

Abstract
Multi-factorial diseases are illnesses that exploit multiple cellular processes, or stages within one process, and thus highly targeted therapies often succumb to the disease, losing efficacy as resistance sets in. Combination therapies have become a mainstay to battle these diseases, however these regimens are plagued with caveats. An emerging avenue to treat multi-factorial diseases is polypharmacology, wherein a single drug is rationally designed to bind multiple targets, and is widely touted to be superior to combination therapy by inherently addressing the latter's shortcomings, which include poor patient compliance, narrow therapeutic windows and spiraling healthcare costs. Through its roles in intracellular trafficking, cell motility, mitosis, protein folding and as a back-up to the proteasome pathway, HDAC6 has rapidly become an exciting new target for therapeutics, particularly in the discovery of new drugs to treat Alzheimer's disease and cancer. Herein, we describe recent efforts to marry together HDAC pharmacophores, with a particular emphasis on HDAC6 selectivity, with those of other targets towards the discovery of potent therapeutics to treat these evasive diseases. Such polypharmacological agents may supercede combination therapies through inherent synergism, permitting reduced dosing, wider therapeutic windows and improved compliance.
Exploiting the tolerance to variability in the capping group of HDAC inhibitors, pharmacophore grafting has spawned polypharmacological co-inhibitors of HDAC6.
Post-translational modifications (PTMs) are covalent modifications to proteins that result in functional diversity, and play key roles in a variety of biological processes.1 Lysine acetylation has fast become one of the more prominent classes of PTMs.2 Histone acetyltransferases (HATs) and histone deacetyltransferases (HDACs) function in opposing roles for the regulation of acetylation: HATs catalyze the addition of an acetyl group to the ε-amino group of lysine residues, while HDACs catalyze their removal.3 The most notable role of HDACs is their deacetylation of lysine residues at histone tails. This affords a significant conformational change in the chromatin, owing to the introduced charge complementary between the negatively-charged DNA and the now-positively-charged histones.4 This process makes the DNA more difficult to access by the transcriptional machinery, resulting in the repression of mRNA synthesis. Due to their function in epigenetic regulation, the HDACs became common targets for diseases involving the dysregulation of gene expression, such as cancer. However, it was later discovered that these enzymes also function on proteins other than histones, thereby expanding their use in a variety of human diseases by controlling post-translational acetylation.2
HDACs are classified according to their function and DNA sequence similarity, and can be broken down into two families: the classical HDACs and the silent information regulator 2 related proteins (sirtuin), which are considered class III HDACs. The classical HDACs are further divided into class I (HDAC1–3 and 8), class IIa (HDAC4, HDAC5, HDAC7 and HDAC9), class IIb (HDAC6 and HDAC10), and class IV (HDAC11).2,4 Class I HDACs are mainly located in the nucleus, while class IIa are shuttled between the nucleus and cytoplasm, and classes IIb and IV are localized to the cytoplasm.4 All of the classical HDACs share sequence similarities and require Zn2+ for deacetylase activity.2,4 Accordingly, binding this zinc ion is exploited in HDAC inhibitor (HDACi) design (Fig. 1).5 Since they function on a wide range of proteins, including those in several pathophysiologic states, the inhibition of HDACs is not only a common practice for the treatment of malignant3,4 (both hematologic and solid tumor cancers) but is also being investigated for nonmalignant diseases including neurodegenerative diseases, such as Alzheimer's disease (AD)4,6,7 and Parkinson's,6,7 as well as inflammation and autoimmune diseases.8,9 Though four pan-HDACis have gained FDA approval, the lack of selectivities in these drugs are associated with adverse side effects including fatigue, nausea, vomiting, and cardiotoxicity.10,11 Selective inhibition of specific isozymes within the family may be a viable strategy to mitigate these unwanted effects.
Fig. 1. A. General HDACi pharmacophore; B. lead HDACis.

As illustrated in Fig. 1, the generic structure of an HDACi consists of three components: a zinc binding group (ZBG) grafted to a capping group via a linking moiety; while the ZBG engages the zinc ion in the active site of the enzyme, the capping group sits on the surface of the cavity.12,13 The hydroxamic acid functional group is the more prevalent ZBG in HDACis; to a lesser extent, carboxylic acids and ortho-amino anilides also feature as the ZBG.4
HDAC6 is a member of class IIb, and the development of drugs targeting this member specifically have increased within recent years due to its unique structure and physiological functions.11 In addition to histones, HDAC6, which is primarily localized to the cytoplasm, is the only HDAC to possess two deacetylase domains, and is responsible for the acetylation of some key non-histones, including α-tubulin, cortactin and heat-shock 90 (Hsp90) function. Thus, HDAC6 is involved in the regulation of intracellular trafficking, cell motility, mitosis, as well as the maturation and maintenance of proteins.14 Indeed, HDAC6 further distances itself from the other family members in regulating the disposal of misfolded proteins, serving as a bridge between the ubiquitin–proteasome system and the aggresome pathway, through its unique ubiquitin-binding domain.15
Recent structural data have shed light on the origins of the serendipitous selectivities in some of the earlier HDAC6 inhibitors (HDAC6is), and it is envisaged this will prove instrumental in the rational design of further HDAC6is with improved selectivities.13,16 Christianson's group deduced from their X-ray co-crystal structures that all three components – the cap, linker and ZBG – of an HDAC6i contribute to affinity and selectivity. In particular, differences in the L1 loop impact cap binding, the unique residue S531 at the entrance to the active site may be targeted by the linking group, and the wider binding pocket permits access by bulkier linkers, such as aryl instead of alkyl, whose attached ZBGs bind the Zn2+ through a monodentate binding mode (the ZBG cannot penetrate deep enough due to the bulkier linker to coordinate through the more typical bidentate binding mode). Therefore, it is proposed that selective inhibition of HDAC6 is possible by modification of a pan-HDACi through increasing steric bulk of the capping and/or linking groups.13,16 Indeed, when comparing the structures of the pan-HDACi SAHA (vorinostat, 1) and the HDAC6i ricolinostat (3), the bulkier capping group of the latter is the sole difference and, therefore, is responsible for the 4-fold increase in selectivity, while the major difference between SAHA and HPOB (2) is the conversion of the alkyl linker to a benzyl linker affording a >16-fold improvement in HDAC6 selectivity (Fig. 1).13
HDAC6 overexpression has been observed in a range of diseases, including cancer, cardiovascular disease, and neurodegenerative diseases, where the aberrant levels may be the cause or a response to the pathophysiology.6,14 Crucially, HDAC6 knockout mice grow and develop without physical or physiological impairments, indicating the selective inhibition of HDAC6 would be tolerated as a therapeutic.17 There have been clinical applications of HDACi (and clinical trials using HDAC6 selective inhibitors) in combination with other agents for the treatment of human disease. Though combination treatments of HDACi with other therapeutic agents have been successful, polypharmacological strategies may provide additional advantages.
Polypharmacology is an emerging field in drug discovery that may revolutionize the treatment of multi-factorial diseases, which are disorders that can exploit alternative parallel and/or redundant pathways to accomplish drug resistance.18,19 In a paradigm shift from conventional drug therapy in which a single drug is designed to bind a single target, polypharmacology is the development of single drugs that have been rationally designed to inhibit multiple targets. This may be achieved in several ways, for example, a single pharmacophore may be fashioned to bind multiple proteins, or two distinct pharmacophores may be ligated to afford a bivalent inhibitor (Fig. 2).18,19 One of the most exciting prospects of this strategy is the anticipated synergistic therapeutic effect, which could allow for lower doses of the therapeutic agent.18,19 The lower dosage could result in less toxic side effects as well as increase the therapeutic window of the drug as opposed to combination therapy. The proposed synergistic effect can be attributed to the simultaneous presence of both target ligands in tissues where they are required for therapeutic effect.18 Additional pharmacologic advantages include a more predictable pharmacokinetic profile and a decrease in drug–drug interactions when compared to combination treatment.18,19 Finally, polypharmacology may increase patient compliance, provide an economic advantage, and be easier to develop.18,19 Dosing schedules when using a combination therapy can often be complex, especially when these agents have significant toxicity; a polypharmacologic agent would make the treatment easier to dose and develop in clinical trials given that it is a single agents and requires less safety and efficacy testing than combination treatments.18
Fig. 2. Two types of polypharmacology.

Given these advantages, this manuscript reviews recent advances in the development of polypharmacological inhibitors that, in addition to binding a secondary target, preferentially inhibit HDAC6 around two-fold or better than the other HDAC family members. Particularly, the tolerance of varied capping groups in the HDACi pharmacophore lends itself to the linked pharmacophore hybridization strategy, and in all the reports that follow, the secondary pharmacophore serves as the understudy to the capping group of HDACi.11 In some cases, the biological activity may be ascribed to other HDAC isoforms, but potent inhibition of HDAC6 suggests some contribution to the observed biological response is likely, and capitalizing on recent structural studies may permit the discovery of more selective HDAC6 co-inhibitors. In the figures that follow, red colouration refers to the HDAC6 pharmacophore whilst blue refers to the co-pharmacophore. Lastly, all the HDAC isoform binding affinity data for the compounds described herein are conveniently located in Table 1.
HDAC IC50 data for the dual inhibitors described in this review.
| Compound | IC50 (nM) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC7 | HDAC8 | HDAC9 | HDAC10 | HDAC11 | |
| 5 | 12 780 ± 110 | 3190 ± 80 | |||||||||
| 7 (CM414) | 310 | 490 | 322 | 91 | |||||||
| 9 | 593 | 3530 | 40 | ||||||||
| 11 (WJ35435) | 16.6 | >1000 | 2.2 | 169.5 | |||||||
| 13 | 11 ± 3 | 9.6 ± 3.2 | 5.6 ± 0.0 | ||||||||
| 15 | 13 ± 1.7 | 24 ± 3.2 | |||||||||
| 17 | 2890 | 2250 | 1830 | 36 | |||||||
| 19 (AUY922) | 130 | 160 | 1150 | 284 | |||||||
| 21 a | 59% | 88% | 20% | ||||||||
| 23 | 107 | 8340 | |||||||||
| 26 (P1) | 27.26 ± 0.59 | 8.21 ± 0.36 | |||||||||
| 29 b | 222 | 49 | 2170 | 8.60% | 16.10% | 2.1 | 1.30% | 740 | 14% | 80 | 930 |
| 31 | 6 ± 2.0 | 1 ± 0.1 | |||||||||
| 34 | 774 ± 30 | 0.1 ± 0.05 | |||||||||
| 36 | 1100 ± 140 | 7472 ± 856 | 234 ± 31 | 46 ± 1.7 | 6065 ± 529 | ||||||
| 38 | 120 | 14.4 | 2470 | ||||||||
| 41 b | 8.7 ± 1.9 | ||||||||||
| 43 (RTS-V5) | 6900 ± 400 | 18 000 ± 1000 | 15 000 ± 2000 | 270 ± 10 | 530 ± 10 | ||||||
| 45 | 281 ± 70 | 19 ± 1 | |||||||||
| 46 (SKLB-23bb) | 422 ± 1 | 386 ± 7 | 439 ± 9 | >10 000 | >10 000 | 17 ± 2 | >10 000 | 3398 ± 487 | >10 000 | 1176 ± 168 | >10 000 |
| 48 | 996 | 470 | 1700 | ||||||||
| 49 | 36.3 | 4.6 | 3880 | ||||||||
| 51 | 467.82 ± 7.80 | 3215.90 ± 31.73 | 275.35 ± 14.47 | ||||||||
Percent inhibition at 1 μM.
Percent inhibition at 10 μM.
Alzheimer's disease
HDAC6 in Alzheimer's disease
Alzheimer's disease (AD), which is characterized by protein aggregate deposits of amyloid-β (Aβ) plaques and tau neurofibrillary tangles, is the most common form of dementia in the elderly population.20 The drug discovery attrition rate for AD is bleak at 99.6%,21 and, although there are drugs available that slow the progression of the disease, there remains no cure, primarily because its molecular pathogenesis is not fully understood.21 While the bulk of the drug discovery efforts into AD has revolved around reducing Aβ levels, alternative targets, including epigenetic targets,22 such as DNA methylation and histone acetylation, are being discovered, testament to its multi-factorial disease infamy.
Since acetylation of histones plays a role in rescuing learning and memory impairment, HDACs are emerging targets for combating the effects of AD,23 in particular class I HDACs and HDAC6, which have been linked to AD-associated memory impairment.24 Furthermore, HDAC6 blockade promotes the removal of Aβ and tau.25 Though HDAC6 as a therapeutic target for neurodegenerative disease was originally controversial,26 more recent studies have reported positive results from selective HDAC6 targeting.6 Tau hyperphosphorylation results in the accumulation of tau aggregates, Aβ one of the hallmarks of the development and progression of AD.27,28 HDAC6 activity, which is elevated in the cortex and hippocampus of AD patients,29 indirectly contributes to this increase in phosphorylation through the deacetylation of serine residues in KXGS motifs within the microtubule-binding domain of tau, rendering them phosphorylatable.30 Recently, the HDAC6 selective inhibitor MPT0G211 was shown to considerably mitigate tau phosphorylation and aggregation. Furthermore, Hsp90 acetylation levels were increased, decreasing Hsp90, which resulted in ubiquitination, hence clearance, of phosphorylated tau proteins.31
GSK-3β/HDAC6 dual inhibitors
GSK-3β is a popular target for AD treatment because it plays a significant role in disease onset and progression; it is a key signaling enzyme that modulates the hyperphosphorylation of tau proteins.28 Both the GSK-3β and HDAC pathways are capable of shifting microglia from the M1 proinflammatory phenotype to the M2 anti-inflammatory; the M1 is responsible for neurotoxic effects while the M2 has neuroprotective effects.32 GSK-3β is also part of the protein complex that is partially responsible for the phosphorylation of HDAC6, which enhances its activity.33
Combination therapies of GSK-3β inhibitors and HDACis have demonstrated synergism in models of AD, as well as enhanced therapeutic selectivities.34–36 Accordingly, De Simone and colleagues developed dual GSK-3β and HDACis through the conjugation of phthalimide, present in several GSK-3β inhibitors, such as 4 (3F8), to the hydroxamic acid ZBG through an alkyl-thiourea linker (Fig. 3).37 Compound 5 was the most potent dual GSK-3β/HDAC6 inhibitor of the library. This dual inhibitor was evaluated both in vitro and in vivo using a zebrafish model, and exhibited the highest affinities (IC50s) for GSK-3β (2.69 μM) and HDAC6 (3.19 ± 0.08 μM), with about four-fold selectivity over HDAC1 (12.78 ± 0.11 μM); these data are similar for SAHA under their assay conditions. When tested in cells, 5 demonstrated a balance in the inhibition of GSK-3β and HDAC6. Furthermore, there was an increase in hyperacetylation of α-tubulin at low treatment levels (<0.1 μM) without an increase in acetylation of histone 3 (H3) at the highest tested concentrations. In vitro studies also confirmed that the inhibitor increased neurogenesis and had the ability to counteract neuronal cell death caused by oxidative stress. This dual inhibitor did not cause any toxic effects up to 100 μM both in cells and in zebrafish. When treating zebrafish embryos, there were no toxic effects seen at 25 μM, though phenotypic changes were shown at 50 μM.
Fig. 3. Conversion of the GSK3β inhibitor 3F8 into a dual GSK3β/HDAC6 inhibitor.

PDE5/HDAC6 dual inhibitors
Phosphodiesterase 5 (PDE5) is upregulated in the brains of AD patients, often with lowered cGMP in the cerebrospinal fluid.38 Inhibition of PDE5 leads to activation of the CREB pathway, crucial for memory formation, and subsequent improvement of AD symptoms in animal models.39 Cuadrado-Tejedor and colleagues previously demonstrated a synergistic effect between the pan-HDACi SAHA and the PDE5 inhibitor tadalafil in terms of mitigating cognitive deficits in AD mice, and diminishing amyloid and tau pathologies, which was greater than either drug alone.39 Motivated by these findings, the authors embarked on the development of a dual PDE5/HDACi: their goal was a moderate class I HDACi (to mitigate side effects observed with pan-HDACis), but a potent HDAC6 and PDE5 inhibitor.40
Accordingly, the largely-solvent exposed (PDB: 1TBF) sulfonylpiperazine motif of the PDE5 inhibitor 6 (sildenafil) was replaced with various linkers to which were grafted hydroxamic acids, while retaining the pyrazolopyrimidinone core (Fig. 4). A library of over 100 compounds was prepared, and after various in vitro screens and ADME studies, compound 7 (CM-414) emerged as a tool to further explore the dual inhibition of PDE5 and HDACs. The cis and trans isomers of CM-414 were prepared and found to be equipotent to the diastereomeric mixture, which was thus used for further studies. CM-414 potently inhibited PDE5 with an IC50 of 60 nM, as well as several HDACs with at least a three-fold selectivity for HDAC6: IC50 = 310, 490, 322 and 91 nM (HDAC1, HDAC2, HDAC3 and HDAC6, respectively). Treatment of Tg2576 mice – a mouse model for AD research in which the mice have a mutant form of amyloid precursor protein (APP695SWE) that produces excess Aβ – with CM-414 resulted in the rescue of cognitive impairment as well as a reduction in Aβ and tau phosphorylation. It is unclear if the dual drug is more efficacious than the corresponding combination therapy as that comparison was not made.
Fig. 4. Conversion of the PDE5 inhibitor sildenafil into a dual PDE5/HDAC6 inhibitor.

Further optimization of CM-414 to enhance selectivity for HDAC6 focused on modifying the linker group;41 aryl hydroxamic acids enhanced the HDAC6/HDAC1 selectivity up to 33-fold, consistent with previous X-ray co-crystal structural data explaining the HDAC6-selectivity of the phenyl hydroxamic acid motif.6 Although CM-414, which functions as a pan-HDACi, achieved greater improvement in memory, the authors acknowledged that it is premature to surmise that pan-HDAC inhibition is more optimal than HDAC6 selective inhibition, and they are engaged in further work here.
PDE9/HDAC6 dual inhibitors
Since elevated levels of phosphodiesterase 9 (PDE9) have also been observed in AD brains,38 Oyzarzabal's research group extended their dual PDE5/HDAC strategy to a dual PDE9/HDAC inhibitor.42 Starting with known PDE9 inhibitors, including 8 (PF-04447943) and the congener PF-04449613 that carries an azetidine ring in place of the pyrrolidine, the authors grafted on various linkers with hydroxamic acid and ortho-amino anilides as ZBG through the pyrrolidine or azetidine rings (Fig. 5). Crucially, the pyrazolopyrimidinone core was retained so that PDE inhibition was sustained, but also the basic nitrogens in the pyrrolidine and azetidine rings were retained since previous work revealed these engage in a water-mediated hydrogen bond with Tyr424, which is a non-conserved residue in the PDE family and believed to be a determinant in PDE9 selectivity.43,44 In all, three families of compounds were prepared: dual PDE9/pan-HDAC, dual PDE9/class I HDAC, dual PDE9/HDAC6 inhibitors. While ortho-amino anilides were essentially inactive against HDAC6, they exhibited triple-digit nanomolar IC50s against HDAC1. In many cases, the hydroxamic acid ZBG elicited greater inhibition of HDAC6 versus HDAC1, typically by a factor of only three or four-fold, although selectivities as high as 32-fold were also reported; the benzyl hydroxamic acid delivered the best selectivity for HDAC6, in keeping with previous crystallographic studies that explained the determinants of HDAC6 selectivity.13 PDE9/HDAC dual inhibitor 9, with a phenyl hydroxamic acid, demonstrated an IC50 for HDAC6 of 40 nM, corresponding to almost 15-fold and 100-fold selectivities over HDAC1 and HDAC2, respectively, and potently inhibited PDE9 with an IC50 of 107 nM, and inactive against PDE5 (IC50 > 10 000 nM). Given these excellent data and satisfactory brain permeability, 9 was selected for further study. In fact, 9 reversed the AD phenotype when aged Tg2576 mice were treated chronically.45
Fig. 5. Conversion of the PDE9 inhibitor PF-04447943 into a dual PDE9/HDAC6 inhibitor.

Cancer
HDAC6 in cancer
HDAC6 overexpression has been reported in several different cancers, including acute myeloid leukemia (AML), ovarian cancer and hepatocellular carcinoma,46–48 and is required for oncogenic transformation, tumor development, and also plays a key role in cancer immunity.44 From a therapeutic perspective, it is particularly noteworthy that knockdown of the enzyme is not detrimental to growth, development, nor overall survival of mice models.15 Indeed, HDAC6 has risen as a prominent target for cancer treatment,11,14 and the more recent efforts to marry together the HDACi pharmacophore with other significant cancer-associated pharmacophores are described below, along with discussions on HDAC6 selectivities.
Topoisomerase poisons/HDAC dual inhibitors
The topoisomerase I and II enzymes are crucial to the regulation of DNA topology; they catalyze the reversible cleavage of single-(type I) and double-strand (type II) DNA, which relieves the torsional stress that develops during replication and transcription. Topoisomerase poisons are small-molecules that stabilize the transient covalent complexes between enzyme and DNA, thereby inhibiting DNA replication and transcription.49–52 As such, topoisomerase poisons have found utility as antineoplastic agents. Their efficacy is directly related to their accessibility to the DNA; the more the histones are acetylated, the less they interact with DNA and the more lethal the DNA damaging agents become.53
Previously, synergy was observed between HDACis and topoisomerase poisons, and this is likely due, at least in part, to prevailing acetylated histones, indicating less condensed chromatin, and greater DNA accessibility.54,55 SAHA is an FDA approved HDACi for cancer treatment; however, this drug has insufficient efficacy on solid tumors when used as a monotreatment.56–58 DACA (10) is a topoisomerase I and topoisomerase II poison that has proven susceptible to resistance, reducing its efficacy in chemotherapy.59 Inspired by the aforementioned synergy, Yu and co-workers replaced the tertiary amino group of DACA with an hydroxamic acid, maintaining the intercalating acridine core intact to arrive at hybrid compound 11 (WJ35435), as illustrated in Fig. 6.60 Synergistic efficacy was observed in both in vitro and in vivo evaluations. The hybrid molecule functioned as both an HDAC6 (IC50 = 2.2 nM) and HDAC1 (IC50 = 16.6 nM) inhibitor, as well as a topoisomerase I inhibitor, apparently losing the topoisomerase II activity of the parent DACA; however, despite proving to be a more potent HDAC6 inhibitor, the therapeutic effects were attributed to HDAC1 inhibitory activity rather than HDAC6. This is because HDAC1 inhibition is known to induce changes in DNA transcription and translation by removing acetyl lysine groups on histones, resulting in conformational changes of chromatin. WJ34534 also displayed characteristics of a more potent topoisomerase poison than DACA.60 It is also important to note that the hybrid molecule was significantly more selective against malignant prostate cancer cells over benign cells.
Fig. 6. Transformation of the topoisomerase poison DACA into a dual topoisomerase I/HDAC6 inhibitor.

WJ35435 was next subjected to anti-proliferation studies, and cell cycle growth at various stages in the human hormone-refractory metastatic prostate cancer cells PC-3 and DU-145 were examined and compared to both SAHA and DACA, although the combination of SAHA and DACA was not explored. The proliferation inhibition of both cell lines was in the sub-micromolar range (IC50 = 390 nM (PC-3) and 330 nM (DU-145)), several fold better than SAHA or DACA, good selectivity was exhibited over benign prostate cells. At low concentrations, there was an arrest at the G1 phase of the cell cycle, consistent with the effects of SAHA. At higher concentrations, cell cycle arrest was observed at the G2/M phase, in line with the effects of DACA. The anti-HDAC activity of the compound was tested by comparing the acetylation of both histone H3 and α-tubulin to controls. There was a drastic increase in acetylation of both proteins at low concentrations. Xenograft nude mice were used to determine the in vivo effects of WJ35435, which resulted in tumor antiproliferation consistent with the in vitro studies, and did not result in weight loss of the animal models.
Structural modifications of the natural product podophyllotoxin (PPT) resulted in the discovery of the FDA-approved anti-cancer drug etoposide (12), which is a potent topoisomerase II poison.61 Given reports of synergy between HDACis and etoposide,62 Zhang et al. hypothesized that more efficacious anti-cancer agents could be realized by the replacement of the glucopyranoside of etoposide with an aromatic capping group coupled to a ZBG through a linker, as exemplified by 13 in Fig. 7.63 The authors investigated the substitution position off the aromatic capping group, the linker length, and two general types of ZBG: the classical hydroxamic acid and the more novel ortho-amino anilide; in general, the latter tends to show greater affinity to HDAC1. Hydroxamate 13 showed high affinity for the HDAC family as well as topoisomerase II, making it the lead compound.
Fig. 7. A dual topoisomerase/HDAC6 inhibitor beginning with etoposide.

When tested against three different HDAC isoforms, compound 13 showed the highest affinity for HDAC6 (IC50 = 5.6 ± 0 nM) over HDAC1 (IC50 = 11 ± 3 nM) and HDAC3 (IC50 = 9.6 ± 3 nM). Topoisomerase II relaxation assays demonstrated that the addition of the ZBG did not affect topoisomerase II inhibition. Compound 13 was also the most potent compound when tested in HCT116 cells with an IC50 value of 3.33 μM, which was four-fold worse than SAHA. While the selectivity for HDAC6 was marginal – and was not the goal of the work – it may be of interest to capitalize on the findings by Christianson's group13 to enhance the HDAC6 selectivity and then investigate the biological ramifications of a dual topoisomerase II/HDAC6 inhibitor.
In later research, Sheng's group prepared the first-in-class triple topoisomerase I/II/HDAC inhibitor. Grafting a SAHA-like linker and hydroxamic acid onto the dual topoisomerase I/II inhibitor 3-amino-10-hdroxylevodiamine 14, their work culminated in 15 (Fig. 8), which potently, and with a similar two-fold selectivity to 13, inhibited HDAC6 and HDAC1 with IC50s of 13 ± 1.7 nM and 24 ± 3.2 nM, respectively.64 As well as retaining dual inhibition of topoisomerases I and II, 15 inhibited the proliferation of HCT116 colorectal cells with sub-micromolar activity (IC50 = 0.41 μM), comparable with the parent topoisomerase I/II inhibitor 14 and almost an order of magnitude better than SAHA; the combination of 14 and SAHA in a molar ratio was not explored.
Fig. 8. A first-in-class triple topoisomerase I/topoisomerase II/HDAC6 inhibitor.

AR/HDAC6 dual inhibitors
The androgen receptor (AR) antagonists enzalutamide (16) and abiraterone are approved treatments for metastatic castration-resistant prostate cancer (mCRPC), but unfortunately they produce no therapeutic effects in a large number of patients; secondary resistance to approved drugs has also been a prevalent issue, as it is with many other cancers.65 It is believed that the lack of therapeutic affect and resistance are due to splice variants of ARs.65 The AR functions as a transcriptional factor that is responsible for growth regulation and development in the prostate; therefore, the AR is a driving force behind prostate cancer.66 Hsp90 controls the activation of the AR, as well as regulating AR stability and nuclear localization.67,68 In order to perform these functions, Hsp90 must be in its active, deacetylated form, which is modulated by HDAC6.68 The relationship between the AR and HDAC6 suggests dual inhibition of these targets may be a promising avenue for the discovery of new mCRPC therapeutics.67–69
Indeed, synergistic increases in the cytotoxicity in preclinical models has been observed with HDACi and AR inhibitor combination therapy, including in models that are treatment resistant.69 And it has been proposed that the AR antagonist of AR/HDAC dual inhibitors, may also function as a homing device, delivering the HDACi to the prostate cancer.70 Several groups have converted the AR antagonist enzalutamide into a dual AR/HDAC6 inhibitor. In 2016, Rosati and colleagues inferred from their cell data that their dual AR/HDAC6 inhibitor was selective for HDAC6 over the other HDACs but no specific binding data were provided to gauge the selectivity.71
In the same year, Jadhavar et al. used the methyl amide of enzalutamide as the grafting point, as depicted in Fig. 9, attaching a wide range of ZBGs, and modifying the linker length as well.72 The authors found that hydroxamate-based dual AR/HDAC inhibitors were the most selective for HDAC6,72 and retained their antiandrogen characteristics. The authors specifically drew attention to compound 17, which had an HDAC6 IC50 of 36 nM, almost 100-fold selectivities over other HDAC isozymes, and an AR binding IC50 of <30 nM. In vitro studies showed that the compound decreased AR levels and increased levels of acetylated tubulin. Control compounds MS-275 (HDAC1 and HDAC3 inhibitor), SAHA (pan-HDACi), and tubastatin A (HDAC6 inhibitor) were probed for changes in AR levels and degrees of tubulin acetylation; only tubastatin A reduced the amount of AR and increased tubulin acetylation, supporting the premise of a link between AR and HDAC6.68 Similarly, 17 lowered AR protein levels and caused the hyperacetylation of tubulin. Finally, given the intrinsic toxicity of the hydroxamic acid functional group coupled with its poor pharmacokinetics, it is noteworthy that the authors observed >100-fold selectivity, albeit moderate affinity (IC50 = 1.12 μM), for HDAC6 with the alternative sulfamide ZBG, which is present in carbonic anhydrase inhibitors; they intend to try to further optimize this, although no further updates have hitherto been reported.
Fig. 9. Discovery of a dual AR/HDAC6 inhibitor.

Hsp90/HDAC6 dual inhibitors
Along with activation of AR, Hsp90 is an ATP-dependent molecular chaperone that is responsible for assisting its substrate proteins in maturation; hence, the inhibition of this protein results in the disruption of many signaling pathways that are exploited by cancers.73 Inhibition of HDAC6 leads to the degradation of survival proteins in human leukemia cells through the acetylation of Hsp90, thereby inhibiting its ATP binding and chaperone function.74,75
Motivated by recent publications describing synergy between Hsp90 inhibitors and HDACis,76–79 Ojha et al. designed dual Hsp90/HDACis by modifying a prior HDACi from their group. Starting with a 5-aminoindoline scaffold, they grafted on various hydroxamic acids at the 5-position to recognize HDACs, and a resorcinol derivative at the 1-position, since this motif is present in several Hsp90 inhibitors, such as 18 (AUY922).80 The most promising dual inhibitor was compound 19 (Fig. 10), with a linker length of six carbons. This small-molecule potently inhibited HDAC6 (IC50 = 1.15 nM) with 113-, 139- and 246-fold selectivities over HDAC1, HDAC3 and HDAC8, respectively (note: there is a conflict between the units in the text and the table in this publication, but the calculated selectivities provided here are consistent throughout the paper), potently inhibited Hsp90 (IC50 = 46 nM) and had the greatest anti-proliferative effects in vitro. Their compounds were evaluated in multiple cell lines, including colorectal cancer (HCT116) and AML (HL60) cells, with GI50 values in the low to sub-micromolar range, largely on a par with SAHA. Notably, those compounds that exhibited potent inhibition of Hsp90 but did not bind the HDACs also demonstrated antiproliferative properties, indicating that the anti-proliferative effect in these instances was due to inhibition of Hsp90. Compound 19 was tested for its effect on Hsp90 client proteins by examining Hsp70, Akt, STAT3, α-tubulin, and histone H3. There was a considerable increase in Hsp70 and decrease in Akt, which is indicative of Hsp90 inhibition. Results also displayed an increase in the acetylation of both H3 and α-tubulin, although the acetylation of α-tubulin occurred at a lower concentration with a more defined increase. These results were augmented when tested over time, along with a decrease in STAT3, another factor characteristic of Hsp90 inhibition.
Fig. 10. A dual Hsp90/HDAC6 inhibitor.

FGFR1/HDAC6 dual inhibitors
Fibroblast growth factors function in many physiological processes, such as embryogenesis, tissue homeostasis and repair, wound healing, and inflammation.81 Fibroblast growth factor receptors (FGFRs), which are tyrosine kinases (TKs), play crucial roles in the cell, including tissue homeostasis, inflammation and wound healing, and can be divided into four isoforms (FGFR1–4), each of which are amplified in many different types of cancer.81 Recently, the combination of HDAC6 inhibitors with tyrosine kinase inhibitors (TKIs) showed efficacy in breast cancer.82,83 Inspired by these reports, Liu and co-workers embarked on the development of dual FGFR1/HDAC6 inhibitors.84 In particular, the solvent-exposed N-ethyl-4-piperazine of the FGFR1 inhibitor 20, which was introduced to improve the compound's physicochemical properties, was replaced with an hydroxamic acid, delivering a phenyl hydroxamic acid moiety, the same motif that furnishes nexturastat A and tubastatin A with HDAC6 selectivity, as shown in Fig. 11.13
Fig. 11. Conversion of the FGFR1 inhibitor 20 into a dual FGFR1/HDAC6 inhibitor.

The library of compounds was first tested for inhibition of HDAC6, followed by anti-proliferative evaluation of breast cancer MCF-7 cells, using SAHA and nexturastat A as controls. It was determined that compound 21 was the most promising compound with the highest affinity for HDAC6 (IC50 = 34 nM) and greatest cytotoxic effects in cells (IC50 = 9 μM). Compounds were also tested for the percent inhibition of FGFR1, HDAC1, HDAC6, and HDAC8 at a concentration of 1 μM. Lead compound 21 had the highest percent inhibition of FGFR1 (64%) as well as excellent inhibition of HDAC6 (88%) with weaker inhibitions of HDAC1 and HDAC8 (59% and 20%, respectively), demonstrating selectivity for the HDAC6 isozyme. The percent inhibitions were determined by a routine HDAC assay with a fluorogenic HDAC peptidic substrate. In general, the HDAC6 inhibition data and the anti-proliferative data were in agreement with one another. Unfortunately, the potent inhibition of FGFR1 by 20 (98% at 1 μM) was lost and could not be recovered, which may be a consequence of a clash of the hydroxamic acid with the FGFR1 active site; it may be prudent to revise the design slightly and re-graft the hydroxamic acid such that it points further into the solvent, for example at the N-ethyl position of the N-ethyl-4-piperazine.
ER/HDAC6 dual inhibitors
The estrogen receptor (ER), which has two sub-types (ERα and ERβ), is a transcription factor that has a regulatory role in many physiological and pathological processes,85 and is itself regulated through ligand binding.86 ERα is found predominantly in the sex organs, whilst ERβ is found more generally about the body. Although both sub-types have highly conserved ligand binding regions (only two amino acid differences), it is thought that they use different ligands and the two subtypes have opposite functions with ERβ inhibiting ERα function. Indeed, in many breast cancers, activation of ERα by estrogens is widely viewed as the culprit for the hyperproliferation,87 and this is directly thwarted by the anti-proliferative activity of Eβ, and thus ERα antagonists and/or ERβ agonists are being considered as sources of new anti-cancer drugs. Some of the more predominant antagonists are hormone agents; the most recently approved class is the selective estrogen receptor modulators (SERMs).88,89 However, these treatments often lead to resistance or are inactive against tumors.90
Recent phase II clinical trials have shown a possible synergistic effect produced by co-treatment of tamoxifen, a SERM, with the HDACi SAHA.91 The ER signaling pathway requires the displacement of repressor proteins as well as the recruitment of activator proteins to transcription complexes with the bound ER.92 It is hypothesized that an interaction occurs between HDAC, the repressor proteins, and the ER in silenced nuclear transcription complexes.92 HDAC1, specifically, was observed to interact with the activation function 2 and DNA binding domain of ERα.93 Additionally, ERα levels were decreased upon knockdown of HDAC1 or inhibition of HDACs.94
Tang and colleagues accordingly developed a library of dual ER/HDACis.95 Their strategy began with the novel SERM oxabicycloheptene sulfonate (OBHS, 22). A range of HDAC binding moieties were attached to these inhibitors, all of which carry a SAHA-like six-carbon methylene linker with a hydroxamic acid, a methyl ester or a carboxylic acid. From these dual inhibitors, 23 (Fig. 12), with a carboxylic acid ZBG, was the most potent antagonist of with a binding affinity ERα (Ki = 25.00 nM) comparable to that of the parent compound OBHS (Ki = 21.53 nM). However, in a dramatic improvement of the two-fold selectivity demonstrated by OHBS, 23 also exhibited the highest ERα/ERβ ratio (28.18), showing that the compound is selective for ERα, an ideal characteristic for cancer treatment. Interestingly, however, when tested for antagonist activity against ERα, compound 23 had a better binding affinity and potency but a reduced efficacy as an antagonist of 25%. 23 also exhibited a nanomolar IC50 value for HDAC1 (0.107 μM) while having a higher value for HDAC6 (8.34 μM). Although the authors were not on a quest for HDAC6 selectivity, it is noteworthy that replacement of the carboxylic acid of 23 with a hydroxamic acid (24) afforded increased selectivity for HDAC6 over HDAC1 (IC50s of 189 nM and 583 nM, respectively). The development of a dual ER/HDAC6 inhibitor is of interest because the overexpression of HDAC6, which is an estrogen-regulated gene, has been observed in ER(+) MCF-7 breast cancer cells.96
Fig. 12. Dual ERα/HDAC1 (23) and ERα/HDAC6 (24) inhibitors.
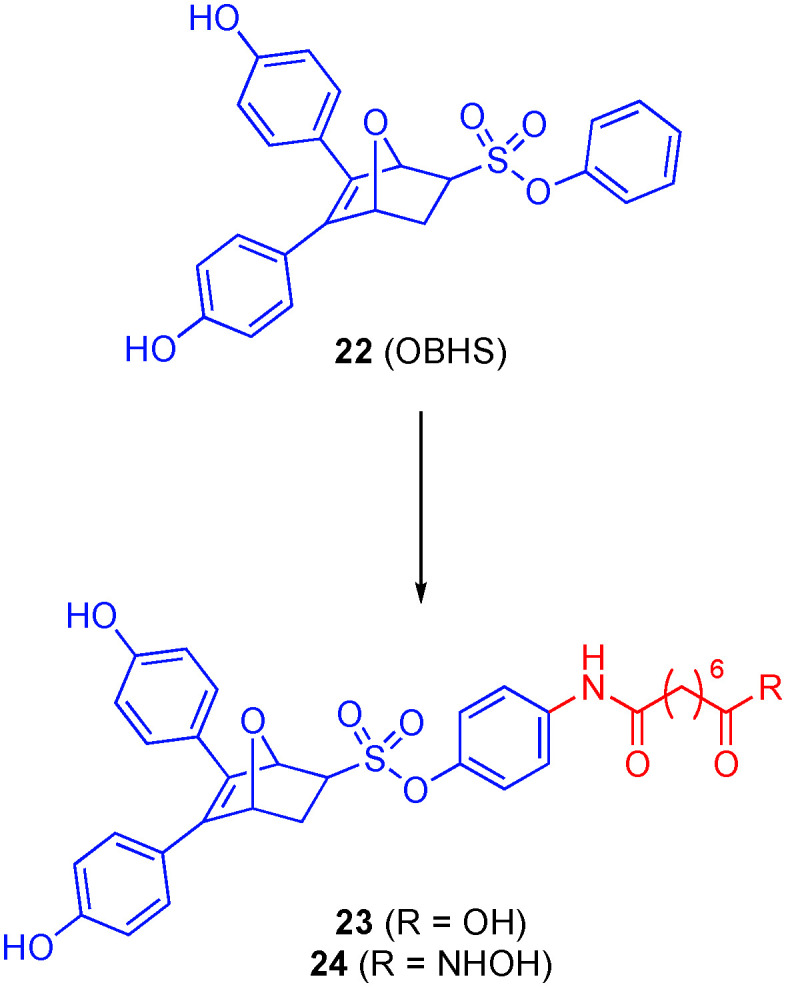
Although 23 was upwards of 30-fold worse in blocking proliferation of two cancer cell lines (MCF-7 (breast) and DU145 (prostate)) than SAHA, when evaluated in healthy VERO cells, 23 was nontoxic with an in vitro therapeutic index (IC50(VERO)/IC50(MCF-7)) > 5.24, which was better than SAHA (1.64). As far as the binding of the polypharmacologic compounds to targets, it is believed that they bind in a serial fashion in which the drug molecule first binds to the ER target, localizing them into the ER(+) tissues followed by dissociation from the target and inhibition of HDAC.
PARP/HDAC6 dual inhibitors
Olaparib (25) is an FDA approved drug for the treatment of BRCA-mutated ovarian cancer.97 This drug inhibits poly(ADP-ribose)polymerase (PARP), which is responsible for multiple cellular processes such as DNA damage response, chromatin remodeling, cell cycle regulation, and cell death.98 It is believed that PARP inhibitors suppress DNA damage repair, therefore sensitizing cancer cells to DNA damaging agents.99 However, olaparib shows limited activity in tumor cells with defective homologous recombination repair (HRR);97 a polypharmacological strategy may help restore the anti-tumor activity of olaparib especially in HRR proficient subtypes.
Both PARP and HDACis sensitize tumor cells to other anticancer agents when delivered as a co-treatment.100,101 Dosing of PARP inhibitors in combination with HDACis produces a synergistic effect both in vitro and in vivo.102–105 It is believed that this is due to HDAC inhibition induces damage in tumor cells while PARP inhibition suppresses the DNA repair mechanisms. HDAC inhibition decreases the levels of DNA damage response as well as downregulates HRR genes, that lead to “BRCAness” and therefore enhancing PARP activity in triple negative breast cancer regardless of BRCA1 mutation, triple negative breast cancer with phosphatase and tensin homolog expression, HRR-proficient ovarian cancer, and prostate cancer.102–105 These previous studies were the basis of creating a small library of polypharmacologic PARP/HDAC inhibitors by grafting hydroxamic acid moieties at the aroyl amide bond location of olaparib.106
When compared to olaparib (values in parentheses), lead compound 26 (P1, Fig. 13) was 19-fold less potent against PARP1 with an IC50 value of 68.15 nM (3.59 nM), and around two-fold less potent against PARP2: IC50 = 5.02 nM (2.81 nM).106 This led to the determination that the piperazine may be important for inhibition of PARP1 and is dispensable for effective inhibition of PARP2. When tested for HDAC1 and HDAC6 inhibition, P1 had values more similar to the control compound, SAHA (values in parentheses), with IC50 values of 27.26 ± 0.59 nM (11.34 nM) for HDAC1 and 8.21 ± 0.36 nM (7.70 nM) for HDAC6. The linker length was directly related to the selectivity for HDAC6; compound 27 (P4) with the shortest linker was the most selective with 30-fold greater affinity towards HDAC6 than HDAC1.
Fig. 13. Transformation of the PARP inhibitor olaparib into a dual PARP/HDAC6 inhibitor.

All four compounds were tested in multiple cell lines in various cancers, including breast, lung and cervical cancers, leukemia, and lymphoma; in general, the in vitro data mirrored the cell data. P1 demonstrated the greatest anti-proliferative activity with IC50 values < 10 μM. In a long-term anti-tumor study using MDA-MB-231 cells, P1 had a higher potency (IC50 = 0.22 μM) than both olaparib (IC50 = 7.92 μM) and SAHA (IC50 = 0.43 μM) controls, although it is unclear if the polypharmacological strategy was superior to the corresponding combination approach since no cell data was provided for the combination of olaparib and SAHA. The lead compound was also less potent than SAHA in healthy MCF-10A cells. Further analysis of P1 revealed that it induced apoptosis in MDA-MD-231 cells and that this was associated with modulation of the HDAC and PARP pathways.
JAK2/HDAC6 dual inhibitors
Activation of Janus kinases (JAKs) – which is a family of non-receptor tyrosine kinases – results in phosphorylation and dimerization of signal transducers and activators of transcription (STAT) protein, leading to gene transcription activation. The JAK/STAT signaling pathway is pivotal in survival, cell proliferation, differentiation and a range of developmental processes. Dysregulation of the pathway is associated with several pathophysiologies, including immune disorders and cancer.107 Hence, JAK inhibitors have been extensively investigated as new therapeutics for the treatment of immune-inflammatory diseases such as rheumatoid arthritis, as well as solid tumors and hematological malignancies.108
The deployment of JAK inhibitors for targeting the JAK/STAT pathway have been effective in the treatment of patients with myeloproliferative neoplasms, which often lead to leukemia.109,110 HDAC inhibition also has implications in T cell lymphoma.111 Recent clinical trials demonstrated the usefulness of the JAK2 inhibitor ruxolitinib and the pan-HDACi panobinostat dosed in combination as a viable treatment for both solid tumors and hematological malignancies; therefore, the combination of these families of inhibitors into a single multi-targeted compound could prove a useful anti-cancer treatment.112–114
Motivated by the observed synergy between the JAK2/FLT3 inhibitor pacritinib (28) and HDACis in cells, Dymock's laboratory embarked on the development of polypharmacologic inhibitors of both JAK2 and, through the grafting of the linker and hydroxamic acid of SAHA, HDACs onto pacritinib.115 Exposed to the solvent, the pyrrolidine ring served as an excellent point of fusion of the two pharmacophores.
Both carboxylic acids and hydroxamic acids were tested as potential ZBG; as anticipated, the carboxylic acids were weak binders, and so the majority of the research centered on hydroxamic acids. In addition, the optimal linker length was six methylenes. Hydroxamic acid 29 (Fig. 14) demonstrated potent HDAC6 inhibitory activity with an IC50 value of 2.1 nM and >100-fold selectivity over HDAC1. This compound also retained potent JAK2 inhibition with IC50 value of 1.4 nM, and was >50-fold selective for JAK2 over a panel of 97 kinases. The library of compounds was also evaluated in both solid tumor and hematological cell lines. The carboxylic acid derivatives displayed no activity in any of these cell lines. However, hydroxamate 29 was potent against all cancer cell lines with IC50s around 1–2 μM, and as low as 0.94 μM in the erythroleukemia cell line HEL92.1.7 that expresses the JAK2V617F mutant. Lastly, the anti-proliferative effects – comparable with that of the controls SAHA and pacritinib – of the lead compounds were consistent with abrogation of the JAK/STAT and HDAC pathways in multiple hematological cell lines.
Fig. 14. A dual JAK2/HDAC6 inhibitor.

Dymock's group also approached the discovery of dual JAK/HDACis through grafting SAHA-like linkers and hydroxamic acids onto the pyrazole portion of the JAK1/2 inhibitor ruxolitinib (30), retaining the crucial pyrrolopyrimidine that is known to bind the hinge region of JAK1/2 (Fig. 15).116 Although favoring HDAC6 ever so slightly, resulting bivalent compound 31 inhibited HDAC1 and HDAC6 equipotently (IC50s of 6.0 ± 2.0 nM and 1.0 ± 0.1 nM, respectively). Compound 31 also potently inhibited JAK1 (IC50 = 11 nM) and JAK2 (IC50 = 65 nM). In a panel of 97 kinases, desmethyl congener 32, which exhibited similar HDAC1/6 binding affinities and selectivities, revealed notable selectivity for the JAK family of kinases. In addition, 32 showed promising anti-tumor activity (high nanomolar/low micromolar) in a panel of cancer cell lines that once again was congruent with blocking the JAK–STAT and HDAC pathways.
Fig. 15. Conversion of the JAK2-selective inhibitor ruxolitinib into a dual JAK2/HDAC6 inhibitor.

More recently, the same group sought a new family of dual JAK2/HDAC inhibitors through capitalizing on the solved co-crystal structure of the JAK2 inhibitor XL019 (33) that revealed regions of the inhibitor that are bound in solvent channels in JAK2.117 A large library of dual inhibitors was synthesized based on XL019 using two different points of attachment of the linking unit and hydroxamic acid of SAHA at positions of XL019 that are solvent-exposed when bound to JAK2.117 Design A projected the HDAC binding moiety from the aniline of XL019 via replacement of the morpholino group, while design B projected these pharmacophores from the acetamide. Design A yielded several potent compounds, one of which is 34 (Fig. 16) with sub-nanomolar activity against both JAK2 and HDAC6 with IC50 values of 0.9 nM and 0.1 ± 0.05 nM, respectively. This compound also demonstrated over 7000-fold selectivity for HDAC6 over HDAC1. It is also of note that the addition of a basic methylpiperazine side chain decreased activity against JAK2 while the activity for the HDAC isozymes remain relatively unchanged. The dual inhibitors demonstrated potent inhibition of a panel of solid tumor and hematological cancer cell lines, mirroring the in vitro data, with anti-proliferative IC50 values as low as 70 nM in the multiple myeloma (MM) cell line KMS-12-BM, considerably more potent than the controls XL019 and SAHA, although in most cancer cell lines, 34 exhibited similar anti-proliferative activities to the parental drugs. Further analysis revealed that the lead compounds induced cell death by apoptosis.
Fig. 16. A dual JAK2/HDAC6 inhibitor based on the JAK2 inhibitor XL019.

Huang and co-workers prepared three series of dual JAK2/HDACis through modifications of the JAK2 inhibitor CYT-387 (35).118 Particularly, the solvent-exposed morpholine was replaced with piperidines substituted in the 4-position with various linkers and (mostly) hydroxamic acids, and a sub-class of this type saw the substitution of the N-cyanomethylene amide with a methyl sulfonamide (also known to elicit JAK2 inhibition),119 and the third class involved replacement of the N-cyanomethylene amide with either an hydroxamic acid or an ortho-amino anilide whilst retaining the morpholine. One of their most potent compounds was 36, depicted in Fig. 17, with IC50s of 8 nM and 46 ± 1.7 nM for JAK2 and HDAC6, respectively. The selectivity of 36 for HDAC6/HDAC1 was about 24-fold, and around 5-fold more selective for HDAC6 over HDAC3. Many of their compounds inhibited the proliferation of hematologic cell lines in the low micromolar range. Significantly, 36 was more potent (IC50 = 340 nM) on the HEL cell line than the combination of CYT-387 and SAHA from which 36 was derived, and that combination exhibited greater anti-tumor activity than either drug alone in K562 cells. Taken together, these data suggest there may be synergy between the JAK2 and HDACis, and that this is more pronounced when the key pharmacophores are present in the same molecule. Leukemia patients are especially vulnerable to Invasive fungal infections (IFIs), and so the authors investigated the efficacy of several of their lead compounds with the anti-fungal drug fluconazole in resistant Candida albicans isolates; several compounds demonstrated synergic effects, including 36. In fact, compound 36 exhibited anti-tumor efficacy in several AML models and synergized with fluconazole in those models infected with resistant C. albicans.
Fig. 17. A dual JAK2/HDAC6 inhibitor.

Liang and colleagues created another dual JAK and HDAC targeting compound by the addition of a hydroxamic acid motif onto their previously reported JAK inhibitor 37.120 The dual inhibitors were fashioned by grafting the hydroxamic acid moiety to the N1 atom of the pyrazole ring, as illustrated in Fig. 18. A library of compounds was synthesized with various substituents on the pyrimidine of the JAK targeting moiety. From this library, compound 38 emerged with the best balance between the two targets and improved anticancer effects in four cell lines compared to ruxolitinib and SAHA controls. Compound 38 was selective for HDAC6 over HDAC2 with IC50s of 14.4 nM and 120 nM, respectively. Most notably, 38 was superior to the combination treatment of the two control compounds. Specifically, 38 exhibited IC50 values ranging from 0.06–0.49 μM while ruxolitinib, SAHA, and a 1 : 1 molar ratio of the two controls ranged from 15.8–>5 μM, 0.27–1.11 μM and 0.30–1.03 μM, respectively. Results indicated the dual inhibitor also possessed good bioavailability using intraperitoneal administration and did not show significant toxic effects against HEL xenografts.
Fig. 18. A dual JAK2/HDAC6 inhibitor.

CRBN/HDAC6 dual inhibitors: PROTACs
Within recent years, protein degradation by proteolysis targeting chimeras (PROTACs) has become a popular strategy in the development of new drugs.121 PROTACs are bifunctional small molecules that contain ligands for both the protein of interest and an E3 ubiquitin ligase.122 Protein degradation begins with the formation of a binary complex between the PROTAC and one of the two target proteins, followed by formation of a ternary complex when bound to both protein targets; this results in the recruitment of the protein of interest to the E3 ligase, causing degradation by the ubiquitin–proteasome system.122
Previously, Tang's laboratory developed the first-in-class HDAC6-sepcific PROTAC employing a pomalidomide derivative as the E3 ligase ligand.123 Although this hybrid was generated by grafting on the alkyl hydroxamic acid of SAHA that is inherently non-HDAC6-selective, the authors observed the selective degradation of HDAC6 in cells, which they note was “unexpected, but not surprising”. Later, the same group focused on improving the HDAC6 selectivity by grafting pomalidomide (39) onto the para-position of the aniline of the HDAC6 selective inhibitor nexturastat-A (Next-A, 40).124 As illustrated in Fig. 19 with a specific example, 18 degraders were thus synthesized using different attachment points and linker chain lengths. When tested using a validated high-throughput in-cell enzyme-linked immunosorbent assay (ELISA), it was determined that the optimal linker length was six methylene units and that the linkage at C4 of pomalidomide's phthalimide ring afforded a more potent inhibitor than at C5. Additionally, there is a phenomenon using pomalidomide analogues that activates CRBN's E3 ligase towards the Ikaros family zinc fingers (IKZFs) and promotes their degradation and this induced degradation is thought to be the cause of their antiproliferative effects in MM.125–127 Though IKZF degradation is usually seen as an undesirable effect in PROTACs, it was hypothesized that HDAC6 degraders maintaining IKZF degradation would have enhanced MM activity.
Fig. 19. An HDAC6-selective PROTAC.

However, only compound 41 was capable of inducing IKZF degradation in the MM cell line MM1S. In-cell ELISA showed 41 had a potency of DC50 = 1.6 nM for HDAC6. In a full dose response experiment, it was seen that HDAC6 was the only member of the HDAC family affected by compound 41; HDAC6 protein levels were reduced starting at 3 nM with maximal effect at 30 nM while IKZF degradation started at 30 nM and was dose dependent. This was interesting given that HDAC6 degradation by the PROTAC requires binding of both ligands while IKZF degradation only requires the binding of pomalidomide. This may be attributed to the turnover rate of the proteins, having tested the re-synthesis rates of the proteins and discerning that IKZF returned to original levels much quicker than did HDAC6.
In the same year, Rao's group also developed an HDAC6-selective PROTAC based on Next-A but this time tethering pomalidomide through the alkyl group of Next-A. However, the antiproliferative effects of this PROTAC were not beneficial over regular inhibition using Next-A.128
Compound 41 was compared to both Next-A and SAHA. In these tests 41 caused increased tubulin acetylation at 100 nM, unlike Next-A and SAHA, indicating that HDAC6 degradation was the primary cause. Additionally, no H3 acetylation was detected, indicating that the degrader had a high selectivity for the HDAC6 isozyme with IC50 of 8.7 nM for HDAC6, similar to Next-A. The role of the pomalidomide moiety in 41 was confirmed to modulate the IKZFs along with HDAC6 degradation being responsible for the marked increased in acetylation. Examination of mRNA levels also showed that the decrease in HDAC6 levels were due to degradation and not transcriptional downregulation. Finally, experiments using modified PROTACs were completed to rule out any possible cell permeability problems. Results indicate that compound 41 functions as an HDAC6 degrader, HDAC6 inhibitor, and IKZF degrader with high selectivity for HDAC6. These characteristics cause the antiproliferative effects of the drug compound in MM cell lines.
Proteasome/HDAC6 dual inhibitor
The ubiquitin–proteasome system introduced in the previous PROTAC section regulates the destruction of cellular proteins. Proteasome inhibitors (PIs), which may be non-covalent or covalent in nature, such as bortezomib, have been clinically validated as anti-cancer therapeutics, in particular for MM, owing to a dependence of the cancer cells on the proteasome for survival.129–131 However, the aggresome pathway, regulated by HDAC6, serves as a back-up, and its upregulation is one particular mechanism attributed to the acquired resistance observed in MM treatment regimens.132 Accordingly, combination studies of HDACis and PIs have been conducted with synergism observed,132 most recently between the HDAC6i ricolinostat and bortezomib.133
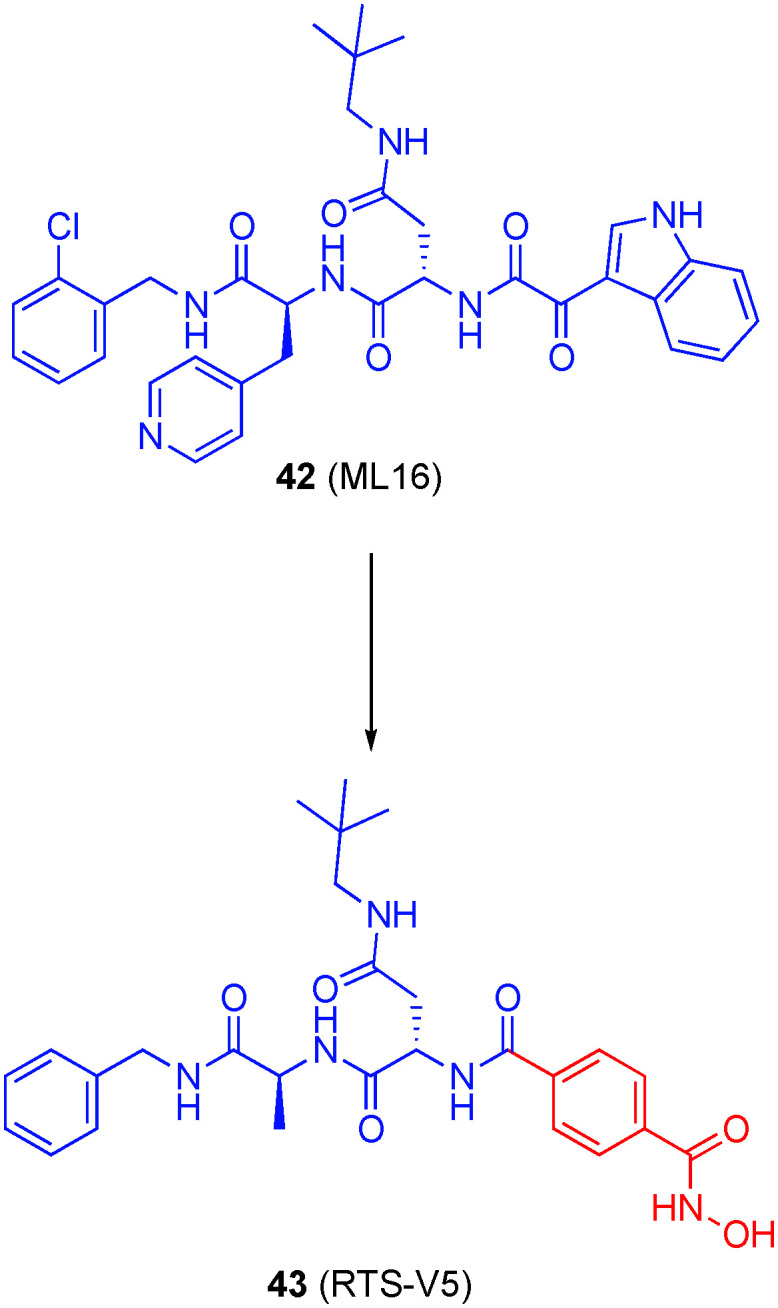
In 2018, Bhatia and colleagues inspected X-ray structures of the peptidomimetic, non-covalent PI ML16 (42) bound to the proteasome and noted that the 3-acylindole portion at the N-terminus was solvent-exposed.134 Accordingly, they replaced this portion with a 4-phenylhydroxamic acid, a motif believed to impart HDAC6-selectivity to furnish the unprecedented dual PI/HDAC 43 (RTS-V5), shown in Fig. 20. This hybrid molecule inhibited HDAC1–3 in the low micromolar range, but much more potently against HDAC8 (IC50 = 0.53 ± 0.01 μM) and HDAC6 (IC50 = 0.27 ± 0.01 μM); a co-crystal structure of 43 with HDAC6 confirmed a monodentate engagement of the zinc ion, as observed with other bulky, phenyl hydroxamates and likely the origin of HDAC6 selectivity. Inhibition of the proteasome was in the high nanomolar range, which is consistent with non-covalent proteasome inhibitors. 43 was cytotoxic to several leukemia and MM cell lines (IC50s ranging from 0.89 to 3.14 μM) – comparable to or slightly outperforming ricolinostat – with limited toxicity towards peripheral blood derived mononuclear cells acquired from healthy patients. In summary, this data and further biological analyses indicated disruption of both the aggresome and proteasome pathways, and heralds an exciting advancement in next-generation treatment for MM patients who become refractory to gold standard treatments like bortezomib due to acquisition of resistance.134 In April 2020, Zhou et al. published dual proteasome/HDAC inhibitors by grafting hydroxamic acid motifs onto the covalent proteasome inhibitor bortezomib.135 However, since these dual inhibitors did not exhibit HDAC6 selectivity, they are outside the scope of this review.
Fig. 20. A non-covalent, dual PI/HDAC6 inhibitor.
Bcl-2/HDAC6 dual inhibitors
Venetoclax (ABT-199, 44), an FDA-approved drug for the treatment of chronic lymphocytic leukemia and is emerging as the standard of care for acute myeloid leukemia, functions through inhibiting the anti-apoptotic BCL-2 protein within the family of the same name.136 Engineered through extensive rounds of medicinal chemistry guided by structural biology, ABT-199 binds with picomolar affinity to the BH3-binding groove on the surface of BCL-2.137 Recently, the combination of BCL-2 inhibitors and HDACis has produced synergistic effect in MM cell lines, as well as rescue sensitivity to bortezomib in resistant cells.138–140
Chen, Zheng and colleagues recognized from the co-crystal structure of ABT-199/BCL-2 that the tetrahydropyran motif makes minimal interactions with the protein and is largely directed to the solvent. Accordingly, and considering the HDACi pharmacophore three component make-up, the authors reasoned that this motif could be replaced with linking units to hydroxamic acid functional groups to accomplish HDAC recognition without mitigating affinity for BCL-2 affinity (Fig. 21).141 In total, seven compounds were prepared with increasing methylene units, from 1 through 7, between the aniline NH and the hydroxamic acid. Compounds were evaluated in vitro for inhibition of HDAC1 and HDAC6. A linker of at least 5 CH2 groups was required for appreciable activity (IC50 < 1 μM), which is consistent with previous findings, and those compounds demonstrated around 10-fold, or better, selectivity for HDAC6. Compound 45 inhibited HDAC1 and HDAC6 with IC50 values of 281 ± 70 and 19 ± 1 nM, respectively, with negligible impact on BCL-2 affinity (relative to ABT-199). 45 was active in cells as well, inhibiting the growth of the MM cell lines RPMI-8226 and U-226 with IC50s of 0.2 μM and 8.7 μM, respectively.
Fig. 21. Conversion of the BCL-2 clinical drug venetoclax into a dual BCL-2/HDAC6 inhibitor.

Tubulin/HDAC6 dual inhibitors
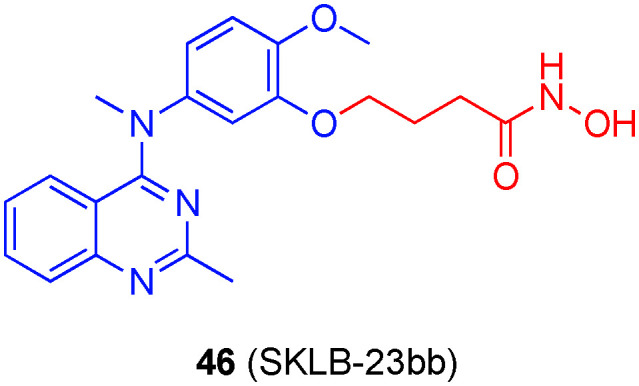
Chen's laboratory previously reported that the HDAC6-selective inhibitor 46 (SKLB-23bb, Fig. 22) outperformed the HDAC6-selective inhibitor ricolinostat in anti-tumor activities both in vitro and in vivo.14246 had an IC50 value of 17 ± 2 nM against HDAC6 with 25-fold and better selectivities over the other isozymes. More recently, the group performed a head to head tumor cell viability study with 46 and ricolinostat, and, while IC50 values for 46 were mostly in the double-digit nanomolar range, in every case, IC50 values for ricolinostat were one to two orders of magnitude worse, typically around the single-digit micromolar range.143 Particularly noteworthy is the observation that 46 was considerably more efficacious against MM cell lines (e.g. MM1S cells: IC50s 67 nM vs. 1.33 μM), Wang and colleagues conducted further research into delineating where this superiority originated. First, it was noted that although 46 inhibited cellular HDAC6, the anti-tumor activity did not correlate with HDAC6 inhibition, and HDAC6 knockout cells remained sensitive. Given that a similar quinazoline motif is present in the microtubule inhibitor MPC6827, the authors considered that 46 might also interfere with the microtubule system. Immunofluorescence staining confirmed that 46 caused disruption of microtubule assembly, coupled with the changes in morphology, the authors were able to infer that 46 is a microtubule polymerization inhibitor, and this was subsequently corroborated with a tubulin polymerization assay. Since correct microtubule assembly is required for cell division, this newly discovered biological activity of 46 explains, in part, the enhanced anti-tumor activity relative to ricolinostat. Further analysis revealed 46 triggered apoptosis. Unusual for HDACis, 46 was effective against solid tumor cell lines. In contrast to ricolinostat, which demonstrates limited anti-tumor activity in vivo against solid tumors when administered alone, orally bioavailable 46 effectively blocked the growth of B-cell lymphoma in vivo.
Fig. 22. The dual tubulin/HDAC6 inhibitor SKLB-23bb.
Several research groups have rationally designed dual tubulin/HDACis to realize especially potent anti-tumor agents, motivated by prior research that indicated anti-tumor synergism between HDACis and microtubule-destabilizing agents.144,145 For example, Zhang et al. grafted linkers with hydroxamic acids onto the scaffold of the tubulin inhibitor colchicine,146 while Hamze's laboratory reported a similar strategy with isocombretastatin.147 While both laboratories discovered potent anti-tumor compounds, either HDAC6 activity was not discussed or the hybrids proved more selective for other HDAC isoforms and will not be discussed further. In 2019, Wang and colleagues functionalized the phenol hydroxyl of combretastatin-like chalcones (for example, 47), which interfere with tubulin assembly through occupying the colchicine binding site, with various linkers and hydroxamic acids to also engage HDACs (Fig. 23).148 Ten hybrid compounds were prepared employing linker lengths between the two pharmacophores of 3 to 7 methylenes. In an initial screen, the optimal linker length was defined as 6–8 carbons, as seen elsewhere. Several compounds demonstrated potent inhibition of HDACs from HeLa cell nucleus extract down to IC50 = 71.5 nM. Although the least potent to HDACs with IC50 values of 996 nM, 470 nM, and 1700 nM for HDAC1, HDAC6, and HDAC8 respectively, 48 was the most potent in cells, inhibiting the proliferation of several cancer cell lines at low to sub-μM, and, in certain cases, outperforming SAHA and the parent combretastatin-like chalcones. However, this data should be treated with caution as the lack of agreement between HDAC and cell data suggests additional target(s) are involved. Moreover, the anti-proliferative studies were not performed with the combination of the parent drugs, and so it is unclear if 48 is demonstrating a synergistic effect. Compound 49, roughly on a par with SAHA in the initial HDAC screening assay, was almost 9-fold selective for HDAC6 over HDAC1 (IC50s 4.6 nM and 36.3 nM, respectively), but proved several-fold less effective in the cell proliferation assays than either of the two parent drugs.
Fig. 23. A dual tubulin/HDAC inhibitor.

Lee and colleagues prepared dual tubulin/HDACis through grafting linkers and hydroxamic acids onto the indole nitrogen of the tubulin inhibitor 50 (SCB01A), as depicted in Fig. 24.149 Their best compound was 51 (MPT0B451),150 which is almost two-fold selective for HDAC6 (IC50 = 275.35 ± 14.47 nM) over HDAC1 (IC50 = 467.82 ± 7.80 nM), retained tubulin polymerization inhibitory activity, and demonstrated double-digit nM IC50 values in a sulforhodamine proliferation assay, was selected for further study. Cell proliferation assays with the AML cell line HL60 and the prostate cancer cell line PC-3, both of which harbor elevated HDAC6 levels, revealed MPT0B451 potently blocked cell growth (IC50s = 42 nM and 1.1 μM, respectively). MPT0B451 caused mitotic arrest and cell death by apoptosis. Lastly, MPT0B451 demonstrated efficacy in HL60 and PC-3 xenograft models.
Fig. 24. A dual tubulin/HDAC6 inhibitor.

Conclusions
Combination therapies are the standard of care for many multi-factorial diseases, particularly cancer. However, this strategy is fraught with challenges, including lack of patient compliance, spiraling healthcare costs, and limited improvement in quality, and extension, of life. Although it is yet to prove itself – indeed, even in this review, the multi-targeting drug was compared side-by-side with the corresponding combination of single-target drugs in only a couple of cases – the emerging field of polypharmacology may succeed where combination therapy fails, with the promise of accomplishing all that combination therapy is purported to do so, without any of the detriment and all in a single drug molecule. This represents a significant paradigm shift in the treatment of multi-factorial disease. Perhaps ideal therapies may lie somewhere in between, for example a dual inhibitor combined with a second drug. Time will tell, but even if polypharmacology does not yield superior results to combination therapy, at the very least, it should expand the drug arsenal. Pan-HDACis are associated with a plethora of side effects that heavily restricts their use in the clinic. Recent structural data has illuminated the binding modes of HDAC6 selective inhibitors, which has been instrumental in advancing the development of such compounds as safer therapies for cancer and AD, among others. In particular, this will facilitate the discovery of polypharmacological inhibitors of HDAC6. While much of the research described herein identified inhibitors that are serendipitously selective for HDAC6 over the other isoforms, it is of interest to further enhance their HDAC6 selectivities in order to better understand the biological ramifications of rationally inhibiting HDAC6, while minimally impacting its family members, and one additional co-target.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
We acknowledge the NIH for funding (NIH/NIGMS T32 GM066706), and thank the University of Maryland School of Pharmacy for supporting our work in this area. We thank PharmD students Anthonia Azubike, Elie Pommier and Andrew Bilodeau for initial literature searches on this review topic.
Notes and references
- Prabakaran S. Lippens G. Steen H. Gunawardena J. Wiley Interdiscip. Rev.: Syst. Biol. Med. 2012;4:565–583. doi: 10.1002/wics.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.-J. Seto E. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Li Y. Seto E. Cold Spring Harbor Perspect. Med. 2016;6:a06831. doi: 10.1101/cshperspect.a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S. Eom G. H. Chonnam Med. J. 2016;52:1–11. doi: 10.4068/cmj.2016.52.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. S. Parmigiani R. B. Marks P. A. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Simões-Pires C. Zwick V. Nurisso A. Schenker E. Carrupt P.-A. Cuendet M. Mol. Neurodegener. 2013;8:7. doi: 10.1186/1750-1326-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijido O. Cacabelos R. Int. J. Mol. Sci. 2018;19:3199. doi: 10.3390/ijms19103199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull E. E. Montgomery M. R. Leyva K. J. BioMed Res. Int. 2016:8797206. doi: 10.1155/2016/8797206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licciardi P. V. Karagiannis T. C. ISRN Hematol. 2012:690901. doi: 10.5402/2012/690901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.-J. Seto E. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Wang X.-X. Wan R.-Z. Liu Z.-P. Eur. J. Med. Chem. 2018;143:1406–1418. doi: 10.1016/j.ejmech.2017.10.040. [DOI] [PubMed] [Google Scholar]
- Zhang L. Zhang J. Jiang Q. Zhang L. Song W. J. Enzyme Inhib. Med. Chem. 2018;33:714–721. doi: 10.1080/14756366.2017.1417274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter N. J. Mahendran A. Breslow R. Christianson D. W. Proc. Natl. Acad. Sci. U. S. A. 2017;114:13459–13464. doi: 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. Zhang C. Hassan S. Liu X. Song F. Chen K. Zhang W. Yang J. J. Hematol. Oncol. 2018;11:111. doi: 10.1186/s13045-018-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez A. Lin T. Ikeda A. K. Simms-Waldrip T. Fu C. Sakamoto K. M. Cancer Res. 2008;68:2557–2560. doi: 10.1158/0008-5472.CAN-07-5989. [DOI] [PubMed] [Google Scholar]
- Osko J. D. Christianson D. W. Bioorg. Med. Chem. Lett. 2020;30:127023. doi: 10.1016/j.bmcl.2020.127023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Kwon S. Yamaguchi T. Cubizolles F. Rousseaux S. Kneissel M. Cao C. Li N. Cheng H.-L. Chua K. Lombard D. Mizeracki A. Matthias G. Alt F. W. Khochbin S. Matthias P. Mol. Cell. Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A. Bajorath J. Rastelli G. J. Med. Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Proschak E. Stark H. Merk D. J. Med. Chem. 2019;62:420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- What is Alzheimer's?, https://alz.org/alzheimers-dementia/what-is-alzheimers, (accessed July 27, 2020)
- Cummings J. L. Morstorf T. Zhong K. Alzheimer's Res. Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J.-Y. Aromolaran K. A. Zukin R. S. Nat. Rev. Neurosci. 2017;18:347–361. doi: 10.1038/nrn.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A. Neuropharmacology. 2014;80:95–102. doi: 10.1016/j.neuropharm.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Vecsey C. G. Hawk J. D. Lattal K. M. Stein J. M. Fabian S. A. Attner M. A. Cabrera S. M. McDonough C. B. Brindle P. K. Abel T. Wood M. A. J. Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Liu C. Wu J. Tao J. J. Sui X. L. Yao Z. G. et al. . J. Alzheimer's Dis. 2014;41:1193–1205. doi: 10.3233/JAD-140066. [DOI] [PubMed] [Google Scholar]
- d'Ydewalle C. Bogaert E. Van Den Bosch L. Traffic. 2012;13:771–779. doi: 10.1111/j.1600-0854.2012.01347.x. [DOI] [PubMed] [Google Scholar]
- Lloret A. Fuchsberger T. Giraldo E. Viña J. Free Radical Biol. Med. 2015;83:186–191. doi: 10.1016/j.freeradbiomed.2015.02.028. [DOI] [PubMed] [Google Scholar]
- Hooper C. Killick R. Lovestone S. J. Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J. Rei D. Guan J.-S. Wang W.-Y. Seo J. Hennig K. M. Nieland T. J. F. Fass D. M. Kao P. F. Kahn M. Su S. C. Samiei A. Joseph N. Haggarty S. J. Delalle I. Tsai L.-H. Nature. 2012;483:222–226. doi: 10.1038/nature10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C. Stankowski J. N. Carlomagno Y. Stetler C. Petrucelli L. Alzheimer's Res. Ther. 2014;6:29. doi: 10.1186/alzrt259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S.-J. Huang F.-I. Liou J.-P. Yang C.-R. Cell Death Dis. 2018;9:655. doi: 10.1038/s41419-018-0688-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Shi Y. Jiang X. Leak R. K. Hu X. Wu Y. Pu H. Li W.-W. Tang B. Wang Y. Gao Y. Zheng P. Bennett M. V. L. Chen J. Proc. Natl. Acad. Sci. U. S. A. 2015;112:2853–2858. doi: 10.1073/pnas.1501441112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. Owens G. C. Makarenkova H. Edelman D. B. PLoS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y. Liang M.-H. Ren M. Marinova Z. Leeds P. Chuang D.-M. J. Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. Taliyan R. Naunyn-Schmiedeberg's Arch. Pharmacol. 2015;388:337–349. doi: 10.1007/s00210-014-1081-2. [DOI] [PubMed] [Google Scholar]
- Lehàr J. Krueger A. S. Avery W. Heilbut A. M. Johansen L. M. Price E. R. Rickles R. J. Short G. F. Staunton J. E. Jin X. Lee M. S. Zimmermann G. R. Borisy A. A. Nat. Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simone A. La Pietra V. Betari N. Petragnani N. Conte M. Daniele S. Pietrobono D. Martini C. Petralla S. Casadei R. Davani L. Frabetti F. Russomanno P. Novellino E. Montanari S. Tumiatti V. Ballerini P. Sarno F. Nebbioso A. Altucci L. Monti B. Andrisano V. Milelli A. ACS Med. Chem. Lett. 2019;10:469–474. doi: 10.1021/acsmedchemlett.8b00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte A. Gil-Bea F. García-Barroso C. Cedazo-Minguez Á. Ramírez M. J. Franco R. García-Osta A. Oyarzabal J. Cuadrado-Tejedor M. Neuropathol. Appl. Neurobiol. 2015;41:471–482. doi: 10.1111/nan.12203. [DOI] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M. Garcia-Barroso C. Sanzhez-Arias J. Mederos S. Rabal O. Ugarte A. Franco R. Pascual-Lucas M. Segura V. Perea G. Oyarzabal J. Garcia-Osta A. Clin. Epigenet. 2015;7:108. doi: 10.1186/s13148-015-0142-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M. Garcia-Barroso C. Sánchez-Arias J. A. Rabal O. Pérez-González M. Mederos S. Ugarte A. Franco R. Segura V. Perea G. Oyarzabal J. Garcia-Osta A. Neuropsychopharmacology. 2017;42:524–539. doi: 10.1038/npp.2016.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabal O. Sánchez-Arias J. A. Cuadrado-Tejedor M. de Miguel I. Pérez-González M. García-Barroso C. Ugarte A. Estella-Hermoso de Mendoza A. Sáez E. Espelosin M. Ursua S. Haizhong T. Wei W. Musheng X. Garcia-Osta A. Oyarzabal J. Eur. J. Med. Chem. 2018;150:506–524. doi: 10.1016/j.ejmech.2018.03.005. [DOI] [PubMed] [Google Scholar]
- Rabal O. Sánchez-Arias J. A. Cuadrado-Tejedor M. de Miguel I. Pérez-González M. García-Barroso C. Ugarte A. Estella-Hermoso de Mendoza A. Sáez E. Espelosin M. Ursua S. Tan H. Wu W. Xu M. Pineda-Lucena A. Garcia-Osta A. Oyarzabal J. ACS Chem. Neurosci. 2019;10:4076–4101. doi: 10.1021/acschemneuro.9b00303. [DOI] [PubMed] [Google Scholar]
- Claffey M. M. Helal C. J. Verhoest P. R. Kang Z. Bundesmann M. W. Hou X. Lui S. Kleiman R. J. Vanase-Frawley M. Schmidt A. W. Menniti F. Schmidt C. J. Hoffman W. E. Hajos M. McDowell L. O'Connor R. E. MacDougall-Murphy M. Fonseca K. R. Becker S. L. Nelson F. R. Liras S. J. Med. Chem. 2012;55:9055–9068. doi: 10.1021/jm3009635. [DOI] [PubMed] [Google Scholar]
- Wang H. Luo X. Ye M. Hou J. Robinson H. Ke H. J. Med. Chem. 2010;53:1726–1731. doi: 10.1021/jm901519f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M. Pérez-González M. García-Muñoz C. Muruzabal D. García-Barroso C. Rabal O. Segura V. Sánchez-Arias J. A. Oyarzabal J. Garcia-Osta A. Front. Aging Neurosci. 2019;11:149. doi: 10.3389/fnagi.2019.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno K. Kanno S. Nitta H. Uesugi N. Sugai T. Masuda T. Wakabayashi G. Maesawa C. Oncol. Rep. 2012;28:867–873. doi: 10.3892/or.2012.1898. [DOI] [PubMed] [Google Scholar]
- Bradbury C. A. Khanim F. L. Hayden R. Bunce C. M. White D. A. Drayson M. T. Craddock C. Turner B. M. Leukemia. 2005;19:1751–1759. doi: 10.1038/sj.leu.2403910. [DOI] [PubMed] [Google Scholar]
- Bazzaro M. Lin Z. Santillan A. Lee M. K. Wang M.-C. Chan K. C. Bristow R. E. Mazitschek R. Bradner J. Roden R. B. S. Clin. Cancer Res. 2008;14:7340–7347. doi: 10.1158/1078-0432.CCR-08-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerno S. Settimo F. D. Taliani S. Simorini F. Motta C. L. Fornaciari G. Marini A. M. Curr. Med. Chem. 2010;17:4270–4290. doi: 10.2174/092986710793361252. [DOI] [PubMed] [Google Scholar]
- Castelli S. Coletta A. D'Annessa I. Fiorani P. Tesauro C. Desideri A. Biol. Chem. 2012;393:1327–1340. doi: 10.1515/hsz-2012-0240. [DOI] [PubMed] [Google Scholar]
- Chen M.-C. Chen C.-H. Chuang H.-C. Kulp S. K. Teng C.-M. Chen C.-S. Hepatology. 2011;53:148–159. doi: 10.1002/hep.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. F. Annu. Rev. Biochem. 1989;58:351–375. doi: 10.1146/annurev.bi.58.070189.002031. [DOI] [PubMed] [Google Scholar]
- Catalano M. G. Fortunati N. Pugliese M. Poli R. Bosco O. Mastrocola R. Aragno M. Boccuzzi G. J. Endocrinol. 2020;191:465–472. doi: 10.1677/joe.1.06970. [DOI] [PubMed] [Google Scholar]
- Ellis L. Hammers H. Pili R. Cancer Lett. 2009;280:145–153. doi: 10.1016/j.canlet.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J. Cubitt C. L. Zhang S. Chiappori A. Cancer Biol. Ther. 2012;13:614–622. doi: 10.4161/cbt.19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. Easley C. Kirkpatrick P. Nat. Rev. Drug Discovery. 2007;6:21–22. doi: 10.1038/nrd2227. [DOI] [PubMed] [Google Scholar]
- Kim H. W. Ko Y. H. Kang S. H. Lee J. G. Taehan Pinyogikwa Hakhoe Chapchi. 2011;52:166–171. [Google Scholar]
- Jakopovic M. Thomas A. Balasubramaniam S. Schrump D. Giaccone G. Bates S. E. Front. Oncol. 2013;3:261. doi: 10.3389/fonc.2013.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alagoz M. Gilbert D. C. El-Khamisy S. Chalmers A. J. Curr. Med. Chem. 2012;19:3874–3885. doi: 10.2174/092986712802002590. [DOI] [PubMed] [Google Scholar]
- Yu C.-C. Pan S.-L. Chao S.-W. Liu S.-P. Hsu J.-L. Yang Y.-C. Li T.-K. Huang W.-J. Guh J.-H. Biochem. Pharmacol. 2014;90:320–330. doi: 10.1016/j.bcp.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Hande K. R. Eur. J. Cancer. 1998;34:1514–1521. doi: 10.1016/S0959-8049(98)00228-7. [DOI] [PubMed] [Google Scholar]
- Kim M. S. Blake M. Baek J. H. Kohlhagen G. Pommier Y. Carrier F. Cancer Res. 2003;63:7291–7300. [PubMed] [Google Scholar]
- Zhang X. Bao B. Yu X. Tong L. Luo Y. Huang Q. Su M. Sheng L. Li J. Zhu H. Yang B. Zhang X. Chen Y. Lu W. Bioorg. Med. Chem. 2013;21:6981–6995. doi: 10.1016/j.bmc.2013.09.023. [DOI] [PubMed] [Google Scholar]
- He S. Dong G. Wang Z. Chen W. Huang Y. Li Z. Jiang Y. Liu N. Yao J. Miao Z. Zhang W. Sheng C. ACS Med. Chem. Lett. 2015;6:239–243. doi: 10.1021/ml500327q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis E. S. Lu C. Luber B. Wang H. Chen Y. Nakazawa M. Nadal R. Paller C. J. Denmeade S. R. Carducci M. A. Eisenberger M. A. Luo J. JAMA Oncol. 2015;1:582–591. doi: 10.1001/jamaoncol.2015.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. E. Li J. Xu H. E. Melcher K. Yong E. Acta Pharmacol. Sin. 2015;36:3–23. doi: 10.1038/aps.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel C. Schnekenburger M. Mazumder A. Teiten M.-H. Kirsch G. Dicato M. Diederich M. Biochem. Pharmacol. 2016;99:31–52. doi: 10.1016/j.bcp.2015.11.005. [DOI] [PubMed] [Google Scholar]
- Ai J. Wang Y. Dar J. A. Liu J. Liu L. Nelson J. B. Wang Z. Mol. Endocrinol. 2009;23:1963–1972. doi: 10.1210/me.2009-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrocco D. L. Tilley W. D. Bianco-Miotto T. Evdokiou A. Scher H. I. Rifkind R. A. Marks P. A. Richon V. M. Butler L. M. Mol. Cancer Ther. 2007;6:51–60. doi: 10.1158/1535-7163.MCT-06-0144. [DOI] [PubMed] [Google Scholar]
- Gryder B. E. Akbashev M. J. Rood M. K. Raftery E. D. Meyers W. M. Dillard P. Khan S. Oyelere A. K. ACS Chem. Biol. 2013;8:2550–2560. doi: 10.1021/cb400542w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati R. Chen B. Patki M. McFall T. Ou S. Heath E. Ratnam M. Qin Z. Mol. Pharmacol. 2016;90:225–237. doi: 10.1124/mol.116.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhavar P. S. Ramachandran S. A. Riquelme E. Gupta A. Quinn K. P. Shivakumar D. Ray S. Zende D. Nayak A. K. Miglani S. K. Sathe B. D. Raja M. Farias O. Alfaro I. Belmar S. Guerrero J. Bernales S. Chakravarty S. Hung D. T. Lindquist J. N. Rai R. Bioorg. Med. Chem. Lett. 2016;26:5222–5228. doi: 10.1016/j.bmcl.2016.09.058. [DOI] [PubMed] [Google Scholar]
- Mahalingam D. Swords R. Carew J. S. Nawrocki S. T. Bhalla K. Giles F. J. Br. J. Cancer. 2009;100:1523–1529. doi: 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L. Lindquist S. L. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Bali P. Pranpat M. Bradner J. Balasis M. Fiskus W. Guo F. Rocha K. Kumaraswamy S. Boyapalle S. Atadja P. Seto E. Bhalla K. J. Biol. Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- Rao R. Fiskus W. Yang Y. Lee P. Joshi R. Fernandez P. Mandawat A. Atadja P. Bradner J. E. Bhalla K. Blood. 2008;112:1886–1893. doi: 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- Kim S. H. Kang J. G. Kim C. S. Ihm S.-H. Choi M. G. Yoo H. J. Lee S. J. Endocrine. 2016;51:274–282. doi: 10.1007/s12020-015-0706-7. [DOI] [PubMed] [Google Scholar]
- George P. Bali P. Annavarapu S. Scuto A. Fiskus W. Guo F. Sigua C. Sondarva G. Moscinski L. Atadja P. Bhalla K. Blood. 2005;105:1768–1776. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- Rahmani M. Yu C. Dai Y. Reese E. Ahmed W. Dent P. Grant S. Cancer Res. 2003;63:8420–8427. [PubMed] [Google Scholar]
- Ojha R. Huang H.-L. HuangFu W.-C. Wu Y.-W. Nepali K. Lai M.-J. Su C.-J. Sung T.-Y. Chen Y.-L. Pan S.-L. Liou J.-P. Eur. J. Med. Chem. 2018;150:667–677. doi: 10.1016/j.ejmech.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Brooks A. N. Kilgour E. Smith P. D. Clin. Cancer Res. 2012;18:1855–1862. doi: 10.1158/1078-0432.CCR-11-0699. [DOI] [PubMed] [Google Scholar]
- Putcha P. Yu J. Rodriguez-Barrueco R. Saucedo-Cuevas L. Villagrasa P. Murga-Penas E. Quayle S. N. Yang M. Castro V. Llobet-Navas D. Birnbaum D. Finetti P. Woodward W. A. Bertucci F. Alpaugh M. L. Califano A. Silva J. Breast Cancer Res. 2015;17:149. doi: 10.1186/s13058-015-0658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. Cai X. Wu C. Liu Y. Zhang J. Gong X. Wang X. Wu X. Zhu T. Mo L. Gu J. Yu Z. Chen J. Thiery J. P. Chai R. Chen L. Int. J. Biol. Sci. 2017;13:505–517. doi: 10.7150/ijbs.18834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Qian C. Zhu Y. Cai J. He Y. Li J. Wang T. Zhu H. Li Z. Li W. Hu L. Bioorg. Med. Chem. 2018;26:747–757. doi: 10.1016/j.bmc.2017.12.041. [DOI] [PubMed] [Google Scholar]
- Jordan V. C. J. Med. Chem. 2003;46:883–908. doi: 10.1021/jm020449y. [DOI] [PubMed] [Google Scholar]
- Jordan V. C. J. Med. Chem. 2003;46:1081–1111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- Jameera Begam A. Jubie S. Nanjan M. J. Bioorg. Chem. 2017;71:257–274. doi: 10.1016/j.bioorg.2017.02.011. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen B. S. Katzenellenbogen J. A. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- Shang Y. Brown M. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Clarke R. Liu M. C. Bouker K. B. Gu Z. Lee R. Y. Zhu Y. Skaar T. C. Gomez B. O'Brien K. Wang Y. Hilakivi-Clarke L. A. Oncogene. 2003;22:7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- Rao R. Balusu R. Fiskus W. Mudunuru U. Venkannagari S. Chauhan L. Smith J. E. Hembruff S. L. Ha K. Atadja P. Bhalla K. N. Mol. Cancer Ther. 2012;11:973–983. doi: 10.1158/1535-7163.MCT-11-0979. [DOI] [PubMed] [Google Scholar]
- Hodges-Gallagher L. Valentine C. D. Bader S. E. Kushner P. J. Breast Cancer Res. Treat. 2007;105:297–309. doi: 10.1007/s10549-006-9459-6. [DOI] [PubMed] [Google Scholar]
- Kawai H. Li H. Avraham S. Jiang S. Avraham H. K. Int. J. Cancer. 2003;107:353–358. doi: 10.1002/ijc.11403. [DOI] [PubMed] [Google Scholar]
- Reid G. Métivier R. Lin C.-Y. Denger S. Ibberson D. Ivacevic T. Brand H. Benes V. Liu E. T. Gannon F. Oncogene. 2005;24:4894–4907. doi: 10.1038/sj.onc.1208662. [DOI] [PubMed] [Google Scholar]
- Tang C. Li C. Zhang S. Hu Z. Wu J. Dong C. Huang J. Zhou H.-B. J. Med. Chem. 2015;58:4550–4572. doi: 10.1021/acs.jmedchem.5b00099. [DOI] [PubMed] [Google Scholar]
- Saji S. Kawakami M. Hayashi S. Yoshida N. Hirose M. Horiguchi S. Itoh A. Funata N. Schreiber S. L. Yoshida M. Toi M. Oncogene. 2005;24:4531–4539. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- Jackson S. E. Chester J. D. Int. J. Cancer. 2015;137:262–266. doi: 10.1002/ijc.28940. [DOI] [PubMed] [Google Scholar]
- Bai P. Mol. Cell. 2015;58:947–958. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- Lord C. J. Ashworth A. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Han Y. Jiang Q. Wang C. Chen X. Li X. Xu F. Jiang Y. Wang Q. Xu W. Med. Res. Rev. 2015;35:63–84. doi: 10.1002/med.21320. [DOI] [PubMed] [Google Scholar]
- Curtin N. J. Szabo C. Mol. Aspects Med. 2013;34:1217–1256. doi: 10.1016/j.mam.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K. Fiskus W. Choi D. S. Bhaskara S. Cerchietti L. Devaraj S. G. T. Shah B. Sharma S. Chang J. C. Melnick A. M. Hiebert S. Bhalla K. N. Oncotarget. 2014;5:5637–5650. doi: 10.18632/oncotarget.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao O. S. Goodman O. B. Mol. Cancer Res. 2014;12:1755–1766. doi: 10.1158/1541-7786.MCR-14-0173. [DOI] [PubMed] [Google Scholar]
- Min A. Im S.-A. Kim D. K. Song S.-H. Kim H.-J. Lee K.-H. Kim T.-Y. Han S.-W. Oh D.-Y. Kim T.-Y. O'Connor M. J. Bang Y.-J. Breast Cancer Res. 2015;17:33. doi: 10.1186/s13058-015-0534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos P. A. Wilson A. J. Saskowski J. Wass E. Khabele D. Gynecol. Oncol. 2014;133:599–606. doi: 10.1016/j.ygyno.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z. Chen S. Sun Q. Wang N. Li D. Miao S. Gao C. Chen Y. Tan C. Jiang Y. Bioorg. Med. Chem. 2017;25:4100–4109. doi: 10.1016/j.bmc.2017.05.058. [DOI] [PubMed] [Google Scholar]
- Harrison D. A. Cold Spring Harbor Perspect. Biol. 2012;4:a011205. doi: 10.1101/cshperspect.a011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz D. M. Kanno Y. Villarino A. Ward M. Gadina M. O'Shea J. J. Nat. Rev. Drug Discovery. 2017;17:78. doi: 10.1038/nrd.2017.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa R. A. Yasothan U. Kirkpatrick P. Nat. Rev. Drug Discovery. 2012;11:103–104. doi: 10.1038/nrd3652. [DOI] [PubMed] [Google Scholar]
- Shreenivas A. Mascarenhas J. Expert Opin. Emerging Drugs. 2018;23:37–49. doi: 10.1080/14728214.2018.1445718. [DOI] [PubMed] [Google Scholar]
- Dickinson M. Prince H. M. Expert Opin. Emerging Drugs. 2014;19:201–213. doi: 10.1517/14728214.2014.896337. [DOI] [PubMed] [Google Scholar]
- Evrot E. Ebel N. Romanet V. Roelli C. Andraos R. Qian Z. Dölemeyer A. Dammassa E. Sterker D. Cozens R. Hofmann F. Murakami M. Baffert F. Radimerski T. Clin. Cancer Res. 2013;19:6230–6241. doi: 10.1158/1078-0432.CCR-13-0905. [DOI] [PubMed] [Google Scholar]
- Novotny-Diermayr V. Hart S. Goh K. C. Cheong A. Ong L.-C. Hentze H. Pasha M. K. Jayaraman R. Ethirajulu K. Wood J. M. Blood Cancer J. 2012;2:e69. doi: 10.1038/bcj.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. Fiskus W. Chong D. G. Buckley K. M. Natarajan K. Rao R. Joshi A. Balusu R. Koul S. Chen J. Savoie A. Ustun C. Jillella A. P. Atadja P. Levine R. L. Bhalla K. N. Blood. 2009;114:5024–5033. doi: 10.1182/blood-2009-05-222133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E. G. Mustafa N. Tan E. C. Poulsen A. Ramanujulu P. M. Chng W. J. Yen J. J. Y. Dymock B. W. J. Med. Chem. 2016;59:8233–8262. doi: 10.1021/acs.jmedchem.6b00157. [DOI] [PubMed] [Google Scholar]
- Yao L. Mustafa N. Tan E. C. Poulsen A. Singh P. Duong-Thi M.-D. Lee J. X. T. Ramanujulu P. M. Chng W. J. Yen J. J. Y. Ohlson S. Dymock B. W. J. Med. Chem. 2017;60:8336–8357. doi: 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- Chu-Farseeva Y. Mustafa N. Poulsen A. Tan E. C. Yen J. J. Y. Chng W. J. Dymock B. W. Eur. J. Med. Chem. 2018;158:593–619. doi: 10.1016/j.ejmech.2018.09.024. [DOI] [PubMed] [Google Scholar]
- Huang Y. Dong G. Li H. Liu N. Zhang W. Sheng C. J. Med. Chem. 2018;61:6056–6074. doi: 10.1021/acs.jmedchem.8b00393. [DOI] [PubMed] [Google Scholar]
- Burns C. J. Bourke D. G. Andrau L. Bu X. Charman S. A. Donohue A. C. Fantino E. Farrugia M. Feutrill J. T. Joffe M. Kling M. R. Kurek M. Nero T. L. Nguyen T. Palmer J. T. Phillips I. Shackleford D. M. Sikanyika H. Styles M. Su S. Treutlein H. Zeng J. Wilks A. F. Bioorg. Med. Chem. Lett. 2009;19:5887–5892. doi: 10.1016/j.bmcl.2009.08.071. [DOI] [PubMed] [Google Scholar]
- Liang X. Zang J. Li X. Tang S. Huang M. Geng M. Chou C. J. Li C. Cao Y. Xu W. Liu H. Zhang Y. J. Med. Chem. 2019;62:3898–3923. doi: 10.1021/acs.jmedchem.8b01597. [DOI] [PubMed] [Google Scholar]
- Lai A. C. Crews C. M. Nat. Rev. Drug Discovery. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd M. S. Testa A. Lucas X. Chan K.-H. Chen W. Lamont D. J. Zengerle M. Ciulli A. Nat. Chem. Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. Song Y. Xie H. Wu H. Wu Y.-T. Leisten E. D. Tang W. Bioorg. Med. Chem. Lett. 2018;28:2493–2497. doi: 10.1016/j.bmcl.2018.05.057. [DOI] [PubMed] [Google Scholar]
- Wu H. Yang K. Zhang Z. Leisten E. D. Li Z. Xie H. Liu J. Smith K. A. Novakova Z. Barinka C. Tang W. J. Med. Chem. 2019;62:7042–7057. doi: 10.1021/acs.jmedchem.9b00516. [DOI] [PubMed] [Google Scholar]
- Krönke J. Udeshi N. D. Narla A. Grauman P. Hurst S. N. McConkey M. Svinkina T. Heckl D. Comer E. Li X. Ciarlo C. Hartman E. Munshi N. Schenone M. Schreiber S. L. Carr S. A. Ebert B. L. Science. 2014;343:301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E. S. Böhm K. Lydeard J. R. Yang H. Stadler M. B. Cavadini S. Nagel J. Serluca F. Acker V. Lingaraju G. M. Tichkule R. B. Schebesta M. Forrester W. C. Schirle M. Hassiepen U. Ottl J. Hild M. Beckwith R. E. J. Harper J. W. Jenkins J. L. Thomä N. H. Nature. 2014;512:49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G. Middleton R. E. Sun H. Naniong M. Ott C. J. Mitsiades C. S. Wong K.-K. Bradner J. E. Kaelin W. G. Science. 2014;343:305–309. doi: 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z. Lv W. Su S. Wu W. Rao Y. Protein Cell. 2019;10:606–609. doi: 10.1007/s13238-018-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck P. Dubiella C. Groll M. Biol. Chem. 2012;393:1101–1120. doi: 10.1515/hsz-2012-0212. [DOI] [PubMed] [Google Scholar]
- Kubiczkova L. Pour L. Sedlarikova L. Hajek R. Sevcikova S. J. Cell. Mol. Med. 2014;18:947–961. doi: 10.1111/jcmm.12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K. Proc. Jpn. Acad., Ser. B. 2009;85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideshima T. Richardson P. G. Anderson K. C. Mol. Cancer Ther. 2011;10:2034–2042. doi: 10.1158/1535-7163.MCT-11-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl D. T. Raje N. Jagannath S. Richardson P. Hari P. Orlowski R. Supko J. G. Tamang D. Yang M. Jones S. S. Wheeler C. Markelewicz R. J. Lonial S. Clin. Cancer Res. 2017;23:3307–3315. doi: 10.1158/1078-0432.CCR-16-2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia S. Krieger V. Groll M. Osko J. D. Reßing N. Ahlert H. Borkhardt A. Kurz T. Christianson D. W. Hauer J. Hansen F. K. J. Med. Chem. 2018;61:10299–10309. doi: 10.1021/acs.jmedchem.8b01487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. Liu X. Xue J. Liu L. Liang T. Li W. Yang X. Hou X. Fang H. J. Med. Chem. 2020;63:4701–4715. doi: 10.1021/acs.jmedchem.9b02161. [DOI] [PubMed] [Google Scholar]
- C. for D. E. and Research, FDA, https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults
- Souers A. J. Leverson J. D. Boghaert E. R. Ackler S. L. Catron N. D. Chen J. Dayton B. D. Ding H. Enschede S. H. Fairbrother W. J. Huang D. C. S. Hymowitz S. G. Jin S. Khaw S. L. Kovar P. J. Lam L. T. Lee J. Maecker H. L. Marsh K. C. Mason K. D. Mitten M. J. Nimmer P. M. Oleksijew A. Park C. H. Park C.-M. Phillips D. C. Roberts A. W. Sampath D. Seymour J. F. Smith M. L. Sullivan G. M. Tahir S. K. Tse C. Wendt M. D. Xiao Y. Xue J. C. Zhang H. Humerickhouse R. A. Rosenberg S. H. Elmore S. W. Nat. Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Chen S. Dai Y. Pei X.-Y. Grant S. Mol. Cell. Biol. 2009;29:6149–6169. doi: 10.1128/MCB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews G. M. Lefebure M. Doyle M. A. Shortt J. Ellul J. Chesi M. Banks K.-M. Vidacs E. Faulkner D. Atadja P. Bergsagel P. L. Johnstone R. W. Cell Death Dis. 2013;4:e798. doi: 10.1038/cddis.2013.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. Zhang Y. Zhou L. Leng Y. Lin H. Kmieciak M. Pei X.-Y. Jones R. Orlowski R. Z. Dai Y. Grant S. Blood. 2014;124:2687–2697. doi: 10.1182/blood-2014-03-564534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R. Fang S. Zhang M. Zhang Q. Hu J. Wang M. Wang C. Zhu J. Shen A. Chen X. Zheng C. Bioorg. Med. Chem. Lett. 2019;29:349–352. doi: 10.1016/j.bmcl.2018.12.052. [DOI] [PubMed] [Google Scholar]
- Yang Z. Wang T. Wang F. Niu T. Liu Z. Chen X. Long C. Tang M. Cao D. Wang X. Xiang W. Yi Y. Ma L. You J. Chen L. J. Med. Chem. 2016;59:1455–1470. doi: 10.1021/acs.jmedchem.5b01342. [DOI] [PubMed] [Google Scholar]
- Wang F. Zheng L. Yi Y. Yang Z. Qiu Q. Wang X. Yan W. Bai P. Yang J. Li D. Pei H. Niu T. Ye H. Nie C. Hu Y. Yang S. Wei Y. Chen L. Mol. Cancer Ther. 2018;17:763–775. doi: 10.1158/1535-7163.MCT-17-0332. [DOI] [PubMed] [Google Scholar]
- Chobanian N. H. Greenberg V. L. Gass J. M. Desimone C. P. Nagell J. R. V. Zimmer T. G. Anticancer Res. 2004;24:539–546. [PubMed] [Google Scholar]
- Chao M.-W. Lai M.-J. Liou J.-P. Chang Y.-L. Wang J.-C. Pan S.-L. Teng C.-M. J. Hematol. Oncol. 2015;8:82. doi: 10.1186/s13045-015-0176-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. Kong Y. Zhang J. Su M. Zhou Y. Zang Y. Li J. Chen Y. Fang Y. Zhang X. Lu W. Eur. J. Med. Chem. 2015;95:127–135. doi: 10.1016/j.ejmech.2015.03.035. [DOI] [PubMed] [Google Scholar]
- Lamaa D. Lin H.-P. Zig L. Bauvais C. Bollot G. Bignon J. Levaique H. Pamlard O. Dubois J. Ouaissi M. Souce M. Kasselouri A. Saller F. Borgel D. Jayat-Vignoles C. Al-Mouhammad H. Feuillard J. Benihoud K. Alami M. Hamze A. J. Med. Chem. 2018;61:6574–6591. doi: 10.1021/acs.jmedchem.8b00050. [DOI] [PubMed] [Google Scholar]
- Wang B. Chen X. Gao J. Su L. Zhang L. Xu H. Luan Y. Bioorg. Med. Chem. Lett. 2019;29:2638–2645. doi: 10.1016/j.bmcl.2019.07.045. [DOI] [PubMed] [Google Scholar]
- Lee H.-Y. Lee J.-F. Kumar S. Wu Y.-W. HuangFu W.-C. Lai M.-J. Li Y.-H. Huang H.-L. Kuo F.-C. Hsiao C.-J. Cheng C.-C. Yang C.-R. Liou J.-P. Eur. J. Med. Chem. 2017;125:1268–1278. doi: 10.1016/j.ejmech.2016.11.033. [DOI] [PubMed] [Google Scholar]
- Wu Y.-W. Hsu K.-C. Lee H.-Y. Huang T.-C. Lin T. E. Chen Y.-L. Sung T.-Y. Liou J.-P. Hwang-Verslues W. W. Pan S.-L. HuangFu W.-C. Front. Pharmacol. 2018;9:205. doi: 10.3389/fphar.2018.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]