for cardiac pharmacological intervention, Fig. 7. Donepezil is a reversible acetylcholinesterase (AChE) inhibitor, and also has high affinity at σ1Rs and is used for the treatment of Alzheimer's disease patients. Donepezil displayed a cardioprotective effect in Alzheimer's disease patients who seemed to have a low cardiovascular mortality risk. However, the mechanism of cardioprotection is unknown and could be caused by AChE inhibition, σ1R binding, or both. However, Horsager et al. speculated that the increased binding and uptake of [11C]donepezil in human heart with age and its cardioprotective effect may be primarily due to the upregulation of σ1Rs.247 Future studies to compare the cardiac uptake of the highly selective σ1R PET ligand, [18F]FTC-146, and blocking studies using the σ1R selective ligand SA4503 and donepezil in different age groups are suggested to confirm the results on elucidating which of the target proteins is upregulated.
A series of radiolabelled spirocyclic piperidine derivatives were reported and some of them are promising PET radioligands for σ1R imaging. Among them are fluspidine and its derivative [18F]7, where its cold ligand demonstrates high σ1R affinity (Kiσ1 = 2.3, σ2/σ1 = 142), Fig. 7. PET imaging evaluation of [18F]7 in rhesus monkeys showed high brain uptake, high specific binding, fast reversible kinetics, and 3 times higher binding potential than that of (S)-fluspidine. Hence, these results suggested the viability of [18F]7 as a σ1R PET radioligand in human brain.248 In contrast, the [18F]8 unlabelled ligand displayed subnanomolar affinity (Kiσ1 = 0.79, σ2/σ1 = 351). Consequently, [18F]8 exhibited slow, irreversible kinetics in monkey brain with no significant washout during a 4 h scan session. This finding made [18F]8 unsuitable for human neuroimaging.248
In order to develop σ1R tumor radioligands and ensure high tumor uptake and low background accumulation, less lipophilic tracers are desired. So, the authors proposed the replacement of the spirocyclic piperidine moiety in [18F]8 (log D7.4 = 2.41) with a more hydrophilic group 1,4-dioxa-8-azaspiro[4.5]decane to reduce the lipophilicity. Accordingly, compound [18F]9 showed low lipophilicity (log D7.4 = 0.81) and was selected as a PET radioligand from the series to image tumors in vitro and in vivo, Fig. 7. [18F]9 displayed high in vitro stability in human plasma at room temperature for 2 h and specific binding to σ1Rs in four different cell lines which were confirmed by blocking studies using SA4503, haloperidol or fluspidine. Furthermore, high accumulation of the radioligand was observed in dynamic PET studies utilizing A431 tumor bearing NMRI nu/nu mice. This accumulation was significantly reduced upon pre-treatment with haloperidol. The results indicated the specific binding of [18F]9 to σ1Rs in the tumors in vivo and that [18F]5 is a promising tumor imaging agent.249
Comp54 is the first σ1R PET radioligand with a 6-hydroxypyridazinone core structure, Fig. 8. It was reported to be a promising σ1R antagonist with high binding affinity (Kiσ1 = 1.4 nmol L−1) and apparently good selectivity (σ2/σ1 = 1365.7).250 Further modification to incorporate the [11C] isotope without significantly changing the main scaffold, and preserving the high affinity and selectivity were described. Lan, Y et al. reported the radiosynthesis and evaluation of two novel [11C] radiolabelled PET tracers as derivatives of comp54: [11C]HCC0923 and [11C]HCC0929. Both unlabelled compounds showed decreased affinity and selectivity at σ1Rs (HCC0923, Kiσ1 = 10.3 nmol L−1, σ2/σ1 = 111.3; HCC0929, Kiσ1 = 5.6 nmol L−1, σ2/σ1 = 272.8).251
Fig. 8. Structure of comp-54 and its derivatives.

However, in PET/CT studies, both radioligands bind to σ1Rs in mouse brain. They demonstrated good selectivity and specificity toward σ1Rs in self-blockade studies using the unlabelled ligands. [11C]HCC0923 exerted high BBB penetration and fast uptake after intravenous bolus injection reaching a maximum uptake within a few minutes, and sustaining binding over the scanning time (60 min). Meanwhile, [11C]HCC0929 showed better affinity, specificity, higher BBB penetration and faster brain clearance kinetic properties.
[11C]HCC0929 was further investigated in PET/CT brain imaging with positive blocking studies to confirm the specificity to σ1R binding using SA4503 (σ1R agonist) and PD144418 (σ1R antagonist).
The radiolabelled uptake of [11C]HCC0929 was extensively decreased in mice brain, with different kinetic uptakes and washout properties. In addition, the biodistribution studies indicated that the major brain functional regions (cortex, cerebellum, brain stem, thalamus, hypothalamus, striatum, hippocampus, and amygdala) were labelled by [11C]HCC0929 and a moderate wash-out rate during the scanning period (60 min) was observed. Also, other organs such as the heart, lungs, and kidneys showed high uptake at 5 min that washed out gradually. However, the maximum uptake in the liver and spleen was behind and peaked at 30 min and 15 min, respectively, and slightly washed out. So, [11C]HCC0929 could be a promising PET imaging agent for σ1R visualization in neurological disorders especially when introducing a new scaffold that might expand the chemical diversity of σ1R PET radioligands which warrants further investigation.251
Conclusion
σ1Rs are attractive targets for the development of pharmacotherapeutic agents for different diseases due to their involvement in many physiological and pathological events. The use of imaging studies has indicated alteration of σ1R expression levels, which have a correlation with the age, disease states, and type of tissue affected. Reduced σ1R densities were noted in certain brain areas of Alzheimer's and Parkinson's disease patients, while increases in expression were seen in areas of nerve damage in chronic pain and different types of tumors. Imaging studies have significantly contributed to the successful identification and classification of σ1Rs as a unique class of chaperone proteins. They also aided in the identification of σ1R expression levels in mammalian brain, assessment of receptor engagement, and quantifying receptor occupancy. This in addition confirms the correlation of σ1R expression with disease progression. Nevertheless, imaging studies continue to give insights about the role of σ1Rs and value their importance in normal and diseased states, which opens new perspectives in pharmacological intervention for many disease state diagnoses and treatments. These studies might also uncover new roles of this receptor in other diseases. Evidently, imaging studies in mice depicted high expression of σ1Rs in salivary glands,238 where the literature revealed no previous reports on σ1R expression in the salivary glands. The only reported information is about the pharmacological effect of σ1R agonists that stimulate salivary gland secretions,252 and the physiological uptake of σ1R radioligands [11C]SA4503 in the submandibular glands.253 Now, after confirming their expression level in mice, it will be interesting to investigate the role of σ1Rs in salivary gland diseases, as they might have a role in the abnormal Ca2+ metabolism or formation of Ca2+ stones. Also, PET imaging studies in humans using [18F]FTC-146 showed significant uptake of [18F]FTC-146 in the human thyroid which confirms high expression of σ1Rs in the thyroid for the first time.65 These findings pique the curiosity of researchers to investigate why there is a high uptake of the σ1R antagonist in the thyroid. What is its role? Since it remains unclear, more studies are warranted. This knowledge could be used to develop future treatments for the salivary glands or thyroid gland. However, more studies are required to assess the role of σ1Rs in these tissues that may envision σ1Rs as a new therapeutic target for new pharmacological intervention and future drug development.
σ1R research efforts are driven by the desire to explore the vast/huge involvement of σ1Rs in many pharmacological activities and diseases with the hope of translating basic research into therapeutic drugs in the future. Therefore, molecular imaging using radiolabelled probes to determine σ1R expression could be used as a diagnostic tool that can guide surgery or treatment, monitor disease progression or exert its therapeutic effect at the targeted tissue. The developed PET and SPECT tracers have a great value especially in the study of CNS diseases due to the inaccessibility of the human brain and the fact that σ1Rs are widely distributed throughout the CNS. In this review, we introduced the recent radiolabelled σ1R targeting probes. The use of imaging studies in drug discovery is inevitable; however, a limited number of successful radioligands are available. Moreover, until now, there have been no successful σ1R tumor imaging agents in clinical trials. Thus, more investigations to synthesize ideal σ1R radioligands with appropriate kinetics and selectivity are still needed for practical clinical translation.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
This project has received funding from The United States Department of Defence DoD PR161310 Contract W81XWH-17-1-0557.
References
- Romero L. Portillo-Salido E. Front. Pharmacol. 2019;10:564. doi: 10.3389/fphar.2019.00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto R. R., in Sigma Receptors: Chemistry, Cell Biology and Clinical Implications, ed. T.-P. Su, R. R. Matsumoto and W. D. Bowen, Springer US, Boston, MA, 2007, pp. 1–23, 10.1007/978-0-387-36514-5_1 [DOI] [Google Scholar]
- Quirion R. Bowen W. D. Itzhak Y. Junien J. L. Musacchio J. M. Rothman R. B. Su T. P. Tam S. W. Taylor D. P. Trends Pharmacol. Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-A. [DOI] [PubMed] [Google Scholar]
- Hellewell S. B. Bowen W. D. Brain Res. 1990;527:244–253. doi: 10.1016/0006-8993(90)91143-5. [DOI] [PubMed] [Google Scholar]
- Hanner M. Moebius F. F. Flandorfer A. Knaus H. G. Striessnig J. Kempner E. Glossmann H. Proc. Natl. Acad. Sci. U. S. A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellewell S. B. Bruce A. Feinstein G. Orringer J. Williams W. Bowen W. D. Eur. J. Pharmacol., Mol. Pharmacol. Sect. 1994;268:9–18. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Lever J. R. Litton T. P. Fergason-Cantrell E. A. Eur. J. Pharmacol. 2015;762:118–126. doi: 10.1016/j.ejphar.2015.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe S. A. J. Culp S. G. De Souza E. B. Endocrinology. 1989;124:1160–1172. doi: 10.1210/endo-124-3-1160. [DOI] [PubMed] [Google Scholar]
- Jansen K. L. R. Faull R. L. M. Dragunow M. Leslie R. A. Brain Res. 1991;559:172–177. doi: 10.1016/0006-8993(91)90303-D. [DOI] [PubMed] [Google Scholar]
- Kekuda R. Prasad P. D. Fei Y.-J. Leibach F. H. Ganapathy V. Biochem. Biophys. Res. Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- Weissman A. D. Su T. P. Hedreen J. C. London E. D. J. Pharmacol. Exp. Ther. 1988;247:29–33. [PubMed] [Google Scholar]
- Mendelsohn L. G. Kalra V. Johnson B. G. Kerchner G. A. J. Pharmacol. Exp. Ther. 1985;233:597–602. [PubMed] [Google Scholar]
- Hayashi T. Su T.-P. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Largent B. L. Gundlach A. L. Snyder S. H. J. Pharmacol. Exp. Ther. 1986;238:739–748. [PubMed] [Google Scholar]
- Vaupel D. B. Eur. J. Pharmacol. 1983;92:269–274. doi: 10.1016/0014-2999(83)90297-2. [DOI] [PubMed] [Google Scholar]
- Seth P. Leibach F. H. Ganapathy V. Biochem. Biophys. Res. Commun. 1997;241:535–540. doi: 10.1006/bbrc.1997.7840. [DOI] [PubMed] [Google Scholar]
- Schmidt H. R. Zheng S. Gurpinar E. Koehl A. Manglik A. Kruse A. C. Nature. 2016;532:527–530. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. Zeng C. Chu W. Pan F. Rothfuss J. M. Zhang F. Tu Z. Zhou D. Zeng D. Vangveravong S. Johnston F. Spitzer D. Chang K. C. Hotchkiss R. S. Hawkins W. G. Wheeler K. T. Mach R. H. Nat. Commun. 2011;2:380. doi: 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pati M. L. Groza D. Riganti C. Kopecka J. Niso M. Berardi F. Hager S. Heffeter P. Hirai M. Tsugawa H. Kabe Y. Suematsu M. Abate C. Pharmacol. Res. 2017;117:67–74. doi: 10.1016/j.phrs.2016.12.023. [DOI] [PubMed] [Google Scholar]
- Alon A. Schmidt H. R. Wood M. D. Sahn J. J. Martin S. F. Kruse A. C. Proc. Natl. Acad. Sci. U. S. A. 2017;114:7160–7165. doi: 10.1073/pnas.1705154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. P. Su T. C. Nakamura Y. Tsai S. Y. Trends Pharmacol. Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu U. B. Ruoho A. E. Mol. Pharmacol. 2016;89:142–153. doi: 10.1124/mol.115.101170. [DOI] [PubMed] [Google Scholar]
- Smith S. B. Adv. Exp. Med. Biol. 2017;964:1–4. doi: 10.1007/978-3-319-50174-1_1. [DOI] [PubMed] [Google Scholar]
- Hayashi T. Su T. P. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Wu Z. Bowen W. D. J. Biol. Chem. 2008;283:28198–28215. doi: 10.1074/jbc.M802099200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekuda R. Prasad P. D. Fei Y. J. Leibach F. H. Ganapathy V. Biochem. Biophys. Res. Commun. 1996;229:553–558. doi: 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- Seth P. Fei Y. J. Li H. W. Huang W. Leibach F. H. Ganapathy V. J. Neurochem. 1998;70:922–931. doi: 10.1046/j.1471-4159.1998.70030922.x. [DOI] [PubMed] [Google Scholar]
- Langa F. Codony X. Tovar V. Lavado A. Gimenez E. Cozar P. Cantero M. Dordal A. Hernández E. Perez R. Monroy X. Zamanillo D. Guitart X. Montoliu L. Eur. J. Neurosci. 2003;18:2188–2196. doi: 10.1046/j.1460-9568.2003.02950.x. [DOI] [PubMed] [Google Scholar]
- Mishra A. K. Mavlyutov T. Singh D. R. Biener G. Yang J. Oliver J. A. Ruoho A. Raicu V. Biochem. J. 2015;466:263–271. doi: 10.1042/BJ20141321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromek K. A. Suchy F. P. Meddaugh H. R. Wrobel R. L. LaPointe L. M. Chu U. B. Primm J. G. Ruoho A. E. Senes A. Fox B. G. J. Biol. Chem. 2014;289:20333–20344. doi: 10.1074/jbc.M113.537993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. P. London E. D. Jaffe J. H. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- Fontanilla D. Johannessen M. Hajipour A. R. Cozzi N. V. Jackson M. B. Ruoho A. E. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. P. Hayashi T. Vaupel D. B. Sci. Signaling. 2009;2:pe12. doi: 10.1126/scisignal.261pe12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. M. Bowen W. D. Walker F. O. Matsumoto R. R. De Costa B. Rice K. C. Pharmacol. Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- Ramachandran S. Chu U. B. Mavlyutov T. A. Pal A. Pyne S. Ruoho A. E. Eur. J. Pharmacol. 2009;609:19–26. doi: 10.1016/j.ejphar.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen W. D. Moses E. L. Tolentino P. J. Michael Walker J. Eur. J. Pharmacol. 1990;177:111–118. doi: 10.1016/0014-2999(90)90260-D. [DOI] [PubMed] [Google Scholar]
- Silva R. R. Parreiras-e-Silva L. T. Pompeu T. E. T. Duarte D. A. Fraga C. A. M. Barreiro E. J. Menegatti R. Costa-Neto C. M. Noël F. Front. Pharmacol. 2019;10:628. doi: 10.3389/fphar.2019.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arena E. Dichiara M. Floresta G. Parenti C. Marrazzo A. Pittalà V. Amata E. Prezzavento O. Future Med. Chem. 2017;10:231–256. doi: 10.4155/fmc-2017-0164. [DOI] [PubMed] [Google Scholar]
- Chien C. C. Pasternak G. W. J. Pharmacol. Exp. Ther. 1994;271:1583–1590. [PubMed] [Google Scholar]
- Chien C. C. Pasternak G. W. Eur. J. Pharmacol. 1993;250:303–308. doi: 10.1016/0014-2999(93)90650-7. [DOI] [PubMed] [Google Scholar]
- Chien C. C. Pasternak G. W. Neurosci. Lett. 1995;190:137–139. doi: 10.1016/0304-3940(95)11504-P. [DOI] [PubMed] [Google Scholar]
- Gregory N. S. Harris A. L. Robinson C. R. Dougherty P. M. Fuchs P. N. Sluka K. A. J. Pain. 2013;14:1255–1269. doi: 10.1016/j.jpain.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto R. R. McCracken K. A. Pouw B. Zhang Y. Bowen W. D. Neuropharmacology. 2002;42:1043–1055. doi: 10.1016/S0028-3908(02)00056-4. [DOI] [PubMed] [Google Scholar]
- Matsumoto R. R. McCracken K. A. Pouw B. Miller J. Bowen W. D. Williams W. De Costa B. R. Eur. J. Pharmacol. 2001;411:261–273. doi: 10.1016/S0014-2999(00)00917-1. [DOI] [PubMed] [Google Scholar]
- Intagliata S. Alsharif W. F. Mesangeau C. Fazio N. Seminerio M. Xu Y. T. Matsumoto R. R. McCurdy C. R. Eur. J. Med. Chem. 2019;165:250–257. doi: 10.1016/j.ejmech.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto R. R. Hemstreet M. K. Lai N. L. Thurkauf A. De Costa B. R. Rice K. C. Hellewell S. B. Bowen W. D. Walker J. M. Pharmacol., Biochem. Behav. 1990;36:151–155. doi: 10.1016/0091-3057(90)90141-4. [DOI] [PubMed] [Google Scholar]
- Matsumoto R. R. Bowen W. D. Tom M. A. Vo V. N. Truong D. D. De Costa B. R. Eur. J. Pharmacol. 1995;280:301–310. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- de la Puente B. Nadal X. Portillo-Salido E. Sánchez-Arroyos R. Ovalle S. Palacios G. Muro A. Romero L. Entrena J. M. Baeyens J. M. López-García J. A. Maldonado R. Zamanillo D. Vela J. M. Pain. 2009;145:294–303. doi: 10.1016/j.pain.2009.05.013. [DOI] [PubMed] [Google Scholar]
- Jose Luis D. Daniel Z. Jordi C. Jose Manuel B. Rafael M. Miquel Angel P. Jose Miguel V. Antoni T. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:172–183. doi: 10.2174/1871524910909030172. [DOI] [PubMed] [Google Scholar]
- Entrena J. M. Cobos E. J. Nieto F. R. Cendán C. M. Gris G. Del Pozo E. Zamanillo D. Baeyens J. M. Pain. 2009;143:252–261. doi: 10.1016/j.pain.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Achenbach T. V. Brunner B. Heermeier K. ChemBioChem. 2003;4:928–935. doi: 10.1002/cbic.200300708. [DOI] [PubMed] [Google Scholar]
- Mei J. Pasternak G. J. Pharmacol. Exp. Ther. 2002;300:1070–1074. doi: 10.1124/jpet.300.3.1070. [DOI] [PubMed] [Google Scholar]
- Cobos E. J. Baeyens J. M. Del Pozo E. Synapse. 2005;55:192–195. doi: 10.1002/syn.20103. [DOI] [PubMed] [Google Scholar]
- Gómez-Soler M. Fernández-Dueñas V. Portillo-Salido E. Pérez P. Zamanillo D. Vela J. M. Burgueño J. Ciruela F. J. Med. Chem. 2014;57:238–242. doi: 10.1021/jm401529t. [DOI] [PubMed] [Google Scholar]
- Hong W. C. Yano H. Hiranita T. Chin F. T. McCurdy C. R. Su T.-P. Amara S. G. Katz J. L. J. Biol. Chem. 2017;292:11250–11261. doi: 10.1074/jbc.M116.774075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos M. Burgueño J. Portillo-Salido E. Plata-Salamán C. R. Vela J. M. Adv. Exp. Med. Biol. 2017;964:85–107. doi: 10.1007/978-3-319-50174-1_8. [DOI] [PubMed] [Google Scholar]
- Villard V. Espallergues J. Keller E. Vamvakides A. Maurice T. J. Psychopharmacol. 2011;25:1101–1117. doi: 10.1177/0269881110379286. [DOI] [PubMed] [Google Scholar]
- The Anavex Life Sciences Corp., https://www.anavex.com/anavex-life-sciences-announces-fast-track-designation-granted-by-u-s-fda-for-anavex2-73-blarcamesine-clinical-development-program-for-the-treatment-of-rett-syndrome/, (accessed February 2020)
- Urfer R. Moebius Hans J. Skoloudik D. Santamarina E. Sato W. Mita S. Muir Keith W. Stroke. 2014;45:3304–3310. doi: 10.1161/STROKEAHA.114.005835. [DOI] [PubMed] [Google Scholar]
- Sahlholm K. Sijbesma J. W. A. Maas B. Kwizera C. Marcellino D. Ramakrishnan N. K. Dierckx R. A. J. O. Elsinga P. H. van Waarde A. Psychopharmacology. 2015;232:3443–3453. doi: 10.1007/s00213-015-3997-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlholm K. Århem P. Fuxe K. Marcellino D. Mol. Psychiatry. 2013;18:12–14. doi: 10.1038/mp.2012.3. [DOI] [PubMed] [Google Scholar]
- Johnston T. H. Geva M. Steiner L. Orbach A. Papapetropoulos S. Savola J.-M. Reynolds I. J. Ravenscroft P. Hill M. Fox S. H. Brotchie J. M. Laufer R. Hayden M. R. Mov. Disord. 2019;34:708–716. doi: 10.1002/mds.27565. [DOI] [PubMed] [Google Scholar]
- Gris G. Portillo-Salido E. Aubel B. Darbaky Y. Deseure K. Vela J. M. Merlos M. Zamanillo D. Sci. Rep. 2016;6:24591. doi: 10.1038/srep24591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B. Behera D. James M. L. Reyes S. T. Andrews L. Cipriano P. W. Klukinov M. Lutz A. B. Mavlyutov T. Rosenberg J. Ruoho A. E. McCurdy C. R. Gambhir S. S. Yeomans D. C. Biswal S. Chin F. T. Theranostics. 2017;7:2794–2805. doi: 10.7150/thno.19378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B. Park J. H. Hjornevik T. Cipriano P. W. Yoon D. Gulaka P. K. Holly D. Behera D. Avery B. A. Gambhir S. S. McCurdy C. R. Biswal S. Chin F. T. Mol. Imaging Biol. 2017;19:779–786. doi: 10.1007/s11307-017-1064-z. [DOI] [PubMed] [Google Scholar]
- Hjørnevik T. Cipriano P. W. Shen B. Park J. H. Gulaka P. Holley D. Gandhi H. Yoon D. Mittra E. S. Zaharchuk G. Gambhir S. S. McCurdy C. R. Chin F. T. Biswal S. J. Nucl. Med. 2017;58:2004–2009. doi: 10.2967/jnumed.117.192641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T.-P. Hayashi T. Curr. Med. Chem. 2003;10:2073–2080. doi: 10.2174/0929867033456783. [DOI] [PubMed] [Google Scholar]
- Weatherspoon J. K. Gonzalez-Alvear G. M. Frank A. R. Werling L. L. Schizophr. Res. 1996;21:51–62. doi: 10.1016/0920-9964(96)00030-8. [DOI] [PubMed] [Google Scholar]
- Horan B. Gifford A. N. Matsuno K. Mita S. Ashby Jr. C. R. Synapse. 2002;46:1–3. doi: 10.1002/syn.10107. [DOI] [PubMed] [Google Scholar]
- Bermack J. E. Debonnel G. J. Pharmacol. Sci. 2005;97:317–336. doi: 10.1254/jphs.CRJ04005X. [DOI] [PubMed] [Google Scholar]
- Yagasaki Y. Numakawa T. Kumamaru E. Hayashi T. Su T.-P. Kunugi H. J. Biol. Chem. 2006;281:12941–12949. doi: 10.1074/jbc.M508157200. [DOI] [PubMed] [Google Scholar]
- Mansur A. Rabiner E. A. Comley R. A. Lewis Y. Middleton L. T. Huiban M. Passchier J. Tsukada H. Gunn R. N. J. Nucl. Med. 2020;61:96–103. doi: 10.2967/jnumed.119.228080. [DOI] [PubMed] [Google Scholar]
- Nguyen L. Lucke-Wold B. P. Mookerjee S. Kaushal N. Matsumoto R. R. Adv. Exp. Med. Biol. 2017;964:133–152. doi: 10.1007/978-3-319-50174-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S. Y. Pokrass M. J. Klauer N. R. De Credico N. E. Su T. P. Expert Opin. Ther. Targets. 2014;18:1461–1476. doi: 10.1517/14728222.2014.972939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y. Mori F. Kon T. Tanji K. Toyoshima Y. Yoshida M. Sasaki H. Kakita A. Takahashi H. Wakabayashi K. Neuropathology. 2014;34:148–158. doi: 10.1111/neup.12080. [DOI] [PubMed] [Google Scholar]
- Weng T. Y. Tsai S. A. Su T. P. J. Biomed. Sci. 2017;24:74. doi: 10.1186/s12929-017-0380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust P. Deuther-Conrad W. Lehmkuhl K. Jia H. Wünsch B. Curr. Med. Chem. 2014;21:35–69. doi: 10.2174/09298673113209990214. [DOI] [PubMed] [Google Scholar]
- Ryskamp D. A. Korban S. Zhemkov V. Kraskovskaya N. Bezprozvanny I. Front. Neurosci. 2019;13:862. doi: 10.3389/fnins.2019.00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penke B. Fulop L. Szucs M. Frecska E. Curr. Neuropharmacol. 2018;16:97–116. doi: 10.2174/1570159X15666170529104323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Volti G. Murabito P. Neural Regener. Res. 2016;11:1392–1393. doi: 10.4103/1673-5374.191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Bhardwaj A. Cheng J. Alkayed N. J. Hurn P. D. Kirsch J. R. Anesth. Analg. 2007;104:1179–1184. doi: 10.1213/01.ane.0000260267.71185.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H. Zhang Y. Huang Y. Neurosci. Lett. 2019;691:3–10. doi: 10.1016/j.neulet.2018.07.033. [DOI] [PubMed] [Google Scholar]
- Jin J.-L. Fang M. Zhao Y.-X. Liu X.-Y. Int. J. Clin. Exp. Med. 2015;8:4808–4820. [PMC free article] [PubMed] [Google Scholar]
- Maurice T. and Goguadze N., in Sigma Receptors: Their Role in Disease and as Therapeutic Targets, ed. S. B. Smith and T.-P. Su, Springer International Publishing, Cham, 2017, pp. 213–233, 10.1007/978-3-319-50174-1_15 [DOI] [Google Scholar]
- Jansen K. L. Faull R. L. Storey P. Leslie R. A. Brain Res. 1993;623:299–302. doi: 10.1016/0006-8993(93)91441-T. [DOI] [PubMed] [Google Scholar]
- Mishina M. Ohyama M. Ishii K. Kitamura S. Kimura Y. Oda K. Kawamura K. Sasaki T. Kobayashi S. Katayama Y. Ishiwata K. Ann. Nucl. Med. 2008;22:151–156. doi: 10.1007/s12149-007-0094-z. [DOI] [PubMed] [Google Scholar]
- Heiss K. Vanella L. Murabito P. Prezzavento O. Marrazzo A. Castruccio Castracani C. Barbagallo I. Zappala A. Arena E. Astuto M. Giarratano A. Volti G. Li. Neurosci. Lett. 2016;626:142–148. doi: 10.1016/j.neulet.2016.05.025. [DOI] [PubMed] [Google Scholar]
- Marra A. Rossi D. Pignataro L. Bigogno C. Canta A. Oggioni N. Malacrida A. Corbo M. Cavaletti G. Peviani M. Curti D. Dondio G. Collina S. Future Med. Chem. 2016;8:287–295. doi: 10.4155/fmc.15.191. [DOI] [PubMed] [Google Scholar]
- Matsuno K. Matsunaga K. Senda T. Mita S. J. Pharmacol. Exp. Ther. 1993;265:851–859. [PubMed] [Google Scholar]
- Grundman M. Morgan R. Lickliter J. D. Schneider L. S. DeKosky S. Izzo N. J. Guttendorf R. Higgin M. Pribyl J. Mozzoni K. Safferstein H. Catalano S. M. Alzheimer's Dementia. 2019;5:20–26. doi: 10.1016/j.trci.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso R. and Navarro X., in Sigma Receptors: Their Role in Disease and as Therapeutic Targets, ed. S. B. Smith and T.-P. Su, Springer International Publishing, Cham, 2017, pp. 235–254, 10.1007/978-3-319-50174-1_16 [DOI] [Google Scholar]
- Mavlyutov T. A. Guo L.-W. Epstein M. L. Ruoho A. E. J. Pharmacol. Sci. 2015;127:10–16. doi: 10.1016/j.jphs.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Saif A. Al-Mohanna F. Bohlega S. Ann. Neurol. 2011;70:913–919. doi: 10.1002/ana.22534. [DOI] [PubMed] [Google Scholar]
- Prause J. Goswami A. Katona I. Roos A. Schnizler M. Bushuven E. Dreier A. Buchkremer S. Johann S. Beyer C. Deschauer M. Troost D. Weis J. Hum. Mol. Genet. 2013;22:1581–1600. doi: 10.1093/hmg/ddt008. [DOI] [PubMed] [Google Scholar]
- Mavlyutov T. A. Epstein M. L. Verbny Y. I. Huerta M. S. Zaitoun I. Ziskind-Conhaim L. Ruoho A. E. Neuroscience. 2013;240:129–134. doi: 10.1016/j.neuroscience.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu A. Gradus T. Altman T. Maimon R. Saraf Avraham N. Geva M. Hayden M. Perlson E. Cell Death Discovery. 2019;10:210. doi: 10.1038/s41419-019-1451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso R. Oliván S. Rando A. Casas C. Osta R. Navarro X. Neurotherapeutics. 2012;9:814–826. doi: 10.1007/s13311-012-0140-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono Y. Tanaka H. Takata M. Nagahara Y. Noda Y. Tsuruma K. Shimazawa M. Hozumi I. Hara H. Neurosci. Lett. 2014;559:174–178. doi: 10.1016/j.neulet.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Mavlyutov T. A., Baker E. M., Losenegger T. M., Kim J. R., Torres B., Epstein M. L. and Ruoho A. E., in Sigma Receptors: Their Role in Disease and as Therapeutic Targets, ed. S. B. Smith and T.-P. Su, Springer International Publishing, Cham, 2017, pp. 255–265, 10.1007/978-3-319-50174-1_17 [DOI] [Google Scholar]
- Mori T. Hayashi T. Su T.-P. J. Pharmacol. Exp. Ther. 2012;341:663. doi: 10.1124/jpet.111.190868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J. Wang L. Zhang T. Zhang B. Chen L. Neurobiol. Aging. 2017;59:171–183. doi: 10.1016/j.neurobiolaging.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Mishina M. Ishiwata K. Ishii K. Kitamura S. Kimura Y. Kawamura K. Oda K. Sasaki T. Sakayori O. Hamamoto M. Kobayashi S. Katayama Y. Acta Neurol. Scand. 2005;112:103–107. doi: 10.1111/j.1600-0404.2005.00432.x. [DOI] [PubMed] [Google Scholar]
- Toyohara J. Sakata M. Ishiwata K. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:190–196. doi: 10.2174/1871524910909030190. [DOI] [PubMed] [Google Scholar]
- Francardo V. Geva M. Bez F. Denis Q. Steiner L. Hayden M. R. Cenci M. A. Neurotherapeutics. 2019;16:465–479. doi: 10.1007/s13311-018-00699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francardo V. Bez F. Wieloch T. Nissbrandt H. Ruscher K. Cenci M. A. Brain. 2014;137:1998–2014. doi: 10.1093/brain/awu107. [DOI] [PubMed] [Google Scholar]
- Bano D. Zanetti F. Mende Y. Nicotera P. Cell Death Discovery. 2011;2:e228. doi: 10.1038/cddis.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J. M. Rego A. C. Eur. J. Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Hyrskyluoto A. Pulli I. Törnqvist K. Ho T. H. Korhonen L. Lindholm D. Cell Death Discovery. 2013;4:e646. doi: 10.1038/cddis.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol'shakova A. V. Kraskovskaya N. A. Gainullina A. N. Kukanova E. O. Vlasova O. L. Bezprozvanny I. B. Bull. Exp. Biol. Med. 2017;164:252–258. doi: 10.1007/s10517-017-3968-7. [DOI] [PubMed] [Google Scholar]
- Squitieri F. Di Pardo A. Favellato M. Amico E. Maglione V. Frati L. J. Cell. Mol. Med. 2015;19:2540–2548. doi: 10.1111/jcmm.12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacham T. Sharma N. Lederkremer G. Z. Front. Mol. Biosci. 2019;6:20. doi: 10.3389/fmolb.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P. Cahill M. E. Jones K. A. VanLeeuwen J.-E. Woolfrey K. M. Nat. Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T. Psychiatry Clin. Neurosci. 2015;69:179–191. doi: 10.1111/pcn.12262. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. J. Pharmacol. Sci. 2015;127:6–9. doi: 10.1016/j.jphs.2014.11.010. [DOI] [PubMed] [Google Scholar]
- Tomihisa N. Masaomi I. Kenji H. Curr. Pharm. Des. 2012;18:875–883. doi: 10.2174/138161212799436476. [DOI] [PubMed] [Google Scholar]
- Hayashi T. Tsai S.-Y. Mori T. Fujimoto M. Su T.-P. Expert Opin. Ther. Targets. 2011;15:557. doi: 10.1517/14728222.2011.560837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino V. Cottone P. Parylak S. L. Steardo L. Zorrilla E. P. Behav. Brain Res. 2009;198:472–476. doi: 10.1016/j.bbr.2008.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skuza G. Rogoz Z. Pharmacol. Rep. 2009;61:1179–1183. doi: 10.1016/S1734-1140(09)70181-1. [DOI] [PubMed] [Google Scholar]
- Lucas G. Rymar V. Sadikot A. Debonnel G. Int. J. Neuropsychopharmacol. 2008;11:485–495. doi: 10.1017/S1461145708008547. [DOI] [PubMed] [Google Scholar]
- Bermack J. Lavoie N. Dryver E. Debonnel G. Int. J. Neuropsychopharmacol. 2002;5:53–62. doi: 10.1017/S1461145701002760. [DOI] [PubMed] [Google Scholar]
- van Broekhoven F. Verkes R. J. Psychopharmacology. 2003;165:97–110. doi: 10.1007/s00213-002-1257-1. [DOI] [PubMed] [Google Scholar]
- Guitart X. Codony X. Ballarín M. Dordal A. Farré A. CNS Drug Rev. 2006;4:201–224. doi: 10.1111/j.1527-3458.1998.tb00065.x. [DOI] [Google Scholar]
- Peeters M. Romieu P. Maurice T. Su T.-P. Maloteaux J.-M. Hermans E. Eur. J. Neurosci. 2004;19:2212–2220. doi: 10.1111/j.0953-816X.2004.03297.x. [DOI] [PubMed] [Google Scholar]
- Sambo D. O. Lin M. Owens A. Lebowitz J. J. Richardson B. Jagnarine D. A. Shetty M. Rodriquez M. Alonge T. Ali M. Katz J. Yan L. Febo M. Henry L. K. Bruijnzeel A. W. Daws L. Khoshbouei H. Nat. Commun. 2017;8:2228. doi: 10.1038/s41467-017-02087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K. Fujita Y. Iyo M. Neuropsychopharmacology. 2007;32:514–521. doi: 10.1038/sj.npp.1301047. [DOI] [PubMed] [Google Scholar]
- Kenji H. CNS Neurol. Disord.: Drug Targets. 2009;8:470–474. doi: 10.2174/187152709789824633. [DOI] [PubMed] [Google Scholar]
- Albayrak Y. Hashimoto K. Psychiatry Invest. 2013;10:417–420. doi: 10.4306/pi.2013.10.4.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx C. E. Keefe R. S. E. Buchanan R. W. Hamer R. M. Kilts J. D. Bradford D. W. Strauss J. L. Naylor J. C. Payne V. M. Lieberman J. A. Savitz A. J. Leimone L. A. Dunn L. Porcu P. Morrow A. L. Shampine L. J. Neuropsychopharmacology. 2009;34:1885–1903. doi: 10.1038/npp.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachshoni T. Ebert T. Abramovitch Y. Assael-Amir M. Kotler M. Maayan R. Weizman A. Strous R. D. Schizophr. Res. 2005;79:251–256. doi: 10.1016/j.schres.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Ishikawa M. Ishiwata K. Ishii K. Kimura Y. Sakata M. Naganawa M. Oda K. Miyatake R. Fujisaki M. Shimizu E. Shirayama Y. Iyo M. Hashimoto K. Biol. Psychiatry. 2007;62:878–883. doi: 10.1016/j.biopsych.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Iyo M. Shirayama Y. Watanabe H. Fujisaki M. Miyatake R. Fukami G. Shiina A. Nakazato M. Shiraishi T. Ookami T. Hashimoto K. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2008;32:1072–1073. doi: 10.1016/j.pnpbp.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Niitsu T. Shirayama Y. Fujisaki M. Hashimoto K. Iyo M. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2010;34:1345–1346. doi: 10.1016/j.pnpbp.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Silver H. Barash I. Aharon N. Kaplan A. Poyurovsky M. Int. Clin. Psychopharmacol. 2000;15:257–261. doi: 10.1097/00004850-200015050-00002. [DOI] [PubMed] [Google Scholar]
- Helmeste D. M. Tang S. W. Bunney Jr. W. E. Potkin S. G. Jones E. G. Eur. J. Pharmacol. 1996;314:R3–R5. doi: 10.1016/S0014-2999(96)00702-9. [DOI] [PubMed] [Google Scholar]
- Weissman A. D. Casanova M. F. Kleinman J. E. London E. D. De Souza E. B. Biol. Psychiatry. 1991;29:41–54. doi: 10.1016/0006-3223(91)90209-5. [DOI] [PubMed] [Google Scholar]
- Hayashi T. Su T.-P. CNS Drugs. 2004;18:269–284. doi: 10.2165/00023210-200418050-00001. [DOI] [PubMed] [Google Scholar]
- Vidal-Torres A. de la Puente B. Rocasalbas M. Touriño C. Andreea Bura S. Fernández-Pastor B. Romero L. Codony X. Zamanillo D. Buschmann H. Merlos M. Manuel Baeyens J. Maldonado R. Vela J. M. Eur. J. Pharmacol. 2013;711:63–72. doi: 10.1016/j.ejphar.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Davis M. P. Expert Opin. Drug Discovery. 2015;10:885–900. doi: 10.1517/17460441.2015.1051961. [DOI] [PubMed] [Google Scholar]
- Romero L. Zamanillo D. Nadal X. Sánchez-Arroyos R. Rivera-Arconada I. Dordal A. Montero A. Muro A. Bura A. Segalés C. Laloya M. Hernández E. Portillo-Salido E. Escriche M. Codony X. Encina G. Burgueño J. Merlos M. Baeyens J. M. Giraldo J. López-García J. A. Maldonado R. Plata-Salamán C. R. Vela J. M. Br. J. Pharmacol. 2012;166:2289–2306. doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wünsch B. J. Med. Chem. 2012;55:8209–8210. doi: 10.1021/jm3011993. [DOI] [PubMed] [Google Scholar]
- Cirino T. J. Eans S. O. Medina J. M. Wilson L. L. Mottinelli M. Intagliata S. McCurdy C. R. McLaughlin J. P. Front. Pharmacol. 2019;10:678. doi: 10.3389/fphar.2019.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh D.-H. Kim H.-W. Yoon S.-Y. Seo H.-S. Kwon Y.-B. Kim K.-W. Han H.-J. Beitz A. J. Na H.-S. Lee J.-H. Anesthesiology. 2008;109:879–889. doi: 10.1097/ALN.0b013e3181895a83. [DOI] [PubMed] [Google Scholar]
- Quadir S. G. Cottone P. Sabino V. Front. Pharmacol. 2019;10:687. doi: 10.3389/fphar.2019.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambo D. O. Lebowitz J. J. Khoshbouei H. Pharmacol. Ther. 2018;186:152–167. doi: 10.1016/j.pharmthera.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz J. L., Hiranita T., Hong W. C., Job M. O. and McCurdy C. R., in Sigma Proteins: Evolution of the Concept of Sigma Receptors, ed. F. J. Kim and G. W. Pasternak, Springer International Publishing, Cham, 2017, pp. 177–218, 10.1007/164_2016_94 [DOI] [Google Scholar]
- Tapia M. A. Lee J. R. Bathe E. L. Rivera L. L. Mason K. L. Cessac M. E. Bodeen J. L. Miller D. K. Will M. J. Behav. Brain Res. 2019;373:112087. doi: 10.1016/j.bbr.2019.112087. [DOI] [PubMed] [Google Scholar]
- Kim F. J. and Maher C. M., in Sigma Proteins: Evolution of the Concept of Sigma Receptors, ed. F. J. Kim and G. W. Pasternak, Springer International Publishing, Cham, 2017, pp. 237–308, 10.1007/164_2017_38 [DOI] [Google Scholar]
- van Waarde A. Rybczynska A. A. Ramakrishnan N. K. Ishiwata K. Elsinga P. H. Dierckx R. A. Biochim. Biophys. Acta. 2015;1848:2703–2714. doi: 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- Crottes D. Rapetti-Mauss R. Alcaraz-Perez F. Tichet M. Gariano G. Martial S. Guizouarn H. Pellissier B. Loubat A. Popa A. Paquet A. Presta M. Tartare-Deckert S. Cayuela M. L. Martin P. Borgese F. Soriani O. Cancer Res. 2016;76:607–618. doi: 10.1158/0008-5472.CAN-15-1465. [DOI] [PubMed] [Google Scholar]
- Happy M. Dejoie J. Zajac C. K. Cortez B. Chakraborty K. Aderemi J. Sauane M. Biochem. Biophys. Res. Commun. 2015;456:683–688. doi: 10.1016/j.bbrc.2014.12.029. [DOI] [PubMed] [Google Scholar]
- Kim F. J. Schrock J. M. Spino C. M. Marino J. C. Pasternak G. W. Biochem. Biophys. Res. Commun. 2012;426:177–182. doi: 10.1016/j.bbrc.2012.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crottès D. Guizouarn H. Martin P. Borgese F. Soriani O. Front. Physiol. 2013;4:175. doi: 10.3389/fphys.2013.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilner B. J. John C. S. Bowen W. D. Cancer Res. 1995;55:408. [PubMed] [Google Scholar]
- Vilner B. J. John C. S. Bowen W. D. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- Ronsisvalle S. Arico G. Cova A. M. Blanco P. Amata E. Pappalardo M. Pasquinucci L. Spadaro A. Ronsisvalle N. Pharmazie. 2016;71:146–151. [PubMed] [Google Scholar]
- Mach R. H. Zeng C. Hawkins W. G. J. Med. Chem. 2013;56:7137–7160. doi: 10.1021/jm301545c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M. Lemaire S. Eur. J. Pharmacol. 1991;209:245–248. doi: 10.1016/0014-2999(91)90176-Q. [DOI] [PubMed] [Google Scholar]
- Abdullah Chowdhury S. Alam S. Aishwarya R. Miriyala S. Panchatcharam M. Alfrad N. B. M. Jonette M. P. Orr A. W. James J. Osinska H. Robbins J. John N. Lorenz. Bhuiyan Md S. J. Am. Heart Assoc. 2018;7:e009775. doi: 10.1161/JAHA.118.009775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai S.-Y. A. Pokrass M. J. Klauer N. R. De Credico N. E. Su T.-P. Expert Opin. Ther. Targets. 2014;18:1461–1476. doi: 10.1517/14728222.2014.972939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banister S. D. Kassiou M. Curr. Pharm. Des. 2012;18:884–901. doi: 10.2174/138161212799436539. [DOI] [PubMed] [Google Scholar]
- Megalizzi V. Le Mercier M. Decaestecker C. Med. Res. Rev. 2012;32:410–427. doi: 10.1002/med.20218. [DOI] [PubMed] [Google Scholar]
- Su T. P. J. Pharmacol. Exp. Ther. 1982;223:284–290. [PubMed] [Google Scholar]
- Brust P. van den Hoff J. Steinbach J. Neurosci. Bull. 2014;30:777–811. doi: 10.1007/s12264-014-1460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierckx R. A., Otte A., De Vries E. F., Van Waarde A., Luiten P. G. and editors, PET and SPECT of Neurobiological Systems, Springer, Berlin, Heidelberg, 2014 [Google Scholar]
- Agdeppa E. D. Spilker M. E. AAPS J. 2009;11:286–299. doi: 10.1208/s12248-009-9104-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintun M. A. Raichle M. E. Kilbourn M. R. Wooten G. F. Welch M. J. Ann. Neurol. 1984;15:217–227. doi: 10.1002/ana.410150302. [DOI] [PubMed] [Google Scholar]
- He Y. Xie F. Ye J. Deuther-Conrad W. Cui B. Wang L. Lu J. Steinbach J. Brust P. Huang Y. Lu J. Jia H. J. Med. Chem. 2017;60:4161–4172. doi: 10.1021/acs.jmedchem.6b01723. [DOI] [PubMed] [Google Scholar]
- James M. L. Shen B. Nielsen C. H. Behera D. Buckmaster C. L. Mesangeau C. Zavaleta C. Vuppala P. K. Jamalapuram S. Avery B. A. Lyons D. M. McCurdy C. R. Biswal S. Gambhir S. S. Chin F. T. J. Nucl. Med. 2014;55:147–153. doi: 10.2967/jnumed.113.120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matte G. Adam M. Lyster D. Nucl. Med. Biol. 2001;28:679–682. doi: 10.1016/S0969-8051(01)00236-0. [DOI] [PubMed] [Google Scholar]
- Weber F. Brust P. Laurini E. Pricl S. Wunsch B. Adv. Exp. Med. Biol. 2017;964:31–48. doi: 10.1007/978-3-319-50174-1_4. [DOI] [PubMed] [Google Scholar]
- Lu F. M. Yuan Z. Quant. Imag. Med. Surg. 2015;5:433–447. doi: 10.3978/j.issn.2223-4292.2015.03.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narra V. Howell R. Harapanhalli R. Sastry K. S. R. Rao D. J. Nucl. Med. 1993;33:2196–2201. [PubMed] [Google Scholar]
- Jeon J. Shim H. E. Mushtaq S. Kang J. A. Nam Y. R. Yoon S. Kim H. R. Choi D. S. Jang B. S. Park S. H. J. Radioanal. Nucl. Chem. 2016;308:23–29. doi: 10.1007/s10967-015-4346-4. [DOI] [Google Scholar]
- Patel N. Duffy B. A. Badar A. Lythgoe M. F. Årstad E. Bioconjugate Chem. 2015;26:1542–1549. doi: 10.1021/acs.bioconjchem.5b00380. [DOI] [PubMed] [Google Scholar]
- Chrastina A. Valadon P. Massey K. A. Schnitzer J. E. J. Vasc. Res. 2010;47:531–543. doi: 10.1159/000313880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrastina A. Schnitzer J. E. Int. J. Nanomed. 2010;5:653–659. doi: 10.2147/IJN.S11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenberger A. G. Majewski S. Saha M. Bradley E. Nucl. Instrum. Methods Phys. Res., Sect. A. 1997;392:299–303. doi: 10.1016/S0168-9002(97)00243-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosson B. Ford A. McGregor K. M. Meinzer M. Cheshkov S. Li X. Walker-Batson D. Briggs R. W. J. Rehabil. Res. Dev. 2009;47:vii–xxxiv. doi: 10.1682/JRRD.2010.02.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambhir S. S. Nat. Rev. Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- Banister S. Roeda D. Dollé F. Kassiou M. Curr. Radiopharm. 2010;3:68–80. doi: 10.2174/1874471011003020068. [DOI] [Google Scholar]
- Levin C. Glover G. Deller T. McDaniel D. Peterson W. Maramraju S. H. J. Nucl. Med. 2013;54:148. [Google Scholar]
- Chang C.-M. Lee B. J. Grant A. M. Groll A. N. Levin C. S. IEEE Trans. Radiat. Plasma Med. Sci. 2018;2:422–431. doi: 10.1109/TRPMS.2018.2852686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano P. W. Lee S. W. Yoon D. Shen B. Tawfik V. L. Curtin C. M. Dragoo J. L. James M. L. McCurdy C. R. Chin F. T. Biswal S. J. Pain Res. 2018;11:2353–2357. doi: 10.2147/JPR.S167839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K. Masuda R. Mishiro K. Wang M. Kozaka T. Shiba K. Kinuya S. Odani A. Bioorg. Med. Chem. 2019;27:1990–1996. doi: 10.1016/j.bmc.2019.03.054. [DOI] [PubMed] [Google Scholar]
- Jadvar H. Chen X. Cai W. Mahmood U. Radiology. 2018;286:388–400. doi: 10.1148/radiol.2017170346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K. Chem. Pharm. Bull. 2019;67:897–903. doi: 10.1248/cpb.c19-00274. [DOI] [PubMed] [Google Scholar]
- van Waarde A. Rybczynska A. A. Ramakrishnan N. Ishiwata K. Elsinga P. H. Dierckx R. A. Curr. Pharm. Des. 2010;16:3519–3537. doi: 10.2174/138161210793563365. [DOI] [PubMed] [Google Scholar]
- Kranz M. Bergmann R. Kniess T. Belter B. Neuber C. Cai Z. Deng G. Fischer S. Zhou J. Huang Y. Brust P. Deuther-Conrad W. Pietzsch J. Molecules. 2018;23:702. doi: 10.3390/molecules23030702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune S. Schepmann D. Lehmkuhl K. Frehland B. Wunsch B. Assay Drug Dev. Technol. 2012;10:365–374. doi: 10.1089/adt.2011.0376. [DOI] [PubMed] [Google Scholar]
- Ogawa K. Shiba K. Akhter N. Yoshimoto M. Washiyama K. Kinuya S. Kawai K. Mori H. Cancer Sci. 2009;100:2188–2192. doi: 10.1111/j.1349-7006.2009.01279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba K. Ogawa K. Mori H. Bioorg. Med. Chem. 2005;13:1095–1099. doi: 10.1016/j.bmc.2004.11.029. [DOI] [PubMed] [Google Scholar]
- Ogawa K. Mizuno Y. Washiyama K. Shiba K. Takahashi N. Kozaka T. Watanabe S. Shinohara A. Odani A. Nucl. Med. Biol. 2015;42:875–879. doi: 10.1016/j.nucmedbio.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Ogawa K. Masuda R. Mizuno Y. Makino A. Kozaka T. Kitamura Y. Kiyono Y. Shiba K. Odani A. Nucl. Med. Biol. 2018;61:28–35. doi: 10.1016/j.nucmedbio.2018.03.005. [DOI] [PubMed] [Google Scholar]
- Avery B. A. Vuppala P. K. Jamalapuram S. Sharma A. Mesangeau C. Chin F. T. McCurdy C. R. Drug Test. Anal. 2017;9:1236–1242. doi: 10.1002/dta.2156. [DOI] [PubMed] [Google Scholar]
- Leitner M. L. Hohmann A. G. Patrick S. L. Walker J. M. Eur. J. Pharmacol. 1994;259:65–69. doi: 10.1016/0014-2999(94)90158-9. [DOI] [PubMed] [Google Scholar]
- de Costa B. R. Bowen W. D. Hellewell S. B. Walker J. M. Thurkauf A. Jacobson A. E. Rice K. C. FEBS Lett. 1989;251:53–58. doi: 10.1016/0014-5793(89)81427-9. [DOI] [PubMed] [Google Scholar]
- Klouz A. Tillement J. P. Boussard M. F. Wierzbicki M. Berezowski V. Cecchelli R. Labidalle S. Onteniente B. Morin D. FEBS Lett. 2003;553:157–162. doi: 10.1016/S0014-5793(03)01011-1. [DOI] [PubMed] [Google Scholar]
- Yous S. Wallez V. Belloir M. Caignard D. H. McCurdy C. R. Poupaert J. H. Med. Chem. Res. 2005;14:158–168. doi: 10.1007/s00044-005-0131-1. [DOI] [Google Scholar]
- Fishback J. A. Mesangeau C. Poupaert J. H. McCurdy C. R. Matsumoto R. R. Eur. J. Pharmacol. 2011;653:1–7. doi: 10.1016/j.ejphar.2010.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen W. D. DeCosta B. R. Hellewell S. B. Walker J. M. Rice K. C. Mol. Pharmacol. 1993;1:117–126. [Google Scholar]
- Weber E. Sonders M. Quarum M. McLean S. Pou S. Keana J. F. Proc. Natl. Acad. Sci. U. S. A. 1986;83:8784–8788. doi: 10.1073/pnas.83.22.8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto R. R. Pouw B. Eur. J. Pharmacol. 2000;401:155–160. doi: 10.1016/S0014-2999(00)00430-1. [DOI] [PubMed] [Google Scholar]
- Schlyer D. J. Volkow N. D. Fowler J. S. Wolf A. P. Shiue C.-Y. Dewey S. L. Bendriem B. Logan J. Raulli R. Hitzemann R. Brodie J. Alavi A. A. MacGregor R. R. Synapse. 1992;11:10–19. doi: 10.1002/syn.890110103. [DOI] [PubMed] [Google Scholar]
- Kawamura K. Ishiwata K. Tajima H. Ishii S.-I. Matsuno K. Homma Y. Senda M. Nucl. Med. Biol. 2000;27:255–261. doi: 10.1016/S0969-8051(00)00081-0. [DOI] [PubMed] [Google Scholar]
- Ishii K. Ishiwata K. Kimura Y. Kawamura K. Oda K. Senda M. NeuroImage. 2001;13:984. doi: 10.1016/S1053-8119(01)92322-5. [DOI] [Google Scholar]
- Matsuno K. Nakazawa M. Okamoto K. Kawashima Y. Mita S. Eur. J. Pharmacol. 1996;306:271–279. doi: 10.1016/0014-2999(96)00201-4. [DOI] [PubMed] [Google Scholar]
- Ishiwata K. Kawamura K. Yajima K. QingGeLeTu Mori H. Shiba K. Nucl. Med. Biol. 2006;33:543–548. doi: 10.1016/j.nucmedbio.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Lever J. R. Gustafson J. L. Xu R. Allmon R. L. Lever S. Z. Synapse. 2006;59:350–358. doi: 10.1002/syn.20253. [DOI] [PubMed] [Google Scholar]
- Kawamura K. Ishiwata K. Shimada Y. Kimura Y. Kobayashi T. Matsuno K. Homma Y. Senda M. Ann. Nucl. Med. 2000;14:285–292. doi: 10.1007/BF02988211. [DOI] [PubMed] [Google Scholar]
- Jun T. Muneyuki S. Kiichi I. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:190–196. doi: 10.2174/1871524910909030190. [DOI] [PubMed] [Google Scholar]
- Sakata M. Kimura Y. Naganawa M. Oda K. Ishii K. Chihara K. Ishiwata K. NeuroImage. 2007;35:1–8. doi: 10.1016/j.neuroimage.2006.11.055. [DOI] [PubMed] [Google Scholar]
- Ishiwata K. Kawamura K. Kubota K. Kobayashi T. Elsinga P. H. Ono M. Maeda M. Ann. Nucl. Med. 2005;19:701. doi: 10.1007/BF02985120. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan N. K. Rybczynska A. A. Visser A. K. D. Marosi K. Nyakas C. J. Kwizera C. Sijbesma J. W. A. Elsinga P. H. Ishiwata K. Pruim J. Dierckx R. A. J. O. van Waarde A. J. Nucl. Med. 2013;54:1377–1383. doi: 10.2967/jnumed.112.115931. [DOI] [PubMed] [Google Scholar]
- Rybczynska A. A. Elsinga P. H. Sijbesma J. W. Ishiwata K. de Jong J. R. de Vries E. F. Dierckx R. A. van Waarde A. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:1167–1175. doi: 10.1007/s00259-009-1076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata M. Kimura Y. Naganawa M. Ishikawa M. Oda K. Ishii K. Hashimoto K. Chihara K. Ishiwata K. Ann. Nucl. Med. 2008;22:143–146. doi: 10.1007/s12149-007-0088-x. [DOI] [PubMed] [Google Scholar]
- Waterhouse R. N. Nobler M. Zhou Y. Chang R. Morales O. Kuwabara H. Kumar A. Van Heertum R. Wong D. Sackeim H. NeuroImage. 2004;22:T29–T30. doi: 10.1016/j.neuroimage.2003.11.016. [DOI] [Google Scholar]
- Zhao J. Chang R. Carambot P. Waterhouse R. N. J. Labelled Compd. Radiopharm. 2005;48:547–555. doi: 10.1002/jlcr.945. [DOI] [Google Scholar]
- Waterhouse R. N. Chang R. C. Zhao J. Carambot P. E. Nucl. Med. Biol. 2006;33:211–215. doi: 10.1016/j.nucmedbio.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Jwad R. S. Pang A. H. C. Hunter L. Read R. W. Aust. J. Chem. 2019;72:213. doi: 10.1071/CH18510. [DOI] [Google Scholar]
- Taro N. Tadashi A. Keiichiro Y. Takeshi N. Reizo S. Masatoshi I. Teiji T. J. Neurosurg. 2004;100:606–610. [Google Scholar]
- Stone J. Årstad E. Erlandsson K. Waterhouse R. Ell P. Pilowsky L. Synapse. 2006;60:109–117. doi: 10.1002/syn.20281. [DOI] [PubMed] [Google Scholar]
- Fischer S. Wiese C. Maestrup E. G. Hiller A. Deuther-Conrad W. Scheunemann M. Schepmann D. Steinbach J. Wunsch B. Brust P. Eur. J. Nucl. Med. Mol. Imaging. 2011;38:540–551. doi: 10.1007/s00259-010-1658-z. [DOI] [PubMed] [Google Scholar]
- Weber F. Brust P. Laurini E. Pricl S. Wünsch B. Adv. Exp. Med. Biol. 2017;964:31–48. doi: 10.1007/978-3-319-50174-1_4. [DOI] [PubMed] [Google Scholar]
- Fischer S. Wiese C. Maestrup E. G. Hiller A. Deuther-Conrad W. Scheunemann M. Schepmann D. Steinbach J. Wünsch B. Brust P. Eur. J. Nucl. Med. Mol. Imaging. 2011;38:540–551. doi: 10.1007/s00259-010-1658-z. [DOI] [PubMed] [Google Scholar]
- Brust P. Deuther-Conrad W. Becker G. Patt M. Donat C. K. Stittsworth S. Fischer S. Hiller A. Wenzel B. Dukic-Stefanovic S. Hesse S. Steinbach J. Wünsch B. Lever S. Z. Sabri O. J. Nucl. Med. 2014;55:1730–1736. doi: 10.2967/jnumed.114.137562. [DOI] [PubMed] [Google Scholar]
- Kranz M. Sattler B. Wüst N. Deuther-Conrad W. Patt M. Meyer P. M. Fischer S. Donat C. K. Wünsch B. Hesse S. Steinbach J. Brust P. Sabri O. Molecules. 2016;21:1164. doi: 10.3390/molecules21091164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig F. A. Fischer S. Houska R. Hoepping A. Deuther-Conrad W. Schepmann D. Patt M. Meyer P. M. Hesse S. Becker G. A. Zientek F. R. Steinbach J. Wunsch B. Sabri O. Brust P. Front. Pharmacol. 2019;10:534. doi: 10.3389/fphar.2019.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James M. L. Shen B. Zavaleta C. L. Nielsen C. H. Mesangeau C. Vuppala P. K. Chan C. Avery B. A. Fishback J. A. Matsumoto R. R. Gambhir S. S. McCurdy C. R. Chin F. T. J. Med. Chem. 2012;55:8272–8282. doi: 10.1021/jm300371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyohara J., Sakata M. and Ishiwata K., in PET and SPECT of Neurobiological Systems, ed. R. A. J. O. Dierckx, A. Otte, E. F. J. de Vries, A. van Waarde and P. G. M. Luiten, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 741–763, 10.1007/978-3-642-42014-6_26 [DOI] [Google Scholar]
- Thomas Lee C. Rikki N. W. Michael K. Curr. Pharm. Des. 2007;13:51–72. doi: 10.2174/138161207779313740. [DOI] [PubMed] [Google Scholar]
- Mach R. Wheeler K. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:230–245. doi: 10.2174/1871524910909030230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K. Ishiwata K. Curr. Pharm. Des. 2006;12:3857–3876. doi: 10.2174/138161206778559614. [DOI] [PubMed] [Google Scholar]
- Wang X. Li D. Deuther-Conrad W. Lu J. Xie Y. Jia B. Cui M. Steinbach J. Brust P. Liu B. Jia H. J. Med. Chem. 2014;57:7113–7125. doi: 10.1021/jm5009488. [DOI] [PubMed] [Google Scholar]
- Ye J. Wang L. Deuther-Conrad W. Chen Y. Zhang X. Zhang J. Huang Y. Brust P. Jia H. J. Labelled Compd. Radiopharm. 2019;62:425–437. doi: 10.1002/jlcr.3738. [DOI] [PubMed] [Google Scholar]
- Henderson F. Hart P. J. Pradillo J. M. Kassiou M. Christie L. Williams K. J. Boutin H. McMahon A. Rapid Commun. Mass Spectrom. 2018;32:721–729. doi: 10.1002/rcm.8090. [DOI] [PubMed] [Google Scholar]
- Moussa I. A. Banister S. D. Giboureau N. Meikle S. R. Kassiou M. Bioorg. Med. Chem. Lett. 2011;21:6820–6823. doi: 10.1016/j.bmcl.2011.09.028. [DOI] [PubMed] [Google Scholar]
- Mattson M. Nat. Rev. Mol. Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Gangangari K. K. Varadi A. Majumdar S. Larson S. M. Pasternak G. W. Pillarsetty N. K. Mol. Imaging Biol. 2020;22:358–366. doi: 10.1007/s11307-019-01369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaert H. Bossuyt A. Flamen P. Mertens J. Franken P. J. Nucl. Med. 1997;38:870–873. [PubMed] [Google Scholar]
- John C. Bowen W. Saga T. Kinuya S. Vilner B. Baumgold J. Paik C. Reba R. Neumann R. Varma V. J. Nucl. Med. 1994;34:2169–2175. [PubMed] [Google Scholar]
- Wilson A. A. Dannals R. F. Ravert H. T. Sonders M. S. Weber E. Wagner Jr. H. N. J. Med. Chem. 1991;34:1867–1870. doi: 10.1021/jm00110a017. [DOI] [PubMed] [Google Scholar]
- Kimes A. S. Wilson A. A. Scheffel U. Campbell B. G. London E. D. J. Med. Chem. 1992;35:4683–4689. doi: 10.1021/jm00103a005. [DOI] [PubMed] [Google Scholar]
- Berardi F. Ferorelli S. Abate C. Pedone M. P. Colabufo N. A. Contino M. Perrone R. J. Med. Chem. 2005;48:8237–8244. doi: 10.1021/jm050654o. [DOI] [PubMed] [Google Scholar]
- Spinelli F. Haider A. Toscano A. Pati M. L. Keller C. Berardi F. Colabufo N. A. Abate C. Ametamey S. M. Am. J. Nucl. Med. Mol. Imaging. 2018;8:32–40. [PMC free article] [PubMed] [Google Scholar]
- Ye T. Liu X. Qu C. Zhang C. Fo Y. Guo Y. Chen X. Shi S. Yang B. Life Sci. 2019;235:116837. doi: 10.1016/j.lfs.2019.116837. [DOI] [PubMed] [Google Scholar]
- Urfer R. Moebius H. J. Skoloudik D. Santamarina E. Sato W. Mita S. Muir K. W. Stroke. 2014;45:3304–3310. doi: 10.1161/STROKEAHA.114.005835. [DOI] [PubMed] [Google Scholar]
- Horsager J. Fedorova T. D. Berge N. V. D. Klinge M. W. Knudsen K. Hansen A. K. Alstrup A. K. O. Krogh K. Gormsen L. Borghammer P. J. Cardiovasc. Pharmacol. Ther. 2019;24:365–370. doi: 10.1177/1074248419838509. [DOI] [PubMed] [Google Scholar]
- Baum E. Cai Z. Bois F. Holden D. Lin S.-F. Lara-Jaime T. Kapinos M. Chen Y. Deuther-Conrad W. Fischer S. Dukic-Stefanovic S. Bunse P. Wünsch B. Brust P. Jia H. Huang Y. J. Nucl. Med. 2017;58:982–988. doi: 10.2967/jnumed.116.188052. [DOI] [PubMed] [Google Scholar]
- Xie F. Bergmann R. Kniess T. Deuther-Conrad W. Mamat C. Neuber C. Liu B. Steinbach J. Brust P. Pietzsch J. Jia H. J. Med. Chem. 2015;58:5395–5407. doi: 10.1021/acs.jmedchem.5b00593. [DOI] [PubMed] [Google Scholar]
- Cao X. Chen Y. Zhang Y. Lan Y. Zhang J. Xu X. Qiu Y. Zhao S. Liu X. Liu B.-F. Zhang G. J. Med. Chem. 2016;59:2942–2961. doi: 10.1021/acs.jmedchem.5b01416. [DOI] [PubMed] [Google Scholar]
- Lan Y. Bai P. Chen Z. Neelamegam R. Placzek M. S. Wang H. Fiedler S. A. Yang J. Yuan G. Qu X. Schmidt H. R. Song J. Normandin M. D. Ran C. Wang C. Acta Pharm. Sin. B. 2019;9:1204–1215. doi: 10.1016/j.apsb.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwald R. D. and Barfknecht C. F., US Pat., 5387614, 1993
- van Waarde A. Jager P. L. Ishiwata K. Dierckx R. A. Elsinga P. H. J. Nucl. Med. 2006;47:150–154. [PubMed] [Google Scholar]