

Abstract
Pyruvate kinase (PK) deficiency is a rare recessive congenital hemolytic anemia caused by mutations in the PKLR gene. This study reports the molecular features of 257 patients enrolled in the PKD Natural History Study. Of the 127 different pathogenic variants detected, 84 were missense and 43 non-missense, including 20 stop-gain, 11 affecting splicing, five large deletions, four in-frame indels, and three promoter variants. Within the 177 unrelated patients, 35 were homozygous and 142 compound heterozygous (77 for two missense, 48 for one missense and one non-missense, and 17 for two non-missense variants); the two most frequent mutations were p.R510Q in 23% and p.R486W in 9% of mutated alleles. Fifty-five (21%) patients were found to have at least one previously unreported variant with 45 newly described mutations. Patients with two non-missense mutations had lower hemoglobin levels, higher numbers of lifetime transfusions, and higher rates of complications including iron overload, extramedullary hematopoiesis, and pulmonary hypertension. Rare severe complications, including lower extremity ulcerations and hepatic failure, were seen more frequently in patients with non-missense mutations or with missense mutations characterized by severe protein instability. The PKLR genotype did not correlate with the frequency of complications in utero or in the newborn period. With ICCs ranging from 0.4 to 0.61, about the same degree of clinical similarity exists within siblings as it does between siblings, in terms of hemoglobin, total bilirubin, splenectomy status, and cholecystectomy status. Pregnancy outcomes were similar across genotypes in PK deficient women. This report confirms the wide genetic heterogeneity of PK deficiency.
1 |. INTRODUCTION
Pyruvate kinase (PK) deficiency is the most frequent enzyme abnormality of the glycolytic pathway, and the most common cause of hereditary non-spherocytic hemolytic anemia. Pyruvate kinase deficiency has a worldwide geographical distribution with an estimated prevalence ranging from 1 to 5 per 100 000 in the Caucasian population. Higher frequencies are present in the Pennsylvania Amish and the Roma communities as a result of a founder effect.1–4 Discrepancies exist between the published estimated prevalence, and the lower prevalence suggested by data from patient registries and clinical practice. This may be explained by a high number of mildly affected undiagnosed patients and challenges in diagnosing PK deficiency.5
Clinical manifestations of PK deficiency reflect the symptoms and complications of lifelong chronic hemolysis, including anemia, jaundice, bilirubin gallstones, splenomegaly, and iron overload.6–9 The severity of symptoms is highly variable, ranging from mild anemia or fully compensated hemolysis to life-threatening anemia necessitating neonatal exchange transfusions and/or subsequent regular transfusion support.10–12
PK deficiency is inherited in an autosomal recessive pattern and is caused by homozygous or compound heterozygous variants in the PKLR gene located on chromosome 1q21.13 To date, more than 300 mutations in the PKLR gene have been reported (https://databases.lovd.nl/shared/genes/PKLR).8,11–15 The majority of these mutations are missense substitutions affecting residues critical to the structure and/or function of the enzyme, followed by frameshift and splicing mutations and premature stop codons; promoter variants and large insertions/deletions (indel) are rare. Recently, compound heterozygous variants with deep intronic mutations as a cause of PK deficiency have been reported.16 The most frequent mutations are distributed regionally: p.R510Q in Northern Europe and the United States, p.R486W in Southern Europe, and p.R490W in Japan.13
Genotype-phenotype correlation in PK deficiency has been investigated through both clinical studies and by in vitro production and characterization of recombinant mutant proteins of human PK.17–19 These studies have suggested that patients with severe hemolytic anemia more commonly have deleterious mutations or missense pathogenic variants which affect the active site or stability of the PK protein. This relationship between disruptive PKLR mutations to more severe symptoms and complications in PK deficiency has also been confirmed by data from the Pyruvate Kinase Deficiency Natural History Study (PKD NHS), a multicenter, international registry.6 With an improved understanding of the relationship between genotype and clinical features, genetic testing has been recommended to assist in determination of expected prognosis and development of an individualized monitoring plan. Recent consensus guidelines on the diagnosis of PK deficiency recommend diagnostic confirmation with genetic testing.5,20
In this manuscript, we report the molecular characterization of 257 patients with PK deficiency enrolled in the PKD NHS. The study highlights the wide molecular heterogeneity of PK deficiency, including 45 novel variants reported in this cohort. The large number of patients enrolled worldwide also allows a description of clinical features of the most common homozygous mutations.
2 |. MATERIALS AND METHODS
2.1 |. Patients
The international PKD NHS was opened at 30 centres (United States (n = 19), Canada (n = 3), Italy (n = 1), Czech Republic (n = 1), Germany (n = 5), Netherlands (n = 1), Table S1). The study protocol was approved by the Institutional Review Board and/or Ethics Committee at each site, and all patients or their legal guardians gave informed consent. Patients were able to participate from afar by signed medical releases or were primarily followed at a centre approved to conduct the study.6
Patients were eligible to be included in the registry study if they had a genetically confirmed diagnosis of PK deficiency with two identified PKLR mutations. If patients had prior genetic testing which confirmed two pathogenic PKLR mutations, this was not repeated. If prior genetic testing was not conducted or the results were not available, samples were sent for molecular analysis by Sanger sequencing (Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico Milan or Yale-New Haven Children’s Hospital).
At the time of enrolment, patients’ medical records were reviewed retrospectively. Data collected included medical history, physical examination, and laboratory and radiologic studies. Clinical complications, including extramedullary hematopoiesis and pulmonary hypertension, were reported within the medical record and defined by individual clinical teams. Medical history missing from the medical records was obtained by patient recall, if known. Transfusion data were collected from medical records. At all sites, except in Lancaster, Pennsylvania, data were only collected if obtained for clinical purposes. In Lancaster, Pennsylvania, in which all enrolled patients were Amish, additional laboratory and radiologic data were collected under a site-specific IRB-approved protocol.
2.2 |. Molecular analysis
For the 119 patients who underwent sequencing through the study, the entire coding region and promoter of the PKLR gene were amplified by polymerase chain reaction (PCR) and automatically sequenced (ABIPRISM 310 Genetic Analyzer, Applied Biosystems, Warrington, UK) using the Big Dye Terminator Cycle Sequencing Kit.21 Large deletions were detected by long range PCR and separation on agarose gel. Mutations were reported using the RPK cDNA sequence of the PKLR gene, with the A of the initiation ATG being assigned number +1 (Transcript refseq ID NM_000298.5). Previously unreported novel mutations were defined as those not described by HGMD, 1000 Genomes, or ExAC databases. The new variants were classified according to the American College of Medical Genetic and Genomics (ACMG) standards and guidelines.22 Expected pathogenicity was assessed by using the online prediction tools Polyphen2, Predict SNP (https://loschmidt.chemi.muni.cz/predictsnp1), and M-CAP (http://bejerano.stanford.edu/mcap).22,23 Prediction analysis for splice site mutations was conducted by the web server tool Human Splice Finder (HSF3.1), using both MaxEnt and HSF algorithms.24
2.3 |. Genotype categories and genotype-phenotype analysis
For this analysis, patients were categorized according to their genotype as follows: (a) missense/missense; (b) missense/non-missense; or (c) non-missense/non-missense mutations. Non-missense pathogenic variants included nonsense, frameshift, in-frame small indels, large deletions, and splicing variants; missense mutations also affecting splicing were categorized as non-missense (i.e., p.R479H). Of the 257 patients with genotype data, 62 patients were excluded from the genotype-phenotype correlation analysis, including patients with three pathogenic variants (n = 2) or promoter variants classified as uncertain significance (n = 3) and patients who did not report clinical and demographical data (n = 2). Patients from the Amish community (homozygous for the splicing mutation p.R479H, n = 55) were also excluded from the main genotype-phenotype analysis. Thus, the genotype-phenotype analysis included 195 patients. The most common homozygous variants, p.R479H and p.R510Q, were analysed separately for an association between genotype and phenotype. Additionally, the correlation between pregnancy outcomes and perinatal course of patients were separately analysed according to genotype groups.
2.4 |. Transfusion status and iron overload
Transfusion needs were classified into the following categories: (a) regularly transfused: ≥6 transfusions within a 12-month period; (b) not regularly transfused: one to five transfusions within a 12-month period; (c) not transfused: no transfusions. Iron overload at enrollment was defined as a maximum ferritin >1000 ng/mL or having received chelation within the 12 months prior to enrolment.
2.5 |. Statistical analysis
Patient demographics, transfusion status, comorbid diagnoses and other disease characteristics were described with frequencies, proportions, medians, means, ranges and inter-quarter ranges (IQR) as appropriate to sample size, overall, by genotype category, and p. R510Q and p.R486W variant subgroups. Differences between genotype categories were identified using a Fisher’s exact test or a Wilcoxon rank-sum test. Sample sizes are presented for those with known data available for each variable. To quantify the degree of similarity between siblings, the intra-cluster correlation coefficient (ICC) was calculated for clinical factors. ICC (range: 0 to 1 in human studies) compares the variance within sibling clusters to the variance between sibling clusters and tends to be larger for small clusters. ICCs and their 95% confidence intervals (CI) were computed using the SAS ICC9 macro.25 Data analyses were performed using SAS software 9.4 (SAS Institute Inc., Cary, NC, USA). Sample sizes are presented for those with known data available for each variable. The P values are two-sided, and P-values <.05 were considered statistically significant.
3. RESULTS
3.1 |. Patients
The PKD NHS enrolled 278 patients with PK deficiency from June 2014 through April 2017. The clinical characteristics of the cohort have been previously described.6,9 After exclusion of 21 ineligible patients in whom two pathogenic PKLR mutations could not be demonstrated, 257 eligible participants are reported herein. Baseline demographic information and clinical complications, available in 255 patients, are presented overall and by genotype category (Table 1).
TABLE 1.
Demographic features and common complications of enrolled patients by PKLR mutation type (n = 255)
| Characteristics | All (N = 255f) |
Missense/Missensea (N = 113) |
Missense/Non-Missense (N = 52) |
Non-Missense/Non-Missense (N = 30) |
P valueb | ||||
|---|---|---|---|---|---|---|---|---|---|
| n | % or median (range) | n | % or median (range) | n | % or median (range) | n | % or median (range) | ||
| Gender, male | 125 | 49% | 66/113 | 58% | 30/52 | 58% | 14/30 | 47% | .3 |
| Age at diagnosis (y) (median, range) | 244e | 0.4 (0-60.3) | 107 | 1.3 (0-60.3) | 50 | 0.7 (0-42.3) | 30 | 0.5 (0-10.9) | .03 |
| Age at enrolment (y) (median, range) | 255 | 19.0 (0.1-69.9) | 113 | 17 (0.1-68.5) | 52 | 17.5 (0.6-69.9) | 30 | 14.6 (0.2-50.3) | .3 |
| Race, Caucasian | 236 | 93% | 103/113 | 91% | 48/52 | 92% | 25/30 | 83% | .1 |
| Ethnicity, hispanic | 18/247 | 7% | 8/108 | 7% | 6/50 | 12% | 4/30 | 13% | .5 |
| Number of lifetime transfusions (median, range) | 191 | 18(1-516) | 81 | 16 (1-486) | 38 | 25(1-516) | 28 | 67 (3-363) | <.001 |
| Transfusion status in the last 12 months | <.001 | ||||||||
| Regularly transused (≥6) | 49/255 | 19% | 22/113 | 19% | 10/52 | 19% | 14/30 | 47% | |
| Not Regularly transfused (1-5) | 43/255 | 17% | 22/113 | 19% | 8/52 | 15% | 8/30 | 27% | |
| Not transfused (0) | 163/255 | 64% | 69/113 | 61% | 34/52 | 65% | 8/30 | 27% | |
| Hemoglobin (g/dL) (median, range) | 252 | 9 (4.3-14.2) | 111 | 9.5 (4.3-13.9) | 51 | 8.7 (6.4-14.2) | 30 | 7.9 (6.2-9.7) | <.001 |
| Hemoglobin post-splenectomy (g/dL) | 148 | 8.8 (4.3-12.8) | 49 | 9.2 (4.3-12.3) | 26 | 8.5 (6.4-12.8) | 21 | 8.1 (6.5-9.7) | .009 |
| Hemoglobin change from pre-splenectomy to post-splenectomy (g/dL) | 72 | 1.6 (-5.5-4.9) | 27 | 1.6 (-3.3-4.5) | 13 | 1.6 (-5.5-3.4) | 13 | 1 (-2.8-2.7) | .3 |
| Transfusion status post-splenectomy | .06 | ||||||||
| Regularly transused (≥6) | 17/146 | 12% | 7/47 | 15% | 4/27 | 15% | 6/20 | 30% | |
| Not Regularly transfused (1-5) | 23/146 | 16% | 9/47 | 19% | 5/27 | 19% | 7/20 | 35% | |
| Not transfused (0) | 106/146 | 73% | 31/47 | 66% | 18/27 | 67% | 7/20 | 35% | |
| Splenectomized | 150/255 | 59% | 50/113 | 44% | 27/52 | 52% | 21/30 | 70% | .03 |
| Gallstones | 112/249 | 45% | 49/109 | 45% | 26/51 | 51% | 12/30 | 40% | .6 |
| Maximum ferritin (mg/dL) (median, range) | 180 | 656 (22-13409) | 75 | 573 (31-9679) | 36 | 604 (22-8220) | 23 | 1700 (423-13409) | <.001 |
| Regularly transused (≥6) | 45 | 1286 (22-9679) | 21 | 979 (73-9679) | 9 | 986 (22-1499) | 12 | 2276 (423-4266) | .02c |
| Not Regularly transfused (1-5) | 31 | 974 (59-13409) | 15 | 648 (59-3890) | 5 | 513 (144-8220) | 8 | 2068 (1065-13409) | .01c |
| Not transfused (0) | 104 | 544 (31-4500) | 39 | 399 (31-4500) | 22 | 566 (73-2644) | 3 | 776 (745-1459) | .1c |
| Iron Overload | 89/186 | 48% | 33/77 | 43% | 19/37 | 51% | 22/26 | 85% | <.001 |
| Regularly transused (≥6) | 32/45 | 71% | 14/21 | 67% | 7/9 | 78% | 9/12 | 75% | 1d |
| Not Regularly transfused (1-5) | 18/33 | 55% | 6/16 | 38% | 3/6 | 50% | 8/8 | 100% | <.004d |
| Not transfused (0) | 39/108 | 36% | 13/40 | 33% | 9/22 | 41% | 5/6 | 83% | .03d |
| Extramedullar/hematopoiesis | 23/213 | 11% | 5/94 | 5% | 0/43 | 0% | 3/26 | 12% | .1 |
| Pulmonary hypertension | 8/238 | 3% | 3/101 | 3% | 1/49 | 2% | 3/29 | 10% | .09 |
| Leg ulcers | 4/241 | 2% | 1/104 | 1% | 2/49 | 4% | 1/29 | 3% | .5 |
| Aplastic crises | 34/246 | 14% | 15/110 | 14% | 9/50 | 18% | 4/28 | 14% | 1 |
| Liver cirrhosis | 8/241 | 3% | 2/107 | 2% | 2/48 | 4% | 1/28 | 4% | .6 |
| Bone fracture | 41/240 | 17% | 18/103 | 17% | 8/49 | 16% | 6/29 | 21% | .6 |
| Endocrine dysfunction | |||||||||
| Thyroid disease | 11/242 | 5% | 5/103 | 5% | 1/51 | 2% | 1/30 | 3% | 1 |
| Growth hormone deficiency | 6/237 | 3% | 4/101 | 4% | 2/48 | 4% | 0/29 | 0% | .6 |
| Hypoparathyroidism | 3/233 | 1% | 1/101 | 1% | 0/46 | 0% | 1/29 | 3% | .3 |
| Diabetes | 2/251 | 1% | 2/109 | 2% | 0/52 | 0% | 0/30 | 0% | 1 |
| Hypogonadal hypogonadism | 0/234 | 0% | 0/101 | 0% | 0/46 | 0% | 0/29 | 0% | NA |
Note: Countries of enrollment: United States (155), Germany (31), Italy (25), Netherlands (23), Canada (14), Czech Republic (7).
Abbreviations: N: Total number of patients enrolled overall; n: number of patients per demographic characteristic; NA, Not applicable. Age expressed as n, median (range) years; all other data expressed as n (%).
Patients with R479H mutation are excluded from the missense/missense cohort.3
P values are obtained by comparing missense/missense and missense/non-missense combined group versus non-missense/non-missense using Wilcoxon rank-sum test for continuous variables (age, hemoglobin, maximum ferritin, number of lifetime transfusions variables), Cochran and Armitage trend test for ordinal variables (transfusion status in the last 12 months, transfusion status post-splenectomy), and Fisher’s exact test for binary variables (gender, race, ethnicity, splenectomized, gallstones, iron overload, extramedullary hematopoiesis, pulmonary hypertension, leg ulcers, aplastic crises, liver cirrhosis, bone fracture, and endocrine dysfunction variables).
Comparing ferritin measurements between the missense/missense and missense/non-missense combined group versus non-missense/non-missense using Wilcoxon rank-sum test in subsets of patients (subsets are: regularly transfused (≥6), not regularly transfused (1-5), not transfused (0) respectively).
Comparing iron overload between the missense/missense and missense/non-missense combined group vs non-missense/non-missense using Fisher’s exact test in subsets of patients (subsets are: regularly transfused (≥6), not regularly transfused (1-5), not transfused (0) respectively).
Eleven patients had unknown age at diagnosis.
Two hundred fifty-five out of 257 PKD patients have reported demographical and clinical data and their data are recorded in the database; the denominators are less than 255 because some patients had unknown data.
3.2 |. Spectrum of mutations
Molecular characterization confirmed the wide genetic heterogeneity of PK deficiency. Overall, 127 different mutations were detected: 84 missense, 20 premature stop codons (seven nonsense and 13 frameshift due to small indels mutations), and 11 splicing mutations; moreover, five large deletions, four in-frame indels, and three promoter variants were detected (Tables S2–S4). Most patients (85%) had at least one missense mutation. Two patients had three different mutations. Excluding the Amish cohort, the two most frequent mutations were p.R510Q and p. R486W accounting for 23% (83/354) and 9% (31/354) of unrelated alleles, respectively. An additional 10 mutations were detected in unrelated patients with a frequency >1.4% each (Figure 1A).
FIGURE 1.
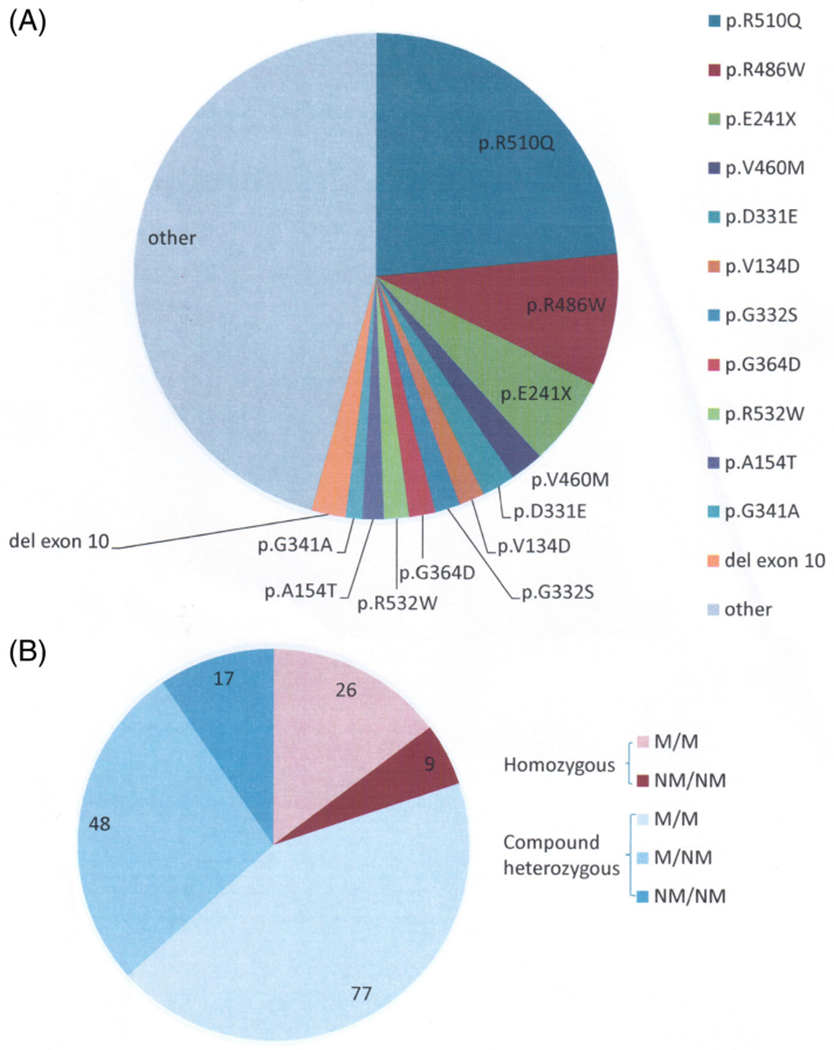
Spectrum of PKLR mutations in the PKD Natural History Study. A, Shows the frequency of all of the detected unrelated mutations. The Amish cohort is excluded from this figure. B, Shows frequency of patients with missense/missense (M/M), missense/non-missense (M/NM), and non-missense/non-missense (NM/NM) mutations
Out of the 177 unrelated patients, 35 had homozygous mutations (19 were homozygous for p.R510Q), 77 patients were compound heterozygous for two different missense mutations, and 48 were compound heterozygous for one missense and one non-missense mutation. Seventeen patients were compound heterozygous for two non-missense mutations (Figure 1B).
Thirteen patients carried large deletions, 11 of them were homozygous. The exact cut-off points were defined for the deletions in only seven of these patients. In six unrelated patients, the reported mutation was c.1437-518_1618+440del, encompassing exons 10 and 11, which has been previously reported in patients with a Roma ethnic background.4 In one patient, the reported mutation was c.283+1914_c.1434del5006, encompassing exons 3-11, which has been previously reported in patients with Asian ancestry.11 In the remaining six patients, reported deletions included exons 10-11, exon 2-3, and another large deletion without precisely reported cut-off points.
Forty-five variants, all predicted to be likely pathogenic or pathogenic following ACMG guidelines, have not been previously reported; among them were 28 missense, two stop codon, six frameshift, three in-frame indel, five splicing, and one promoter mutation (Figure 2, Table S5). Of the 257 patients, 55 (21%) were found to have at least one new molecular variant.
FIGURE 2.
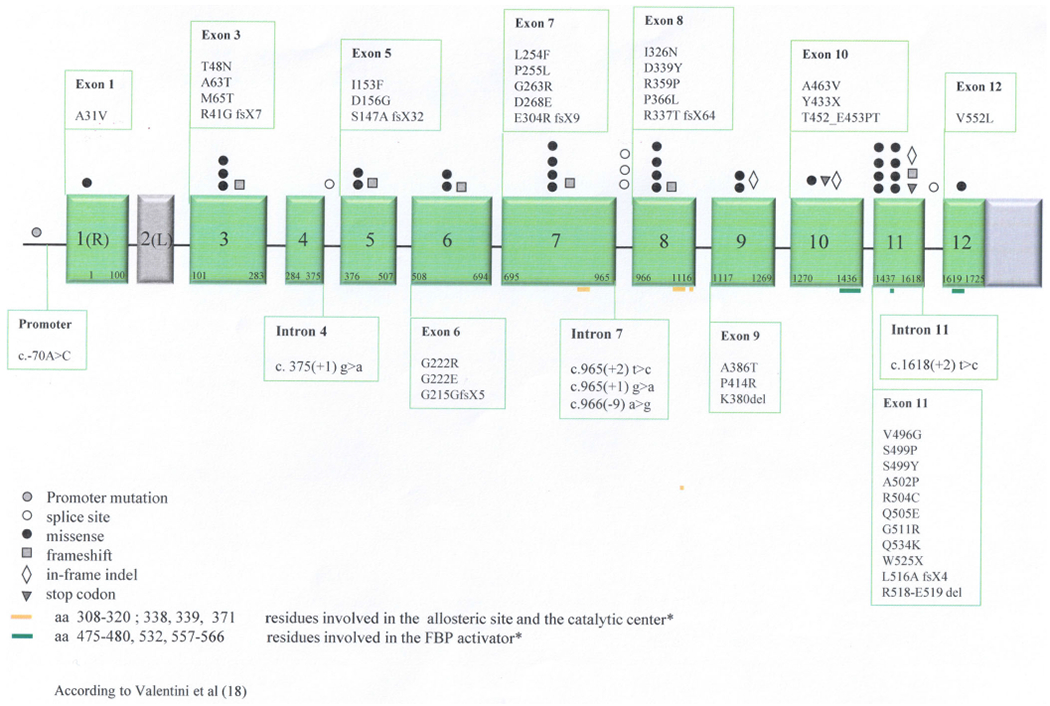
Novel PKLR gene variants identified in the PKD NHS cohort17
3.3 |. Genotype-phenotype associations
Of the 195 patients in the genotype-phenotype analytic cohort, patient PKLR gene mutation types included: 113 (58%) missense/missense, 52 (27%) missense/non-missense, and 30 (15%) non-missense/non-missense mutations (Table 1). As compared with patients with missense/missense (M/M) or missense/non-missense (M/NM) PKLR mutations, patients with non-missense/non-missense (NM/NM) mutations have a lower hemoglobin level post-splenectomy (M/M and M/NM: 8.7 g/dL range: 4.3-12.8, n = 75; NM/NM: 8.1 g/dL, range: 6.5-9.7, n = 21; P = .009), higher number of lifetime transfusions (M/M and M/NM: 20, range: 1-516; NM/NM: 67, range: 3-363; P = .001), higher rate of iron overload (M/M and M/NM: 46%; NM/NM: 85%; P = .001), and higher rate of splenectomy (M/M and M/NM: 47%; NM/NM: 70%; P = .03). The frequency of pulmonary hypertension and extramedullary hematapoiesis was highest in patients with non-missense/non-missense mutations (M/M and M/NM: 3.6% versus NM/NM 12%; and M/M and M/NM: 2.7% versus NM/NM 10%, respectively). Other complications were of similar frequency between the groups including gallstones, leg ulcers, bone fractures, and endocrinopathies.
3.4 |. Sibling-phenotype correlation
Of the 257 patients, 88 had one or more siblings who were also in this study. These 88 comprised a total of 38 families, with a minimum of two siblings and a maximum of five siblings per family. There were 29 families of two affected siblings; seven with three affected siblings; one with four affected siblings, and one with five affected siblings.
With ICCs ranging from 0.4 to 0.61, about the same degree of similarity exists within sibling clusters as it does between sibling clusters, in terms of hemoglobin, total bilirubin, splenectomy, and cholecystectomy (Table S6).
In most of the families (28/38, 74%), siblings were homogeneous in terms of their splenectomy and transfusion status. Of these 28 homogeneous families, 10 families were not splenectomized and not on regular transfusion, 17 families were splenectomized but not on regular transfusion, one family was splenectomized and receiving regular transfusion.
3.5 |. Genotype and perinatal and pregnancy outcomes
The in utero and perinatal course of patients with PK deficiency was not significantly different based on genotype (Table 2). Of patients documented to have had perinatal complications and/or treatment, the rate of in utero transfusions was not significantly different between the mutation groups: M/M 46% (12/26), M/NM 56% (8/15), and NM/NM 40% (4/10). Among patients for whom data were available, the rate of treatment with exchange transfusions was also not different between the groups: M/M 42% (34/80), M/NM 43% (17/40), and NM/NM 43% (9/21). There were 29 pregnancies in women with M/M mutations, 13 with M/NM, and seven with NM/NM. The rate of full-term healthy births was not different between the groups (Table 2). However, management during pregnancy was significantly different by genotype. Only 42% of pregnant women with M/M mutations were transfused during pregnancy, while 100% of those with NM/NM mutations were transfused during pregnancy.
TABLE 2.
Perinatal course and pregnancy outcomes of patients by PKLR mutation typea
| Characteristics | Missense/Missense N = 113 patients n |
Missense/Non-Missense N = 52 patients n |
Non-Missense/Non-Missense N = 30 patient n |
P valueb |
|---|---|---|---|---|
| Perinatal Complications/Treatment | 28/100 (28%) | 15 /48 (31%) | 11/28 (39%) | .26 |
| In utero transfusions | 12/26 (46%) | 8/15 (53%) | 4/10 (40%) | 1.0 |
| Hydrops | 3/26 (12%) | 5/14 (36%) | 1/11 (9%) | 1.0 |
| Exchange transfusion | 34/80 (42%) | 17/40 (43%) | 9/21 (43%) | 1.0 |
| Phototherapy | 69/78 (88%) | 29/37 (78%) | 17/20 (85%) | .22 |
| Pregnancy Outcomes/Management (data presented per pregnancy of affected woman with PKD)a | n = 29 pregnanciesa from 16 pregnant women | n = 13 pregnanciesa from 6 pregnant women | n = 7 pregnanciesa from 3 pregnant women | |
| Normal birth - full-term | 20/29 (69%) | 8/13 (62%) | 5/7 (71%) | 1.0 |
| Pre-maturity | 3/29 (10%) | 0/13 (0%) | 2/7 (29%) | .24 |
| Transfusions during pregnancy | 9/21 (43%) | 3/6 (50%) | 5/5 (100%) | .043 |
Patients with known and available clinical data. For each proportion calculated, the denominator is the number of patients or pregnancies with known data for that characteristic.
Comparing Missense/Missense to non-Missense/non-Missense group using Fisher exact test.
3.6 |. Homozygous p.R479H variant
All 55 Amish patients were homozygous for the p.R479H PKLR variant. Most patients were diagnosed soon after birth based on clinical symptoms in the setting of the high prevalence of PK deficiency in the Amish community, with a median age of diagnosis of two days (range 0 days-10 years, n = 52), and 53% required exchange transfusion after birth (Table S7). In the 12-month period prior to enrolment, one Amish patient (2%) was regularly transfused, four patients (7%) received 1-5 transfusions, and 50 (91%) were not transfused. Of the 13 Amish women who had been pregnant, 46% (6/13) required transfusions during pregnancy, similar to the M/M group.
Most patients were regularly transfused until splenectomy; the rate of splenectomy was higher in this subgroup [93% (51/55)] than it was in patients without a homozygous p.R479H variant [50% (99/200); P<.001]. The median post-splenectomy hemoglobin was 9.4 g/dL (range: 8-11.2, n = 51). The median maximum ferritin was 616 ng/mL (range 126-3258) with 33% (14/42) of patients meeting criteria for iron overload at enrolment. Although the rate of cholecystectomy in this subgroup [27% (15/55)] was lower than it was in patients without a homozygous p.R479H variant [43% (86/200); P = .04], the rate of gallstones [44% (24/54)] was similar to those without the homozygous p. R479H variant [45% (86/195)]. No differences were observed in homozygous p.R479H patients and patients without homozygous p.R479H variant regarding the rate of pulmonary hypertension [2% (1/54) versus 3% (7/184); P = .7], whereas 33% of the group had extramedullary hematopoiesis, significantly higher than patients without the homozygous p.R479H variant [5% (8/168); P < .001].
3.7 |. Homozygous p.R510Q variant
Within the homozygous p.R510Q group (n = 19), the median age at diagnosis was 0.2 years (range 0-48 years; n = 18, Table S8). The median hemoglobin of those splenectomized and not regularly transfused was 11.3 g/dL (range: 9.9-11.5, n = 5) consistent with a high rate of complete hemoglobin response to splenectomy in patients with this genotype. However, the rate of splenectomy in this cohort (37%) was also lower than in patients without the homozygous p. R510Q variant [61% (143/236); P = .053]. Within the 12-month period prior to enrolment, two patients (11%) received regular transfusions, six patients (32%) received 1-5 transfusions, and 11 patients (58%) were not transfused.
The 37% (7/19) rate of cholecystectomy was similar to patients without the homozygous p.R510Q variant [40% (94/236)]. The median maximum ferritin level was 896 ng/mL (range 64-5698, n = 8), similar to other patients in the M/M group [573 ng/mL (range 31-9679, n = 67); P = .6]. The rate of iron overload [38% (3/8)] was also similar to other patients in the M/M group [43% (30/69)].
Patients with a p.R510Q variant in the compound heterozygote state with a non-missense mutation (n = 19) had a more severe clinical presentation than the homozygous p.R510Q cohort and were similar to the NM/NM cohort.
4 |. DISCUSSION
Pyruvate kinase deficiency is a rare congenital hemolytic anemia with a diverse phenotype and wide spectrum of severity. The PKD NHS is an international registry of patients with PK deficiency which has led to an improved understanding of the clinical spectrum and current management strategies for patients with this disorder.6 In the present study, we report the highly diverse molecular features of this cohort and an in depth description of the observed genotype-phenotype correlations.
This study confirms the wide genetic heterogeneity of PK deficiency with 127 different pathogenic variants in this cohort, 45 of which have not been previously reported. There are now 371 identified pathogenic PKLR gene variants associated with PK deficiency (https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php). Of the 45 new variants, 43 have never been reported in HGMD or other databases. The remaining two, p.A31V and p.D339Y, are reported with a MAF ExAc frequency lower than 10−4 to 10−5, drastically below the cut-off of the definition of polymorphism. All of the missense variants were predicted as possibly or likely pathogenic using Polyphen2, M-CAP or other in silico tools, and were associated with reduced PK enzyme activity. All of the splicing mutations, except one, affected the invariant dinucleotides of the intron bordering sequences. The variant c.966(−9)A>G was also predicted to affect splicing by Human Splicing Finder 3.124 and classified as likely pathogenic, because it was identified at homozygous level in a patient with reduced PK activity and congenital hemolytic anemia.
The mutations were distributed throughout the PKLR gene and affected all exons; however, there was a higher frequency of variants in exons 5, 8, 9 and 11 with 9.1%, 9.3%, 10.5% and 16.4% of the nucleotides affected, respectively. Exon 11 had the highest number of mutated alleles due to presence of the common mutations p.R486W and p.R510Q. Both these variants involve an arginine residue and are located in C domain responsible for tetramer stability. Of note, 10 out of the 23 amino acid substitutions identified in this domain involve an arginine residue.10 Despite the increased frequency of variants in certain exons, the molecular diversity of PK deficiency underlines the need to analyze the entire PKLR gene, without limiting the analysis to the most common variants that cover no more than 30% of the total number of mutated alleles.
Among the 257 cases studied in this cohort, two unrelated patients carried three different mutations; two of them were non-missense and present in both patients (p.E241X and p.V276WfsX45), and the third in each was a missense mutation (p.R510Q and p.A495V). Although parent samples were not available for these two patients to define the inheritance of the variants, we hypothesize that both non-missense mutations were located on the same allele, in trans, to the missense mutation, thus resulting, at clinical level, as a missense/non-missense genotype. Given the lack of parent studies, these patients were not included in the genotype-phenotype analysis. In one of the two patients, anemia had been observed since birth with a post-splenectomy hemoglobin of 10.4 g/dL. The other patient also presented with anemia at birth and then has received regular transfusions with an intact spleen to date. The presence of two different variants transmitted on the same allele has already been reported in other patients,26–29 and contributes to the wide molecular heterogeneity observed in PK deficiency. This finding might also lead to a possible misdiagnosis of an affected patient with compound heterozygous mutations rather than a patient in the heterozygote state.
Despite sequencing all exons, intronic flanking regions, and the erythroid promoter of PKLR gene, patients with a clinical phenotype of PK deficiency and low PK enzyme levels are sometime found to have only one PKLR mutation or no mutations at all.5 Among the 278 initially enrolled participants in the PKD NHS, 21 were considered ineligible for this study due to the inability to demonstrate two pathogenic variants after the exclusion of large deletion performed by long range PCR. For some cases, unknown mutations may be in intronic or regulatory regions that are not covered by conventional diagnostic molecular techniques or that cannot be considered as pathogenic in the absence of functional tests.5,16 An example of such a case was published in which a deep intronic mutation (c.283+109C>T, intron 2) was coupled with missense mutation p.G332S.30 Since an increasing number of pathogenic deep intronic mutations are being described across different disease conditions (α spectrinLEPRA (SPTA1 c.4339-99C>T31–33) and Androgen Insensitivity Syndrome (c2450-118A>G)),34 clinical molecular testing algorithms may have to be revised to include extended intronic regions, especially when only one missense mutation is identified in the first pass.
The structural architecture of the PK molecule contributes to the heterogeneity of biochemical properties of the abnormal variants. Pyruvate kinase is a tetramer with four identical subunits, each consisting of four structural domains. Enzyme activation is thought to involve a combination of domain and subunit rotations coupled to alterations in the active site geometry.17 Patients with compound heterozygote variants of the same genotype may have different combinations of tetramers, resulting from differences in the assembly of the molecule. This is due to a variable amount of one monomer resulting from one mutated allele with respect to the other one. This hypothesis, together with the possible effect of yet unidentified genetic modifiers of the expression of PK, may explain the clinical variability seen among siblings with the same variants.
In this cohort, 142 patients were compound heterozygotes, making genotype-phenotype correlations challenging. Given the number of variants and patients who are compound heterozygotes, patients were stratified according to their type of mutations (missense/missense, missense/non-missense, non-missense/non-missense). However, there are some limitations to this categorization. For example, some missense mutations may result in marked protein instability or functional inactivity, or, although not directly involved in splicing consensus sequences, may result in altered splicing. On the other hand, some non-missense variants may not have a significant impact on the protein structure.
The study showed that patients with two non-missense mutations had lower hemoglobin levels post-splenectomy, higher numbers of lifetime transfusions, higher rates of iron overload, and higher rates of splenectomy. Although patient and provider management decisions determine transfusion and splenectomy status, these data confirm previous observations that patients with two non-missense mutations tend to have a more severe clinical phenotype, including serious but rare complications, such as extramedullary hematopoiesis and pulmonary hypertension. These complications were seen more frequently in patients with non-missense mutations or with missense mutations characterized by severely decreased protein stability, suggesting that these complications may be associated with absence or severely decreased PK protein.18
The observed genotype-phenotype associations are consistent with previous reports.12 Due to the intrinsic difficulties of examining the relationship between genotype-phenotype in PK deficiency, future studies should consider studying the relationship between residual PK protein level and clinical phenotype. Indeed, studies have shown that there is a relationship between residual PK protein and hemoglobin response to an oral PK activator in clinical development.35
Surprisingly, there was no association identified between PKLR genotype and the frequency of complications in utero or in the newborn period, suggesting that the clinical presentation at birth is not predictable, even among siblings. Pregnancy was similarly tolerated across genotypes in women with PK deficiency. Pregnancy outcomes were not different among genotype groups; however, all women with two non-missense PKLR variants required transfusion support during pregnancy, whereas only 43% of women with two missense mutations required transfusions. This is consistent with the general finding that patients with two non-missense variants tend to require more transfusions and have more iron loading than the other variant groups.
The clinical features of the two largest groups of patients with homozygous genotypes were analysed separately, homozygote p.R510Q (n = 19) and homozygote p.R479H (n = 55). Most of the patients homozygous for the p.R479H variant, seen in the Amish population, presented with symptoms soon after birth leading to a rapid diagnosis in early infancy. The rate of splenectomy, 93%, was the highest in the homozygous p.R479H cohort, likely related to an attempt to limit the time period of patients’ requirement for regular transfusions. These patients also had the highest rate of certain complications, including extramedullary hematopoiesis. The severity of the clinical phenotype correlates with the severity of the effect of this variant, causing abnormal splicing, consistent with what has been observed in prior studies.3,36
The p.R510Q is considered the most common variant in the Caucasian population with a reported allelic frequency of 0.7 × 103 (Gnom AD https://gnomad.broadinstitute.org/) and was detected in 23% of the mutated unrelated alleles in this series of patients. The observation of its association with the haplotype [(ATT)14/(T)10/C1705/C1738/C1992] supports the hypothesis of a single origin of this variant.10 The homozygous p.R510Q cohort had clinical features similar to the M/M group with prior splenectomy in only 37% but generally with a robust post-splenectomy hemoglobin response to the treatment (Hb level after splenectomy 11.3 g/dl in homozygous p.R510Q group vs. 9.2 g/dl in the M/M group). Of note, patients with a p.R510Q in the compound heterozygote state with a non-missense mutation (n = 19) had a more severe clinical presentation than the homozygous p.R510Q cohort and were similar to the NM/NM cohort. This finding suggests that the drastic protein instability caused by the p.R510Q mutation may result in worsening of severity when associated with variants with a different drastic effect on the protein.17,18
In addition to the limitations of the mutation characterization groups, other limitations of this study include its retrospective observational design, the lack of genome-wide data to account for relatedness of patients, and the limited number of patients despite the global effort and number of sites. In addition, clinical, laboratory, and radiologic monitoring and treatments, including transfusions and splenectomy, varied and may have been biased by provider, hospital, and region. The genotype-phenotype relationships in PK deficiency described here underscores the importance of genetic testing not only to confirm the diagnosis but also to discuss prognosis and establish a monitoring plan for patients.5 The findings from the in depth evaluation of the cohorts with common homozygous variants as well as siblings suggest that non-genotype factors, such as degree of splenomegaly, splenic conditioning, other red cell variants, and co-inherited variants in iron and bilirubin metabolism and different treatment approaches may also be significant contributors to the clinical phenotype. Future analyses aimed at describing this variation may reduce the clinical overlap among patient groups.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge all the patients with pyruvate kinase deficiency who contributed to this Natural History Study data. The study was supported by Fondazione IRCCS Ca’ Granda Policlinico Milano, Project number RC 175/04, 2015-2017. The Pyruvate Kinase Deficiency Natural History Study was supported by research funding from Agios Pharmaceuticals.
Funding information
Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico Milano, Grant/Award Number: Project number RC 175/04 2015-2017
CONFLICTS OF INTEREST
RFG, PB, WB, BG, EJvB, KK, JLK, SC have served as scientific advisors Agios Pharmaceuticals. RFG, BG, EJvB, WB received research funding from Agios Pharmaceuticals. The remaining authors declare no competing financial interests.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood. 2000;95(11):3585–3588. [PubMed] [Google Scholar]
- 2.Carey PJ, Chandler J, Hendrick A, et al. Prevalence of pyruvate kinase deficiency in northern European population in the north of England. Northern Region Haematologists Group. Blood. 2000;96(12):4005–4006. [PubMed] [Google Scholar]
- 3.Kanno H, Ballas SK, Miwa S, Fujii H, Bowman HS. Molecular abnormality of erythrocyte pyruvate kinase deficiency in the Amish. Blood. 1994;83(8):2311–2316. [PubMed] [Google Scholar]
- 4.Baronciani L, Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J Clin Invest. 1995;95(4):1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bianchi P, Fermo E, Glader B, et al. Addressing the diagnostic gaps in pyruvate kinase (PK) deficiency: Consensus recommendations on the diagnosis of PK deficiency. Am J Hematol. 2019;94(1):149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: data from the pyruvate kinase deficiency natural history study. Blood. 2018;131(20):2183–2192. [DOI] [PubMed] [Google Scholar]
- 7.Grace RF, Cohen J, Egan S, et al. The burden of disease in pyruvate kinase deficiency: patients’ perception of the impact on health-related quality of life. Eur J Haematol. 2018;101(6):758–765. [DOI] [PubMed] [Google Scholar]
- 8.Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015;90(9):825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Beers EJ, van Straaten S, Morton DH, et al. Prevalence and management of iron overload in pyrvuate kinase deficiency: report from the pyruvate kinase deficiency natural history study. Haematologica. 2018;104(2):e51–e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: from genetics to clinical manifestations. Baillieres Best Pract Res Clin Haematol. 2000. March;13(1):57–81. [DOI] [PubMed] [Google Scholar]
- 11.Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130(1): 11–25. [DOI] [PubMed] [Google Scholar]
- 12.Zanella A, Fermo E, Bianchi P, et al. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21(4):217–231. [DOI] [PubMed] [Google Scholar]
- 13.Satoh H, Tani K, Yoshida MC, et al. The human liver-type pyruvate kinase (PKL) gene is on chromosome 1 at band q21. Cytogenet Cell Genet. 1988;47(3):132–133. [DOI] [PubMed] [Google Scholar]
- 14.Canu G, De Bonis M, Minucci A, Capoluongo E. Red blood cell PK deficiency: an update of PK-LR gene mutation database. Blood Cells Mol Dis. 2016;57:100–109. [DOI] [PubMed] [Google Scholar]
- 15.Bianchi P, Zanella A. Hematologically important mutations: red cell pyruvate kinase (Third update). Blood Cells Mol Dis. 2000;26(1):47–53. [DOI] [PubMed] [Google Scholar]
- 16.Lezon-Geyda K, Rose MJ, McNaull MA, et al. Pklr intron splicing-associated mutations and alternate diagnoses are common in pyruvate kinase deficient patients with single or no PKLR coding mutations. Blood. 2018;132(suppl 1):3607. [Google Scholar]
- 17.Valentini G, Chiarelli LR, Fortin R, et al. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002;277(26):23807–23814. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Chiarelli LR, Bianchi P, et al. Human erythrocyte pyruvate kinase: characterization of the recombinant enzyme and a mutant form (R510Q) causing nonspherocytic hemolytic anemia. Blood. 2001; 98(10):3113–3120. [DOI] [PubMed] [Google Scholar]
- 19.Zanella A, Bianchi P, Fermo E, et al. Molecular characterization of the PK-LR gene in sixteen pyruvate kinase-deficient patients. Br J Haematol. 2001;113(1):43–48. [DOI] [PubMed] [Google Scholar]
- 20.Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184(5): 721–734. [DOI] [PubMed] [Google Scholar]
- 21.Zanella A, Bianchi P, Baronciani L, et al. Molecular characterization of PK-LR gene in pyruvate kinase-deficient Italian patients. Blood. 1997; 89(10):3847–3852. [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48(12):1581–1586. [DOI] [PubMed] [Google Scholar]
- 24.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hertzmark E, Spiegelman D. The SAS ICC9 Macro (2010); https://cdn1.sph.harvard.edu/wp-content/uploads/sites/271/2012/09/icc9.pdf [Google Scholar]
- 26.Percy MJ, van Wijk R, Haggan S, et al. Pyruvate kinase deficient hemolytic anemia in the Northern Irish population. Blood Cells Mol Dis. 2007;39(2):189–194. [DOI] [PubMed] [Google Scholar]
- 27.Christensen RD, Yaish HM, Nussenzveig RH, Agarwal AM. Siblings with severe pyruvate kinase deficiency and a complex genotype. Am J Med Genet A. 2016;170(9):2449–2452. [DOI] [PubMed] [Google Scholar]
- 28.Raphaël MF, Van Wijk R, Schweizer JJ, et al. Pyruvate kinase deficiency associated with severe liver dysfunction in the newborn. Am J Hematol. 2007;82(11):1025–1028. [DOI] [PubMed] [Google Scholar]
- 29.Pissard S, Max-Audit I, Skopinski L, et al. Pyruvate kinase deficiency in France: a 3-year study reveals 27 new mutations. Br J Haematol. 2006;133(6):683–689. [DOI] [PubMed] [Google Scholar]
- 30.Bagla S, Bhambhani K, Gadgeel M, et al. Compound heterozygosity in PKLR gene for a previously unrecognized intronic polymorphism and a rare missense mutation as a novel cause of severe pyruvate kinase deficiency. Haematologica. 2019;104(9):e428–e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wichterle H, Hanspal M, Palek J, Jarolim P. Combination of two mutant alpha spectrin alleles underlies a severe spherocytic hemolytic anemia. J Clin Invest. 1996;98(10):2300–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chonat S, Risinger M, Dagaonkar N, et al. The Spectrum of Alpha-Spectrin Associated Hereditary Spherocytosis. Blood. 2015;126:941. [Google Scholar]
- 33.Gallagher PG, Maksimova Y, Lezon-Geyda K, et al. Aberrant splicing contributes to severe a-spectrin-linked congenital hemolytic anemia. J Clin Invest. 2019;30:2878–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Känsäkoski J, Jääskeläinen J, Jääskeläinen T, et al. Complete androgen insensitivity syndrome caused by a deep intronic pseudoexonactivating mutation in the androgen receptor gene. Sci Rep. 2016;6: 32819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grace RF, Rose C, Layton DM, et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N Engl J Med. 2019;381(10):933–944. [DOI] [PubMed] [Google Scholar]
- 36.Rider NL, Strauss KA, Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old-order Amish cohort: longitudinal risk and disease management. Am J Hematol. 2011;86(10):827–834. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.