

Abstract
Purpose:
The methylation status of the O6-methylguanine DNA methyltransferase (MGMT) gene promoter is predictive for benefit from temozolomide in glioblastoma (GBM). A clinically optimized cutoff was sought allowing patient selection for therapy without temozolomide, while avoiding to withhold it from patients who may potentially benefit.
Experimental Design:
Quantitative MGMT methylation-specific PCR data were obtained for newly diagnosed patients with GBM screened or treated with standard radiotherapy and temozolomide in four randomized trials. The pooled dataset was randomly split into a training and test dataset. The unsupervised cutoff was obtained at a 50% probability to be (un)methylated. ROC analysis identified an optimal cutoff supervised by overall survival (OS).
Results:
For 4,041 patients valid MGMT results were obtained, whereof 1,725 were randomized. The unsupervised cutoff in the training dataset was 1.27 (log2[1,000 × (MGMT+1)/ACTB]), separating unmethylated and methylated patients. The optimal supervised cutoff for unmethylated patients was −0.28 (AUC = 0.61), classifying “truly unmethylated” (≤−0.28) and “gray zone” patients (>−0.28, ≤1.27), the latter comprising approximately 10% of cases. In contrast, for patients with MGMT methylation (>1.27) more methylation was not related to better outcome. Both methylated and gray zone patients performed significantly better for OS than truly unmethylated patients [HR = 0.35, 95% confidence interval (CI), 0.27–0.45, P < 0.0001; HR = 0.58, 95% CI, 0.43–0.78, P < 0.001], validated in the test dataset. The MGMT assay was highly reproducible upon retesting of 218 paired samples (R2 = 0.94).
Conclusions:
Low MGMT methylation (gray zone) may confer some sensitivity to temozolomide treatment, hence the lower safety margin should be considered for selecting patients with unmethylated GBM into trials omitting temozolomide.
Introduction
A predictive role has been shown for the methylation status of the O6-methylguanine DNA methyltransferase (MGMT) gene promoter for benefit from temozolomide (TMZ) in patients with newly diagnosed glioblastoma (GBM) (1–4). Consequently, the MGMT methylation status is used as a stratification factor in trials comprising temozolomide treatment. All contemporary trials have confirmed the strong prognostic role of the MGMT status in patients with GBM treated with the combination of radiation concurrent with temozolomide, followed by maintenance temozolomide (TMZ/RT ⟶ TMZ; refs. 1, 5–10). The lack of efficacy of temozolomide in MGMT unmethylated GBM warrants replacement with an agent with a different mechanisms of action, or omission of temozolomide to avoid futile therapy and associated toxicity. Trials specifically designed to selecting only patients with MGMT unmethylated GBM and replacing temozolomide in the experimental arm are becoming a common strategy in clinical research (refs. 11–14; CheckMate 498, NCT02617589 and N2M2, NCT03158389). However, the best way of assessing the MGMT promoter methylation status remains strongly debated (15). It remains unclear which pattern and extent of MGMT promoter methylation is required to prevent MGMT-mediated DNA repair that sensitizes patients with GBM to alkylating agent chemotherapy. A correlation of the extent of MGMT promoter methylation with outcome in patients treated with temozolomide chemo-radiotherapy has been suggested (16, 17). Accurate and reproducible assays with clinically relevant cutoffs are required, in order not to withhold temozolomide from patients who may potentially benefit, while sparing others from unnecessary toxicity and cost.
In this analysis we aim at revisiting the MGMT methylation cutoff using the pooled datasets of recent prospective randomized clinical trials, which had used the same quantitative, methylation-specific PCR (qMSP) MGMT assay (18,19) and had delivered the identical backbone treatment of TMZ/RT ⟶ TMZ to patients with newly diagnosed GBM. These combined datasets provide the unique opportunity to explore and validate the relationship between the extent of MGMT promoter methylation and overall survival (OS).
The specific goals of this research project are (i) reevaluation of the technical (unsupervised) cutoff that discriminates methylated and unmethylated patients, whereby patients have a 50% probability to be methylated or unmethylated, (ii) definition of an optimal cutoff for patients with GBM, supervised by OS, (iii) validation of the findings in an independent test dataset, (iv) evaluation of the assay reproducibility, and finally (v) comparison with the current assay-based classification used in routine diagnostics. The overarching goal is to provide one or more cutoffs that allow treatment decisions for personalized therapy and appropriate selection of patients into clinical trials omitting temozolomide.
Materials and Methods
Data selection
Quantitative MGMT promoter methylation data was obtained from four trials for newly diagnosed GBM, with central MGMT testing by the same qMSP assay, applying the same cutoff [1 in log2 space; ] (18,19), and using the standard TMZ/RT⟶TMZ schedule as backbone treatment (5). Patients with available MGMT classification (n = 4,458) had been randomized into (i) the control arm of the phase III AVAGlio trial (n = 472, NCT00943826; ref. 8), (ii) the control or experimental arm of the RTOG 0825 phase III trial (n = 621, NCT00884741; ref. 7), (iii) the control or experimental arms of the CENTRIC (phase III) or CORE (phase II) trials that selected patients with a methylated or unmethylated MGMT promoter, respectively (n = 545, CENTRIC NCT00689221; n = 265, CORE NCT00813943; refs. 18, 20); or (iv) patients who were screened, but neither randomized in CENTRIC (n = 2,328) nor CORE (n = 227). All four selected trials failed to demonstrate improvement in OS of the experimental arm based on HRs as reported. For randomized patients, survival data and baseline information with respect to age, extent of surgery, and performance status were available. Data can be applied for via the following weblink: http://www.eortc.be/services/forms/erp/request.aspx.
Constitution of training and test cohort
For this analysis, only samples passing the quality threshold for providing a “valid” test result (≥1,250 copies of the normalizer gene β-actin, ACTB) were considered. This all patients (all-P) population included both randomized and screened patients, whereas the randomized patients (rand-P) population was a subset of the all-P population. The data was randomly split into a training and a test cohort, stratified for trial, extent of resection (complete resection, partial resection, biopsy only, other), and performance status (PS = 0, PS ≥ 1). The all-P training cohort was used for the unsupervised analyses, while the rand-P training cohort was used for the supervised analyses of the relationship between the extent of MGMT methylation and OS. Validation of the findings was performed in the all-P and rand-P test cohorts, respectively.
Retest dataset
A cohort of patients was selected randomly among patients screened but not randomized for CENTRIC. Retest tissue sections had been set aside for this purpose as of protocol, if enough tissue was available. Sample identifiers of retest tissue sections were blinded (relabeled). The initial MGMT testing was performed at the certified MDxHealth site in Liège, Belgium and retesting took place at their laboratory in Irvine, CA. Only samples with valid ACTB results in both the original and retest data were selected.
All protocols were approved by the local ethics committees or institutional review boards and competent authorities, and patients provided written informed consent for trial participation and/or participation in marker screening including retesting. The trials were performed according to the guidelines of Helsinki (21).
qMSP MGMT assay
The qMSP MGMT test was performed and evaluated essentially as described previously (18, 19) and is commercially available (PredictMDx test). In brief, DNA was isolated from sections of macro-dissected formalin-fixed, paraffin-embedded (FFPE) tumor tissue. After bisulfite treatment the copy numbers of methylated MGMT and the reference gene ACTB were determined by quantitative PCR. A valid test required a minimum of 1,250 ACTB copies measured.
For this study the calculation of the ratio of the MGMT and ACTB copy numbers was slightly modified as compared with the original procedure (19) by adding one MGMT copy to the numerator: . The result is termed corrected MGMT log2 ratio hereafter. Samples with zero MGMT copies would otherwise be lost after logarithmic transformation. For the calculations the original MGMT values were used, ignoring the technical limit of detection of the assay set at ≥10 copies of methylated MGMT.
Determination of the unsupervised cutoff and MGMT methylation status
We applied a bimodal Gaussian mixture distribution to model the corrected MGMT log2 ratio. The unsupervised cutoff in the all-P training cohort, defined as the 50% probability to be (un)methylated, was used to classify patients as unmethylated (≤cutoff point) or methylated (>cutoff point). The same cutoff was used to classify the patients in the test cohort.
Determination of the optimal cutoff supervised by OS
To identify an optimal cutoff point supervised by OS in both unmethylated and methylated patients in the rand-P training cohort, time-dependent ROC analysis with nearest neighbor estimation was used (22). OS predictions at 2 years were made in both groups. The optimal supervised cutoff point was chosen as the value that maximized the Youden index, if the AUC was >0.6, otherwise no cutoff point was determined. The optimal supervised cutoff point was used to classify patients further, both in the training and test populations.
Statistical analysis
The all-P and rand-P training and test cohorts were compared using descriptive statistics. Categorical variables are presented as frequencies and percentages. Continuous variables are described by their median and interquartile range. Initial comparison of OS by MGMT status was performed using Kaplan–Meier plots accompanied by a log-rank test.
A univariate Cox model assessed the effect of MGMT methylation status on OS, whereas a multivariate Cox model was used for sensitivity purposes. All survival analyses were stratified by trial. Statistical significance was determined at the two-sided 5% significance level.
Assay MGMT methylation status reproducibility and comparison with the original procedure was quantified using Cohen Kappa coefficient. The original procedure uses the uncorrected MGMT log2 ratio with a cutoff of one and a lower safety margin of −0.75. This cutoff was based on 602 patient samples from CENTRIC, and the lower safety margin was set at the lower bound 95% confidence interval (CI) of being unmethylated as described previously (12). A limit of detection of the diagnostic assay was also applied that sets <10 methylated MGMT copies to unmethylated.
All analyses were carried out in R version 3.3.0.
Results
Descriptive analyses
Valid qMSP MGMT results were available for 4,041 patient samples (all-P population;90.6% of all available samples), consisting of 2,316 patients screened only (57.3%) and 1,725 randomized patients (rand-P population, 42.7%;Fig. 1). Only for the latter, full treatment and survival outcome data were available. The all-P population was randomly split into training and test cohort stratified for trial and clinical factors, respectively, comprising 2,021 and 2,020 patients. The rand-P training and test cohorts contained 863 and 862 patients, respectively. The origin of the patients (trial) and baseline characteristics are summarized in Table 1 and were balanced between cohorts.
Figure 1.
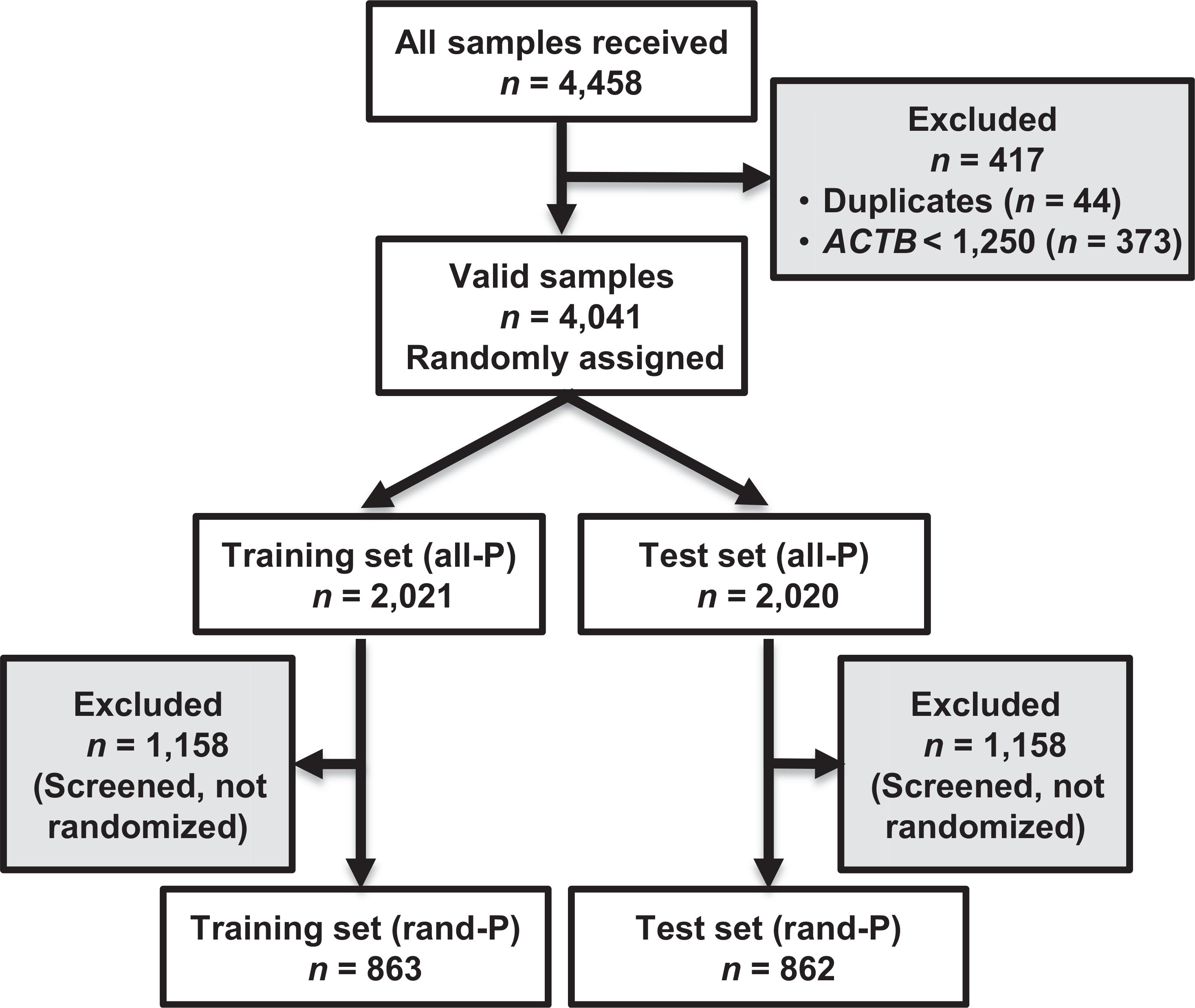
Flow of patient samples through the study. all-P, All patients population; rand-P, randomized patient population; ACTB, β-actin gene.
Table 1.
Patient origin (trial) and baseline characteristics of split datasets
| Training cohort (%) |
Test cohort (%) |
|||
|---|---|---|---|---|
| all-P (n = 2,021) | rand-P (n = 863) | all-P (n = 2,020) | rand-P (n = 862) | |
| Trial | ||||
| CENTRIC (randomized) | 264 (13.1) | 264 (30.6) | 262 (13.0) | 262 (30.4) |
| CENTRIC (screened) | 1,058 (52.4) | NA | 1,058 (52.4) | NA |
| CORE (randomized) | 132 (6.5) | 132 (15.3) | 133 (6.6) | 133 (15.4) |
| CORE (screened) | 100 (4.9) | NA | 100 (5.0) | NA |
| AVAGlio | 170 (8.4) | 170 (19.7) | 170 (8.4) | 170 (19.7) |
| RTOG 0825 | 297 (14.7) | 297 (34.4) | 297 (14.7) | 297 (34.5) |
| Baseline characteristics | ||||
| Performance status (randomized patients only) | ||||
| PS = 0 | NA | 516 (59.8) | NA | 514 (59.6) |
| PS ≥ 1 | NA | 347 (40.2) | NA | 345 (40.0) |
| Missing | NA | 0(0) | NA | 3 (0.4) |
| Extent of resection (randomized patients only) | ||||
| Complete resection | NA | 454 (52.6) | NA | 453 (52.6) |
| Partial resection | NA | 394 (45.7) | NA | 392 (45.5) |
| Biopsy only | NA | 6 (0.7) | NA | 7 (0.8) |
| Other | NA | 8 (0.9) | NA | 8 (0.9) |
| Missing | NA | 1 (0.1) | NA | 2 (0.2) |
| Age in years (randomized patients only) | ||||
| Median (Q1, Q3) | NA | 57 (50, 63) | NA | 57.5 (50, 64) |
| Corrected log2 MGMT ratio | ||||
| Median (Q1, Q3) | −0.58 (−2.00, 3.24) | 0.63 (−1.97, 4.77) | −0.58 (−1.96, 3.54). | 1.06 (−1.79, 4.94) |
Assay reproducibility
The reproducibility of the assay was evaluated in 218 paired sample sets with ACTB copies ≥ 1,250 in both the original and retest data. Retest values for the corrected MGMT log2 ratio were plotted in function of the original values (Fig. 2). The coefficient of determination (R2) was >93%, indicating that most of the variability in the retest data could be explained by the original data.
Figure 2.
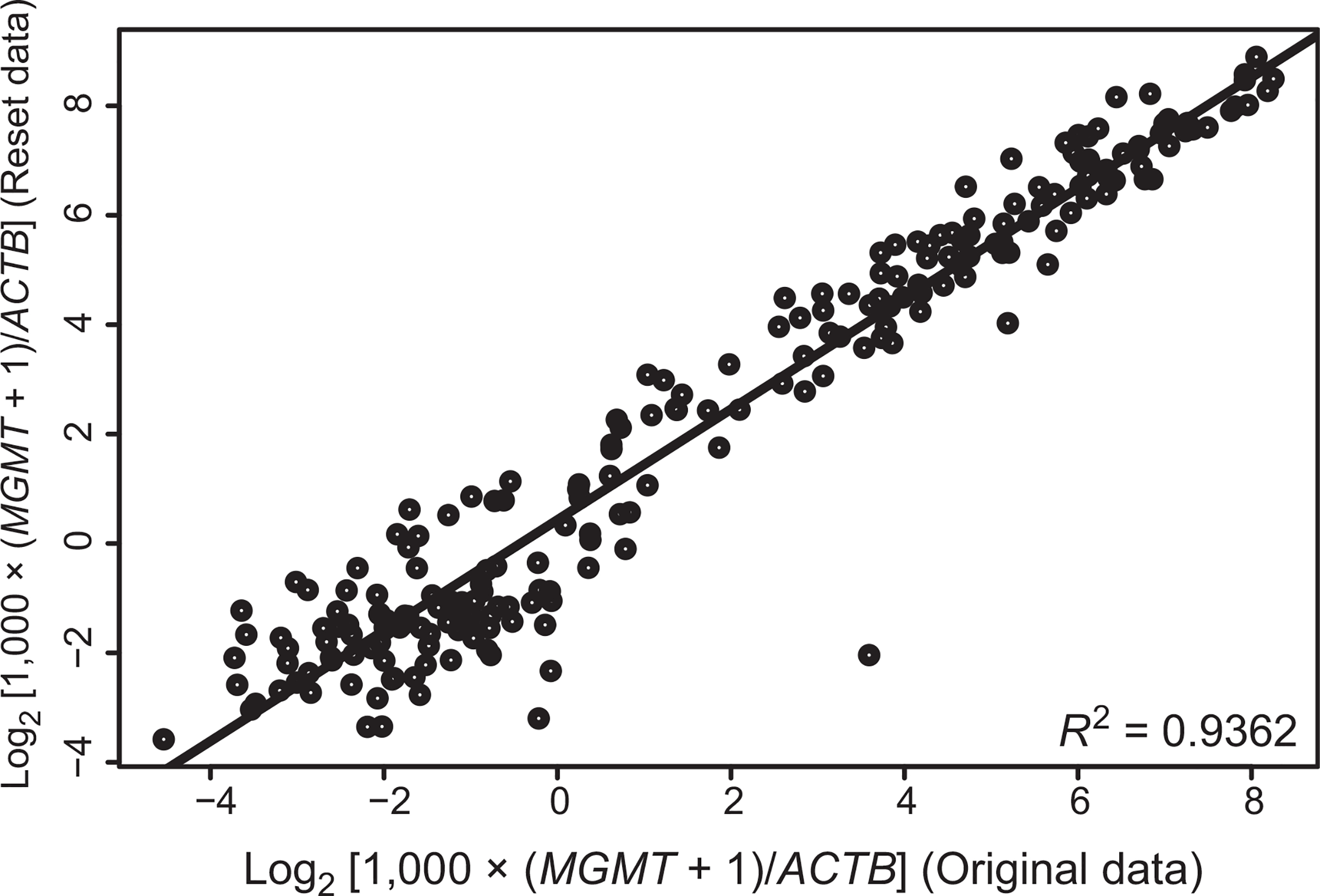
Reproducibility of qMSP MGMT assay. The original and retest dataset (corrected MGMT log2 ratios, ) from 218 paired samples are visualized in a scatter plot. The R2 was 93%. Retests were performed using a second set of FFPE tumor sections in a different laboratory blinded to the original results.
Unsupervised technical cutoff
The unsupervised cutoff for the corrected MGMT log2 ratio, separating methylated and unmethylated samples, was equal to 1.27 on the log2 scale (Fig. 3A). After assignment of the MGMT methylation status there were 1,332 unmethylated patients (65.9%) and 689 methylated patients (34.1%) in the all-P training cohort.
Figure 3.
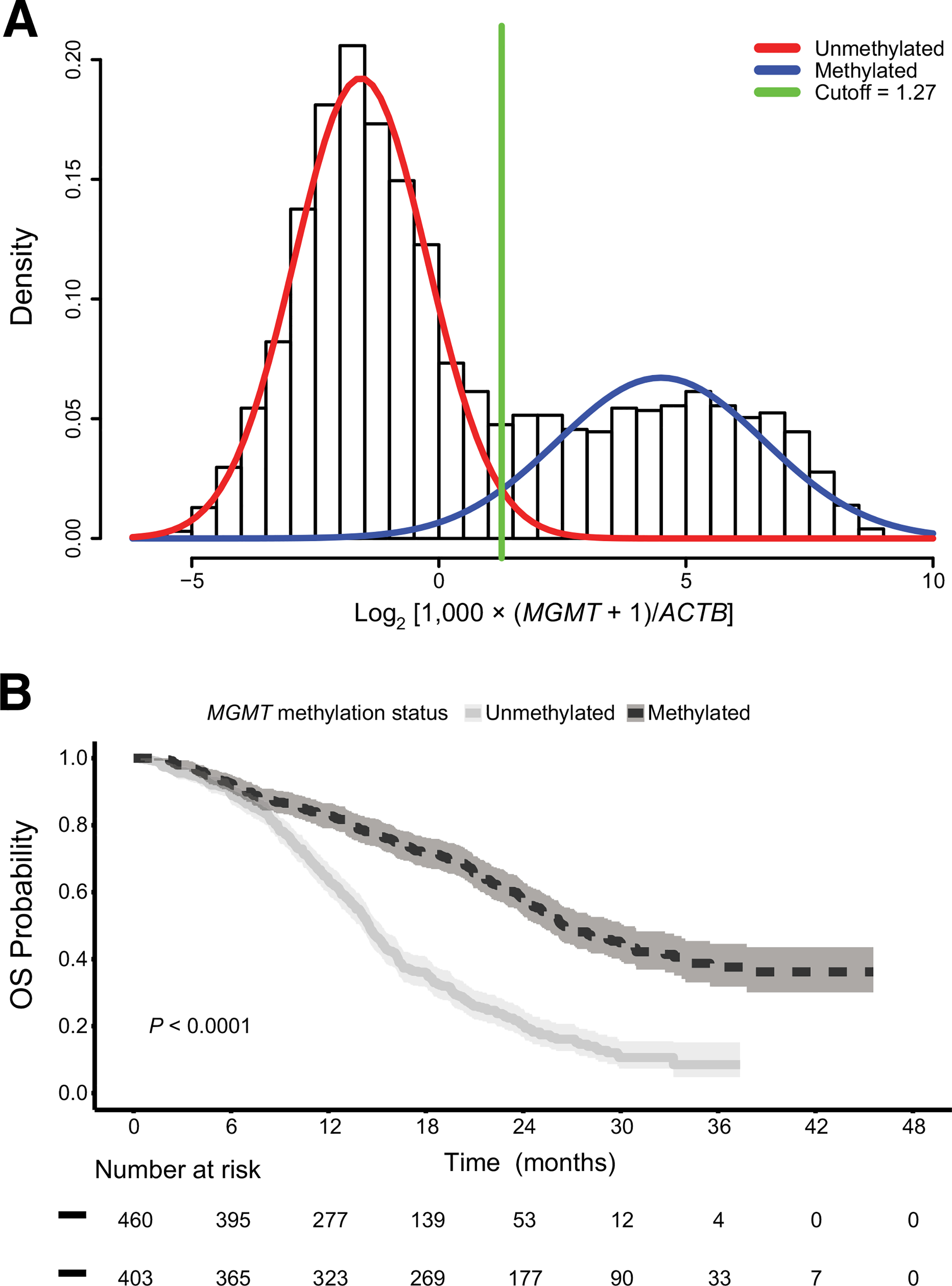
A, Unsupervised MGMT promoter methylation cutoff and OS. The unsupervised cutoff of 1.27 obtained in the all-P training cohort is indicated in green in the bimodal distribution of the corrected MGMT log2 ratio values . B, The Kaplan-Meier plot visualizes OS in the rand-P training cohort separated into patients with MGMT promoter-methylated and -unmethylated tumors (P < 0.0001, log-rank test). The shaded area represents the 95% CI.
Association between MGMT methylation status and OS
The median OS from randomization in the whole rand-P training cohort was 19.3 months (95% CI, 17.5–20.7). Baseline characteristics were balanced between the 460 MGMT unmethylated patients (53.3%) and 403 methylated patients (46.6%; Supplementary Table S1). Median OS was 14.5 months (95% CI, 14.0–15.3) and 26.5 months (95% CI, 25.1–30.2), respectively (Fig. 3B). MGMT-methylated patients had a significantly longer OS compared with unmethylated patients (HR = 0.39; 95% CI, 0.30–0.50; P < 0.0001, log-rank test). Similar results were obtained in the multivariate analysis (Supplementary Table S2).
Supervised optimal cutoff
For the unmethylated patients in the rand-P training cohort a time-dependent ROC curve with an AUC equal to 0.61 was obtained, resulting in an optimal cutoff point of −0.28 on the log2 scale (Supplementary Fig. S1A). This corresponds to a 96% probability of being unmethylated as visualized in Fig. 4A and B. In contrast, for the methylated patients the ROC curve yielded an AUC of 0.50, suggesting no association between extent of methylation and outcome (Supplementary Fig. S1B).
Figure 4.
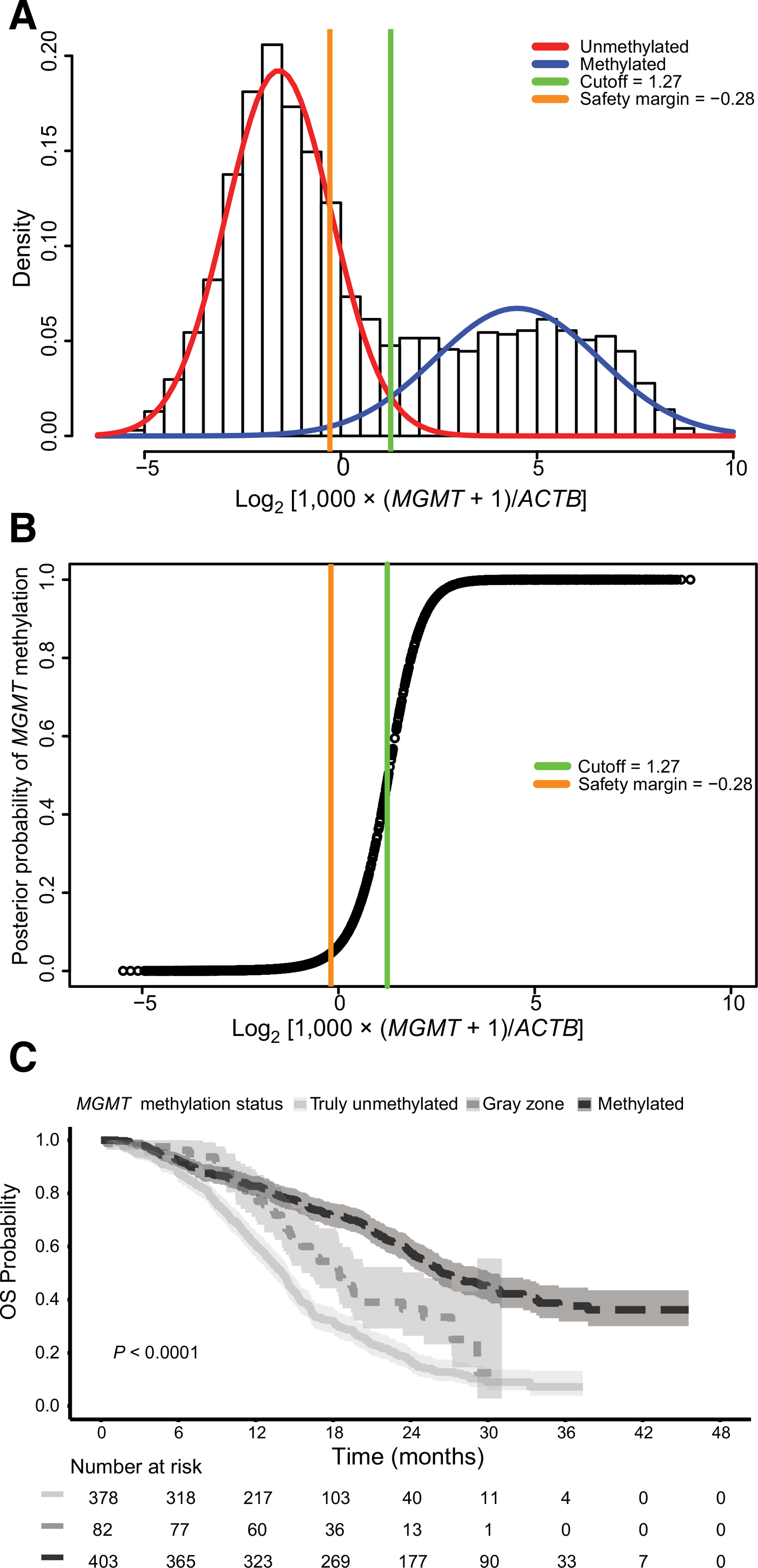
Optimal MGMT promoter methylation cutoff and OS. A, The position of the optimal cutoff point of −0.28 [corrected MGMT log2 ratio value, log2 ] is indicated in orange in the bimodal distribution of the entire all-P training cohort. It corresponds to a 96% chance to be unmethylated (4% chance to be methylated) as illustrated in the posterior probability plot (B) and defines the lower bound of the “gray zone” (−0.28, and ≤1.27). C, The Kaplan-Meier plot visualizes the outcome of patients in the randomized patient (rand-P) training cohort separated into MGMT promoter-methylated (<1.27), gray zone (—0.28, and <1.27), and truly unmethylated patients (<−0.28; P < 0.0001, log-rank test). The shaded area represents the 95% CI.
Lower safety margin and OS
The optimal supervised cutoff of −0.28 obtained in the unmethylated rand-P training subset was applied as a lower safety margin in the entire rand-P training cohort. The gray zone comprised of 82 patients (9.5%), while 378 patients (43.8%) were labeled as truly unmethylated. The Kaplan-Meier plot is displayed in Fig. 4C and the survival curves differed significantly according to the log-rank test (P < 0.0001). Univariate Cox regression analysis resulted in a HR of 0.35 (95% CI, 0.27–0.45; P < 0.0001) for the methylated patients, and a HR of 0.58 for patients in the gray zone (95% CI, 0.43–0.78; P < 0.001), respectively, when compared with the truly unmethylated patients. Similar results were obtained in the multivariate analysis (Table 2).
Table 2.
Outcome by MGMT promoter methylation status in the rand-P training and test cohorts
| Methylation status | N (%) | Observed events | Median survival (months) | HR (95% CI) | P | Adj. HR (95% CI)a | Padj |
|---|---|---|---|---|---|---|---|
| Training cohort | |||||||
| Truly unmethylated | 378 (43.8) | 302 | 14.0 (13.1–14.7) | 1.00 | – | 1.00 | – |
| Gray zone | 82 (9.5) | 50 | 18.5 (16.2–25.0) | 0.58 (0.43–0.78) | <0.001 | 0.64 (0.47–0.87) | <0.01 |
| Methylated | 403 (46.7) | 203 | 26.5 (25.1–30.2) | 0.35 (0.27–0.45) | <0.0001 | 0.32 (0.25–0.42) | <0.0001 |
| Test cohort | |||||||
| Truly unmethylated | 375 (43.5) | 299 | 13.6 (12.9–14.7) | 1.00 | 1.00 | ||
| Gray zone | 70 (8.1) | 46 | 16.5 (13.8–20.6) | 0.70 (0.51–0.96) | 0.03 | 0.71 (0.52–0.97) | 0.03 |
| Methylated | 417 (48.4) | 219 | 25.6 (23.2–28.4) | 0.38 (0.29–0.49) | <0.0001 | 0.36 (0.28–0.46) | <0.0001 |
Adjusted for age, ECOG performance status, and extent of resection.
Validation of unsupervised cutoff and supervised safety margin and OS in the independent test cohort
There were 375 truly unmethylated patients (43.5%), 70 gray zone patients (8.1%), and 417 methylated patients (48.4%) in the rand-P test cohort. The median OS in the whole rand-P test cohort was 17.7 months (95% CI, 16.7–19.3). MGMT-methylated patients had a significantly longer OS compared with unmethylated patients (Supplementary Table S2; Supplementary Fig. S2A). When including the lower safety margin, the survival curves differed significantly (log-rank test, P < 0.0001; Supplementary Fig. S2B). The univariate Cox model resulted in a HR of 0.38 (95% CI, 0.29–0.49; P < 0.0001) for the methylated patients, and a HR of 0.70 for patients in the gray zone (95% CI, 0.5l-0.96; P = 0.03), both compared with the truly unmethylated patients. Similar results were obtained in the multivariate model (Table 2).
Good classification in retest dataset
Application of the 1.27 unsupervised cutoff to the retest dataset of 218 paired samples yielded 8 methylation status mismatches (3.7%; Supplementary Table S3). Cohen Kappa coefficient for inter-rater agreement was 0.93 (95% CI, 0.88–0.98) indicating almost perfect agreement between the original and retest methylation status. After also applying the lower safety margin the Kappa value was 0.80 (95% CI, 0.73–0.88), still indicating almost perfect agreement (Supplementary Table S4). A value of 0.89 (95% CI, 0.83–0.95) was obtained, when in addition applying the limit of detection of the diagnostic qMSP assay that considers <10 copies of methylated MGMT below the limit of detection and classifies them as unmethylated by default (Supplementary Table S5).
Comparison of the validated new classification with the original procedure
When comparing our classification method to the original procedure and cutoff (18, 19) in both the all-P training and test cohort, Cohen Kappa coefficients of, respectively, 0.93 (95% CI, 0.92–0.94) and 0.95 (95% CI, 0.94–0.96) were obtained, indicating almost perfect agreement (Supplementary Table S6). When the limit of detection of the diagnostic assay was applied, the comparison between the original and new classification method (Supplementary Table S7) resulted in a Cohen Kappa value of 0.98 (95% CI, 0.97–0.99) in both the all-P training and test cohorts.
Discussion
We aimed at determining a clinically relevant cutoff for the qMSP MGMT assay that is most widely used in clinical trials for patient stratification and, more importantly, for treatment strategies omitting temozolomide in patients with unmethylated GBM. The technical cutoff of the MGMT assay used has been defined as the value where the probability of being methylated or unmethylated is 50% (18, 19). The uncertainty regarding the methylation status close to the cutoff is high. Our pooled analysis from four randomized trials allowed determination and validation of the technical cutoff as well as a clinically relevant cutoff, supervised by OS in a large pooled dataset of patients treated uniformly with the current standard of care (TMZ/RT ⟶ TMZ).
This supervised optimal cutoff (−0.28, corrected MGMT log2 ratio) was situated below the technical cutoff obtained (1.27) and represents a lower safety margin which defines a gray zone of “low” methylation (Fig. 2). Patients whose MGMT value was situated in this gray zone did significantly better than those classified as “truly” MGMT unmethylated (<−0.28). Application of the lower safety margin in trials comparing schedules of TMZ/RT ⟶ TMZ to RT only (4) may shed new light on the interpretation of the apparent “low benefit” from temozolomide in the “MGMT-unmethylated” population. Consequently, grayzone patients may benefit from temozolomide treatment and should not be considered for treatments withholding temozolomide.
In contrast, among patients classified as MGMT methylated (>1.27, above the technical cutoff), a higher extent of methylation was not associated with a further gain in OS. This may suggest that detection of MGMT methylation in GBM using this assay is indicative of the second hit, completely inactivating MGMT. The first hit is the GBM characteristic loss of one copy of chromosome 10 on which MGMT resides (10q26; ref. 23). For tumor types retaining both copies of MGMT other clinical cutoffs may apply predicting sensitivity to temozolomide/alkylating agents as we have recently reported for IDH mutated grade II glioma treated with temozolomide or radiotherapy in the EORTC-22033 randomized phase III trial (24, 25).
Comparison of the here presented OS-supervised MGMT classification (methylated, gray zone, or unmethylated) with the original classification and cutoffs (12, 18) revealed a high level of agreement. In the original classification procedure we had defined the safety margin as the 95% probability to be unmethylated (12) on the basis of theoretical considerations as it is unknown which methylation pattern and how much methylation is required for complete silencing of MGMT expression in GBM (26). This safety margin was applied for patient selection into trials omitting temozolomide (12, 13). This boundary is very similar to the safety margin determined with the OS supervised analysis in this study that corresponds to a 96% chance of being unmethylated. Thus, our study now demonstrates the clinical importance of respecting a gray zone by providing the respective supporting outcome data. Implementation of the safety margin essentially groups methylated and gray zone results into the temozolomide-requiring patient population and selects the truly unmethylated patients as suitable for treatment without temozolomide. This needs to be taken into account for clinical trial planning.
Despite the large dataset and high reproducibility of the assay (R2 = 0.94) our study suffers from some limitations. All analyses were retrospective, which might have caused patient selection and cannot guarantee that training and test cohorts were balanced in terms of unmeasured confounders. In addition, no survival data was available for screened patients only, reducing the sample size for supervised and subgroup analyses. Yet, no better datasets to address this important issue for clinical practice and future clinical trial design is likely to become available.
It is important to note that the extent of methylation as measured and quantified by different MGMT tests may not necessarily have the same biological significance. Distinct tests use different principles (15) and/or interrogate different CpGs that do not all have the same impact on MGMT silencing (26–28), which is the principle mechanism for sensitizing patients to temozolomide. Consequently, cutoffs and corresponding safety margins need to be determined and validated for each assay (17, 29–31).
In conclusion, this analysis demonstrates that the qMSP assay is robust and technically reproducible, and confirms the strong impact of MGMT methylation on outcome in a large clinical trial population treated with TMZ/RT ⟶ TMZ. The reestablishment of the cutoffs in a large dataset with a slightly different calculation model and using outcome information, yielded almost identical classification into methylated, gray zone, and truly unmethylated patients as compared with the original procedure described previously (12). The clinically relevant cutoff informed by OS defined a gray zone with a safety margin that identifies patients who perform significantly better than truly unmethylated patients and may have some benefit from temozolomide. This gray zone could be validated in an independent dataset indicating that these patients should not be selected for treatment schemes avoiding temozolomide.
With this study we aim to encourage stratified temozolomide treatment for patients with GBM implementing a safety margin for guiding treatment decisions. This should facilitate testing new treatment paradigms without temozolomide in patients with MGMT unmethylated GBM who direly need better treatments.
Supplementary Material
Translational Relevance.
MGMT testing is disputed, which hinders stratified therapy and clinical trials omitting temozolomide. It is therefore of importance to determine the clinically relevant cutoff(s) defining the MGMT promoter methylation status for glioblastoma (GBM) that allows safe clinical decision making and patient selection into trials omitting temozolomide. The pooled analysis of quantitative MGMT methylation-specific PCR data from 4,041 patients with GBM screened or randomized in four clinical trials allowed determination and validation of an unsupervised cutoff and a lower cutoff supervised by outcome. The latter defines a “gray zone” comprising patients with low MGMT methylation who performed significantly better than truly unmethylated patients. This lower safety margin is suitable for selecting truly unmethylated patients for stratified therapy to spare patients unnecessary toxicity.
Acknowledgments
Written on behalf of the European Organisation for Treatment and Research of Cancer (EORTC) Brain Tumor Group and the AVAGlio, CENTRIC, CORE, and RTOG 0825 Clinical Trial Groups. E. Genbrugge’s fellowship at EORTC (Brussels, Belgium) was supported by a grant from the EORTC Brain Tumor Group.
Disclosure of Potential Conflicts of Interest
O.L. Chinot reports receiving commercial research grants from Roche and speakers bureau honoraria from Celdex and Immatics, and is a consultant/advisory board member for Abbvie. L.B. Nabors is a consultant/advisory board member for Abbvie. W. van Criekinge is an employee of MDxHealth. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 2.Malmstrom A, Gronberg BH, Marosi C, Stupp R, Frappaz D, Schultz H, et al. Temozolomide versus standard 6-week radiotherapy versus hypofractionated radiotherapy in patients older than 60 years with glioblastoma: the Nordic randomised, phase 3 trial. Lancet Oncol 2012;13:916–26. [DOI] [PubMed] [Google Scholar]
- 3.Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol 2012;13:707–15. [DOI] [PubMed] [Google Scholar]
- 4.Perry JR, Laperriere N, O’Callaghan CJ, Brandes AA, Menten J, Phillips C, et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl J Med 2017;376:1027–37. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol 2013;31:4085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 2014;370:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014;370:709–22. [DOI] [PubMed] [Google Scholar]
- 9.Stupp R, Taillibert S, Kanner A, Read W, Steinberg DM, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 2017;318:2306–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373–85. [DOI] [PubMed] [Google Scholar]
- 11.Hegi ME, Stupp R. Withholding temozolomide in glioblastoma patients with unmethylated MGMT promoter-still a dilemma? Neuro Oncol 2015;17:1425–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wick W, Gorlia T, Bady P, Platten M, van den Bent MJ, Taphoorn MJ, et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMTpromoter hypermethylation (EORTC 26082). Clin Cancer Res 2016;22:4797–806. [DOI] [PubMed] [Google Scholar]
- 13.Herrlinger U, Schafer N, Steinbach JP, Weyerbrock A, Hau P, Goldbrunner R, et al. Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6-Methylguanine-DNA methyltransferase nonmethylated glioblastoma: the randomized GLARIUS Trial. J Clin Oncol 2016;34: 1611–9. [DOI] [PubMed] [Google Scholar]
- 14.Weller M Where does O(6) -methylguanine DNA methyltransferase promoter methylation assessment place temozolomide in the future standards of care for glioblastoma? Cancer 2018;124:1316–8. [DOI] [PubMed] [Google Scholar]
- 15.Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, et al. MGMT testing–the challenges for biomarker-based gliomatreatment. Nat Rev Neurol 2014;10:372–85. [DOI] [PubMed] [Google Scholar]
- 16.Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer 2009;101: 124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quillien V, Lavenu A, Ducray F,Joly MO, Chinot O, Fina F, et al. Validation of the high-performance of pyrosequencing for clinical MGMT testing on a cohort of glioblastoma patients from a prospective dedicated multicentric trial. Oncotarget 2016;7:61916–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071–22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 2014;15:1100–8. [DOI] [PubMed] [Google Scholar]
- 19.Vlassenbroeck I, Califice S, Diserens AC, Migliavacca E, Straub J, Di Stefano I, et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J Mol Diagn 2008;10:332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol 2015;17:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.World Medical Association General Assembly. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. J Int Bioethique 2004;15:124–9. [PubMed] [Google Scholar]
- 22.Heagerty PJ, Lumley T, PepeMS. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000;56:337–44. [DOI] [PubMed] [Google Scholar]
- 23.Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016;164:550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bady P, Kurscheid S, Delorenzi M, Gorlia T, van den Bent MJ, Hoang-Xuan K, et al. The DNA methylome of DDR genes and benefit from RT or TMZ in IDH mutant low-grade glioma treated in EORTC 22033. Acta Neuropathol 2018;135:601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumert BG, Hegi ME, van den Bent MJ, von Deimling A, Gorlia T, Hoang-Xuan K, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033–26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol 2016;17: 1521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sciuscio D, Diserens AC, van Dommelen K, Martinet D, Jones G, Janzer RC, et al. Extent and patterns of MGMT promoter methylation in glioblastoma- and respective glioblastoma-derived spheres. Clin Cancer Res 2011;17: 255–66. [DOI] [PubMed] [Google Scholar]
- 27.Malley DS, Hamoudi RA, Kocialkowski S, Pearson DM, Collins VP, Ichimura K. A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol 2011;121:651–61. [DOI] [PubMed] [Google Scholar]
- 28.Bady P, Sciuscio D, Diserens AC, Bloch J, van den Bent MJ, Marosi C, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol 2012;124: 547–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reifenberger G, Hentschel B, Felsberg J, Schackert G, Simon M, Schnell O, et al. Predictive impact of MGMT promoter methylation in glioblastoma of the elderly. Int J Cancer 2012;131:1342–50. [DOI] [PubMed] [Google Scholar]
- 30.Xia D, Reardon DA, Bruce JL, Lindeman NI. The clinical implications of inconsistently methylated results from glioblastoma mgmt testing by replicate methylation-specific PCR. J Mol Diagn 2016;18:864–71. [DOI] [PubMed] [Google Scholar]
- 31.Ida CM, Butz ML, Jenkins RB, Sarkaria JN, Kitange GJ, Giannini C, et al. Real-time methylation-specific polymerase chain reaction for MGMT promoter methylation clinical testing in glioblastoma: an alternative detection method for a heterogeneous process. Am J Clin Pathol 2017;148:296–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.