

Abstract
Robust methods are critical for testing the in vivo regulatory mechanism of RNA binding proteins. Here we report improvement of a protein–mRNA tethering assay to probe the function of an RNA binding protein in its natural context within the C. elegans adult germline. The assay relies on a dual reporter expressing two mRNAs from a single promoter and resolved by trans-splicing. The gfp reporter 3′UTR harbors functional binding elements for λN22 peptide, while the mCherry reporter 3′UTR carries mutated nonfunctional elements. This strategy enables internally controlled quantitation of reporter protein by immunofluorescence and mRNA by smFISH. To test the new system, we analyzed a C. elegans Nanos protein, NOS-3, which serves as a post-transcriptional regulator of germ cell fate. Unexpectedly, tethered NOS-3 enhanced reporter expression. We confirmed this enhancement activity with a second reporter engineered at an endogenous germline gene. NOS-3 enhancement of reporter expression was associated with its amino-terminal intrinsically disordered region, not its carboxy-terminal zinc fingers. RNA quantitation revealed that tethered NOS-3 enhances stability of the reporter mRNA. We suggest that this direct NOS-3 enhancement activity may explain a paradox: Classically Nanos proteins are expected to repress RNA, but nos-3 had been found to promote gld-1 expression, an effect that could be direct. Regardless, the new dual reporter dramatically improves in situ quantitation of reporter expression after RNA binding protein tethering to determine its molecular mechanism in a multicellular tissue.
Keywords: RNA binding protein, NOS-3, Nanos, C. elegans, germline
INTRODUCTION
Protein–mRNA tethering is a well-established method to investigate the direct regulatory effects of RNA binding proteins (RBPs). These assays rely on two components: an RBP tagged with a λN22 peptide or MS2 coat protein domain and a reporter mRNA harboring binding sites for λN22 or MS2 in its 3′UTR (Baron-Benhamou et al. 2004; Coller and Wickens 2007). The RBP is thus tethered to reporter mRNA with high affinity and specificity, and its regulatory effect inferred from changes in expression or stability of the reporter RNA compared to a control. Tethering assays have proven tremendously useful. They have revealed that some RBPs down-regulate mRNA expression by promoting RNA turnover (Bhandari et al. 2014; Raisch et al. 2016) or repressing translation (Pillai et al. 2004), while others up-regulate mRNA expression by stabilizing RNA (Coller et al. 1998; Gray et al. 2000) or enhancing translation (De Gregorio et al. 1999). Tethering assays are thus an exceptional tool to dissect the molecular functions of RBPs.
The regulatory effect of a tethered RBP is deduced from measurements of both the reporter RNA's stability and translation. When assays are done in cultured cells or yeast, bulk detection methods are sufficient, but when conducted in a multicellular tissue or organism, cell-specific methods become essential. Here we report the development of a new tethering assay for use in the C. elegans germline, a richly patterned tissue in which mRNAs are dynamically regulated as cells develop from dividing stem cells to differentiating gametes (Hubbard and Schedl 2019). This assay takes advantage of a dual reporter that includes an internal control for better quantitation, a PEST domain to restrict reporter protein half-life, and high affinity epitope tags for each reporter protein. We test our assay with NOS-3, an RBP belonging to the conserved Nanos family (Kraemer et al. 1999; Subramaniam and Seydoux 1999). Nanos RBPs regulate germline development from nematodes to mammals (Tsuda et al. 2003; Suzuki et al. 2012; Kusz-Zamelczyk et al. 2013). Tethering assays of human and Drosophila Nanos orthologs, performed in HEK293 or S2 cells, respectively, demonstrate that both are repressors of reporter expression and identified a Not1-interacting domain responsible for that repression (Bhandari et al. 2014; Raisch et al. 2016). We selected NOS-3 to test our new assay, and expected it would repress reporter expression. However, we found instead that tethered nematode NOS-3 enhances reporter expression, a surprising result that implies that NOS-3 may promote expression of its target mRNAs in the C. elegans germline.
RESULTS AND DISCUSSION
We set out to improve the tethering assay in the C. elegans germline. Our starting point was a λN22-boxB tethering system previously used in this tissue (Wedeles et al. 2013; Aoki et al. 2018, 2021). The earlier system tagged the RBP of interest with λN22 peptide, henceforth λN, and recruited RBP::λN to germline-expressed, reporter mRNA via boxB RNA hairpins in its 3′-UTR (Fig. 1A; Aoki et al. 2018, 2021). λN binds to boxB hairpins with high affinity and specificity (Baron-Benhamou et al. 2004). The GFP-tagged, histone H2B reporter protein localizes to the nucleus of the expressing cell for straightforward visualization. While functional, the previous system lacked internal controls to compare expression between animals. Histone reporters can also persist and mark cells or their daughters, expanding perceived protein expression boundaries (Merritt et al. 2008). Thus, more accurate measurement of reporter expression required additional revisions to the assay.
FIGURE 1.

Tethered NOS-3 increases GFP reporter expression. (A) Previous tethering assay used in the C. elegans germline (Aoki et al. 2018, 2021). GFP reporter mRNA is under control of a germline-expressed, mex-5 promoter and has three boxB stem–loops in its 3′UTR. The RNA-binding protein (RBP) is tagged with λN. (B) Dual reporter tethering assay (this work). The nascent transcript driven by mex-5 promoter is resolved by trans-splicing into two mRNAs that encode distinct reporters. The gfp reporter RNA has three functional boxB stem–loops in its 3′UTR; the mCherry reporter 3′UTR has three mutated boxB stem–loops that do not bind λN and therefore provides an internal control. Addition of an OLLAS tag to GFP and a V5 tag to mCherry enables sensitive immunostaining and immunoblotting. (C) Schematic of C. elegans germline. Germline stem cells (GSCs) reside in the progenitor zone (yellow); GSC daughters enter and progress through meiotic prophase (green) and finally differentiate into oocytes (pink); red box, mid-pachytene region imaged in Figure 1D; bracket, region quantitated in several figures begins at distal end and goes 310 µm, stopping near the proximal end of the pachytene region. (D) Confocal images (max projection) of germlines stained to detect NOS-3::FLAG and NOS-3:: λN::FLAG (αFLAG, column 1); GFP::H2B::OLLAS::PEST (αOLLAS, columns 2 and 3); V5 antibodies to detect mCherry::H2B::V5::PEST (αV5, column 4), and DNA (DAPI, column 5). All images were acquired with the same microscope settings. All images in each column were visualized at the same contrast in Image J; the lower OLLAS contrast in column 2 was optimized for reporter expression with NOS-3:: λN::FLAG, while the higher constrast in column 3 was optimized for expression with the dual reporter only and NOS-3::FLAG. Wild-type has no tagged proteins and serves as a negative control. Note that images for wild-type, reporter only, NOS-3::FLAG and NOS-3:: λN::FLAG are replicated in Figure 3B. (E) In situ quantitation of GFP (αOLLAS) normalized to mCherry (αV5) reporter proteins, as a function of germline position. Bold lines show averages, and dashed lines and shading show one standard deviation (see Materials and Methods). Wild-type serves as a negative control. Wild-type (N2, no dual reporter; n = 54), NOS-3::FLAG (n = 34), NOS-3:: λN::FLAG (n = 52) worms were analyzed for D and E. Same curves and values are included in Figure 3D; Supplemental Figure S1D,E and Supplemental Figure S2. (F) Immunoblot to assay GFP (αOLLAS) and mCherry (αV5) reporter protein expression. Actin serves as a loading control, and wild-type worms as the negative control (last lane). Whole worm lysate (n = 60 young adult worms) used for all samples.
The improved reporter incorporates three new components into the previous system, all with the goal of enhancing quantitation of expression in specific cell types (Fig. 1B; Supplemental Fig. S1A). First, we generated a reporter operon that introduces a second mRNA reporter as an internal control, as conceived by others (Merritt et al. 2008). A single nascent transcript generates two reporter mRNAs by trans-splicing: One encodes GFP-H2B and can bind RBP::λN via boxB RNA hairpins in its 3′-UTR; the second encodes mCherry-H2B and cannot bind RBP::λN because mutated boxB RNA hairpins in its 3′-UTR abrogate λN binding (Chattopadhyay et al. 1995). Second, we added a mouse ornithine decarboxylase (MODC) PEST domain to the carboxyl terminus of both reporter proteins. The PEST degron shortens the half-life of the reporter protein to restrict it to cells expressing reporter RNA (Frand et al. 2005; Farley and Ryder 2012; Kaymak et al. 2016). Third, we added high affinity epitope tags with commercially available antibodies based on successful immunoblot and immunostaining in worms: OLLAS into GFP::H2B::PEST (Smith et al. 2016) and V5 into mCherry::H2B::PEST (Aoki et al. 2021). A construct encoding the dual reporter was integrated into the C. elegans genome as a Mos single copy insertion (MosSCI, see Materials and Methods). The C. elegans germline is patterned along its U-shaped distal-proximal axis with germline stem cells (GSCs) at the distal end, progressive differentiation of their daughters more proximally, and differentiated gametes at the proximal end (Fig. 1C; Hubbard and Schedl 2019). Immunostaining against OLLAS and V5 tags revealed that both GFP and mCherry histone H2B reporter proteins expressed and colocalized with DNA in germ cell nuclei (Fig. 1D). This dual reporter thus allows for precise quantitation of expression from both tethered and untethered mRNA in individual cells within the C. elegans germline (Fig. 1B).
We tested the dual reporter with NOS-3, an mRNA binding protein and germ cell fate regulator. NOS-3 regulates two fate choices that occur along the germline axis, the sperm/oocyte and mitosis/meiosis decisions (Kraemer et al. 1999; Eckmann et al. 2004; Hansen et al. 2004). Genetic analyses of how NOS-3 functions in those two decisions poses a paradox. NOS-3 decreases expression of FEM-3 (Arur et al. 2011), a positive regulator of the sperm fate, but increases expression of GLD-1 (Hansen et al. 2004; Brenner and Schedl 2016), a positive regulator of meiotic entry. However, these genetic results do not address whether NOS-3 regulation is direct or indirect. If NOS-3 were primarily a repressor, like other members of the Nanos family (see Introduction), its enhancement of GLD-1 expression might be indirect, perhaps by repressing a gld-1 mRNA repressor.
To ask how NOS-3 regulates expression when tethered, we inserted a 3xFLAG epitope tag with or without λN into the endogenous nos-3 gene, using CRISPR/Cas9 gene editing (Supplemental Fig. S1B). Both NOS-3::FLAG and NOS-3::λN::FLAG proteins were expressed and localized similarly to the the germline cytoplasm (Fig. 1D, first column), as reported previously for endogenous NOS-3 (Kraemer et al. 1999). Moreover, the λN::FLAG-tagged NOS-3 behaved like wild-type NOS-3 in a genetic assay (Supplemental Fig. S1C), validating its use to test NOS-3 function. We proceeded to ask how tethered NOS-3 affects reporter expression. Expression of GFP and mCherry reporters was assayed by immunostaining against their OLLAS and V5 tags, respectively. GFP expression decreased along the distal-proximal germline axis in both NOS-3::FLAG and NOS-3:: λN::FLAG strains (Fig. 1E; Supplemental Fig. S1D), indicating that this pattern is inherent to the dual reporter. ImageJ was used to optimize readability of OLLAS staining by differentially adjusting contrast. At low contrast, OLLAS staining was only faintly detectable with the dual reporter only or the reporter plus NOS-3::FLAG (Fig. 1D, second column). However, at higher contrast, OLLAS staining was easily observed in those germlines but became saturated in germlines with NOS-3:: λN::FLAG (Fig. 1D, third column). Thus, addition of the λN tag to NOS-3 dramatically increased expression of the GFP reporter (Fig. 1D, second and third columns, and Supplemental Fig. S1D). In contrast, expression of the mCherry internal control reporter protein was observed at comparable levels in all germlines carrying the dual reporter (Fig. 1D, fourth column; Supplemental Fig. S1E). We used this mCherry control to normalize for variable reporter protein expression across worms (Fig. 1B). Ratios of GFP to mCherry reporter expression were quantified as a function of germline position (Fig. 1E; see Supplemental Fig. S1D,E for graphs of the two reporter proteins on their own). In NOS-3::FLAG germlines, the ratio was modest, but in NOS-3:: λN::FLAG germlines, the ratio increased significantly throughout the distal germline arm. Immunoblots of the reporter proteins confirmed enhancement of GFP reporter expression specifically (Fig. 1F). We conclude that NOS-3 can enhance reporter expression when tethered.
The NOS-3 enhancement of reporter expression was unexpected because other Nanos homologs repress expression when tethered (Bhandari et al. 2014; Raisch et al. 2016). To further test the effect of tethered NOS-3, we engineered an endogenous germline gene to function as a tethering reporter. Three boxB hairpins were inserted into the 3′UTR region of the pgl-1 gene, which encodes a SNAP-tagged PGL-1 (Fig. 2A). The SNAP tag permits straightforward visualization of PGL-1 protein in P granules without affecting its function (Aoki et al. 2021). As with the dual reporter, NOS-3:: λN::FLAG dramatically increased PGL-1::SNAP abundance when compared to NOS-3::FLAG by imaging (Fig. 2B,C) and immunoblot (Fig. 2D). Tagged NOS-3 was expressed similarly in the NOS-3::FLAG and NOS-3:: λN::FLAG pgl-1 reporter strains (Fig. 2D). We conclude that tethered NOS-3 enhances expression of two distinct reporter mRNAs.
FIGURE 2.

Tethered NOS-3 increases expression of pgl-1 endogenous gene. (A) Tethering assay using SNAP-tagged pgl-1 gene as the reporter. Endogenous pgl-1 includes a SNAP tag to visualize its protein product and 3× boxB hairpins to recruit NOS-3:: λN::FLAG to its 3′UTR. (B) Tethered NOS-3 increases expression of PGL-1 protein in the worm germline. Confocal images (max projection) of adult gonads stained for PGL-1::SNAP (SNAP ligand), NOS-3 (αFLAG), and DNA (DAPI). Wild-type worms serve as a negative control. (C) Quantitation of PGL-1::SNAP. SNAP ligand signal was averaged across the distal 5–105 µm of germlines. Wild-type (N2, no PGL-1::SNAP reporter; n = 44), NOS-3::FLAG (n = 33), NOS-3:: λN::FLAG (n = 41) worms analyzed. (***) P < 0.0001. (D) Immunoblot to assay PGL-1 (αSNAP) and NOS-3 (αFLAG) protein expression. Actin serves as the loading control, and wild-type worms as a negative control (first lane). Whole worm lysate (n = 30 young adult worms) used for all samples.
To probe the region or regions within NOS-3 responsible for enhancing expression, we generated a battery of NOS-3:: λN::FLAG variants by CRISPR/Cas9 gene editing of the endogenous locus. Full length NOS-3 protein consists of a large amino-terminal region predicted to be intrinsically disordered (analyzed by IUPred2A [Erdos and Dosztanyi 2020]), and a Nanos-like zinc finger near the carboxyl terminus (Fig. 3A, top; Kraemer et al. 1999; Subramaniam and Seydoux 1999). Variants were made with in-frame deletions or a premature stop codon (Fig. 3A, bottom). All protein variants described were expressed, albeit at differing expression levels (Fig. 3B, first column; 3C, bottom panel). Of note, full length NOS-3::FLAG with or without λN expressed comparably (Fig. 3C). Reporter expression was examined in all variants by imaging (Fig. 3B) and calculating ratios of GFP to mCherry reporter protein abundance in germlines (Fig. 3D; Supplemental Fig. S2), as well as by immunoblot (Fig. 3C). All variants enhanced GFP reporter expression relative to the internal control to some extent (Fig. 3D; Supplemental Table S1; Supplemental Fig. S2). Of note, the enhancement does not require the carboxy-terminal Zinc finger but instead relies on the intrinsically disordered region constituting the large amino terminus (Fig. 3D). Intriguingly, this region also includes the FBF-1 interaction domain (Kraemer et al. 1999) and ERK/MAP Kinase (MPK) docking site (Arur et al. 2011). FBF-1 is an RNA binding protein belonging to the Pumilio and FBF (PUF) family (Wickens et al. 2002), which are conserved Nanos binding partners (Goldstrohm et al. 2018). Our results with NOS-3 variants in the tethering assay suggest that NOS-3 binding to FBF-1 is not necessary for its enhancement function, although likely critical for its endogenous target specificity (Murata and Wharton 1995; Zamore et al. 1997; Kraemer et al. 1999; Sonoda and Wharton 1999; Weidmann et al. 2016; Arvola et al. 2017; Malik et al. 2019). Site-specific phosphorylation of NOS-3 via its MPK docking site occurs in the proximal germline for oocyte maturation (Arur et al. 2011). The tethered NOS-3 enhancement of reporter expression is observed throughout the distal arm of the germline, which includes the late pachytene region where NOS-3 is phosphorylated. Therefore, the ability of tethered NOS-3 to increase protein expression is likely independent of its phosphorylation state. Regardless, the variant results suggest that enhancement activity is likely distributed across NOS-3 protein. The NOS-3 deletions may affect protein stability, or NOS-3 may have multiple protein binding sites responsible for enhanced GFP expression. An intriguing idea is that evolution has given NOS-3 multiple enhancement regions to robustly increase protein expression.
FIGURE 3.
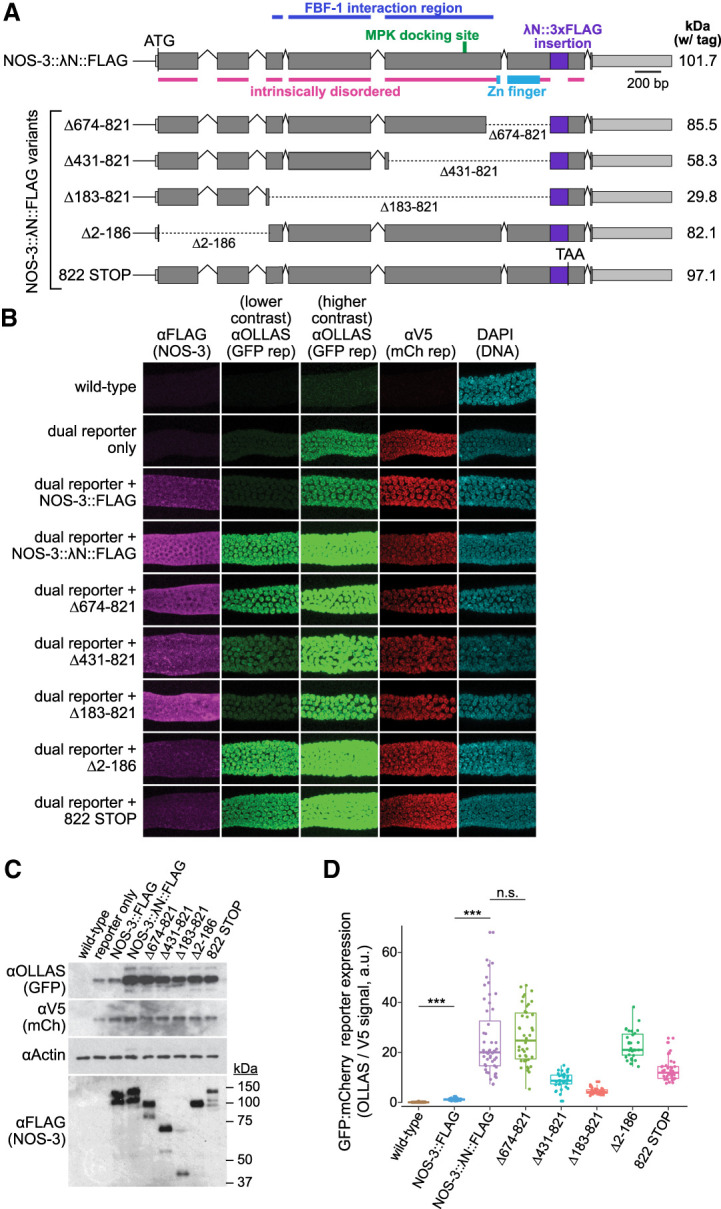
Analysis of tethered NOS-3 deletion mutants for enhancement of reporter expression. (A) nos-3 locus and variants used to identify the region necessary for reporter expression enhancement. Variants include four in-frame deletions and a premature STOP codon that removes amino acids carboxy-terminal to the tag insert. Variant protein sizes (kilodaltons, kDa) shown at right. Coding regions, gray; untranslated regions, light gray; λN::FLAG insert, purple. Intrinsically disordered regions were predicted by IUPred2A (Erdos and Dosztanyi 2020). FBF-1 interacting region (Kraemer et al. 1999) and MPK docking site (Arur et al. 2011) labeled as previously identified. (B) Confocal images (max projection) of adult germlines. Conventions as detailed in Figure 1D. See Figure 1B for germline image location. Note that images for wild-type, reporter only, NOS-3::FLAG, and NOS-3:: λN::FLAG are replicated from Figure 1D. (C) Immunoblot to assay expression of reporter proteins (above) and NOS-3 variant proteins (below). Conventions as in Figure 1F. SDS-PAGE sizes (kDa) shown to right for the NOS-3 immunoblot. Whole worm lysate (n = 60 young adult worms) used for all samples. (D) Relative abundance of GFP and mCherry reporter proteins. Ratio of signals from GFP (αOLLAS) and mCherry (αV5) reporter proteins, averaged across distal 100 µm of germline and normalized to wild-type negative control. Wild-type (N2, no dual reporter; n = 54), NOS-3::FLAG (n = 34), NOS-3:: λN::FLAG (n = 52), Δ183–821 (n = 37), Δ431–821 (n = 36), Δ674–821 (n = 42), Δ2–186 (n = 31), 822 STOP (n = 37) worms analyzed for B and D. Curves and values of wild-type, NOS-3::FLAG and NOS-3:: λN::FLAG are same as reported in Figure 1E and Supplemental Figure S2. (***) P-value <0.0001. n.s., not significant (P-value = 0.5735). All P-values for comparisons between strains can be found in Supplemental Table S1.
To investigate how tethered NOS-3 enhances expression of the gfp reporter RNA, we assessed the abundance of the two dual reporter mRNAs in germlines harboring NOS-3:: λN::FLAG or controls. The gfp and mCherry reporter mRNAs were visualized using distinct smFISH probe sets, which permitted simultaneous imaging of both reporter mRNAs in the same animal (Fig. 4A; see Materials and Methods). We predicted that tethered NOS-3 affects either stability or translation of the gfp reporter mRNA. If tethered NOS-3 stabilizes its target RNA, the gfp mRNA levels should be higher in animals carrying NOS-3 tagged with λN than those without (Fig. 4B, top). If tethered NOS-3 solely promotes translation, the gfp mRNA levels should be similar with or without λN (Fig. 4B, bottom). mCherry mRNA levels serve as a normalizing control for transcription.
FIGURE 4.

Tethered NOS-3 enhances stability of gfp reporter mRNAs. (A) Experimental design. Reporter mRNAs from dual reporter visualized in dissected gonads (top) with differentially labeled smFISH probes to gfp and mCherry (mCh) mRNAs (bottom). (B) Tethered NOS-3 might increase reporter expression by affecting either stability or translation of the reporter RNA. These two mechanisms have distinct predictions for effects on reporter mRNA abundance. More or less RNA abundance is depicted in the figure as larger or smaller letters, respectively, in animals possessing tethered NOS-3:: λN::FLAG (left column) or untethered NOS-3::FLAG (right column). (C) Representative images of smFISH in wild-type, reporter only, NOS-3::FLAG and NOS-3:: λN::FLAG germlines. Images are average projections of five slices (0.3 µm) from confocal microscopy. gfp mRNAs (gfp smFISH, green), mCherry mRNAs (mCherry smFISH, magenta), GFP protein (fluorescence signal [488], yellow), or DNA (DAPI, light blue). (D) Ratios of gfp:mCherry mRNAs from smFISH quantitation. (***) P-value <0.0001. n.s., not significant (P-value = 0.6352). All reporter strains were significantly different than wild-type (P-value <0.0001). Wild-type (N2, no dual reporter; n = 19), dual reporter only (n = 32), NOS-3::FLAG (n = 37), NOS-3:: λN::FLAG (n = 30) from two combined experiments. (E) Model that NOS-3 directly enhances expression of gld-1 mRNA, perhaps together with the GLD-2 poly(A) polymerase. Repression of FEM-3 is proposed to be indirect through enhancement of an unidentified inhibitory factor.
smFISH detected gfp and mCherry mRNAs in all germlines carrying the dual reporter (Fig. 4C, columns 1 and 2), but not in wild-type germlines without the reporter. mRNA signals appeared as cytoplasmic spots, as established in other studies (Raj et al. 2008; Lee et al. 2017). The gfp mRNAs were more abundant than mCherry mRNAs (Fig. 4C, columns 1 and 2), which could result from differences in probe binding efficiency or differential mRNA processing. Fluorescence of the GFP protein was higher in reporter expressing germlines with NOS-3 tagged with λN (Fig. 4C, column 3), consistent with immunostaining of its epitope tag. The number of gfp mRNAs was normalized to mCherry mRNA counts in the same germlines, and gfp:mCherry mRNA abundance ratios were compared between strains (Fig. 4D). This analysis revealed that the normalized gfp mRNA levels were significantly different between tethered and untethered NOS-3 (Fig. 4D). As expected, all germlines carrying the dual reporter had more detectable reporter mRNA signal than the wild-type control. More importantly, the gfp:mCherry mRNA ratios were similar for NOS-3::FLAG and wild-type NOS-3, but increased for tethered NOS-3:: λN::FLAG (Fig. 4D). This result fits the prediction that NOS-3 enhances gfp expression by increasing mRNA stability, though we cannot exclude an additional effect on enhancing translation.
The enhancement activity of tethered NOS-3 was surprising given that its homologs act as repressors when tethered (Bhandari et al. 2014; Raisch et al. 2016). Yet the NOS-3 enhancement activity may help explain a puzzling genetic result. Removal of NOS-3 increases FEM-3 protein abundance (Arur et al. 2011), consistent with repressive activity, but its removal also lowers GLD-1 protein abundance (Hansen et al. 2004; Brenner and Schedl 2016). One explanation for the GLD-1 decrease, based on dogma that Nanos proteins are repressors, is that NOS-3 represses a gld-1 repressor and the GLD-1 change is indirect. However, here we find that tethered NOS-3 directly enhances expression of two distinct mRNAs, the gfp mRNA of the dual reporter and the endogenous pgl-1 mRNA that we engineered for tethering. The data supports a model whereby NOS-3 promotes GLD-1 expression directly and FEM-3 repression indirectly (Fig. 4E). NOS-3 enhancement may work in parallel with the GLD-2 poly(A) polymerase, which also increases expression of gld-1 mRNA through elongation of its poly(A) tail (Suh et al. 2006, 2009). We emphasize that a NOS-3 repressive activity remains possible. MPK phosphorylation may have modulated a switch between NOS-3 acting as a repressor and activator (Arur et al. 2011). However, that MPK phosphorylation is spatially restricted to one region of the germline, whereas NOS-3 enhancement activity is not similarly restricted. Clearly, further investigation of NOS-3 enhancement activity is warranted as is investigation of classical Nanos orthologs in their natural context.
The enhancement activity of tethered NOS-3 also expands the genetic toolkit for analyzing protein function. Methods to decrease protein abundance are readily available with classical genetics, RNAi and gene editing, but methods to increase protein abundance have been less robust, though examples exist. Expression of specific genes can be increased by modifying CRISPR/Cas9 (Konermann et al. 2015) or tethering proteins associated with translation, like poly(A) binding protein (PABP) (Coller et al. 1998), eIF4E (De Gregorio et al. 2001), or eIF4G (De Gregorio et al. 1999). We can now add NOS-3 tethering as a method to increase gene expression, at least in the C. elegans germline. More broadly, pairing the dual reporter tethering assay with smFISH allows direct investigation of mechanistic functions of RNA binding proteins in complex multicellular tissues. By analyzing the ratios of tethered versus untethered transcripts, one can assess mechanisms of mRNA turnover and translational modulation. This mechanistic dissection performed within the natural context of a multicellular organism allows regulatory mechanisms to be analyzed in different tissues and cells and at different stages of development. The dual reporter tethering-smFISH pairing is adaptable to other organisms with single gene editing and shows promise to elucidate the post-transcriptional regulatory mechanisms of a wide variety of mRNA binding proteins.
MATERIALS AND METHODS
Worm maintenance and strains
Strains were maintained at 20°C or 25°C, as previously described (Brenner 1974). Supplemental Table S2 lists strains analyzed, which are available upon request.
CRISPR–Cas9 gene editing
CRISPR–Cas9 genome editing was used to modify endogenous nos-3 and pgl-1 gene loci, following an established protocol (Paix et al. 2015). Briefly, worms were injected with a ribonucleoprotein complex consisting of recombinant Cas9 protein, tracrRNA, gene-specific crRNAs, and a single stranded DNA repair template. The injection mix included a crRNA and repair template designed to induce a dominant loss of function mutation in unc-58 as a marker for successful editing (Arribere et al. 2014). Unc progeny from injected animals were singled and PCR screened for edits at the intended locus. Animals homozygous for edits were isolated and proper editing confirmed by Sanger sequencing; confirmed homozygotes were outcrossed with N2 a minimum of two times before analysis. Supplemental Table S3 lists guide RNAs and repair DNA oligos.
MosSCI insertion of the dual reporter gene
The dual reporter construct included a mex-5 promoter (Merritt et al. 2008), a region encoding eGFP::Histone H2B tagged with a mouse ornithine decarboxylase (MODC) PEST domain (Li et al. 1998) and a 1×OLLAS epitope tag (Park et al. 2008), a tbb-2 3′UTR harboring a 3× boxB (Wang et al. 2011), a gpd-2/gpd-3 transplice site (Huang et al. 2001), an mCherry::Histone H2B fusion tagged with MODC PEST and 1×V5 epitope tag (Hanke et al. 1992), a tbb-2 3′UTR harboring a mutated 3x boxB sequence, and a tbb-1 intergenic region (Supplemental Fig. S1A). This construct was inserted into the C. elegans genome as a Mos1-mediated single copy transgene insertion (MosSCI) (Nance and Frokjaer-Jensen 2019). To this end, plasmids encoding the Mos1 transposon, injection markers, and dual reporter were injected into the gonad of EG8081 worms (Frokjaer-Jensen et al. 2008) containing the targeted oxTi177 site on chromosome IV. Progeny of injected animals were screened for integration of the dual reporter and loss of extrachromosomal arrays with injection marker. The inserted construct was verified by Sanger sequencing. Animals with the insertion were outcrossed two times and propagated as homozygotes at 25°C, to enhance reporter expression.
Gonad immunostaining, imaging, and fluorescence quantitation
Germline staining was performed following standard protocols (Aoki et al. 2021). For immunostaining, gonads were extruded and fixed. In Figures 1, 3, and 4, and Supplemental Figures S1, S2, gonads were fixed with 1% paraformaldehyde in 100 mM K2HPO4 for 20 min and permeabilized with ice-cold methanol for 10 min. For Figure 2, gonads were fixed with 3% paraformaldehyde in 100 mM K2HPO4 for 10 min and permeabilized with 0.2% Triton-X. Fixed, permeabilized gonads were subsequently incubated with primary antibodies overnight at 4°C (rat anti-OLLAS [1:1000, L2 clone, Novus Biologicals, #NBP1-96713], rabbit anti-V5 [1:1000, Novus Biologicals, #NB600-381], mouse anti-FLAG [1:1000, M2 clone, Sigma, #F3165]). They were then incubated with secondary antibodies for 1 h at room temperature (donkey Alexa 488 anti-rat [1:1000, Invitrogen, #A21208], goat Alexa 555 anti-rabbit [1:1000, Invitrogen, #A21429]; or donkey Alexa 647 anti-mouse [1:1000, Invitrogen, #A31571]). DAPI (0.5 ng/µL) was also included in the secondary antibody incubation to visualize DNA. For PGL-1::SNAP protein imaging, gonads were incubated with 30 nM SNAP JF 549 ligand (Grimm et al. 2015) for 1 h at room temperature, as previously performed (Aoki et al. 2021). Samples were mounted on glass slides with Vectashield (Vector Laboratories). All staining experiments were performed a minimum of two times and yielded comparable results.
Images were captured on a Leica TCS SP8 scanning laser confocal microscope running LAS X software (version 3.5.2.18963; Leica Microsystems CMS GmbH.) with a 40x oil-immersion objective and 1–1.2× zoom. Image slices (1.5 µm) were taken in sequence using identical microscope capture settings. Brightness and contrast were adjusted linearly and identically across all samples in FIJI/ImageJ (Schindelin et al. 2012). For Figures 1D, 3B, columns 2 and 3 were generated using different ImageJ contrast ranges. To quantify fluorescent signal, image stacks were z-projected by average intensity in ImageJ. Gaussian-blurred, DAPI fluorescence was used to set a threshold and mask the germlines and multiplied through the GFP and mCherry channels. Intensity of the antibody fluorescent signal was measured and averaged in 1 µm bins from the distal end of the gonad along the distal-proximal axis. For the box plot graphs, germlines were quantitated at the distal end (5–105 µm), the region where the detected reporter protein signal was strongest (Supplemental Fig. S2). The same region was measured in the SNAP ligand quantitation. Samples from at least two independent replicates were analyzed together after normalizing to a common control sample. All analysis was done using FIJI and automated using Python. Student t-tests were performed in Prism9 (version 9.0.0).
Immunoblots
For immunoblots, young adults (18–20 h past mid-L4 at 25°C; 60 worms per sample in Figs. 1F, 3C, 30 worms per sample in Fig. 2D) were boiled in 5× SDS sample buffer (250 µM Tris pH 6.8, 25 µM EDTA, 25% glycerol [v/v], 5% SDS, 70 mM 2-mercaptoethanol) for 5 min, analyzed by SDS-PAGE using a 10% stacking gel, transferred to a polyvinylidene difluoride (PVDF) membrane, and blocked with 5% powdered milk + PBS-T (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.1% Tween-20). Blots were incubated in 5% powdered milk + PBS-T with primary antibodies (rat anti-OLLAS [1:1000, L2 clone, Novus Biologicals, #NBP1-96713], mouse anti-V5 [1:3000, Bio-Rad, #MCA1360], mouse anti-FLAG [1:1000, M2 clone, Sigma, #F3165], rabbit anti-SNAP [1:1000, polyclonal, New England Biolabs, P9310S], or mouse anti-tubulin [1:40,000, Sigma, #T5168]) overnight at 4°C. After washing with PBS-T, blots were incubated with Horseradish peroxidase (HRP)-conjugated antibodies (donkey anti-mouse [H + L] [1:10,000, Jackson ImmunoResearch], goat anti-rat [H + L] [1:10,000, Jackson ImmunoResearch], or goat anti-rabbit [H + L] [1:10,000, Jackson ImmunoResearch]) in 5% powdered milk + PBS-T, washed with PBS-T, and developed with a combination of SuperSignalTM West Pico Sensitivity substrate (Thermo Scientific, #34080) and SuperSignalTM West Femto Sensitivity substrate (Thermo Scientific, #34095). Blots were imaged on Carestream Kodak BioMax light film (Sigma-Aldrich, #1788207). The film was developed using a Konica Minolta SRX-101A film processor. Immunoblots were stripped and reprobed with western blot stripping buffer (Thermo Scientific (Pierce), #21059).
Genetic assay for NOS-3 biological function
nos-3 and gld-2 single null mutants enter meiosis normally, but gld-2(ø);nos-3(ø) double mutants produce a synthetic germline tumor (Eckmann et al. 2004; Hansen et al. 2004). To test the function of λN-tagged NOS-3, we DAPI-stained worms of three genotypes: (1) gld-2(q497); nos-3(q902), (2) gld-2(q497), and (3) gld-2(q497); nos-3(q650), and scored by compound microscopy for presence of germline tumors (Supplemental Fig. S1C). For staining, worms were fixed with 1% paraformaldehyde in 100 mM K2HPO4 and stained with DAPI (0.5 ng/µL).
RNA staining, imaging, and quantitation
smFISH probe sets were designed (https://www.biosearchtech.com/support/education/stellaris-rna-fish) and synthesized commercially with conjugated fluorophores by Stellaris/Biosearch Technologies (Supplemental Table S4). The probe set directed against gfp exons contained 38 unique oligonucleotides labeled with CAL Fluor Red 610. The probe set against mCherry exons contained 39 unique olignucleotides labeled with Quasar 570. A 250 µM stock concentration of each probe set was made by dissolving lyophilized probes in RNase-free TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). A final concentration of 0.05 µM was used for hybridization of both probe sets.
Samples were prepared, stained, and imaged for smFISH as previously described (Lee et al. 2016). Briefly, animals were grown at 25°C for 18–20 h past the mid-L4 stage, and their gonads extruded in PBS-T + 0.25 mM levamisole. Samples were then fixed in 3.7% formaldehyde (Fisher Scientific, F79-500) for 13–15 min and permeabilized in RNase-free PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4) + 0.1% Triton-X for 10–12 min. Samples were incubated at room temperature in PBS-T for 30–45 min, equilibrated in smFISH wash buffer (2× SSC [Thermo Fisher Scientific, AM9763], 10% Formamide, DEPC water, 0.1% Tween-20) for 15–20 min, and incubated in hybridization buffer plus gfp and mCherry smFISH exon probes (0.05 µM) at 37°C for 46–48 h. Samples were then washed with smFISH wash buffer and DAPI (1 µg/µL) at 37°C for 40–50 min. Finally, samples were resuspended in 18 µL Prolong Glass mounting medium (Life Technologies Corporation), mounted on glass slides, and cured in the dark for at least 24 h before imaging.
Germlines were imaged on a Leica TCS SP8 laser scanning confocal microscope equipped with a Leica HC PL APO CS2 63×/1.40 NA oil immersion objective, sensitive detectors (HyDs), standard Photomultipliers (PMTs), and LAS software (version 3.5.2.18963; Leica Microsystems CMS GmbH.). Since the molecular effects of NOS-3 tethering were observed broadly in the germline, gonads were imaged in the mid-pachytene region (Fig. 4A). The zoom factor was set to 3.0 (300% zoom), the pinhole to 95.5 µm, the window to 1024 × 512 pixels, and gonads were imaged at full depth with a z-step size of 0.3 µm. Images were taken using bidirectional scan at 8000 Hz. Channels were imaged sequentially between stacks. The mCherry mRNA probed with Quasar 570 was excited with 561 nm wavelength (3.5%, HeNe) and signal collected on a HyD detector from 564–588 nm with gain set to 80. Line scans were averaged 16 times, and frames were accumulated four times. The GFP mRNA probed with Cal Fluor 610 was excited with 594 nm wavelength (3.5%, HeNe) and signal was collected on a HyD detector from 600–680 nm with gain set to 80. Line scans were averaged 32 times, and frames were accumulated twice. The native fluorescence of fixed GFP was excited with 488 nm wavelength (0.75%, Argon [25%]) and signal collected on a HyD detector from 495–555 nm with gain set to 80. Line scans were averaged 16 times, and frames were accumulated twice. Nuclear signal from DAPI was excited at 405 nm (1.0%, UV) and signal was collected on a PMT detector from 412–508 nm with gain set to 500–575 and an offset of −2.0%. Lines were averaged six times, and frames accumulated twice. Representative images were created using ImageJ. Partial maximum intensity projections were created and brightness adjusted in ImageJ. All images were treated identically.
Confocal smFISH images were analyzed by Imarisx64 (Imaris, version 9.3.1) and ImarisFileConverterx64 (Imaris, version 9.2.1) on a Dell Precision 5820 with a 64-bit Windows 10 Education operating system, an Intel Xeon W-1245 CPU @3.70GHz processor, and 128 GB of RAM. To count the number of signals detecting GFP and mCherry mRNAs in each image, “Spots” were identified by the software Creation Wizard for the gfp and mCherry mRNA image channels. The same framework was used for all Spots algorithms, though the filter thresholds differed between gfp and mCherry mRNA, and also from replicate set to replicate set. The general Spots Creation algorithm is as follows: Enable Region Of Interest = false; Enable Tracking = false; Source Channel Index = 1; Estimated XY Diameter = 0.250 µm; Estimated Z Diameter = 0.600 um; Background Subtraction = true. The different thresholds for the (Classify Spots) “Quality” filters are as follows: gfp experiments 1 and 2: 4.1530; mCherry experiment 1: 12.922; mCherry experiment 2: 8.1280. The different thresholds for the (Classify Spots) “Intensity Mean” are as follows: gfp experiments 1 and 2: between 16.342 and 62.617; mCherry experiment 1: between 41.252 and 87.664; mCherry experiment 2: between 26.791 and 114.70. Manual thresholds were determined by eye. Identical gfp or mCherry mRNA thresholds were applied to all gfp or mCherry images within the same experimental replicate. Student t-tests were performed in Prism9 (version 9.0.0). Data from two independent experiments was combined before statistical tests were run.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Lavis for supplying the SNAP JF 549 ligand, C. Lee for smFISH advice, S. Crittenden for Nanos advice, L. Vanderploeg for help with figure revisions, and members of the Kimble and Wickens laboratories for helpful discussions. T.R.L. was supported by the NSF (National Science Foundation); this material is based upon work supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1747503. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not reflect the views of the National Science Foundation. J.K. was an Investigator of the Howard Hughes Medical Institute and is now supported by National Institutes of Health (NIH) R01 GM134119. S.T.A. was supported by NIH K99 HD081208.
Author contributions: J.D. and T.R.L. conceived and performed the experiments and helped write the paper; J.K. and S.T.A. conceived the experiments and wrote the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078693.121.
Freely available online through the RNA Open Access option.
REFERENCES
- Aoki ST, Porter DF, Prasad A, Wickens M, Bingman CA, Kimble J. 2018. An RNA-binding multimer specifies nematode sperm fate. Cell Rep 23: 3769–3775. 10.1016/j.celrep.2018.05.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki ST, Lynch TR, Crittenden SL, Bingman CA, Wickens M, Kimble J. 2021. C. elegans germ granules require both assembly and localized regulators for mRNA repression. Nat Commun 12: 996. 10.1038/s41467-021-21278-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, Fire AZ. 2014. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198: 837–846. 10.1534/genetics.114.169730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arur S, Ohmachi M, Berkseth M, Nayak S, Hansen D, Zarkower D, Schedl T. 2011. MPK-1 ERK controls membrane organization in C. elegans oogenesis via a sex-determination module. Dev Cell 20: 677–688. 10.1016/j.devcel.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvola RM, Weidmann CA, Tanaka Hall TM, Goldstrohm AC. 2017. Combinatorial control of messenger RNAs by pumilio, nanos and brain tumor proteins. RNA Biol 14: 1445–1456. 10.1080/15476286.2017.1306168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron-Benhamou J, Gehring NH, Kulozik AE, Hentze MW. 2004. Using the λN peptide to tether proteins to RNAs. Methods Mol Biol 257: 135–154. 10.1385/1-59259-750-5:135 [DOI] [PubMed] [Google Scholar]
- Bhandari D, Raisch T, Weichenrieder O, Jonas S, Izaurralde E. 2014. Structural basis for the Nanos-mediated recruitment of the CCR4-NOT complex and translational repression. Genes Dev 28: 888–901. 10.1101/gad.237289.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. 10.1093/genetics/77.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner JL, Schedl T. 2016. Germline stem cell differentiation entails regional control of cell fate regulator GLD-1 in Caenorhabditis elegans. Genetics 202: 1085–1103. 10.1534/genetics.115.185678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Garcia-Mena J, DeVito J, Wolska K, Das A. 1995. Bipartite function of a small RNA hairpin in transcription antitermination in bacteriophage lambda. Proc Natl Acad Sci 92: 4061–4065. 10.1073/pnas.92.9.4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller J, Wickens M. 2007. Tethered function assays: an adaptable approach to study RNA regulatory proteins. Methods Enzymol 429: 299–321. 10.1016/S0076-6879(07)29014-7 [DOI] [PubMed] [Google Scholar]
- Coller JM, Gray NK, Wickens MP. 1998. mRNA stabilization by poly(A) binding protein is independent of poly(A) and requires translation. Genes Dev 12: 3226–3235. 10.1101/gad.12.20.3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gregorio E, Preiss T, Hentze MW. 1999. Translation driven by an eIF4G core domain in vivo. EMBO J 18: 4865–4874. 10.1093/emboj/18.17.4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gregorio E, Baron J, Preiss T, Hentze MW. 2001. Tethered-function analysis reveals that elF4E can recruit ribosomes independent of its binding to the cap structure. RNA 7: 106–113. 10.1017/S1355838201000577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmann CR, Crittenden SL, Suh N, Kimble J. 2004. GLD-3 and control of the mitosis/meiosis decision in the germline of Caenorhabditis elegans. Genetics 168: 147–160. 10.1534/genetics.104.029264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdos G, Dosztanyi Z. 2020. Analyzing protein disorder with IUPred2A. Curr Protoc Bioinformatics 70: e99. 10.1002/cpbi.99 [DOI] [PubMed] [Google Scholar]
- Farley BM, Ryder SP. 2012. POS-1 and GLD-1 repress glp-1 translation through a conserved binding-site cluster. Mol Biol Cell 23: 4473–4483. 10.1091/mbc.e12-03-0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frand AR, Russel S, Ruvkun G. 2005. Functional genomic analysis of C. elegans molting. PLoS Biol 3: e312. 10.1371/journal.pbio.0030312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40: 1375–1383. 10.1038/ng.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Hall TMT, McKenney KM. 2018. Post-transcriptional regulatory functions of mammalian pumilio proteins. Trends Genet 34: 972–990. 10.1016/j.tig.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NK, Coller JM, Dickson KS, Wickens M. 2000. Multiple portions of poly(A)-binding protein stimulate translation in vivo. EMBO J 19: 4723–4733. 10.1093/emboj/19.17.4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. 2015. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12: 244–250. 10.1038/nmeth.3256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke T, Szawlowski P, Randall RE. 1992. Construction of solid matrix-antibody-antigen complexes containing simian immunodeficiency virus p27 using tag-specific monoclonal antibody and tag-linked antigen. J Gen Virol 73: 653–660. 10.1099/0022-1317-73-3-653 [DOI] [PubMed] [Google Scholar]
- Hansen D, Wilson-Berry L, Dang T, Schedl T. 2004. Control of the proliferation versus meiotic development decision in the C. elegans germline through regulation of GLD-1 protein accumulation. Development 131: 93–104. 10.1242/dev.00916 [DOI] [PubMed] [Google Scholar]
- Huang T, Kuersten S, Deshpande AM, Spieth J, MacMorris M, Blumenthal T. 2001. Intercistronic region required for polycistronic pre-mRNA processing in Caenorhabditis elegans. Mol Cell Biol 21: 1111–1120. 10.1128/MCB.21.4.1111-1120.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard EJA, Schedl T. 2019. Biology of the Caenorhabditis elegans germline stem cell system. Genetics 213: 1145–1188. 10.1534/genetics.119.300238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaymak E, Farley BM, Hay SA, Li C, Ho S, Hartman DJ, Ryder SP. 2016. Efficient generation of transgenic reporter strains and analysis of expression patterns in Caenorhabditis elegans using library MosSCI. Dev Dyn 245: 925–936. 10.1002/dvdy.24426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517: 583–588. 10.1038/nature14136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer B, Crittenden S, Gallegos M, Moulder G, Barstead R, Kimble J, Wickens M. 1999. NANOS-3 and FBF proteins physically interact to control the sperm-oocyte switch in Caenorhabditis elegans. Curr Biol 9: 1009–1018. 10.1016/S0960-9822(99)80449-7 [DOI] [PubMed] [Google Scholar]
- Kusz-Zamelczyk K, Sajek M, Spik A, Glazar R, Jedrzejczak P, Latos-Bielenska A, Kotecki M, Pawelczyk L, Jaruzelska J. 2013. Mutations of NANOS1, a human homologue of the Drosophila morphogen, are associated with a lack of germ cells in testes or severe oligo-astheno-teratozoospermia. J Med Genet 50: 187–193. 10.1136/jmedgenet-2012-101230 [DOI] [PubMed] [Google Scholar]
- Lee C, Sorensen EB, Lynch TR, Kimble J. 2016. C. elegans GLP-1/Notch activates transcription in a probability gradient across the germline stem cell pool. Elife 5: e18370. 10.7554/eLife.18370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Seidel H, Lynch T, Sorensen E, Crittenden S, Kimble J. 2017. Single-molecule RNA fluorescence in situ hybridization (smFISH) in Caenorhabditis elegans. Bio Protoc 7. 10.21769/BioProtoc.2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR. 1998. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem 273: 34970–34975. 10.1074/jbc.273.52.34970 [DOI] [PubMed] [Google Scholar]
- Malik S, Jang W, Park SY, Kim JY, Kwon KS, Kim C. 2019. The target specificity of the RNA binding protein Pumilio is determined by distinct co-factors. Biosci Rep 39: BSR20190099. 10.1042/BSR20190099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G. 2008. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18: 1476–1482. 10.1016/j.cub.2008.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Wharton RP. 1995. Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos. Cell 80: 747–756. 10.1016/0092-8674(95)90353-4 [DOI] [PubMed] [Google Scholar]
- Nance J, Frokjaer-Jensen C. 2019. The Caenorhabditis elegans transgenic toolbox. Genetics 212: 959–990. 10.1534/genetics.119.301506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. 2015. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics 201: 47–54. 10.1534/genetics.115.179382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Cheong C, Idoyaga J, Kim JY, Choi JH, Do Y, Lee H, Jo JH, Oh YS, Im W, et al. 2008. Generation and application of new rat monoclonal antibodies against synthetic FLAG and OLLAS tags for improved immunodetection. J Immunol Methods 331: 27–38. 10.1016/j.jim.2007.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Artus CG, Filipowicz W. 2004. Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA 10: 1518–1525. 10.1261/rna.7131604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisch T, Bhandari D, Sabath K, Helms S, Valkov E, Weichenrieder O, Izaurralde E. 2016. Distinct modes of recruitment of the CCR4-NOT complex by Drosophila and vertebrate Nanos. EMBO J 35: 974–990. 10.15252/embj.201593634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. 2008. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879. 10.1038/nmeth.1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Calidas D, Schmidt H, Lu T, Rasoloson D, Seydoux G. 2016. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife 5: e21337. 10.7554/eLife.21337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Wharton RP. 1999. Recruitment of Nanos to hunchback mRNA by Pumilio. Genes Dev 13: 2704–2712. 10.1101/gad.13.20.2704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam K, Seydoux G. 1999. nos-1 and nos-2, two genes related to Drosophila nanos, regulate primordial germ cell development and survival in Caenorhabditis elegans. Development 126: 4861–4871. [DOI] [PubMed] [Google Scholar]
- Suh N, Jedamzik B, Eckmann CR, Wickens M, Kimble J. 2006. The GLD-2 poly(A) polymerase activates gld-1 mRNA in the Caenorhabditis elegans germ line. Proc Natl Acad Sci 103: 15108–15112. 10.1073/pnas.0607050103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh N, Crittenden SL, Goldstrohm A, Hook B, Thompson B, Wickens M, Kimble J. 2009. FBF and its dual control of gld-1 expression in the Caenorhabditis elegans germline. Genetics 181: 1249–1260. 10.1534/genetics.108.099440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Saba R, Miyoshi K, Morita Y, Saga Y. 2012. Interaction between NANOS2 and the CCR4-NOT deadenylation complex is essential for male germ cell development in mouse. PLoS ONE 7: e33558. 10.1371/journal.pone.0033558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, Kobayashi S, Saga Y. 2003. Conserved role of nanos proteins in germ cell development. Science 301: 1239–1241. 10.1126/science.1085222 [DOI] [PubMed] [Google Scholar]
- Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, et al. 2011. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472: 120–124. 10.1038/nature09819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedeles CJ, Wu MZ, Claycomb JM. 2013. Protection of germline gene expression by the C. elegans Argonaute CSR-1. Dev Cell 27: 664–671. 10.1016/j.devcel.2013.11.016 [DOI] [PubMed] [Google Scholar]
- Weidmann CA, Qiu C, Arvola RM, Lou TF, Killingsworth J, Campbell ZT, Tanaka Hall TM, Goldstrohm AC. 2016. Drosophila Nanos acts as a molecular clamp that modulates the RNA-binding and repression activities of Pumilio. Elife 5: e17096. 10.7554/eLife.17096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, Parker R. 2002. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet 18: 150–157. 10.1016/S0168-9525(01)02616-6 [DOI] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, Lehmann R. 1997. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3: 1421–1433. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.