

Standfirst
Myositis is a group of conditions that vary greatly in risk factors, clinical manifestations, laboratory markers, presumed pathogenetic mechanisms, treatment responses and prognoses. Approaches to divide myositis into mutually exclusive and stable phenotypes are being considered, but are we thinking comprehensively enough in our attempts at classification?
The term myositis covers a heterogeneous spectrum of autoimmune connective tissue diseases that range in severity from asymptomatic to life-threatening disorders, and in extent from the involvement of many organ systems to only a single organ (that might not even include muscle pathology, as occurs with dermatomyositis, interstitial lung disease or arthritis with myositis autoantibodies and minimal to no evidence of myositis)1. Over the past century, the heterogeneity of myositis and its classification criteria have increased, and the latter have created difficulties by causing a lack of consistency in patient populations among investigations, a problem highlighted in a new study by Loarce-Martos et al.2 that took a retrospective look the accuracy of a polymyositis diagnosis using a variety of classification criteria.
From the first attempts to divide myositis into useful subgroups on the basis of clinical features, through the inclusions of pathology, division by age of onset and autoantibody profiles, to the most recent inclusion of serum cytokines and chemokines using machine learning techniques, more and more phenotypes have been identified3. Whereas most myositis classification criteria have been formed based on expert opinion or group consensus, a major international effort resulted in the publication of the first data-driven, validated, robust EULAR–ACR classification criteria for myositis4. This important accomplishment standardized classification criteria so that future studies could be more easily compared because they would be investigating similar patients. Although the EULAR–ACR criteria are a major improvement over prior efforts, their approach has limitations in that the phenotypes identified are still heterogeneous and data regarding the necrotizing myopathies and many autoantibodies were not available for inclusion owing to the retrospective nature of the study.
In their investigation, Loarce-Martos et al. attempted to address some of these limitations by carefully reassessing a cohort of 37 patients previously classified as having polymyositis according to both the EULAR–ACR criteria and expert opinion2. The authors included considerations of available autoantibody profiles, muscle tissue analysis, and other features, and used different sets of previously published classification criteria for inclusion body myositis (IBM), immune-mediated necrotizing myopathy (IMNM), antisynthetase syndrome (either by Connor criteria or Solomon criteria), connective tissue disease overlap myositis and dermatomyositis (according to the presence of typical skin rash and/or the presence of dermatomyositis-specific autoantibodies). Of the 37 patients, only nine (24%) remained classified as having polymyositis2. The classifications of the other patients included: IMNM (14%); connective tissue disease overlap myositis (19%); unspecified myopathy (16%); dermatomyositis (5%); cancer-associated myopathy (8%); and non-inflammatory myopathy (3%). In addition, four patients (10%) had insufficient data to confidently reclassify. The authors concluded that polymyositis should be considered a rare myositis subgroup.
These results are not surprising given the very different definitions and approaches used in the various classification criteria that were chosen to define the patients in this report2. For example, dermatomyositis and IBM would also be rarer entities if patients with these conditions were separated according to their expression of specific autoantibodies and considered as separate phenotypes. Furthermore, the myositis subgroups the authors describe are still heterogeneous; for example, classification as antisynthetase syndrome only required the presence of an antisynthetase autoantibody and one or two clinical findings out of a whole variety, so this subgroup could include patients who were previously classified as either polymyositis or dermatomyositis.
The expansion in the number of proposed and discordant myositis classification schemes, which have little data-based evidence to support them and no documented reproducibility or consistency in their derivation, now seriously threatens the capacity to advance the field5. A further shortcoming of most of these newer classification criteria is the lack of validation or evidence to suggest that they perform any better than the EULAR–ACR criteria for determining optimal treatment or prognosis. Going forward, these multiple incompatible classification criteria might result in the inability to compare many clinical, epidemiologic, or molecular studies, as well as trials of different therapies or even of the same therapy, because patients with different kinds of myositis could have been investigated depending upon the classification criteria used. Another negative outcome could be decreased interest by the pharmaceutical industry, funding agencies, and regulatory agencies in supporting future myositis investigations as the reliability and capacity to reproduce the studies would be suspect when using different criteria.
No simple solution exists to this current undesirable state, but perhaps we are not considering the problem appropriately? Current findings suggest that interactions between genetic and environmental risk factors alter gene expression and induce molecular and immune pathway changes that over time result in pathologic and clinical features that lead to myositis and similar autoimmune diseases6,7. Like the fabled blind men of India, who by feeling different parts of an elephant each described a different entity, we might similarly be focusing on different and limited manifestations of the whole myositis beast. New comprehensive approaches are needed to assess the totality of myositis. Novel technologies and artificial intelligence-augmented data analytics could be used to synthesize assessments of genomics, exposomics, clinical features, pathology, epigenetics, immunology, metabolomics, transcriptomics, proteomics, microbiomics, outcomes, and other aspects into a unified whole that would expand on previous proposals for dissecting complex diseases8. This approach would require large, multicentre, multispecialty international studies that include analogous comparisons with confounding conditions to define and validate new phenotypes and classification criteria.
As the current early attempts to identify additional homogeneous myositis phenotypes suggest9, it seems likely that a comprehensive, multidimensional analytical approach could result in the identification of extremely homogeneous phenotypes that could be called ‘elemental disorders”10. Each elemental disorder would be defined as a mutually exclusive and stable phenotype that consists of cardinal clinical signs, symptoms, pathology, laboratory findings, and molecular features that result from the interactions of the necessary and sufficient genetic and environmental risk factors, in the relative absence of protective factors, through a distinct pathogenic mechanism (Figure 1). Defining elemental disorders should result in the need for far fewer patients in a study given their high homogeneity, and could result in greatly improved treatments via the discovery of targeted, individualized therapies. Ultimately, it is even possible that defining elemental disorders could enable the prevention of disease by avoidance of environmental risk factors in genetically predisposed individuals, or possibly via targeted gene therapy of risk alleles when unavoidable environmental triggers exist.
Figure 1. The elemental disorder hypothesis.
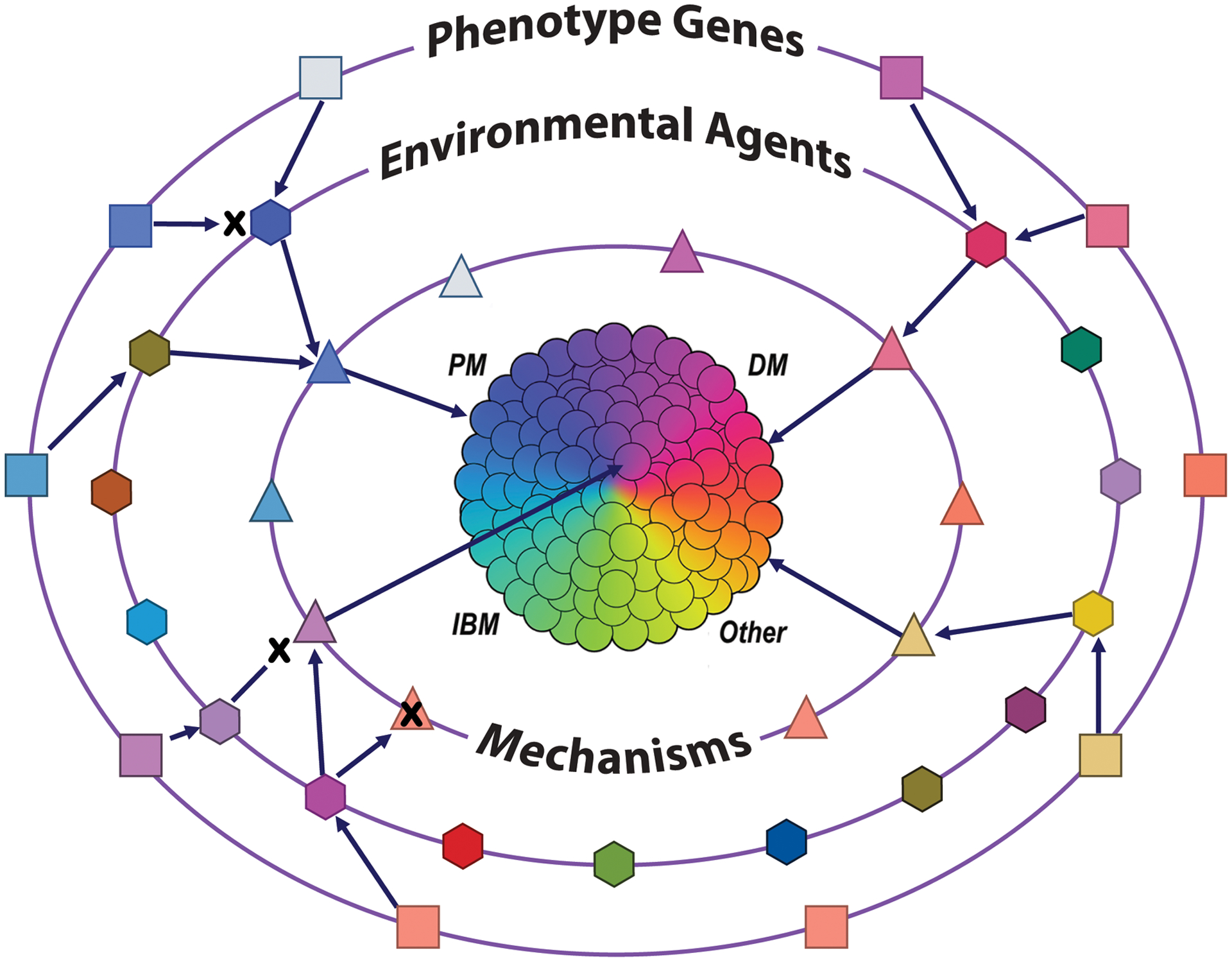
Disease phenotypes as currently classified (i.e., PM, polymyositis; DM, dermatomyositis; IBM, inclusion body myositis, others) are heterogeneous and composed of multiple elemental disorders (centre circles). Elemental disorders are defined as mutually exclusive and stable phenotypes that result from the interaction of the necessary and sufficient genetic (outer ring of boxes) and environmental (middle ring of hexagons) risk factors, in the relative absence of protective factors, through a distinct pathogenic mechanism (inner circle of triangles). For some elemental disorders, multiple genes are required (indicated by arrows), and for others, multiple environmetnal exposures are needed to induce the mechanisms that lead to elemental disorders. Protective genetic, environmental or mechanistic factors (indicated by an X), can prevent progression to disease in otherwise permissive circumstances.
The current limited attempts at developing new classifications for myositis are a beginning that needs to be greatly enhanced. It is time to think much more expansively about the classification of the myositis spectrum and other conditions, and to conduct comprehensive deep phenotyping investigations to identify their elemental disorders and allow for optimized treatments, cures, and prevention.
Acknowledgements
This work of the author was supported by the Intramural Research Program of the US National Institutes of Health, National Institute of Environmental Health Sciences. T. O’Hanlon and E. Shamim are acknowledged for their useful comments on the manuscript.
Footnotes
Competing interests
The author declares no competing interests.
References
- 1.Barsotti S & Lundberg IE Myositis an evolving spectrum of disease. Immunol Med 41, 46–54, doi: 10.1080/13497413.2018.1481571 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Loarce-Martos J, Lilleker JB, Parker M, McHugh N & Chinoy H Polymyositis: is there anything left? A retrospective diagnostic review from a tertiary myositis centre. Rheumatology (Oxford), doi: 10.1093/rheumatology/keaa801 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Lilleker JB & Chinoy H Can machine learning unravel the complex IIM spectrum? Nat Rev Rheumatol 16, 299–300, doi: 10.1038/s41584-020-0412-6 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Lundberg IE et al. 2017 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Adult and Juvenile Idiopathic Inflammatory Myopathies and Their Major Subgroups. Arthritis Rheumatol 69, 2271–2282, doi: 10.1002/art.40320 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundberg IE, de Visser M & Werth VP Classification of myositis. Nat Rev Rheumatol 14, 269–278, doi: 10.1038/nrrheum.2018.41 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Miller FW, Lamb JA, Schmidt J & Nagaraju K Risk factors and disease mechanisms in myositis. Nat Rev Rheumatol 14, 255–268, doi: 10.1038/nrrheum.2018.48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deane KD et al. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol 31, 3–18, doi: 10.1016/j.berh.2017.08.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun YV & Hu YJ Integrative Analysis of Multi-omics Data for Discovery and Functional Studies of Complex Human Diseases. Adv Genet 93, 147–190, doi: 10.1016/bs.adgen.2015.11.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eng SWM, Olazagasti JM, Goldenberg A, Crowson CS, Oddis CV, Niewold TB, Yeung RS & Reed AM A clinically and biologically based subclassification of the idiopathic inflammatory myopathies using machine learning. ACR Open Rheumatol. 2, 158–166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gourley M & Miller FW Mechanisms of disease: Environmental factors in the pathogenesis of rheumatic disease. Nat.Clin.Pract.Rheumatol 3, 172–180 (2007). [DOI] [PubMed] [Google Scholar]