

Abstract
The rising interest in Kv7 modulators originates from their ability to evoke fundamental electrophysiological perturbations in a tissue-specific manner. A large number of therapeutic applications are, in part, based on the clinical experience with two broad-spectrum Kv7 agonists, flupirtine and retigabine. Since precise molecular structures of human Kv7 channel subtypes in closed and open states have only very recently started to emerge, computational studies have traditionally been used to analyze binding modes and direct the development of more potent and selective Kv7 modulators with improved safety profiles. Herein, the synthetic and medicinal chemistry of small molecule modulators and the representative biological properties are summarized. Furthermore, new therapeutic applications supported by in vitro and in vivo assay data are suggested.
This review describes the synthetic and medicinal chemistry of small molecule modulators of the voltage-gated Kv7 (KCNQ) potassium channels and the available data of their biological and clinical properties.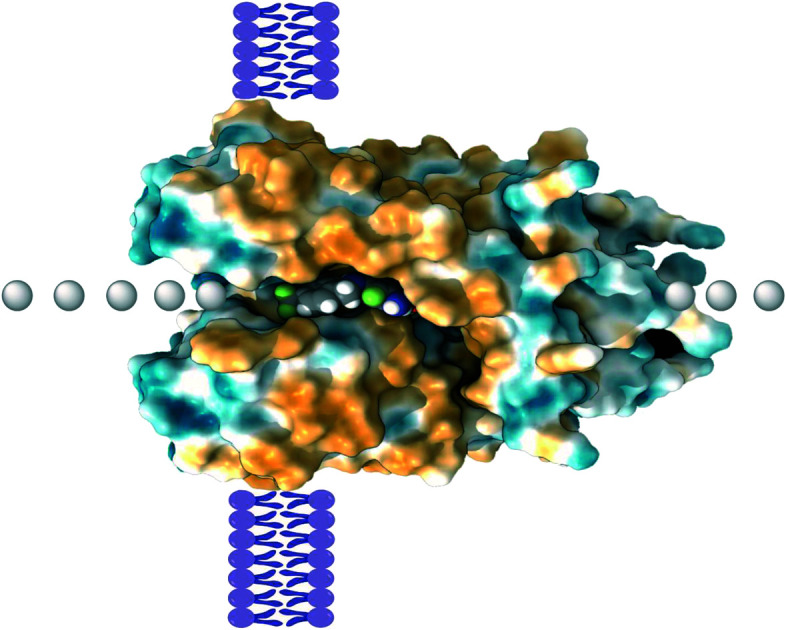
1. Introduction
This review describes the synthesis of small molecule modulators of the voltage-gated potassium channels and their biological and clinical properties. It extends several earlier medicinal chemistry reviews on these subjects.1–6
Kv7 (KCNQ) channels are non-inactivating voltage-gated potassium channels, originally called M-channels, and are expressed by KCNQ genes. They have slow gating kinetics compared to other voltage-gated potassium channels.7,8 Once activated, the resulting outward potassium flow generates an outward current, leading to a more negative cell membrane potential, and raising the threshold for firing an action potential. This current, commonly referred to as M-current (IM) due to its strong inhibition by the activation of muscarinic acetylcholine receptors, was first detected in bullfrog sympathetic neurons and subsequently identified in neuronal, cardiovascular, and epithelial human tissues.9–14 The M-current exerts neuronal excitability control of both the central (CNS) and peripheral nervous system (PNS) (sympathetic,15 parasympathetic,16 and somatosensory neurons17), affecting excitatory as well as inhibitory pathways in the brain.18 Upon Kv7 channel activation at a subthreshold potential, hyperpolarization of the neuronal membrane has been observed, setting the neuronal resting potential and shaping the action potential profile, as well as contributing to medium (50–100 ms) and slow (0.1–2 s) after hyperpolarizations during refractory periods.19–23Fig. 1 summarizes the known electrophysiological effects of the M-current.
Fig. 1. Summary of M-current electrophysiological effects in neurons. Following Kv7 channel opening, the outward K+ flow hyperpolarizes the membrane potential, requiring a greater stimulus to trigger an action potential. Spike frequency adaptation was observed after train of action potentials were induced by depolarizing current injection, limiting the ability of the neurons to fire repetitively. Theta waves were recorded in CA1 pyramidal neurons in the hippocampus, and were noted to disappear after M-current suppression.

In neurons, Kv7 channel activation differs in outcome based on subcellular distribution. During high frequency bursts of the action potential, activation of perisomatic Kv7 channels increases the spike interval, leading to a phenomenon called spike frequency adaptation and counterbalancing the fluctuations derived from multi-stimuli that reach dendrimers and contribute to the refractory period.24,25 The axon initial segment (AIS), located at the proximal axon/soma interface, is involved in the transformation of soma stimuli into action potentials. The AIS is also a subcellular neural portion in which Kv7 channels are highly expressed. They are connected to Na+ channels via the ankyrin G protein, which reduces the likelihood of spontaneous neuronal firing.26 By means of AIS Kv7 channel inhibition, in fact, it has been demonstrated that the resting membrane potential can be set to more depolarized values and that the threshold for the action potential can be negatively shifted (to more negative values).27 Kv7 channels localized in the axon terminus may also regulate neurotransmitter release.28
Another property of the M-current is its ability to generate electric resonance, called M-resonance, at theta (Θ)-frequencies (2–7 Hz). In neurons, electric resonance consists of an increase in the oscillation amplitude at certain frequencies. The M-resonance, detected in pyramidal neurons in the hippocampus, is important for spatial navigation and memory functions.29 Not all factors governing its occurrence are well understood, but it is involved in the pathophysiology of ion-channels.30 In addition to the M-current, the h-current (mediated by HCN channels) is also known to generate electric resonance, contributing to olfactory sensation, rhythm of neuronal spiking, postsynaptic neuron firing rate, and pathological conditions such as epilepsy.31–34 In a single cell system, M-resonance allows the passage of signals with a higher frequency than a cutoff frequency (acting as a high-pass filter), determining the speed of the response to sensory stimuli. For instance, the M-resonance in retina rod cells accelerates the response to light in dark environments.35
At the neuronal circuit level, resonance induced at Θ-frequencies was shown to be a necessary component of neuroplasticity, learning, and memory, inducing synchronous activity within the local circuit. In confirmation of this observation, knocking out the KCNQ2 gene in pyramidal neurons in the mouse hippocampus suppressed Θ-resonance and resulted in spatial cognitive dysfunction.14,25
An important structural feature of the Kv7 channel subfamily is the preferential formation of homo- and hetero-tetramers of the five Kv7 proteins (Kv7.1–Kv7.5, also reported as KCNQ1–KCNQ5 belonging to the Q subfamily of voltage-gated potassium channels), predominantly in a 2 + 2 stoichiometry.36,37 These Kv7 channel protein sequences comprise of 650 to 940 amino acids that are organized in six transmembrane domains and an ion-selective pore structure (6TM/1P). The S1–S4 segments constitute the voltage-sensitive domain (VSD), whereas the S5–S6 segments in combination with the enclosed loop form the pore domain. The short N-terminus and the longer C-terminal tail are both extending into the cytosol and contain a multitude of co-factor interaction sites. The four C-terminal helices are responsible for the affinity between Kv7 proteins during tetramer assembly (Fig. 2).38–42
Fig. 2. Schematic representation of Kv7 channel structures. Helix-A binding sites: AKAP79/150, CaM, PIP2, PKC, PKA, and syntaxin A (Kv7.2 only). Helix-B binding sites: AKAP79/150, PKC, PKA, CaM. p38 MAPK, CDK5. Helix-C binding sites: site for tetramerization and PIP2. Helix-D binding sites: KCNE1 (for Kv7.1), AKAP yotiao (Kv7.1 only), and PIP2. Helix-D/chain terminal binding sites: ankyrin G (Kv7.2 and Kv7.3 only) and need 4–2 (Kv7.1 only).

Kv7.1 (also known as KvLQT1) is widely expressed in cardiac myocytes as a homomer or in association with KCNE1 (minK protein) or KCNE3 channels, and plays a critical role in the repolarization of the cardiac potential through the generation of the slow, delayed rectifier current (IKs current).43,44 Kv7.1 is also expressed in the inner ear, thyroid gland, lung, ovaries, proximal and distal tubule of the nephron, pancreas, intestinal system, and vascular smooth muscles.45,46 Upon membrane depolarization, the outward shift of the S4 segment in Kv7.1 triggers a pore opening in the fully activated-open conformation (AO) via the intermediate-open (IO) state.47 Both AO and IO exhibit differences in gating properties, in addition to their modulation by the auxiliary KCNE1 protein (which can suppress the IO state and enhance AO). Moreover, in contrast with IO, AO is fostered at a more depolarizing potential, resulting in slower current kinetics and channel inactivation at certain voltage stimuli.48,49
It is noteworthy that the evaluation of the affinity of small molecules to Kv7.1 in in vitro ion channel electrophysiology assays is required by the Comprehensive in vitro Proarrhythmia Assay (CiPA). CiPA was developed as an in vitro paradigm for cardiac safety evaluation (prevention of drug-induced arrhythmias) of drug candidates to improve specificity compared to preclinical hERG and clinical TQT studies.50 In fact, during the evaluation of drug effects on ventricular repolarization, several cardiac currents are monitored, including IKs (Kv7.1), IKr (Kv11.1), ICa,L (CaV1.2), INa (NaV1.5), IKur (Kv1.5), Ito,f (Kv4.3),and IK1 (Kir2.1–2.3). Based on CiPA results, a candidate molecule could be stopped in the pre-clinical phase if there is sufficient evidence for its possible involvement in proarrhythmia side effects.51
The Kv7.2 channel is located both in central and peripheral nervous systems,52 showing a high expression level in neocortex and hippocampus. High current amplitude Kv7.2/7.3 heteromers are the most abundant Kv7.2 channel assembly compared to the less abundant low current amplitude homomeric channels and Kv7.4 or Kv7.5 heteromers.53 Particular attention has been given to Kv7.2/7.3 channels for their M-current contributions in neuronal tissues.17
The Kv7.4 channel variant is predominantly expressed in homomeric forms in dopaminergic neurons, auditory, and vestibular systems, and as Kv7.4/7.5 heteromer in the smooth muscle cells (e.g. gastrointestinal tract, bladder, arteries, airways, uterus, blood vessels, corpus cavernosum penis).54–62 In contrast, the Kv7.5 protein is expressed in various regions of the brain, both in homomeric forms as well as Kv7.3/7.5 heteromer, and in skeletal muscles as Kv7.4/7.5 heteromer.63
Kv7 channel activity is negatively influenced by neurotransmitter activated Gq/11-coupled receptors,64 altering co-factor and/or accessory protein affinity with Kv7 subunits such as PIP2,65 calmodulin (CaM),66 A-kinase anchoring protein (AKAP79/150),67 and ankyrin G,68 and via the calcium-free/calcium-bound CaM equilibrium (Fig. 3).64 The open-state and the voltage-conductance of Kv7 channels are phosphatidylinositol-4,5-bisphosphate (PIP2) dependent. Several PIP2 binding sites have been identified in Kv7 proteins. Among them, PIP2 interacts with a cluster of basic amino acids in the C-terminus (overlap with the calmodulin binding site)69 and in the S4–S5 linker region.70 Activation of muscarinic acetylcholine receptors (especially M1 and M3) promotes PIP2 hydrolysis through phospholipase C activation, resulting in PKC phosphorylation. Activated PKC phosphorylates the C-terminus in the CaM binding site of the Kv7.2 subunit assisted by AKAP79/150, which allows a binding between PKC and the Kv7 channel complex. In the phosphorylated state, dissociation of CaM occurs, destabilizing the Kv7 channel/PIP2 complex and consequently PIP2 hydrolysis, resulting in M-current suppression.70–73 This negative regulation pathway of the Kv7 channels is shared with the activation mechanism of other G protein-coupled receptors (GPCRs), such as histamine H1, 5-hydroxytryptamine 5-HT2C, P2Y1, P2Y2, P2Y4, and P2Y6 nucleotide receptors, mGluR1 and mGluR5 metabotropic glutamate receptors, and opioid receptors. Moreover, several peptide receptors are able to modulate Kv7 channel activity, including angiotensin, GnRH, substance P and bradykinin.74 The M-current inhibition of bradykinin, in fact, is determined by an increment of intracellular Ca2+ shifting the calcium-free/calcium-bound CaM equilibrium to the right side. Since calcium-free CaM is fundamental for PIP2 affinity to Kv7 channels, increasing the intracellular Ca2+ level is recognized by CaM bonded to the Kv7 channel, which is converted to calcium-bound CaM, and decreasing Kv7 channel/PIP2 affinity.75 The calcium-free/calcium-bound CaM equilibrium modulation was observed for Kv7.2, Kv7.4, and Kv7.5 but not for Kv7.1 and Kv7.3.76
Fig. 3. Overview of Kv7 channel negative modulation through activation of Gq/11-coupled receptors (green box) and the calcium-CaM pathway (blue box). BKR: bradykinin receptor; PLC: phospholipase-C; PIP2: phosphatidylinositol-4,5-bisphosphate; CaM: calmodulin; AKAP79/150: A-kinase anchoring protein 79/150; PKC: protein kinase C; ER: endoplasmic reticulum; DAG: diacylglycerol; IP3: inositol-1,4,5-triphosphate; IP3R: inositol-1,4,5-triphosphate receptor.

To date, many diseases have been attributed to mutations of KCNQ genes encoding Kv7 proteins with a higher or lower ability to hyperpolarize cell membranes (Fig. 4).77–82 Not surprisingly, the development of Kv7 modulators (both agonists and antagonists) has triggered further studies of therapeutic utility, specificity of Kv7 tissue expression, and cellular response to selective Kv7 subtype modulators. However, the major challenges in the development of safe and effective drugs in this field have been attributed to Kv7 modulator subtype selectivity, a possible link to undesired on-target effects, and metabolic properties.83 The formation of reactive redox metabolites and the lack of ion-channel selectivity have hampered the development of drugs which can augment or supersede already existing therapies. However, in some cases, a Kv7 modulator might very well emerge as a first-in-class treatment of diseases for which only palliative or symptomatic care is currently available.
Fig. 4. Overview on inherited diseases emerging from KCNQ gene mutations. BFNE: benign familial neonatal epilepsy; KCNQ2-DEE: KCNQ2-encephalopathy; BFIE: benign familial infantile epilepsy; RE: rolandic epilepsy; ASD: autism spectrum disorder; EE: epileptic encephalopathy; IGE: idiopathic generalized epilepsy; KS: Kleefstra syndrome; ID: intellectual disability; DFNA2: autosomal dominant hearing loss 2; RWS: Romano–Ward syndrome (LQTS); JLNS: Jervell and Lange-Nielsen syndrome (LQTS + deafness); AF: atrial fibrillation.

2. Small molecule Kv7.2–Kv7.5 agonists in the market or in use in advanced clinical studies
2.1. Flupirtine
Flupirtine (Chart 1) was the first compound that was identified as a Kv7 activator, even though its development predates the discovery of these channels. The drug was initially introduced as a non-opioid nonsteroidal anti-inflammatory analgesic in the 1980's by the German pharmaceutical company Chemiewerk Homburg, and later marketed by TEVA and MEDA in the European market.84Flupirtine is a pan-Kv7.2–Kv7.5 agonist able to exert analgesic, muscle relaxation, and neuroprotective effects, and it can also interact with other receptors (Table 1).85 Its binding interaction in Kv7 proteins is strongly depending on the electron density of the carbamate moiety, combined with an overall prevalence of hydrophobic interactions. A potential binding pose was predicted for a homology model based on a Kv1.2/Kv2.1 X-ray structure.86 The carbamate group of flupirtine might engage in a H-bond with the NH-indole of Trp265 as revealed by molecular modeling studies with Kv7.3.87
Chart 1. Chemical structures of Kv7.2–Kv7.5 activators.

Kv7 agonist affinity at non-Kv7 ion-channels and modulation of non-IKs currents.
| Affinity at non-Kv7 ion-channels | Effects on ligand-gated receptors | Modulation of non-IKs currents | |
|---|---|---|---|
| Flupirtine 85,102–104 | No effects at ≤10 μM on voltage-gated Na+ or Ca2+ channels | - Indirect inhibition of NMDA | - Activation of Kir current (EC50 = 0.6 μM) |
| -No affinity at α1-, α2-adrenoceptors, 5-HT1-, 5-HT2-, AMPA/kainite, glycine, nicotinic receptors and TRPV1 | |||
| - Activation of GABAA (EC50 = 21 μM in dorsal root ganglions; EC50 = 13 μM in hippocampal neurons) | |||
| RTG 95,105–110 | - Inhibition of Kv1–Kv9 and Kv11 subfamilies at concentration >100 μM | - No effect at NMDA, AMPA, and kainate receptors | - Inhibition of Kv2.1 current (IC50 = 22 μM) |
| - IC50 >50 μM against Kv1.5 and hERG (IC50 = 59 μM) | - Selective subtype modulator of GABAA receptors with preference for extrasynaptic δ-subunit containing GABA receptors | ||
| - IC50 >100 μM against CaV (29% inhibition at 100 μM) and NaV (25% inhibition at 100 μM) | |||
| - Lacks activity against Kir2.1, Kir3.1, and K2P1.1 at concentrations >100 μM | |||
| BMS-204352 105,106,111 | - Activation of BK channels (EC50 = 0.35 μM for KCa channels with 1 μM Ca2+; EC50 >5 μM for KCa channels with 50 nM Ca2+) | - No effect on dopamine D3 receptor | - No effect on hIK, hSK1, and CFTR currents |
| - No effect on Kv1.3, Kv1.5, Kv2.1, and Ca2+-activated chlorine channels | - Negative modulator of GABAA receptors | ||
| ML277 112,113 | - Weak inhibition of NaV1.5 and CaV1.2; hERG: IC50 >30 μM | - No activation of IKs | |
| ICA-27243 124 | - No interaction with high voltage-activated calcium channels and NaV1.2 | - No effects on GABA receptors | |
| ML213 11,114 | No interaction with hERG and Kir2.1 at 10 μM |
Flupirtine became the first clinically approved Kv7 channel agonist in 1984 in Europe on the basis of its unique analgesic and muscle relaxation properties compared to opioids and non-steroidal anti-inflammatory drugs. It was widely administered to treat NSAID-refractory pain in patients for whom opioid analgesics were contraindicated. However, the escalation in the use of this prescription drug also triggered an increase in the occurrence of severe liver injuries, leading in some cases to liver transplantation or death.88,89 Despite the ensuing restriction in 2013 to manage this risk, liver problems continued to emerge. For this reason, the marketing authorization for flupirtine was withdrawn by the European Medicines Agency (EMA).90 Despite this significant setback, flupirtine remained a compound of great interest for its potential therapeutic properties and as a starting point for the development of more potent and selective Kv7 activators with different metabolic pathways, minimizing undesirable toxicity.89,91,92
2.2. Retigabine
Epileptic seizures are generated by abnormal neuronal activity in the cerebral cortex and high-frequency bursts of action potential. The resulting discrepancy between inhibitory and excitatory neuronal pathways in the CNS in favor of the latter can arise as a result of alterations in circuit levels (e.g. abnormal synaptic connectivity) and receptor and ion-channel functions.93,71 In several epilepsy phenotypes, mutations of K+ channel genes have been observed. Kv7.2 and Kv7.3 mutations also contribute to epilepsy insurgence, as in the case of a benign form of neonatal epilepsy (BFNS), KCNQ2-encephalopathy, and rolandic epilepsy.81 Accordingly, a flupirtine analog, retigabine (RTG), was developed as an adjuvant for the treatment of particular forms of seizures in epilepsy, and commercialized by GSK in 2011 as Trobalt® in Europe and Potiga® in the US (Chart 1).94RTG was identified as a pan-Kv7.2–Kv7.5 agonist with a greater affinity for heteromeric Kv7.2/7.3 channels, antagonizing Kv7.1 at high concentrations during positive potentials (see Table 2 for a comparison of Kv7 agonists potencies) as well as other ion-channels (Table 1).95
Potency of Kv7 agonists based on current improvement (I/I0) or half-maximal voltage negative shifts(ΔV1/2) in cells expressing Kv7 channels.
| ML213 | RTG | BMS-204352 | ICA-27243 | ML277 | R-L3 | ||
|---|---|---|---|---|---|---|---|
| Kv7.1 | I/I0 and related EC50 | CHO cells: 2.66 fold; 0.26 ± 0.02 μM112 | Xenopus oocyte: 0.68 fold; 0.96 μM113 | ||||
| ΔV1/2 and related EC50 | CHO cells: −0.7 mV; 100.1 ± 6.5 μM (IC50)95 | ||||||
| Kv7.1/KCNE1 | I/I0 and related EC50 | CHO cells: 1.61 fold; ∼0.15 μM113 | |||||
| ΔV1/2 and related EC50 | |||||||
| Kv7.2 | I/I0 and related EC50 | CHO cells: 4.45 fold; 0.23 ± 0.04 μM114 | HEK293 cells: 1.6 ± 0.1 fold (10 μM); EC50 NA105 | HEK293 cells: 2.3 ± 0.3 fold (10 μM); EC50 NA105 | CHO cells: >30 μM112 | ||
| ΔV1/2 and related EC50 | CHO cells: −37.4 ± 3.0 mV; 0.34 ± 0.07 μM114 | CHO cells: −24.2 mV; 2.5 ± 0.6 μM95 | |||||
| Kv7.2/7.3 | I/I0 and related EC50 | HEK293 cells: 1.4 ± 0.03 fold (10 μM); EC50 NA105 | HEK293 cells: 1.3 ± 0.03 fold (10 μM); EC50 NA105 | CHO cells: 0.4 ± 0.1 μM; (I/I0 NA)124 | |||
| CHO cells: 0.34 ± 0.05 μM; (I/I0 = NA)130 | |||||||
| ΔV1/2 and related EC50 | CHO cells: −30.4 mV; 1.9 ± 0.2 μM95 | CHO cells: ⋍19 mV; (10 μM) 4.8 ± 1.6 μM124 | |||||
| Kv7.3 | I/I0 and related EC50 | ||||||
| ΔV1/2 and related EC50 | CHO cells: −42.8 mV; 0.6 ± 0.3 μM95 | ||||||
| Kv7.3/7.4 | I/I0 and related EC50 | HEK293 cells: 2.0 ± 0.2 (10 μM); EC50 NA105 | HEK293 cells: 1.6 ± 0.1 fold (10 μM); EC50 NA105 | ||||
| ΔV1/2 and related EC50 | |||||||
| Kv7.3/7.5 | I/I0 and related EC50 | CHO cells: ∼3.25 fold; 1.4 ± 0.17 μM107 | |||||
| ΔV1/2 and related EC50 | CHO cells: −22.0 mV; 1.4 ± 0.14 μM107 | ||||||
| Kv7.4 | I/I0 and related EC50 | HEK293 cells: 2.2 ± 0.3 fold; 1.4 μM105 | HEK293 cells: 2.0 ± 0.2 fold; 2.4 μM105 | CHO cells: 9.7 ± 0.1 μM; (I/I0 = NA)124 | CHO cells: >30 μM112 | ||
| ΔV1/2 and related EC50 | A7r5 cells: −25.0 ± 2.5 mV; 1.6 ± 0.2 μM11 | CHO cells: −24.6 mV; 5.2 ± 0.9 μM105 | HEK293 cells: −6.4 ± 1.8 mV (10 μM); EC50 NA105 | ||||
| HEK293 cells: −11 ± 1.9 mV (10 μM); EC50 NA105 | |||||||
| Kv7.4/7.5 | I/I0 and related EC50 | ||||||
| ΔV1/2 and related EC50 | A7r5 cells: −34.2 ± 3.3 mV; 3.8 ± 1.2 μM11 | ||||||
| Kv7.5 | I/I0 and related EC50 | HEK293 cells: 3 ± 0.4 fold (10 μM); EC50 NA106 | HEK293 cells: >12 fold; 2.4 μM106 | ||||
| ΔV1/2 and related EC50 | A7r5 cells: −43.9 ± 7.7 mV; 4.5 ± 2.0 μM11 | HEK293 cells: −1.0 ± 0.1 (10 μM); EC50 NA106 | HEK293 cells: −1.1 ± 0.1 mV (10 μM); EC50 NA106 |
The anti-seizure properties of RTG result from neuronal excitability dampening; the compound acts preferentially on high-frequency firing neurons, slowing deactivation or speeding the rate of activation of Kv7 channels.96 These effects arise from a hyperpolarization shift in the voltage dependence of the channel activation process, and channel conductance improvements.97
The precise binding interactions of RTG on Kv7 channels have not yet been fully elucidated. However, studies on channel mutants and extensive molecular docking suggested several amino acid residues that are critical for RTG's interactions, such as Phe240, Ala265, Leu268, Leu292, Ala295, Gly301, Leu243 (S5 helix), Leu275 and Leu299 (S6 helix). Trp236 (Kv7.2 numbering) and Trp265 (in Kv7.3) in the pore domain strongly influence RTG's potency and Kv7.2–5 versus Kv7.1 selectivity.98–100 Moreover, through amino acid mutagenesis studies, Thr271 (S5 helix), Leu272 (S5 helix), Leu338 (S6 helix), and Leu314 (pore domain) were demonstrated to be involved in the binding of RTG to the Kv7.3 channel, where the agonist molecule appears to adopt two different binding poses, with the carbamate group either in the vicinity of Leu314 or Trp265.87 Furthermore, homology models of the closed and open conformations of the Kv7.3 pore module (PM) suggested that π–π interactions of Trp265 in S5 and Phe343 in S6 were major determinants of the stability of the closed conformation of the PM. Docking with RTG destabilized this π–π interaction in favor of the open channel conformation.101
Molecular docking and dynamics studies using the open state of KCNA2 (Kv1.2) as a starting point for generating a Kv7.2 homology model revealed that the NH–carbamate and the amino group in the 2-aminophenyl moieties of the RTG structure were key interaction sites. Moreover, electrostatic potential and average local ionization energy analyses revealed contributions of van der Waals and hydrophobic interactions, a halogen bond, and π–π stacking to its Kv7.2 affinity.100 The recently reported cryo-electron microscopy (cryo-EM) structures of human Kv7.2 in the apo state and in complex with RTG (pdb code 7cr2) demonstrate that RTG binds at the pore domain and activates the channel by an allosteric modulation.115 Analogously, cryo-EM structures of human Kv7.4 and its complexes with the opener RTG and the blocker linopirdine are now available.116 These structures reveal new functional group recognition sites and ligand activation mechanisms, and therefore provide a vastly improved opportunity for structure-guided rational drug design and optimization.
Although the majority of direct contacts between the small molecule and the pore region are based on hydrophobic interactions, a H-bond can form between the NH of the indole of Trp265 in Kv7.3 and the carbamate oxygen atom of RTG. Furthermore, the results of this and other studies are likely also relevant for Kv7.2, Kv7.4, and Kv7.5 proteins, as well as other Kv7.3 activators such as BMS-204352 and ML213 (vide infra).87
In spite of its promising mechanism of action and early success in the market, RTG was discontinued by GSK in 2017 after the manifestation of severe side effects in a small number of patients, and after receiving a black-box warning by the FDA.117,118 In 2016, these side effects had already led EMA to limit RTG use.119 Further studies on RTG safety after long-term exposure in patients affected by partial-onset seizures have been reported, resulting in the disclosure of new ophthalmological and dermatological complications.120
2.3. BMS-204352
In a related effort, Bristol Myers Squibb launched the Kv7 positive modulator BMS-204352 (Flindokalner, Maxipost®) as a clinical candidate for acute ischemic stroke (Chart 1). The mechanism of action of the (S)-enantiomer consists of pan Kv7.2–Kv7.5 activation, inhibiting Kv7.1 with comparable potency and promoting the positive modulation of Ca2+-activated K+ channels (BK channels), which are located in brain regions such as cortex, hippocampus, and thalamus. The (R)-enantiomer showed the same properties as BMS-204352 against Kv7.1 and BK channels but exerted a negative modulation on Kv7.2–Kv7.5 channels.121
BMS-204352 showed promise in the clinic since it was well tolerated in phase I (single and multiple i.v. doses; 0.001 to 0.2 mg kg−1) and phase II clinical trials (multiple i.v. doses; 0.1 to 2 mg kg−1) when administered within 48 h after stroke onset. However, BMS-204352 failed phase III studies due to the lack of efficacy in acute stroke patients.122 Recently, a new clinical trial aimed to clarify a possible use of BMS-204352 in headache/migraine, but at this time trial results have yet to be published (ClinicalTrials.gov identifier: NCT03887325).123
2.4. ICA-27243
The structurally simplified amide ICA-27243 (Chart 1) is a Kv7.2/7.3-selective agonist with a marked capability to differentiate between Kv7.4 (20 fold) and Kv7.3/7.5 (>100 fold).124,125 This selectivity is probably a function of the interaction with a low-similarity VSD region in Kv7 proteins (at the C-terminal end of S2 and the N-terminus of S3). In contrast to the non-selective Kv7.2–7.5 opener RTG, the activity of ICA-27243 is not affected by the mutation of the key Trp residue in the S5 domain and does not appear to be linked to the S5–S6 domain. Difficulties in identifying the specific amino acid residues responsible for the binding of ICA-27243 appeared to be due to the presence of multiple, non-contiguous residues forming the binding pocket in the folded channel protein.126
In comparison, ICA-27243 and RTG displayed a differential functional effect on Kv7.2 and Kv7.4 channels. While augmentation of the current amplitude by ICA-27243 was observed in a voltage-independent manner over the whole range of the membrane potential, RTG increased the current amplitude in a more narrow potential range, as the effect switched from an activatory to an inhibitory phase at a more depolarised, voltage-dependent potential. A combination of ICA-27243 and RTG on Kv7.4 resulted in an additive increase in the current amplitude and shift in V1/2 which indicated activation of Kv7.4 by both compounds occurred through separate but compatible mechanisms, and the channel was able to accommodate both compounds simultaneously. A combined treatment with both compounds on Kv7.2 also resulted in an augmentation of current amplitude, dominated by the activity of ICA-27243. It was suggested that the Kv7.2 channel was able to bind both compounds, but a conformational change due to ICA-27243 masked the effect of RTG.
ICA-27243 was developed as an antiepileptic drug, with the advantage to not require dose escalation after subchronic treatment in a rodent model of seizures. ICA-27243 also has a high therapeutic index for anticonvulsant activity (lower seizure protection dose at 1 mg kg−1 p.o. in rodents) versus locomotor impairment.127
2.5. ML213
Only few positive modulators selectively enhance the outward K+ current of Kv7.2 and Kv7.4 proteins.128 Among them, the ICA-27243 analogue ML213 (Chart 1) showed a unique selectivity profile for Kv7.2, Kv7.4, Kv7.5 and heteromeric Kv7.4/7.5 channels, interacting with an essential Trp residue in S5 analogous to RTG and BMS-204352.11
In CYP450 metabolic stability assays, ML213 was inactive (>30 μM) against all CYP enzymes (CYP3A4, 2D6, 2C9, 1A2) tested. Unfortunately it displayed a high clearance in human and rat liver microsomes. In in vivo PK studies, ML213 had a moderate brain to plasma distribution ratio of 0.37 (brainAUC/plasmaAUC = 0.37), which was considered acceptable for a first-generation Molecular Libraries Screening Center Network (MLSCN) probe molecule.129
3. Pharmacological applications of Kv7.2–Kv7.5 activators supported by in vivo data
3.1. Neuropathic pain and analgesia
Neuropathic pain (NP) is a sensation described as persistent or sporadic dysesthesia (painful discomfort in limbs) or allodynia (pain caused by normally neutral stimuli) resulting from alteration of the somatosensory nervous system, and caused by events such as spinal cord injury, metabolic disease, viral infection, cancer, immune disorder, or multiple sclerosis.131,132 The insurgence of NP can be classified by two distinct mechanisms: peripheral sensitization (PS) and central sensitization (CS). Among other factors, a prolonged exposure of spinothalamic tract (STT) neurons to neurotransmitters can trigger spontaneous firing and CS.133,134 Overactivation of astrocytes and microglia can also play a role in CS due to abnormal release of proinflammatory cytokines.135 At the molecular level, NP can be generated by channelopathies resulting in overexpression and/or upregulation of voltage-gated sodium and calcium channels, as well as loss of voltage-gated potassium channels. These result in a lower threshold for an action potential and/or a higher firing rate in peripheral or central nociceptive neurons, hyperexciting nociceptive fibers and promoting neurotransmitter release.136 Moreover, PKA/PKC-driven gene transcription is induced after damage to nociceptive neurons, further contributing to neuronal hyperexcitability and therefore to NP.137 Over- or down-responsiveness to neurotransmitter stimuli can also underlie NP insurgence, including downregulation of Kv7 channel expression in primary afferent nociceptive fibers.138
The role of Kv7 channels in the somatosensory nervous system (especially Kv7.2 in peripheral and central terminals of nociceptive primary afferents, dorsal root ganglia and dorsal horn neurons) suggested a possible use of Kv7 agonist for NP treatment as well as the treatment of refractory pain.139 The analgesic properties of Kv7 agonists are thought to be due to primary afferent fiber hyperpolarization and reduction of Aδ-and C-fiber stimulation at the dorsal horn. In vivo studies were primarily conducted in rats, where administration of RTG suppressed allodynia and hyperalgesia induced by trigeminal nerve injury (RTG doses: 0.19 to 15 mg kg−1; i.p.) and injured paws (RTG doses: 5 to 20 mg kg−1; p.o.).140,141
Fibromyalgia is often associated with channelopathies due to mutations in Kv7 channels, and is characterized by NP, sleep disturbance and cognitive dysfunctions.142 Since flupirtine was shown to reduce the symptoms of fibromyalgia in an open-label trial,143RTG was also investigated in this model. RTG promoted a reduction in NP during treatment of post-herpetic neuralgia in a phase IIa trial (ClinicalTrials.gov identifier: NCT00612105).144 Additional evidence of the analgesic efficacy of RTG (dosed 7.5–15 mg kg−1; i.p.) and flupirtine (7.5–20 mg kg−1; i.p.) was found in a rat model of gouty arthritis, which is a common rheumatic disease caused by deposits of monosodium urate crystals in joints and periarticular tissues.145 Moreover, hyperalgesia was reduced by RTG (dosed 5 mg kg−1; p.o.) in diabetic NP rats, derived from a downregulation and activity reduction of Kv7.2, Kv7.3, and Kv7.5 in dorsal root ganglion neurons.146
NP can also be caused by heavy and prolonged pharmacological therapy, such as cancer treatment. The nociceptor hyperexcitability resulting from paclitaxel-induced peripheral neuropathy (PIPN) generated an acute pain refractory to the common analgesic drugs.147,148 Early-stage administration of RTG attenuated PIPN without an observed interference with paclitaxel's anticancer properties.149
Brain injury, gliosis, astrocytosis and neuronal damage after exposure to the irreversible cholinesterase inhibitor sarin were mitigated in rats by RTG in combination with atropine, reducing seizure-induced brain damage.150 The analgesic effects of RTG and flupirtine were further investigated for the treatment of musculoskeletal pain, postoperative pain, and migraine.84,151,152
3.2. Depression
In the prefrontal cortex, excitatory glutamatergic neurons, also known as pyramidal cells, are involved in cognitive processing, whereas locomotor function is controlled by pyramidal neurons in the corticospinal tract.153
The evidence of M-current activity in the pyramidal neurons of the prefrontal cortex suggested a possible involvement in diseases such as depression, schizophrenia, and bipolar disorder.154 Notably, hyperactivation of ventral tegmental area dopaminergic neurons, linked to major depressive disorders, was significantly reduced by a daily administration of RTG in mice (1 mg kg−1 d−1; i.p.).155 An open-label study was performed on medication-free individuals affected by major depressive disorder with a dose of 900 mg RTG per day for ten weeks. Results showed a marked reduction of depression and anhedonic symptoms with a good tolerability, and an increase in reward learning.156
Treatment-resistance major depressive disorders are often associated with neuroinflammation, and in some cases triggered by obesity due to hyperexpression of pro-inflammation cytokines such as TNF-α, IL-6, IL-1β, and an increase in the glutamatergic pathway.157,158 A consistent reduction in anxious/depressive-like behavior was observed in a male mice model of obesity after RTG treatment (1 mg kg−1 day−1; i.p.). Interestingly, the reversal of depression, achieved after RTG treatment, was not observed for other antidepressants, including desipramine (40 mg kg−1, p.o.), fluoxetine (18 mg kg−1, p.o.), and ketamine (6 mg kg−1, i.p.).159
3.3. Neurodegenerative diseases
Neurodegenerative diseases (NDs) include movement and cognitive disorders in which a progressive degeneration of the structure and function of CNS and PNS are observed. Currently, available therapies are mainly focused on symptomatic treatments since only very few drugs are able to delay the neurodegenerative process.160,161 Several simultaneous ion-channel aberrations have been identified in neuronal tissue during NDs, and the resulting hyper- or hypo-excitability conditions seem to be a fundamental feature of the pathology (Table 3).5 Kv7 modulators were shown to control the altered neural excitability in in vivo models of NDs.
Correlation between aberrant ion-channels and drugs targeting neurodegeneration pathways.
| Ref. | |||
|---|---|---|---|
| Alzheimer's disease | Aberrant ion-channels | Kv3.4, Kv4.2, NaV1.1, Kv7.2, VDAC1, Kv1.3, Kv1.5, KCa3.1, CLIC1, TRP channel, NA+/K+ ATPase pump, CaV1.1, N-, P/Q-, R-type Ca2+ channels, VDAC1, HCN channels | 184 |
| 185 | |||
| 186 | |||
| 187 | |||
| Aberrant ligand-gated ion-channels | AMPARs, NMDARs, nAChRs | 188 | |
| 189 | |||
| 190 | |||
| Drug treatments | - Acetylcholinesterase inhibitors: donepezil, rivastigmine, and galantamine | 191 | |
| - NMDA receptor antagonists: memantine | 192 | ||
| Parkinson's disease | Aberrant ion-channels | Kv4.3, KCa2.3, KCa1.1, CaV1.2, CaV1.3, T-type Ca2+ channel, Hv1 proton channel, VDAC1, HCN channels | 184 |
| 189 | |||
| Aberrant ligand-gated ion-channels | Kir3.1, Kir3.2, NMDARs, KATP channels (mutations both in Kir6.2 and SUR1) | 193 | |
| 194 | |||
| Drug treatments | - Dopamine precursors: levodopa (in association with dopa decarboxylase inhibitors: carbidopa, benserazide) | 195 | |
| 196 | |||
| - COMT inhibitors: tolcapone, entacapone | |||
| - MAO-B inhibitors: safinamide, selegeline, and rasagiline | |||
| - Dopamine agonists: bromocriptine, pergolide, pramipexole, ropinirole, piribedil, cabergoline, apomorphine, lisuride, and rotigotine | |||
| - Anticholinergic: trihexyphenidyl, benztropine, ethopropazine, and selegiline | |||
| Huntington's disease | Aberrant ion-channels | Kv2.1, Kv7.2/7.3, KCa2.3, Kv4, HVA Ca2+ channels, CLIC1 channel | 184 |
| 197 | |||
| Aberrant ligand-gated ion-channels | GABAA receptors, Kir2.1, Kir2.3, Kir4.1 | 198 | |
| 199 | |||
| Drug treatments | - Tetrabenazine | 200 | |
| - Valproic acid | 201 | ||
| - Dopamine receptors activators: levodopa, pramipexole | 202 | ||
| Multiple sclerosis | Aberrant ion-channels | NaV1.2, NaV1.5, NaV1.6, NaV1.8, Kv1.1, Kv1.2, Kv1.3, Kv1.4, Kv2.1, Kv3.1, CaV1.2, CaV1.3, CaV1.4, CaV2.2, TRPM4 and ASIC1a channels | 184 |
| 203 | |||
| Aberrant ligand-gated ion-channels | AMPA, NMDA, kainate receptors, Kir4.1 | 204 | |
| 205 | |||
| Drug treatments | During acute clinical manifestations: corticosteroids: methylprednisolone | 206 | |
| Drugs used to reduce relapse rate in relapsing remitting multiple sclerosis: | 207 | ||
| - Interferon β-1a, interferon β-1b, peginterferon β-1a | |||
| - Glatiramer acetate | |||
| - Type II topoisomerase inhibitor: mitoxantrone | |||
| - Monoclonal antibodies: natalizumab, alemtuzumab, ocrelizumab | |||
| - Sphingosine 1-phosphate receptors modulators: fingolimod, siponimod, ozanimod | |||
| - Dihydroorotate dehydrogenase inhibitor: teriflunomide | |||
| - Dimethyl fumarate | |||
| - Purine analogue: cladribine | |||
| Drugs used to reduce relapse rate in progressive multiple sclerosis: | |||
| - Ocrelizumab, alemtuzumab, siponimod, clabridine, and mitoxantrone | |||
| Amyotrophic lateral sclerosis | Aberrant ion-channels | NaV1.3, NaV1.5, NaV1.6, Kv1.1, Kv1.2, Kv1.6, Kv7.2, L-, N-, P/Q-, T-type Ca2+ channels, VDAC1, HCN channels | 184 |
| 189 | |||
| Aberrant ligand-gated ion-channels | AMPA, GABA receptors, Kir4.1 | 208 | |
| 209 | |||
| Drug treatments | - Riluzole | 210 | |
| - Edaravone | 211 |
Studies in rat models of Parkinson's disease (PD) with lesions in the substantia nigra compacta demonstrated that the intra-lateral habenula (Lhb) injection of RTG (1.5 to 6 μg per rat) reversed the working memory impairment, reducing the neuronal firing rate and increasing the dopamine and serotonin levels in the ventral medial prefrontal cortex, thus highlighting the role of Kv7.2 channels in this disease.162,163
As a continuation of studies in the rat model, an application of the experimental drug ICA-27243 was suggested for an l-DOPA induced dyskinesias (LID) treatment in patients affected by PD. In fact, the evidence for a reduction of spontaneous involuntary dystonic and choreatic movements after RTG treatment was mainly attributed to an excitability modulation of striatal projection neurons through Kv7.2/Kv7.3 channel activation.164
ICA-27243 ameliorated LID symptoms in hemiparkinsonian rats at a dose of 5–10 mg kg−1 i.p., but did not exhibit this effect at a higher dose. Moreover, the abnormal involuntary movement reduction extended to 180 min from treatment onset but no further, probably due to the Kv7 channel expression on striatal interneurons that antagonize striatum outcome.165–167 Although further investigations of the involvement of Kv7 channels in LID are needed, it is interesting to note that ICA-27243 did not interfere with the anti-PD effect of l-DOPA.165
Based on evidence of the high similarity between the M-current in rat native cortical neurons and human induced pluripotent stem cells (hiPSC) derived neurons,168 several studies evaluated the role of a Kv7 agonist in amyotrophic lateral sclerosis (ALS),169,170 identifying RTG as a clinical trial candidate in patients affected by ALS.171
ALS is characterized by muscle stiffness with gradual muscle weakness, leading to death or need for respiratory support, generally after 3–5 years from the appearance of symptoms, including progressive degeneration of cerebral cortex motor neurons and/or brain stem- and spinal cord motor neurons.172,173 In a phase II clinical trial, RTG oral doses of 600 or 900 mg per day were used in ALS subjects (ClinicalTrials.gov identifier: NCT02450552), resulting in inhibition of the spontaneous firing of motor neurons in a dose–response manner.171 To support the hypothesis of the utility of Kv7.2–Kv7.5 activators in ALS treatment, clinical trials have also been carried out with flupirtine: an oral flupirtine maleate treatment trial in multiple sclerosis (FLORIMS) was performed in order to investigate safety and efficacy. After a dose of 300 mg per day for 12 months, in combination with interferon β-1b, good tolerability with an asymptomatic increase of liver enzymes and an absence of hepatotoxicity were found. However, further studies are needed to confirm the ability of flupirtine maleate to reverse multiple sclerosis conditions.174Flupirtine also completed a phase II trial for relapsing remitting multiple sclerosis (RRMS) (ClinicalTrials.gov identifier: NCT00623415), but results from this study have also not yet been published.
Huntington's disease (HD) is an inherited neurodegenerative disorder of striatal output neurons (SONs) characterized by early stage symptoms (motor and cognitive dysfunctions) and progressive neuronal decay. In the first phase of the disease, the hyperexcitation of SONs leads to excitotoxic molecule release, triggering biochemical cascades and culminating in neuronal death.175,176 A daily i.p. administration of 10 mg kg−1 of RTG in R6/2 mice (a transgenic mice model which expresses truncated human mutated huntingtin with expanded CAG repeats)177 was able to ameliorate the depressed M-current in SONs and prevent locomotor impairment, suggesting an opportunity for further studies of the use of Kv7 agonists for motor dysfunction treatment in the early stages of HD.178
3.4. Neuroprotection
A major unmet need in neuroprotective therapy is the treatment of traumatic brain injury (TBI) since currently there is no pre- or post-TBI pharmacological treatment available.
Cerebral vasospasm is characterized by a prolonged and severe constriction of the cerebral artery due to abnormal spasmogenic substance released under hyperexcitability conditions. Although the pathogenesis remains poorly understood, cerebral vasospasm is quite often triggered during rupture of intracranial aneurysms, resulting in extravasation of blood into the subarachnoid space and causing subarachnoid hemorrhage (SAH).179
M-Current activators were found to repristinate the membrane potential in spasmogens-treated cells and to relax the pre-constricted arteries. This evidence was confirmed through in vivo studies with RTG (7.5 mg kg−1 i.p. twice per day) in a rat model of SAH-induction, wherein cerebral vasospasm was alleviated.180
The low vascular perfusion during and after an ischemic stroke triggers a neurotoxic biochemical cascade, resulting in neuronal hyperexcitability which underlies the insurgence of seizure and epilepsy.181,182
Recently, the increased excitatory activity in a TBI animal model was found to promote Kv7.2 overexpression in specific regions of the hippocampus without affecting Kv7.3 levels.183
An in vitro study demonstrated the neuroprotective ability of flupirtine and RTG in an oxygen/glucose deprivation (OGD) model in hippocampal neurons.212 Subsequently, in vivo studies confirmed the in vitro protective effects. A single dose of RTG (10.5 mg kg−1) was administered via tail vein in mice affected by induced acute cerebral infarction or a catastrophic stroke model, resulting in a significant decrease in necrotic lesions if the Kv7 channel agonist was administered 0–3 h after neuronal hypoxia onset.213 The effectiveness of RGT in post-TBI disease prevention was also demonstrated at a lower dose (1.2 mg kg−1; i.p. single dose 30 min post TBI induction) in mice subjected to controlled cortical impact (CCI) as a model of brain injury.214 However, the efficacy of neuroprotection by RTG remains controversial: in mice cortical neurons, RTG exhibited an inverse neuroprotective behavior during OGD-reperfusion, augmenting presynaptic glutamatergic release and consequently overstimulation of NMDA receptors. In this scenario, conversely, the Kv7 blocker XE991 was found to exert neuroprotection.215
A recent in vivo assay was performed with the purpose of exploring a neonatal hypoxia-ischemia (HI) injury treatment with flupirtine. HI-induced male rat pups were treated with flupirtine maleate (25 mg kg−1 day−1) post-induction. The results showed a significantly reduction in HI-induced hippocampal and cortical tissue loss, preserving motor and cognitive functions.216
Anti-manic properties were also addressed with Kv7 channel openers, especially with heteromeric Kv7.2/7.3 activation.217 In a mice model for mania, RTG (10 mg kg−1 i.p.) and ICA-27243 (5.0 mg kg−1 i.p.) provided a basal 2-deoxyglucose uptake reduction in brain tissue and consequently a recovery of the cerebral metabolic activity that was altered in the bipolar disorder.218–221 Moreover, Kv7 agonists and ICA-27243 increased the phosphoserine-9 levels of GSK3β in the prefrontal cortex and hippocampus, a region that is fundamental for the emotional and cognitive aspects of bipolar disorders.222
3.5. Auditory diseases
Tinnitus is a widespread pathology presented as a sporadic or continuous perception of phantom background noise that can affect one or both ears. Its etiology is not well understood yet, but it appears to be triggered by the insurgence of a neuronal hyperexcitability status. The development of tinnitus can be due to a multitude of possible reasons, including noise-induced hearing loss, ear infections, aging, neurodegeneration, and brain tumors, and commonly it is accompanied by depression and anxiety.223 Administration of BMS-204352 (10 mg kg−1 i.p.) or RTG (10 mg kg−1 i.p.) in a rat model of tinnitus induced with sodium salicylate (SS)224,225 prevented hearing damage at high frequencies (≥20 KHz) as well as low frequencies (≤8 KHz).226 Interestingly, both enantiomers of the experimental drug BMS-204352 suppressed tinnitus symptoms in vivo in a dose-dependent manner, suggesting the involvement of other mechanisms or polypharmacology effects.227
The RTG analog SF0034 (vide infra) was found to prevent the development of tinnitus in a model of noise-induced hearing loss without affecting temporal processing or hearing thresholds.271
Deafness non-syndromic autosomal dominant 2 (DFNA2) is a disease induced by altered Ik in type I hair and outer hair cells in the vestibular apparatus. It is thought to be due to Kv7.4 mutations. Identification and possible modulation of new Kv7.4 variants responsible for DFNA2 has grown in interest in the last decade.228 Except for channels with mutations in the pore region, RTG restored the heterogeneous Kv7.4 function in vitro, laying the foundation for a new possible approach to DFNA2 treatment.229,230BMS-204352 was also investigated in cells expressing DFNA2-derived mutated Kv7.4 channels, and was found to revert the low-activity fast-deactivation kinetics resulting from these mutations.231
3.6. Smooth muscle disorders
Activation of M-current resulted in a relaxation of the thoracic aorta segment, renal artery, and mesenteric artery. This effect was explained by a hyperpolarization of the smooth muscle cells through Kv7.4 and Kv7.5 activation, laying the foundation of a possible therapeutic application of Kv7 modulators in smooth muscle disorders.4,232 In this context, it is interesting to note that ML213 induced the relaxation of detrusor smooth muscle (DSM) in rats,233,234 guinea pig,235,236 pigs,234 and humans;60 overall leading to a hyperpolarization of the membrane potential and a decrease in global intracellular Ca2+ concentration, and thus highlighting a possible use in the management of overactive bladder syndrome.237
3.7. Erectile dysfunction
Erectile dysfunction (ED) is often an early indicator of a potential risk of diabetes and cardiovascular diseases.238 Treatment options include nitric oxide donors or PDE5 inhibitors, penile artery dilators and trabecular smooth muscle relaxing agents.239 However, the administration of the aforementioned drugs can be contraindicated due to cardiovascular complications.240 Evidence for Kv7.3–Kv7.5 expression in penile arteries (Kv7.5 > Kv7.3 and Kv7.4)62 and Kv7.1, Kv7.3, Kv7.4 and Kv7.5 in corpus cavernosum (predominance of Kv7.4) stimulated investigations of the role of Kv7 agonists in ED.241 Indeed, BMS-204352 and ML213 were effective in smooth muscle tone regulation in penile artery and corpus cavernosum strips of a rat model of human metabolic syndrome, revealing a down-regulation of Kv7.3, Kv7.4, and Kv7.5, and a Kv7.1 up-regulation in corpus cavernosum.62 Moreover, the down-regulation of Kv7.4 in corporal smooth muscle (CSM) in rats affected by diabetes mellitus underlined its role in ED. Relaxation of CSM strips induced by ML213 in a non-endothelium-dependent manner in human corpus cavernosum, presumably through activation of Kv7.4, Kv7.5 and Kv7.4/7.5 due the absence of Kv7.2 in this tissue, suggested Kv7.4 as a potential target for ED treatment.241
3.8. Muscle hyperexcitability diseases
Muscle hyperexcitability is the ultimate outcome of several channelopathies, and some of these are still lacking in efficacious pharmacological treatments. For example, myotonic congenita (MC) is a channelopathy characterized by muscle stiffness derived from a CIC-1 muscle chlorine channel mutation. To date, the treatment of MC is based on hyperexcitability reduction by Na+ current blockers, but with poor clinical results.242 Kv7 agonists were found to attenuate muscle myotonia in in vitro systems,243 enhancing K+ current during trains of action potentials. RTG (30 mg kg−1; i.p.) was also effective in preventing depolarization in MC mice models.244
3.9. Airway diseases
Evidence of Kv7 channel expression in mouse nodose neurons and in lung-specific nodose neurons (mainly Kv7.3) suggested a modulation of the airway vagal sensory C-fiber outcome. RTG induced a shift toward negative potential of neuronal membranes and promoted a reduction in spontaneous and agonist-evoked firing in lung nodose C-fibers in mice. The antitussive properties of RTG in mice were confirmed through a brief (15 min) administration of 250 μM of aerosolized RTG. These results lend evidence to the potential application of Kv7 agonists in inflammatory airway diseases such as chronic coughing, dyspnea, bronchoconstriction, and hypersecretion disorders.245
4. Properties of Kv7.1 selective agonists
Since the binding region of most known Kv7.2–Kv7.5 agonists is located in a pore domain that is distinctly different in the Kv7.1 protein, it is not surprising that positive Kv7 modulators demonstrate the ability to discriminate between Kv7.1 and other Kv7 channels. Phenylboronic acid (PBA), 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS), hexachlorophene (HCP), mallotoxin, and 3-ethyl-2-hydroxy-2-cyclopenten-1-one (CPT1) are representative for the positive Kv7.1 modulator class which can interact both with Kv7.1 homomers and/or Kv7.1/KCNE1 heteromers.246–250 Although, in most cases, Kv7.1 agonists are able to activate only this specific Kv7 subtype, the challenge in this category is their pronounced ability to modulate other ion-channels, such as hERG, Cav1.2, and Nav1.5.249–253 The preferred Kv7.1 expression in heart and its contribution to cardiac electrophysiology (ventricular repolarization through IKs current activation) ensured that Kv7.1 agonists were evaluated in anti-long QT syndrome (LQTS). However, a Kv7 enhancer's ability to modulate other ion channels represents a major obstacle for this purpose.
ML277 (Chart 2) is a potent and selective Kv7.1 activator (>100 fold selectivity over Kv7.2–Kv7.5 proteins)112 with a low capability to interact with the above mentioned cardiac ion-channels (Tables 1 and 2).113 Its binding site has not yet been clearly defined, but based on molecular dynamics studies and Kv7.1 and Kv7.1/KCNE1 docking simulations, ML277 was suggested to bind between the intracellular part of the S2–S3 loops and the S4–S5 linker, and the pore region.254ML277 has also been suggested as anti-LQTS since it showed high ion-channel selectivity, shortening the action potential duration in both human-iPSC-derived cardiomyocytes and guinea pig cardiomyocytes.113,254,255 Moreover, it enhanced the current of the AO state, leaving unchanged the IO state-derived current in Kv7.1 protein. This unique feature of ML277 could be an innovative aspect of a LQTS treatment, since Kv7.1/KCNE1 association generates IKs current in the AO state.256 In addition to a LQTS treatment, other types of arrhythmia could potentially be treated with a Kv7.1 agonist. ML277 demonstrated a shortening of the action potential duration (APD) and a suppression of cytosolic Ca2+ transient (CaT) in stimulated single rabbit atrial myocytes via IKs current activation.257 This pharmacological behavior could be useful in the prevention or treatment of atrial fibrillation, a type of atrial arrhythmia that can precede cardiac stroke and heart failure, and for which an efficient treatment is still unknown.258,259
Chart 2. Chemical structures of Kv7.1 activators.

L-364,373 (R-L3) is a benzodiazepine type Kv7.1 agonist that was found to activate IKs current at μM concentrations in ventricular myocytes of the guinea pig, mimicking LQTS1 conditions252 and shortening APD suppressing early after depolarization in a rabbit model of LQTS2 (Chart 2).260 However, R-L3 failed when tested in dog ventricular myocytes; possibly due to the opposing effect of the two enantiomers in activating ((R)-enantiomer) and blocking ((S)-enantiomer) IKs current.261,262 As found for Kv7 agonists, the predicted binding site of R-L3 was located in S5–S6 in Kv7.1, supported by modeling studies.263
Alterations in IKr current demonstrated an induction in severe arrhythmias, including torsade de pointes and LQTS2. These findings have been observed after IKr inhibition for high dose and/or prolonged exposure to class III antiarrhythmic drugs and channelopathies such as the loss-of-function mutation in hERG. In this scenario, the compensation of decreased IKr current by IKs improvement has been evaluated in guinea pig ventricular myocytes using enantiopure R-L3. The results showed both a shortened APD and a decreased duration in the late stage of repolarization (reduced triangulation of AP), typical in LQTS2.264
5. Design, synthesis, and properties of Kv7 agonists
5.1. Flupirtine, RTG, and analogs
A general synthetic route for the preparation of flupirtine and structurally related analogs started with commercially available 2,6-dichloro-3-nitropyridine (1), which, after nucleophilic aromatic substitution with ammonia in methanol, was converted to 6-chloro-3-nitropyridin-2-amine (2). Base-mediated nucleophilic substitution of the chloride with suitably substituted benzyl amines 3 afforded 6-amino-2-benzylamino-5-nitropyridines 4, and reduction of the nitro group gave triaminopyridines 5. The electron-rich 3-amino substituent on the pyridine could be selectively reacted with chloroformates to yield the targeted carbamates 6 (Scheme 1).265,266
Scheme 1. Overview of the synthesis of flupirtine and structural analogs.

An analogous synthetic approach toward RTG commenced with a reductive amination of 2-nitro-1,4-phenyldiamine (7) with substituted benzaldehydes to form intermediates 8 (Scheme 2). Carbamoylation of the more nucleophilic secondary amine was followed by addition of a second chloroformate to provide dicarbamate 10. Target analogs 11 were obtained after concomitant reduction of the nitro group and hydrogenolysis of the Cbz carbamate.266
Scheme 2. Overview of the synthesis of RTG and structural analogs.

While acting as trailblazers for the clinical utility of Kv7 channel agonists, both flupirtine and RTG were withdrawn from the market, among other problems due to hepatotoxicity and tissue discoloration side effects, respectively. Metabolism or (photo)chemical degradation of these drugs might play a role and produce reactive intermediates which can covalently modify endogenous nucleophiles.83 Most likely, flupirtine forms the electrophilic intermediate 12 upon oxidation, which reacts with bio-nucleophiles (Scheme 3). Along these lines, an in vitro study reported oxidation of flupirtine by peroxidase followed by bioconjugation with glutathione.267
Scheme 3. Proposed toxification pathways of flupirtine and RTGvia quinone diimines 12 and 14 and the RTG dimer 16.

In related work, it was proposed that upon oxidation, RTG was converted to quinone diimine 14; this species might undergo dimerization followed by further oxidation to form phenazinium ions (e.g.15), potentially causing the blue skin pigmentation observed in some patients after prolonged treatment with RTG.268 In addition, RTG, on exposure to hypervalent iodine reagent as a xenobiotic oxidative metabolism model, underwent dimerization to form the poorly soluble dimer 16 which was also identified as an RTG manufacturing impurity.269,270
In order to increase potency while improving channel selectivity and chemical stability, as well as reducing the rate of oxidation or dimerization of RTG, structure–activity relationship (SAR) analyses mainly pursued an optimization of zones 2 and 3 of the lead compound (Chart 3).
Chart 3. Structures of selected analogs of flupirtine and RTG, and a medicinal chemistry zone model for SAR studies.

Analogs were tested in a functional cell-based assay for Kv7.2/7.3 channel activation. Fluorine substitution at positions 5 and 6, or at the 3,5-position of the triaminophenyl ring, did not significantly affect potency. A 3-fluoro substituted analog of RTG, SF0034, demonstrated a significant increase in affinity at Kv7.2/7.3 channels, about 5-fold compared to the reference.271 Like RTG, SF0034 also required W236/W265 tryptophan residues in the intracellular end of the S5 helix for exerting its gating effects on Kv7.2/7.3 channels. SF0034 was more potent than RTG in the reduction of the anticonvulsant activity in human partial epilepsy and in preventing seizures (SF0034, ED50: 3.96 ± 1.55 mg kg−1; RTG, ED50: 43.13 ± 22.26 mg kg−1).
For the synthesis of SF0034, a regioselective nucleophilic aromatic substitution of 2,3-difluoro-6-nitroaniline (17) with 4-fluoro benzylamine followed by one pot reduction of the nitro group in 18 and a chemoselective acylation with ethyl chloroformate were used (Scheme 4).271
Scheme 4. Synthesis of SF0034.

In addition to chemical lability, another drawback of RTG is its relatively low brain-to-plasma distribution (0.16), which may reduce its anti-epileptic activity and could be responsible for potential off-target effects (Table 4). Nan et al. reported a novel Kv7 activator, P-retigabine (P-RTG), incorporating a propargyl group at the N-position of the RTG linker moiety,272 that improved the brain-to-plasma ratio (2.3).273P-RTG was equivalent in terms of Kv7 channel subtype selectivity and potency compared to RTG. To determine the suppression of neuronal excitability, recombinant Kv7.2/7.3 channels were expressed in CHO cells. In vitro studies showed that P-RTG (EC50 0.99 ± 0.74 μM) was comparable to RTG (EC50 0.64 ± 0.49 μM) in terms of potentiation of the outward neuronal M-current of Kv7.2/7.3 channels. In an in vivo MES-induced seizure mouse model, P-RTG (ED50 6.5 mg kg−1) was 2.5 times more potent than RTG (ED50 15.0 mg kg−1) in suppressing epileptic activity. P-RTG also displayed a better overall PK profile than RTG. Even though the clearance rate of P-RTG in blood was much higher (t1/2 0.89 h), this did not significantly affect its half-life in brain (t1/2 values in the brain: 2.6 h and 3.0 h for P-RTG and RTG, respectively). In addition, the higher brain distribution of P-RTG did not produce a more severe toxicity effect in the nervous system based on the rotarod test. The protective index (P = TD50/ED50) was defined as the ratio given by the toxic dose divided by the therapeutic dose. A high P value is preferable over a lower one. Accordingly, the higher P value of P-RTG (15.9) than RTG (4.9) signifies a larger safety window for P-RTG.
PK parameters of P-RTG and RTG for oral administration in mice (20 mg kg−1).
| Compound | Sample | T max (h) | C max ng mL−1 g−1 | AUC0−t ng h mL−1 g−1 | AUC0−α ng h mL−1 g−1 | MRT (h) | t 1/2 (h) | Ratio of AUC0−t brain/plasma |
|---|---|---|---|---|---|---|---|---|
| P-RTG | Plasma | 0.25 | 1849 | 1935 | 1943 | 2.09 | 0.89 | |
| Brain | 0.25 | 7635 | 4466 | 4839 | 1.95 | 2.61 | 2.30 | |
| RTG | Plasma | 4.00 | 2788 | 21 088 | 34 353 | 10.90 | 6.99 | |
| Brain | 0.25 | 849 | 3460 | 3849 | 5.20 | 3.02 | 0.16 |
For the synthesis of P-RTG, diamine 19 was converted to triamino-benzene 20 by catalytic hydrogenation (Scheme 5). Selective protection of the primary 1,2-diamines with Boc2O followed by alkylation of the secondary amine with propargyl bromide furnished biscarbamate 21. Boc-deprotection and treatment with ethyl chloroformate led to P-RTG.
Scheme 5. Synthesis of P-RTG.

Dalby-Brown et al. introduced a flupirtine/RTG analog, NS15370 (Scheme 6), which activated recombinant homo- and heteromeric Kv7.2–Kv7.5 channels in HEK293 cells at sub-micromolar concentrations (EC50 ∼ 100 nM).274 Most significantly, NS15370 did not have any detectable effect on the GABAA receptor combinations most abundantly expressed in the brain (α1β2γ2; α3β2γ2; α5β2γ2) at concentrations up to 30 μM, suggesting that the activation of Kv7 channels in itself was sufficient to elicit its antiepileptic activity in rodents. NS15370 displayed a complex concentration-dependent mode of action as revealed by voltage-clamp analysis. An equivalent (∼20 mV) hyperpolarizing shift of V1/2 was achieved with NS15370 at much lower concentration (<0.03 μM) than with RTG (10 μM) suggesting that NS15370 was 100–300 times more potent than RTG. Moreover, NS15370 gradually and reversibly decreased evoked firing frequency and increased spike frequency adaptation in hippocampal CA1 neurons, and also reduced the autonomous firing of dopaminergic neurons in the substantia-nigra pars compacta. In addition, NS15370 reduced locomotor hyperactivity induced by the N-methyl-d-aspartate receptor antagonist MK-801, as well as in combination with chlordiazepoxide (CDP) and d-amphetamine (AMP). NS15370 also showed a potent, dose-dependent anticonvulsant activity in the mouse 6 Hz seizures and the rat amygdala kindling model of partial seizures.275
Scheme 6. Synthesis of NS15370.

Aromatic nucleophilic substitution of 6-chloro-3-nitropyridin-2-amine (1) with morpholine provided nitro-pyridine 22, and subsequent nucleophilic substitution with 4-fluoro benzylamine furnished 23 (Scheme 6). Reduction of the nitro group in 23 followed by selective coupling with acyl chloride 25 and subsequent salt formation afforded NS15370 in good yield.274
In another series of SAR studies on RTG, Wang et al. reported novel analogs as selective modulators of Kv7.4 and Kv7.5 channels without activating neuronal Kv7.2 and Kv7.3 (Chart 4).276
Chart 4. Structures of RTG analogs as selective Kv7.4 and Kv7.5 modulators.

In this study, the subtype specificity of RTG was improved upon altering the substituents at N-1 and N-3 positions. The synthetic routes for these N-1 and N-3 substituted RTG analogues were similar to P-RTG (Scheme 5).
Initially, substituents at the N-1 position were varied to generate a series of compounds 26. Among them, the N-1 propargyl substituted 26a was found to have the highest agonist potency for Kv7.4 and Kv7.5 channels (Chart 4). To determine the specificity of 26a on Kv7 channels, Kv7.1–Kv7.5 channels were individually expressed in CHO-K1 cells and then tested using a whole-cell voltage-clamp technique, determining dose–response curves. Analog 26a had an EC50 = 0.89 μM on Kv7.4 channels, yet it did not affect homomeric Kv7.2 channels at similar concentrations. The specificity of 26a decreased in the following order: Kv7.4 > Kv7.5 > Kv7.3, and the compound was inactive at Kv7.1 and Kv7.2 channels in this assay.276
Subsequently, two new series of N-2 and N-3 substituted analogues, 27 and 28, were prepared while maintaining the N-1 substituent as a propargyl group. For 27, the R2 group at N-2 and N-3 carbamates was varied from alkyl and cycloalkyl to aryl. In general, alkyl-substituted analogues of 27 showed better potency than the corresponding cycloalkyl and aryl analogues. Among them, the bis-tert-butyl-substituted 27a displayed the highest agonist potency and subtype selectivity. In series 28, N-2 substitution was kept constant as an ethyl carbamate, and substituents at the N-3 carbamate were varied. Similar to 26 and 27, 28 lacked agonist activity on Kv7.2 but displayed significant activation on Kv7.4 and Kv7.5 channels. Among these analogues, the N-2 ethylated, N-3 tert-butyl-substituted 28a showed the highest agonist potency with EC50 values of 0.78 and 1.68 μM on Kv7.4 and Kv7.5 channels, respectively, whereas for RTG, the respective EC50 values were 5.90 and 3.45 μM. In contrast, compound 28a showed a maximal 1.5-fold increase of the outward potassium current on Kv7.3 and Kv7.2/7.3 channels.276
In order to probe the mechanism of action and toxicity of flupirtine, Link and co-workers reported novel analogs of flupirtine with varying redox properties, designed in such a way that the substituents should interfere with azaquinone diimine formation by inductive electronic effects. The hypothesis was tested by introducing N-methylation at the carbamate position or at N-2/N-6 positions.277
Due to the higher acidity of the carbamate group vs. the amines, regioselective mono N-methylation of the carbamate nitrogen atom was feasible and provided analog 29 (Scheme 7). The fused bicyclic flupirtine derivative 31 was prepared by elimination of ethanol by heating to 200 °C to give 30, followed by carbamoylation. The N-methylated flupirtine analogs 32–35 and deaminoflupirtines 36 and 37 (Chart 5) were prepared using analogous procedures to those described in Scheme 1.
Scheme 7. Synthesis of flupirtine derivatives 29 and 31.

Chart 5. N-Methylated flupirtine derivatives 32–35 and deaminoflupirtine analogs 36 and 37.

Based on cyclic voltammetry studies, it was suggested that substitution of either the primary or the secondary amines of flupirtine [32, 33 and 34; anodic peak potential (Ep.a) 294–385 mV] had little effect on the Ep.a relative to flupirtine (Ep.a 350 mV) (Table 5). However, compounds with substitution at the carbamate nitrogen (29, 31 and 35) as well as those lacking the amino group at C-2 (36·HCl and 37·HCl) caused a substantial shift in the Ep.a by about +300 mV.277
Comparisons of anodic peak potentials (Ep.a) of flupirtine and analogs, and EC50 values on Kv7.2/7.3 channels in HEK293 cells.
| Compound | E p.a [mV] | EC50a [μM] |
|---|---|---|
| Flupirtine | 350 | 3.6 |
| RTG | 299 | 1.9 |
| 29 | 652 | >100 |
| 31 | 612 | >100 |
| 32 | 294 | 1.9 |
| 33·HCl | 320 | 2.6 |
| 34·HCl | 385 | 3.5 |
| 35 | 667 | >100 |
| 36·HCl | 642 | >100 |
| 37·HCl | 653 | >100 |
EC50 values were obtained in HEK293 cells overexpressing Kv7.2/7.3 channels.
These analogues were tested for their modulator activity (EC50) against Kv7.2/7.3 channels using a fluorescence-based thallium-flux assay in HEK293 cells (Table 5).277 Among all flupirtine derivatives, compounds with methyl substitution at N-2 and N-6 (32, 33, 34) acted as Kv7.2/7.3 channel openers, whereas analogs with N-carbamate substitutions were ineffective at opening channels at up to 100 μM concentration. The C-2 deaminoflupirtine derivatives 36·HCl and 37·HCl were notably less active than flupirtine maleate. Thus, the correlation between Ep.a and EC50 values signified that more easily oxidized (with lower Ep.a values) flupirtine derivatives were more potent than those that were less easily oxidized. Based on hepatotoxicity (TAMH cells) and cytotoxicity (MTT assay) studies, it was concluded that there was no direct correlation between Ep.a and LD50 values. Therefore, Kv7.2/7.3 channel opening activity and hepatotoxicity are likely not linked.277
Many recent studies of flupirtine analogs have focused on improving the safety profile of this chemotype. Link and co-workers proposed that the ease of oxidation of the electon-rich arene in flupirtine as well as RTG could be reduced by replacing the secondary amine with a thioether linkage.86,278 By removing one of the three nitrogen atoms connected to the arene, the primary oxidation was likely to occur at the sulfur atom, converting it to a sulfoxide or sulfone, and, thus, an electron-withdrawing group that further decreased electron density on the conjugated benzene ring. Also, a sulfoxide or sulfone metabolite should be less electrophilic and potentially less toxic than a quinone-imine. A series of sulfides were prepared in order to test their modulator activity on Kv7.2/7.3 channels as well as toxicity. Furthermore, the Ep.a of these analogs was calculated from cyclic voltammetry data to predict a correlation between ease of oxidation and activity.
Nucleophilic substitution of the 6-chloro group in 6-chloro-3-nitro pyridine 38 with a thiol was followed by alkylation of 39 with a variety of alkyl bromides under basic conditions to provide 40 (Scheme 8).92 Subsequent reduction of the nitro group followed by selective carbamoylation afforded flupirtine thioether derivatives 41.
Scheme 8. Synthesis of sulfide analogs of flupirtine.

In the RTG series, the preparation of sulfide analog 49 started from commercially available 2-fluoro 4-bromo aniline 42 (Scheme 9). Oxidation of the aromatic amine to the nitro-compound 43 followed by selective replacement of the fluoride with NH3 in ethanol provided 44. Introduction of a thiol into 44 was problematic as the disulfide 45 was formed as the major product. Alkylation of thiol 46 with 4-fluorobenzyl bromide provided thioether 47. Reduction of the S–S bond using NaBH4 followed by subsequent S-alkylation also gave 47 in good yield. Di-Boc protection of the primary amine followed by nitro group reduction, carbamoylation with ethyl chloroformate and final Boc-deprotection afforded 49.
Scheme 9. Synthesis of an RTG thioether analog.

Thioether analogues were expected to be metabolically labile and undergo oxidation to sulfoxide and sulfones catalyzed by flavin monooxygenase (FMO) or CYP P450 enzymes. In order to test the activity and toxicity profile of the putative metabolites, a variety of sulfoxide and sulfones were also prepared from the corresponding sulfides by mCPBA oxidation.
A fluorescence-based thallium-flux assay in HEK293 cells with overexpressed Kv7.2/7.3 channels was used to determine the potency (EC50) of these modulators. The efficacy of the analogues was calculated relative to the maximal flupirtine-induced fluorescence signal. RTG was found to be six times more potent than flupirtine (Table 6). The sulfide analogue 41a was slightly less potent than flupirtine (EC50 = 1.28 μM vs. 0.92 μM) but had similar efficacy. In analogy to the data reported by Kumar et al.,279 if substituents at the pyridine core were modified, for example, replacing the fluoro group with a trifluoromethyl or pentafluorosulfanyl group (41b, 41c), the potency increased up to six-fold; the main drawback of 41c was its lower efficacy (78%).
Comparison of sulfide analogs 41a–i and 49–52 to flupirtine and RTG.
| Compound | R1 | R2 | R3 | EC50a [μM] | Efficacyb (%) |
|---|---|---|---|---|---|
| Flupirtine | 0.92 | 100 | |||
| RTG | 0.25 | 134 | |||
| 41a | NH2 | 4-Fluorobenzyl | Ethoxy | 1.28 | 93 |
| 41b | NH2 | 4-Trifluoromethylbenzyl | Ethoxy | 0.24 | 115 |
| 41c | NH2 | 4-(Pentafluoro-λ6-sulfanyl)benzyl | Ethoxy | 0.24 | 78 |
| 41d | NH2 | 1.1′-Biphenyl | Ethoxy | 0.25 | 69 |
| 41e | NH2 | 4-Fluorobenzyl | Butyl | 2.13 | 140 |
| 41f | NH2 | 4-Fluorobenzyl | Isobutyl | 4.54 | 118 |
| 41g | 4-Morpholinyl | 4-Fluorobenzyl | 3,5-Difluorobenzyl | 0.0009 | 127 |
| 41h | 1-Pyrrolidinyl | Isobutyl | (3,5-Difluorophenyl)ethyl | 0.0014 | 136 |
| 41i | CH3 | 4-Fluorobenzyl | Ethoxy | 0.27 | 129 |
| 49 | 0.17 | 128 | |||
| 50 | 0.45 | 72 | |||
| 51 | >10 | — | |||
| 52 | 1.02 | 122 |
EC50 values were obtained in HEK293 cells overexpressing Kv7.2/7.3 channels.
Calculated percentage relative to the maximum flupirtine-induced fluorescence signal.
Introduction of a second phenyl group (41d) increased potency but reduced efficacy. However, addition of more hydrophilic groups than 4-fluorobenzyl, e.g. pyridinomethyl and piperidinoethyl, led to inactive analogs.
The carbamate moieties were also exchanged with amide groups. Amides derived from pentanoic acid (41e) and isopentanoic acid (41f) yielded products with potencies in the low micromolar range (EC50 = 2.13–4.54 μM) (Table 6).
However, sulfide 41g containing a 3,5-difluorophenylacetic acid amide and a morpholine moiety was highly potent on Kv7.2/7.3 channels, with an EC50 = 0.0009 μM and high efficacy (127%). When the 3,5-difluorophenylacetic acid amide part was kept constant and the remainder of the molecule was altered, a positive correlation between the lipophilicity and the channel opening activity was found, with 41g continuing to represent the most potent analog as well as the one with the highest lipophilic character (log D7.4 = 4.7). Extending the amide side chain by a methylene group, followed by substitution of the 4-fluorobenzyl residue with an isobutyl group and replacement of the NH2 group by pyrrolidine (Table 6), yielded another very potent compound, 41h (EC50 = 0.0014 μM).86
Since the 5-position of the pyridine ring was found to be susceptible to substitution reactions with endogenous proteins or peptides, it was blocked in analogs containing methyl (50) or bromo (51) substituents, or by replacement of the pyridine with a pyrimidine heterocycle (52) (Chart 6).
Chart 6. Flupirtine sulfide, sulfoxide and sulfone analogues.

For analog 50, the potency doubled (EC50 = 0.45 μM), whereas the efficacy decreased (72%). A stronger effect was observed after shifting the methyl group to the 2-position, replacing the amino group (41i, EC50 = 0.27 μM, efficacy 129%). In contrast, the 5-bromo analog 51 was completely inactive, probably due to the steric bulk and the electron-withdrawing effects of the bromine substituent. Just as with the conversion of flupirtine to RTG, replacing the pyridine with a phenyl ring in 49 improved the potency about eight-fold, while the pyrimidine-substituted sulfide analog 52 remained moderately active (Table 6). A variety of sulfoxide (53) and sulfone (54) analogs were also prepared to obtain information about the biological activity of the putative metabolites. However, none of these showed detectable channel opening activity at concentrations up to 10 μM.86
The toxicity of these compounds was analyzed in an MTT assay with two cell lines of hepatic origin, TAMH and Hep-G2.
Due to the poor solubility of the compounds in water, the toxicity was expressed as an LD25 value (concentration which decreased cell viability to 75%). Even though a general trend in toxicity was not found after replacing the secondary amine with the sulfide, oxidizing the sulfides to the sulfoxides or sulfones led to lower toxicity in the hepatic cell lines.86
Homology modeling of Kv7.2 and Kv7.3 based on the structure of the crystallized Kv1.2/2.1 channel97,101 placed the 4-fluorobenzyl moiety and the carbamate residues in extended hydrophobic pockets, with Leu and Phe residues representing the predominant binding site species. This observation was consistent with the experimental data that confirmed that the exchange of the benzyl motif for the more hydrophilic pyridyl or piperidyl scaffolds diminished Kv7 channel opening activity. The model suggested the presence of a H-bond between the primary amino group of 41a and Leu299 of Kv7.2. In addition, hydrogen bonds between the hydroxy group of Ser303 and the primary amine as well as the carbonyl moiety of 41a were also found. The docking simulations of 41h exhibited a different binding mode compared to the carbamate derivatives (e.g.41avs.41h). The preferred pose was found to be a flipped orientation of the molecule, with the amide chain pointing toward Leu314 and the small sulfide chain being positioned in a larger cleft near Phe254.86
Using a retro-metabolic drug design approach, new analogues of flupirtine and RTG were synthesized in five iterative cycles to improve pharmacological activity while reducing liver toxicity.91 Replacement of the primary amino group with a morpholine or a bulky ether group dramatically increased the Kv7.2/7.3 channel opening activity. Replacement of the ethyl carbamate with substituted aromatic groups also increased positive channel modulation. An amide at the 3-position of the central pyridine ring was most effective in increasing activity. Among these analogs, 55, 56, 57 and 58 were the most potent positive modulators of Kv7.2/7.3 channels (Chart 7).
Chart 7. Structures of flupirtine/RTG analogues 55–58 and Lu AA41178.

Analog 57 was highly potent (EC50 3.7 nM) and demonstrated a superior efficacy of 165% (Table 7), but also resulted in higher hepatotoxicity than flupirtine. The dimorpholino analog 58 was found to be the most balanced compound in terms of lipophilicity (log D7.4 = 3.58), water solubility, and hepatoxicity profile in both TAMH (LD25 = 212 μM) and HEP-G2 (LD25 = 231 μM) cell lines.
Potency and efficacy of flupirtine and RTG and selected analogs.
| Compound | EC50a [μM] | Efficacyb (%) |
|---|---|---|
| Flupirtine | 0.918 | 100 |
| RTG | 0.147 | 134 |
| 55 | 0.0038 | 115 |
| 56 | 0.0042 | 154 |
| 57 | 0.0037 | 165 |
| 58 | 0.0113 | 111 |
EC50 values were obtained in HEK293 cells overexpressing Kv7.2/7.3 channels.
Calculated percentage relative to the maximum flupirtine-induced fluorescence signal.
Recently, Grupe et al. reported a related flupirtine analog, morpholine Lu AA41178, as a potent pan-opener of Kv7.2–Kv7.5 (EC50 range of 3–10 μM for increasing currents at −40 mV potential) without binding to common cardiac ion channels or effecting a positive modulation of GABAA channels (Chart 7).280 At minimum effective plasma levels of <1000 ng mL−1 and a brain/plasma ratio of 1.2, Lu AA41178 demonstrated efficacy in assays of epilepsy, schizophrenia and depression.
In 2008, Vernier et al. disclosed a series of RTG analogs bearing indoline and indole substituents that acted as Kv7 activators.281 However, detailed pharmacology, metabolism and photostability studies of these conformationally restricted compounds were only more recently reported by Ostacolo et al.282 SAR studies were performed on three different types of analogs; indolines (59), indoles (60) and tetrahydronaphthalenes (61) (Chart 8).
Chart 8. Structures of RTG and analogs 59, 60 and 61.

The activity of these analogs on Kv7.2 channels was studied using a whole-cell patch clamp electrophysiology technique in CHO cells transiently transfected with Kv7.2 cDNA. Ramp-evoked currents were measured at both −40 and 0 mV membrane potentials (at 10 μM compound concentration) to predict a wider spectrum of pharmacological effects on neuronal excitability. In the indoline series, although compounds 62a–d were able to increase Kv7.2 currents more significantly than RTG at 0 mV, no analog was improved over RTG at −40 mV. Interestingly, in the indole series, 63 and 64 (Scheme 10) were able to increase Kv7.2 currents more strongly than RTG at 0 mV as well as at −40 mV. It was suggested that the slower on/off kinetics on Kv7.2 of compounds 63 and 64 compared to RTG might be a consequence of their increased hydrophobicity.
Scheme 10. Synthesis of indoline and indole analogs of RTG.

The synthesis of indolines 62 utilized a reductive amination of 5-nitro indoline (65a) with appropriately substituted benzaldehydes to give intermediates 66 (Scheme 10).
Continuous flow hydrogenation of the nitro group in 66 followed by sulfonation of the amine 67 afforded indolines 62a–c. Due to the lability of the 2-nitrobenzyl group under hydrogenation conditions, analog 62d was synthesized by a different synthetic route where hydrogenation was performed prior to the reductive amination step. The indole amide analogues 63 and 64 were prepared from 5-nitroindole (69) by alkylation with 4-trifluoromethyl benzyl bromide in the presence of a strong base, followed by reduction and amidation with the appropriate acyl chloride (Scheme 10). Compound 63 proved to be 12 times more potent than RTG in activating Kv7.2 current, while 64 was equipotent to RTG (Table 8).
EC50 of RTG, 63 and 64 at different Kv7 channels.
| Subtype channel | EC50a (μM) | ||
|---|---|---|---|
| RTG | 63 | 64 | |
| Kv7.2 | 0.93 ± 0.43 | 0.08 ± 0.04 | 0.63 ± 0.07 |
| Kv7.2/7.3 | 1.72 ± 0.41 | 0.21 ± 0.14 | 0.34 ± 0.06 |
| Kv7.3 | 3.90 ± 1.70 | 10.5 ± 6.00 | 0.92 ± 0.64 |
| Kv7.4 | 1.98 ± 0.84 | 0.10 ± 0.02 | 0.08 ± 0.20 |
| Kv7.5 | 0.80 ± 0.24 | 1.07 ± 0.12 | 0.35 ± 0.14 |
EC50 values were obtained in CHO cells overexpressing Kv7 channels.
At steady state and in 0.03 μM concentration, indole amide 63 doubled Kv7.2 maximal conductance and caused a leftward shift of about 18 mV in the V1/2 of Kv7.2 currents, while 0.03 μM RTG failed to show this effect. It was also suggested that Trp236 in the S5 transmembrane domain on Kv7.2 exerts a critical influence on binding 63 and 64 within the hydrophobic pore pocket, similar to RTG binding at this site. Compounds 63 and 64 were also more potent than RTG on heteromeric Kv7.2/7.3 and homomeric Kv7.4 channels, whereas only 64 was more potent than RTG in activating homomeric Kv7.5 channels (Table 8). In conclusion, the subtype selectivity profile for 63 was Kv7.2 = Kv7.4 = Kv7.2/7.3 > Kv7.5 ≫ Kv7.3 ≫ Kv7.1, while for 64 it was Kv7.4 > Kv7.5 = Kv7.2 = Kv7.2/7.3 = Kv7.3 ≫ Kv7.1. Finally, the tetrahydronaphthalene 61 was completely inactive at Kv7.2 channels.
Pharmacophore models of sulfonamide 62a and amide 64 contained the following features: a hydrogen bond acceptor (HBA), a hydrogen bond donor (HBD), a common hydrophobic region (HY1), and two aromatic regions (Ar1 and Ar2); HY2 (for 64) and HY3 (for 62a). HY2 and HY3 would interact with a larger, flexible pocket on Kv7.2, where flexible ligands could bind in different conformations while interacting with additional amino acid residues. Homology modeling of the Kv7.2 region also supported the presence of two hydrophobic pockets surrounding two sides of Trp236 that could be the counterpart of HY1 and HY2/3 on the channel protein.
In a photostability assay, sulfonamides 62a–c were found to be less stable than RTG under exposure to natural light. After 6 h, the degradation of 62a–c was almost complete (by HPLC), whereas for RTG the degradation was about 47%. This faster degradation could be due to the sulfonyl activation of the p-quinone diimine intermediate,283 representing the starting point in the degradation toward RTG-like byproducts. Finally, the amide derivatives 63 and 64 were the most photostable RTG analogs, showing less than 10% degradation after a 6 h time course. In in vitro metabolism assays using S9 fraction human liver microsomes, compound 63 was found to undergo extensive (48.7 ± 2.5%) phase I metabolism compared to RTG (3.1 ± 0.2%).279 With decreasing lipophilicity of the side chain, analog 64 gained an improved phase I metabolic stability (17.3 ± 1.7% degradation). Promisingly, compound 64 also displayed increased stability toward phase II metabolism (in UGT and NAT activity assays) compared to RTG.
After pioneering studies by Fried and Sabo in 1954 on CH–F substitutions in corticosteroids,284 the fluorine atom has increasingly been employed as a bioisosteric replacement for the hydrogen atom. Fluorine distinguishes itself by chemical inertness, ability to modulate physicochemical parameters such as acidity and lipophilicity, and increased metabolic stability.285,286RTG analogs NR561-29, NR561-45 and RL673-02 featured an SF5-function in zone 1 of the pharmacophore model, a new bioisostere of the CF3O- and CF3-groups that sterically resembles a tert-Bu-group but has similar electron-withdrawing properties to a NO2-group (Charts 9 and 10).287,288
Chart 9. Highly fluorinated analogs of RTG.

Chart 10. Zone SAR model of RTG analogs.
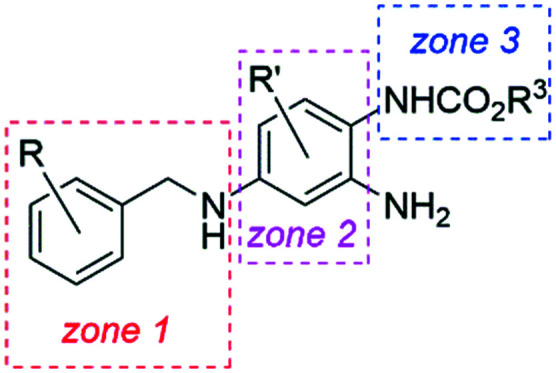
A POC (proof-of-concept) SAR study explored the CF3-substituted analogs NR561-40, NR561-50, NR579-04, RL648-81 (RL-81), RL648-73, RL702-18, and RL740-73 to determine the effects of fluorination on positive Kv7 channel modulation. Additionally, in zone 3, the ethyl group was replaced with iso-propyl (RL702-32) and cyclo-propyl (RL702-56, RL844-36, RL880-12). Finally, pyridines RL702-24 and RL880-12 were representative selections for fluorinated heterocycles in zone 1.270
The synthesis of these compounds followed the general experimental strategies summarized in Scheme 11. For most of the target compounds, a reductive amination was used to link the benzylic amine with the aniline moiety to give intermediate 72. After selective introduction of the benzyloxycarbonyl (Cbz) protecting group, the o-nitroaniline 73 was acylated and the Cbz protecting group was removed concomitantly with the reduction of the nitro group to generate the desired analogs.
Scheme 11. Reductive amination approach to fluorinated RTG analogs.

In an alternative approach, suitably substituted benzylic amines were added to para-nitrofluorobenzenes under SNAr conditions, followed by reduction of the nitro groups with zinc and ammonium chloride, and selective N-acylation of the resulting para-aminoaniline 75 (Scheme 12). In a late-stage modification, RL648-81 was treated with Togni's trifluoromethylating agent in acetonitrile to obtain the bistrifluoromethylated RL740-73.
Scheme 12. SNAr approach to fluorinated RTG analogs.

Pyridine-containing bioisosteres in zone 1 were prepared in an SNAr fashion from the corresponding pyridyl alkylamines and fluorinated anilines. For example, condensation of (3-fluoro-5-(trifluoromethyl)pyridin-2-yl)methanamine with 2,3,5-trifluoro-6-nitroaniline in the presence of triethylamine and catalytic iodine, followed by reduction of the nitro group with zinc and selective carbamoylation, provided RL880-12 (Scheme 13).
Scheme 13. Preparation of 2-pyridylmethylamine analog RL880-12.

The effect of the fluorinated analogs, including RTG and flupirtine as positive standards, on Kv7 channel activation was tested as part of the Lilly Open Innovation Drug Discovery (OIDD) program. Ion channel agonistic potency was determined in the automated electrophysiology IonWorks Barracuda (IWB) HTS platform in HEK293 cells (Table 9).289 The resulting 10-point concentration–response curves provided a measure of compound efficacy; the agonist concentration that doubled the conductance at the voltage leading to 15% channel activation was expressed as EC2×. Therefore, the lower the value of EC2× for a particular compound, the higher its efficacy as a positive channel modifier. Compared to flupirtine and RTG EC2×, RL648-81 demonstrated ca. 8–50 times higher (vs.flupirtine) or slightly higher (vs.RTG) efficacy at all tested Kv7 channels in this assay. A meta-positioned CF3 isomer, RL648-73, was twice as potent at Kv7.2/7.3, with substantially decreased activity at Kv7.4/7.5 vs.RL648-81. The corresponding SF5-analog, RL673-02, acted indeed as a bioisostere of the CF3-analog RL648-73, showing a very similar channel activity profile. Interestingly, installing a CF3-group at the aniline ring in RL740-73 completely abrogated activity at all channel types. This position appears to be very sensitive to steric bulk in the Kv7.2 channel pore region. The hepta-fluorinated RL844-50 also lost all activity at these channels. Slightly reducing the steric bulk next to the carbamate, such as in the hexa-fluorinated RL844-36, increased channel subtype selectivity, since this compound still lacked agonist activity at Kv7.3/7.5, Kv7.4, and Kv7.4/7.5, but now demonstrated a sub-micromolar EC2× (0.93 μM) at Kv7.2/7.3. Supporting this SAR trend, the 2-fluoro-4-trifluoromethylbenzylamine analog RL702-18 showed an EC2× = 0.18 ± 0.11 μM, reflecting high potency. In most cases, changing the ethyl to the cyclo-propyl carbamate increased potency, and, in particular, selectivity of the analogs, i.e.RL702-32. RL702-56 was the most active analog on Kv7.2/7.3 prepared in this series, with an EC2× of 0.11 ± 0.02 μM.
Efficacy of fluorinated RTG analogs in human Kv7 channel conductance studies.
| Analog | Kv7.2/7.3 | Kv7.3/7.5 | Kv7.4 | Kv7.4/7.5 |
|---|---|---|---|---|
| EC2× ± SD [μM] | EC2× ± SD [μM] | EC2× ± SD [μM] | EC2× ± SD [μM] | |
| Flupirtine | 2.00 ± 0.69 | 1.58 ± 0.69 | 5.62 ± 1.86 | 3.50 ± 2.68 |
| RTG | 0.33 ± 0.08 | 0.34 ± 0.04 | ND | ND |
| RL648-81 | 0.26 ± 0.13 | 0.29 ± 0.06 | 0.10 ± 0.04 | 0.09 ± 0.04 |
| RL648-73 | 0.14 ± 0.05 | 0.20 ± 0.08 | 0.40 ± 0.13 | 0.66 ± 0.01 |
| RL673-02 | 0.36 ± 0.25 | 0.21 ± 0.11 | 0.65 ± 0.39 | 0.65 ± 0.28 |
| RL648-73 | >10 | >10 | >10 | >10 |
| RL702-32 | 0.16 ± 0.11 | 0.16 ± 0.10 | 0.19 ± 0.10 | 4.98 ± 1.12 |
| RL702-56 | 0.11 ± 0.02 | 0.23 ± 0.10 | 0.38 ± 0.30 | 0.28 ± 0.03 |
| RL702-18 | 0.18 ± 0.11 | 0.34 ± 0.01 | 0.11 ± 0.04 | 0.30 ± 0.26 |
| RL844-36 | 0.93 ± 0.21 | >10 | >10 | >10 |
| RL880-12 | 1.00 ± 0.50 | >10 | >10 | >10 |
| RL702-24 | 0.61 ± 0.34 | 0.69 ± 0.35 | 0.95 ± 0.13 | 1.27 ± 0.05 |
Trifluoromethylated pyridines mainly exhibited lower selectivity or lower potency. The trifluorinated RL880-12 remained an effective agonist with a 1.00 μM EC2× at Kv7.2/7.3, and high subtype selectivity; the 3-pyridyl analog RL702-24 was essentially equipotent, thus suggesting that the fluorination pattern and not the core arene in zone 1 was the dominant determinant of selectivity and activity.270
The effects of fluorine substitution on analog activity were analyzed computationally as a function of the electron density at the carbamate oxygen and the ortho-aniline nitrogen (Fig. 5). The electron-density surface encoded with the electrostatic potential of RL648-81vs.RL844-50 showed a significantly decreased electron density at both sites as a result of the two additional fluorine atoms in RL844-50. Accordingly, it was hypothesized that Kv7.2–Kv7.5 channel potency was strongly influenced by electrostatic and H-bonding interactions at the carbamate oxygen and the ortho-aniline, with decreased electron density being detrimental and therefore leading, in the case of RL844-50, to an inactive analog. Based on mutation and modeling studies, the tryptophan residue W236 in Kv7.2 (or, analogously and respectively, W265, W242, W235 in Kv7.3, Kv7.4, Kv7.5) was thought to be critical for binding, and this residue was likely to participate in H-bonding interactions with carbamate or amide groups of these agonists (Fig. 6).
Fig. 5. Electron-density surface encoded with electrostatic potential for RL648-81 and RL844-50. Colors reflect a property range of +170 kJ mol−1 (blue) to −310 kJ mol−1 (red).

Fig. 6. Docking model of RL648-81 in the Kv7.2 channel pore region. PyMOL 2.3.3 generated hydrophobic regions are shown as yellow/orange patches on the protein surface, and hydrophilic regions are green and blue (see also Fig. 7).

It should also be noted that RL648-81 (log P = 3.4) is more lipophilic than RTG (log P = 2.5), which, due to the highly hydrophobic nature of the pore region, might be another factor why RL648-81 proved to be a stronger agonist than RTG.270
5.2. Acrylamide series
In addition to RTG and flupirtine analogs, many other positive Kv7 modulators have been reported in the literature. Bristol Myers Squibb applied a high-throughput screening (HTS) assay using thallium(i) influx detection to identify acrylamide 78 as a lead candidate. Further optimization led the acrylamide so called (S)-1 as an orally bioavailable Kv7.2 opener (Chart 11).290
Chart 11. Structures of acrylamides (S)-1, BMS-568274 and selected analogs.

Coupling of 3-phenylacrylic acid (83) with (S)-1-(3-methoxyphenyl)ethylamine 84 in the presence of the EDAC (EDCI) carbodiimide coupling reagent provided amide 85 (Scheme 14). BBr3-Mediated demethylation followed by conversion of the resulting phenol to a triflate and subsequent Pd-catalyzed coupling with morpholine afforded acrylamide (S)-1.
Scheme 14. Synthesis of acrylamide (S)-1.

A two-electrode voltage clamp technique was used to determine the effect of (S)-1 on the outward potassium current at a single membrane voltage (−40 mV) in Xenopus oocytes expressing cloned mouse Kv7.2 (mKv7.2) channels. Based on a concentration–response curve, an EC50 value of 6.0 μM on mKv7.2 channels was determined. To rule out any oocyte-specific endogenous expression of other Kv7 subtypes,291(S)-1 was also examined in HEK293 cells expressing mKv7.2 channels, and the obtained EC50 value (3.3 ± 0.6 μM) was in general agreement with the prior data in the oocyte expression system.
Similar to RTG on hKv7.2 channels expressed in oocytes, current–voltage (I–V) relationship studies (−100 to +40 mV) on compound (S)-1 showed that it produced a significant increase in outward current from approximately −70 to +10 mV along with a hyperpolarizing shift in the threshold for current activation. At higher voltage, it resulted in a decrease in the amplitude of the currents. Similarly, compound (S)-1 inhibited mKv7.2 conductance at more positive test potentials in HEK293 cells (≥15 mV), indicating a “crossover” voltage in the conductance–voltage relationship curve.
Later, Olesen and co-workers found that the combined activation/blocking of Kv7.2 and Kv7.2/7.3 by (S)-1 in Xenopus oocytes was strongly voltage-dependent, whereas application of (S)-1 on Kv7.4 and Kv7.5 channels profoundly increased the current amplitude at all voltages.98 Moreover, contrary to the positive effects on the Kv7.2–Kv7.5 channels, (S)-1 inhibited the currents through Kv7.1 and Kv7.1/KCNE1 channels. Similar to RTG, the tryptophan residue 242 in the fifth transmembrane segment S5 was shown to be crucial for the positive modulating effect of (S)-1 on Kv7.4 channels expressed in Xenopus oocytes.
In vivo studies using a cortical spreading depression (CSD) rat model of migraine found that (S)-1 significantly reduced the incidence of cortical depolarizations as well as depolarizing stimuli. The oral bioavailability of (S)-1 was excellent in both rat and dog; 93% and 100%, respectively. Acrylamide (S)-1 was also found to have moderate brain penetration (B/P = ∼0.8) in both rat and dog.290
SAR studies of (S)-1 covered the acrylamide pharmacophore, introducing substitutions (fluorine, methoxy) at the aromatic rings as well as changing the 3′-amino substituent to 2,6-cis-dimethylmorpholinyl, homomorpholinyl, dimethylamino, 4-methyl-piperazin-1-yl, etc., led to an improved set of derivatives (Chart 12).292 This study further indicated the need for the (S)-configuration at the (1-phenyl)ethyl moiety and the α,β-unsaturated acrylamide functionality with a free N–H on the acrylamide.
Chart 12. Structures of acrylamides (S)-2, (S)-3, (S)-4, (S)-5, (S)-6, (S)-7, (S)-8, and SMB-1.

In vitro isoform-selective chemical inhibition studies in human liver microsomes revealed that (S)-1 was metabolized by CYP3A4, a major isoform of CYP P450, to produce a reactive intermediate which covalently bound to the active site of the enzyme, leading to irreversible metabolism-dependent inactivation (MDI). Structure CYP3A4-MDI relationship studies were then carried out to identify the possible site(s) responsible for the generation of the reactive intermediate during metabolism of (S)-1.290
Reduction of the α,β-unsaturated double bond (80) or introduction of an aryl fluoride (79a–c) did not eliminate the CYP3A4-MDI activity. Since the initial lead compound 78 did not show CYP3A4-MDI activity, the morpholine group was most likely responsible for the formation of reactive quinone-type intermediates such as 81 or 82via hydroxylation at C-4′ of the electron-rich N-phenylmorpholine moiety (Chart 11). In order to block this potential hydroxylation, the H-atom at C-4′ was exchanged to a fluoro group, and a series of 4-fluoro acrylamides were prepared (Chart 11). Among them, BMS-568274 was found to have comparable Kv7.2 opener activity (EC50 1.2 μM in HEK293 cells) to (S)-1 (EC50 3.3 μM), and lacked CYP3A4-MDI activity.293
In a subsequent pharmacokinetics (PK) study, the oral absorption of BMS-568274 in rats was found to be fast, with a peak concentration within 0.7 h. Upon i.v. dosing, the compound displayed a moderate plasma clearance and a relatively long elimination half-life. The oral bioavailability of BMS-568274 was determined to be 84% in rats. BMS-568274 also demonstrated significant in vivo activity in a cortical spreading depression model of migraine.293
The concept of restricting bond rotation to improve potency has been widely used in medicinal chemistry. In order to increase the potency of (S)-1 and according to this principle, the morpholine group was fused into the aromatic ring, restricting rotation of the C3′–N bond while decreasing the overall entropy of the resulting analog (Chart 11). This strategy led to the identification of (S)-2 as a potent and efficacious Kv7.2 opener (Chart 12).294
Further exploration of bioisosteric replacements of the 3,4-dihydro-2H-benzo[1,4]oxazine moiety of (S)-2 led to the identification of acrylamide (S)-3 as highly potent Kv7.2 opener, as well as acrylamides (S)-4 and (S)-5 as highly efficacious analogs (Chart 12).295
Additional optimizations of both potency and efficacy proved to be more challenging, as the newly developed acrylamide (S)-6 was twice as potent as (S)-3 but less efficacious. After extensive SAR studies, the two biaryl ether acrylamides (S)-7 and (S)-8 were identified to meet the desired potency and efficacy criteria (Table 10).296
Whole-cell patch-clamping data (−40 mV) for acrylamide analogs.
| Compound | EC50a (nM) | E/Erefb |
|---|---|---|
| (±)-(S)-3 | 72 ± 2 | 1.0 ± 0.1 |
| RTG | 1300 ± 40 | 1.6 ± 0.1 |
| (S)-1 | 3200 ± 600 | 1.4 ± 0.1 |
| (S)-2 | 60 ± 10 | 1.8 ± 0.1 |
| (S)-3 | 20 ± 1 | 1.1 ± 0.1 |
| (S)-4 | 330 ± 10 | 2.4 ± 0.1 |
| (S)-5 | 430 ± 20 | 2.4 ± 0.2 |
| (S)-6 | 10 ± 0.1 | 0.85 ± 0.01 |
| (S)-7 | 2.4 ± 0.1 | 2.2 ± 0.1 |
| (S)-8 | 0.9 ± 0.1 | 2.9 ± 0.1 |
EC50 values were obtained in HEK 293 cells overexpressing Kv7.2 channels.
The efficacy of the test compound relative to (±)-(S)-3.
The synthesis of (S)-2 started with a reaction of hydroxylamine hydrochloride and commercially available 6-acetyl-4H-benzo[1,4]-oxazin-3-one (87) to form oxime 88 (Scheme 15). RANEY®-Ni reduction of the oxime followed by a reduction of the amide bond in (±)-89 formed diamine (±)-90. Coupling of the primary amine group of (±)-90 with 3-(2-fluoro-phenyl)acrylic acid led to acrylamide (±)-91 which after chiral HPLC resolution provided (S)-91. Alkylation of the secondary amine in (S)-91 with the appropriate alkyl halide furnished acrylamides (S)-2. Acrylamides (S)-3, (S)-4, (S)-5, (S)-6, (S)-7 and (S)-8 were synthesized using a similar strategy. SMB-1 was prepared from (S)-2 by a base-mediated alkylation of the amide NH with methyl iodide.
Scheme 15. Synthesis of acrylamide (S)-2 and SMB-1.

The modulatory effects of these acrylamides on the outward potassium current were tested using whole-cell patch clamp recordings from HEK293 cells stably expressing mKv7.2 channels. The rate control in Kv7.2 activation kinetics produced by (S)-2 was similar to (S)-1; however, unlike (S)-1, there was no significant inhibition of Kv7.2 current at positive membrane potentials. Further studies showed that (S)-2 also activated human homomeric Kv7.2–Kv7.5 and heteromeric Kv7.2/7.3 channels expressed in Xenopus oocytes.297
Unlike neuronal Kv7 channels, the homomeric Kv7.1 channels were weakly inhibited by (S)-2. Application of (S)-2 resulted in a decrease of the deactivation kinetics of Kv7.2 and Kv7.4 channels, which might indirectly influence Kv7 channels bound to (S)-2 to remain in the open state for a longer period of time upon activation.
Inactivation has been described as a key regulatory mechanism of Kv7.4 and Kv7.5 channels.298 Steady-state inactivation is operational for >30% of channels at physiologically relevant membrane potential. (S)-2 was found to reduce the inactivation of Kv7.4, presumably due to increased current amplitudes. However unlike RTG, apart from the pore site Trp residue, (S)-2 also appeared to interact with other residues. Surprisingly, by replacing the Trp residue with Leu, the mutant channel Kv7.2-W236L was inhibited by (S)-2 as seen in a reduction of current amplitude without affecting the voltage-dependence of activation. This suggested the existence of a subtype-specific additional inhibitory binding site, or a mode of binding not dependent on W236.295
After some SAR studies, the dioxolane (S)-3 (EC50 = 20 ± 1 nM) was determined to be a three-fold more potent analog of (S)-2. Moreover, a dihydrobenzofuran analog, (S)-4, and a dihydroindole derivative, (S)-5, showed improved efficacy over (S)-3, RTG, and even (S)-2 by approximately 140%, 50%, and 31%, respectively (Table 10). Finally, acrylamides (S)-7 and (S)-8 were very potent and highly efficacious openers of Kv7.2 channels. As shown in Table 10, (S)-7 was more potent than (S)-2 and RTG by a factor of 25 and 542, respectively. Acrylamide (S)-8 was the most potent analog identified in this series, with an EC50 in the 1 nM range. In terms of efficacy (E/Eref value), compound (S)-7 was 22% and 38% more efficacious in opening Kv7.2 channels than (S)-2 and RTG, respectively. Furthermore, compound (S)-8 was also 33% more efficacious than (S)-7 (Table 10).
Compounds (S)-2, (S)-4 and (S)-5 demonstrated significant reduction of neuronal hyperexcitability in rat hippocampal slices. (S)-7 was also efficacious in attenuating NP in a spinal nerve ligation (Chung model)299 and a diabetic neuropathy model.300
In order to develop subtype-specific Kv7 based analogs, a methylated version of (S)-2, SMB-1, was synthesized (Chart 12).301(S)-2 was found to act as an inhibitor of Kv7.2 channels (IC50 = 7.4 μM in Xenopus oocytes), which suggested that upon methylation of the amide nitrogen the activating function of (S)-2 was disabled, while its inhibitory component was preserved. SMB-1 produced a weak activation of Kv7.4 currents compared to (S)-2, without affecting the voltage-dependence activation of Kv7.4. The EC50 value was measured to be 5.7 μM in Xenopus oocytes expressing Kv7.4 channels. In binding studies with mutant channel proteins, Kv7.2-W236L and Kv7.4-W242L, the activation of SMB-1 on Kv7.4 was found to be critically dependent on the Trp residue in S5, while the inhibitory effect on Kv7.2 was independent of this residue. Moreover, in mutant Kv7.2-L275V channel proteins, the inhibitory efficacy of 10 μM SMB-1 was reduced, suggesting that binding to Kv7.2 also occurred at or close to this binding pocket.
5.3. Benzamides
A high-throughput screening campaign resulted in the identification of 3,4-dichloro-N-(pyridine-3-yl) benzamide (92), as a weak agonist (EC50 = 6 μM, Rb+ assay, CHO cells) at Kv7 channels (Chart 13).302,303
Chart 13. Structures of benzamides, ICA-069673 and analogs.

Subsequent SAR studies included modifications to the amide (reduction and alkylation), substitutions on both aromatic rings, as well as replacements of the substituted benzene with heterocycles, and led to the N-(6′-substituted pyridin-3-yl)-benzamide core 93. Evaluation criteria were based on selective potency at Kv7.2/7.3 versus Kv7.3/7.5 as well as Kv7.1 antagonist activity. For a difluorinated benzene analog (R3 = R4 = F), the optimal substituent at the 6′-position (X) of the pyridine was found to be a chlorine atom (ICA-27243, EC50 = 0.38 μM, Kv7.2/7.3) (Chart 1).303
When substituents on the benzene portion were varied, it was found that in general R2-substitution was not tolerated, whereas electron-withdrawing functions (e.g. Cl, F, CF3, except CN) at the R3/R4 positions had comparable activity to ICA-27243.
The most potent compound in this series was the 3-fluoro-4-chloro derivative 94 with a Kv7.2/7.3 EC50 = 0.08 μM, but 94 was also weakly active against Kv7.1 (IC50 = 15 μM). Compounds having a fluorine substituent on the R3-position and various heteroatom substituents at the R4-position of the benzene ring had Kv7.2/7.3 activity in the nanomolar to micromolar range. Among these, the most active derivative, 95 (R4 = OiPr), had an EC50 = 0.17 μM, while its Kv7.1 activity was >100 μM (Chart 13). In the pyridine series, ICA-27243 demonstrated the best overall in vitro and in vivo profile.
A typical synthesis of these compounds used 2-chloro-5-nitropyridine (96) to form the aminopyridine 97 after catalytic hydrogenation, and coupling with 3,4-difluorobenzoyl chloride provided ICA-27243 (Scheme 16).303ICA-069673 was prepared from 2-amino-5-nitropyrimidine (98), which, after conversion to the corresponding 2-chloro derivative using tert-butylnitrite followed by cupric chloride and subsequent nitro reduction using iron in acetic acid, resulted in 2-chloro-5-aminopyrimidine (99). Finally, amide coupling of 99 with 3,4-difluorobenzoylchloride provided ICA-069673.
Scheme 16. Synthesis of benzamides ICA-27243 and ICA-06973.

In toxicity studies, ICA-27243 was found to induce nonhemolytic anemia in rats and cynomolgus monkeys. A metabolite derived from the susceptibility of the 2-chloro-pyridine moiety toward SNAr reactions with endogenous nucleophiles was found to be responsible for this toxicity. A further search for a structurally different analog with a similar in vitro profile but devoid of toxicity led to the identification of the pyrimidine derivative ICA-069673 (Chart 13).303 This compound was 20-fold more selective towards Kv7.2/7.3 over Kv7.3/7.5 (Table 11) and was almost inactive in a panel of cardiac ion channels (hERG, Nav1.5, L type channels, and Kv7.1) and on GABAA gated channels at 10 μM. Even though ICA-069673 was less potent than ICA-27243, it was more efficacious and selective towards heteromeric Kv7.2/7.3 and did not engage Kv7.1. ICA-069673 also showed slightly improved activity over the pyridine analog ICA-27243 in rat MES and PTZ-induced seizure assays (Table 11). ICA-069673 was absorbed well and bioavailable (63%, Tmax 0.5 h; after 2 mg kg−1 i.v. administration t1/2 was 1.2 h, clearance was 1039 mL h−1 kg−1, and volume of distribution (Vz) was 1804 mL kg−1). After passing GLP safety studies, this compound was advanced to investigational new drug (IND) status and progressed into phase I clinical trials.303
Comparison of potency and antiseizure activity of ICA-27243/069673.
| Compound | Kv7.2/7.3 EC50a (μM) | Kv7.3/7.5 EC50a (μM) | Kv7.1 IC50 (μM) | Rat MES ED50 (mg kg−1) | Rat PTZ ED50 (mg kg−1) |
|---|---|---|---|---|---|
| ICA-27243 | 0.38 | ND | 41 | 1.5 | 2.2 |
| ICA-069673 | 0.69 | 14.3 | >100 | 1.5 | <1 |
EC50 values were obtained in CHO cells overexpressing Kv7 subtype channels. ND; not determined; MES: maximal electroshock seizure; PTZ: pentylenetetrazol.
In A7r5 vascular smooth muscle cells, ICA-069673 selectively activated Kv7.4 over Kv7.5 channels.11 Mutation of the Phe residue at position 143 to Ala in Kv7.4 significantly reduced the activity of ICA-069673. The mutant Kv7.4-F143A channel also caused a 9 mV negative shift of the Kv7.4 channel activation curve, thereby suggesting Phe143 was involved in voltage sensing.
In order to investigate the functional roles of Kv7.2- and Kv7.3-containing channels in guinea pig DSM excitability and contractility, ICA-069673 was used to inhibit contractions in DSM isolated strips in a concentration-dependent manner.235 Consistent with the inhibitory activity of ICA-069673, Ca2+ influx through L-type Cav channels was blocked, thereby causing relaxation of DSM isolated strips.235
The related benzamide derivative ICA-110381 was found to be a voltage-independent opener of Kv7 channels, with an efficacy for Kv7.2 > Kv7.4 > Kv7.3/7.5 in CHO cells (Chart 14).304
Chart 14. Structures of benzamides ICA-110381, ztz240 and analogs.

Based on the positive effect of ICA-110381 on the RTG-insensitive Trp mutant Kv7.2 W236L, it was suggested that the target binding site of ICA-110381 might be different from that of RTG. ICA-110381 showed anticonvulsive properties in amygdala-kindled rats by elevating the threshold of induction for afterdischarges (AD), and also reduced seizure severity and duration. In CA1 pyramidal cells, the IM current enhancement in the presence of ICA-110381 resulted in a damping of neuronal excitability by membrane hyperpolarization and a decrease in membrane resistance. ICA-110381 induced a significant enhancement of somatic resonance frequency on the single cell level without affecting the synaptic transmission.
In a high-throughput screen of 20 000 compounds from the ChemBridge Diverset™ library using the Rb+ flux assay, the closely related benzamide 5337031 was identified as a lead that potentiated homomeric Kv7.2 channels with an EC50 value of 9.8 ± 0.2 μM (Chart 14).305 A redesign of the lead was guided by a positional screen and a “Topliss scheme”, leading to the discovery of ztz240 (Chart 14). At 10 μM concentration, ztz240 significantly increased Kv7.2 currents with an EC50 value of 5.8 ± 0.9 μM and also showed noticeably slowed channel deactivation. Compound ztz240 potentiated Kv7.4 and Kv7.5 more strongly than Kv7.2 based on the increase in current amplitude. Similar to ICA-27243, the mutant Kv7.2 W236L channel was sensitive to ztz240. Further SAR studies on ztz240 were carried out to identify critical pharmacophore features responsible for the decrease in deactivation kinetics. The Tl+ assay results revealed that a nitrogen atom at the 3-position was important for this activity. Fluorine substitution at the 4′-position contributed more highly than at the 3′-position toward the slow-down of the deactivation current. Also, a chlorine or bromine atom at the 4-position of the pyridine ring played an important role in the left shift of V1/2 and also in mediating slow deactivation kinetics, which implied that the two modes of potentiation were due to different substituent effects.305
A comprehensive study involving electrophysiology, mutagenesis, homology modeling, molecular docking, and molecular dynamics simulation was conducted to determine the pocket and binding mode of ztz240 at Kv7.2 channels. The mutagenesis studies suggested that the binding pocket of ztz240 was located at the gating charge pathway of the VSD.306 Further electrophysiological experiments also supported ztz240's attachment to the open-state VSD. F233 was a key residue of the charge-transfer center in the Kv1.2–2.1 chimera.306,307 Similarly, the corresponding conserved F137 in Kv7.2 was suggested to be present at the occluded site of the gating charge pathway. The aromatic moieties of ztz240 may have hydrophobic interactions with F137. Indeed, a mutation of Kv7.2's F137 to Ala (F137A) significantly decreased ztz240's activity. In addition, the Kv7.2 F137A mutation did not affect the activity of other activators, such as RTG, ZnPy and NH29, which suggested that F137 was critical for activity of ztz240. Additional functional mutations, E130A, I134A, G138A, R207W and R210A, also significantly reduced the potentiation activity of ztz240, indicative of the presence of multiple essential residues in VSD responsible for ztz240 activity. The binding ability of the Kv7.2 VSD for chemical ligands suggested that the gating charge pathway might be structurally different from other Kv channels. Particularly, the discordant phenotypes of the F233X and F137X mutations indirectly supported a different gating charge pathway at this charge-transfer center.306 A recent cryo-EM structure of human Kv7.2 in complex with ztz240 confirmed that this compound activated the channel through a different mechanism and binding mode than RTG.115 In fact, ztz240 bound at the VSD and directly stabilized it in the activated state, whereas RTG bound at the pore domain and activated Kv7.2 by allosteric modulation.
Electrophysiological data obtained for two derivatives of ztz240, YGP1 and YGP2, also suggested that ztz240 preferably bound to the VSD in a conformation placing the chloropyridine at the bottom of the pocket and the fluorophenyl portion at its entrance. The ligand-VSD interactions were predicted by MD simulations, suggesting a hydrophobic interaction between ztz240, I134 and F137. The linker amide nitrogen formed a H-bond with the side chain of E130, the pyridine ring was involved in a CH–π interaction with the α-H atom of R207, and the chlorine atom underwent an electrostatic interaction with the guanidyl group of R210 and formed a halogen bond with the carbonyl oxygen of R207. On the basis of this model, a virtual screening campaign with the SPECS database, covering ∼200 000 compounds targeting the newly identified binding pocket, identified 25 hits that were selected for bioassays.306 Among them, 9 compounds with 5 different chemotypes were found to significantly potentiate Kv7.2 outward current. The most active 3 compounds, i.e.YG002 (EC50 = 4.64 ± 0.14 μM), YG007 (EC50 = 1.60 ± 0.16 μM) and YG025 (EC50 = 5.92 ± 0.21 μM), (Chart 14), contained two aryl groups connected by a thiourea (YG002), hydrazide (YG025), or urea (YG007) linker. The placement of the aromatic moieties in ztz240 and other activators in the gating charge pathway occupied the space between R207 and R210, thereby preventing a downward movement of the first 4 arginines (R1–R4) to the resting state.308,309 In the murine MES seizure model, ztz240, YG007 and YG018 exhibited in vivo anti-epileptic activity and protection rates comparable to that of RTG.
A related selective Kv7.2/7.3 and Kv7.3/7.5 opener, PF-04895162 (ICA-105665), discovered by Icagen Inc., showed promising preclinical data and advanced to clinical studies for the treatment of epilepsy. However, it failed a phase I clinical trial due to hepatotoxicity as a result of bile salt protein export and mitochondrial inhibition.310
5.4. Fenamates
Meclofenamic acid (meclofenamate) and diclofenac are widely used nonsteroidal anti-inflammatory drugs (NSAIDs), acting as nonselective inhibitors of COX-1 and COX-2 enzymes (Chart 15).311 Both were also found to be potent activators of heteromeric Kv7.2/7.3 channels, shifting the voltage activation curve leftward and slowing deactivation kinetics.312
Chart 15. Structures of meclofenamic acid, diclofenac and their analogs.

Upon external application, meclofenamate caused a concentration-dependent increase in the Kv7.2/7.3 current amplitude with EC50 values of 25 ± 2 μM and 11.7 ± 5.2 μM in CHO cells and X. oocytes, respectively. Similarly, diclofenac had an EC50 value of 2.6 ± 1.3 μM in potentiating Kv7.2/7.3 current in CHO cells.312 Interestingly, neither compound affected the current amplitude of other delayed rectifier channels, e.g. Kv1.2, Kv1.5, Kv2.1 and also Kv7.1, Kv7.1/KCNE1. In A7r5 rat aortic smooth muscle cells, diclofenac activated Kv7.4 channels with an EC50 of ∼100 μM, whereas it inhibited Kv7.5 currents with an IC50 ∼20 μM.313 However diclofenac was less sensitive at heteromeric Kv7.4/7.5 channels at −20 mV, probably due to the voltage-dependence of its actions. It was suggested that diclofenac might be useful as a pharmacological probe capable of distinguishing among Kv7.4 homomers, Kv7.5 homomers, and Kv7.4/7.5 heteromers.313
In cultured cortical rat neurons, both compounds enhanced the M-current and drastically reduced the number of evoked and spontaneous action potentials that exhibited regular spiking patterns with no significant spike adaptation. Co-application studies suggested that RTG and meclofenamate acted independently on Kv7.2/7.3 channels. Interesting differences between RTG and meclofenamate or diclofenac included that RTG was found to exert its strongest opener action on Kv7.3 homomeric channels, whereas meclofenamate was more potent on Kv7.2 homomeric channels. Diclofenac exhibited in vivo anticonvulsant activity in a MES seizure mouse model with an ED50 value of 43 mg kg−1.312
In order to dissociate the M-channel (Kv7.2/7.3) property from COX inhibition, SAR studies on meclofenamate and diclofenac were performed, keeping the diphenylamine moiety and a terminal hydroxyl group constant (Chart 15).314 Converting the carboxylate into an amide abolished COX-inhibition while preserving M-channel modulating activity. In contrast, all ester derivatives displayed COX-inhibition along with M-channel activation. The opener potency was measured by the extent of left-shift (ΔV50) of the activation curve of Kv7.2/7.3 channels. It was found that substituting the aromatic rings with NO2 groups significantly increased potency; e.g. ester 100 (ΔV50 = −4.1 mV) was less potent than ester 101 (ΔV50 = −31.0 mV); whereas for amides, tri-nitro analog NH29 was a stronger channel opener (ΔV50 = −31.3 mV) than mono-nitro analog 102 (ΔV50 = −12.2 mV). The diphenylamine moiety itself was also important for M-channel activity, as NSAIDs devoid of this group but substituted with a carboxylate, e.g.ibuprofen, fenoprofen or naproxen, did not exhibit M-channel activities. This effect indicated that the bridging secondary amine function was crucial for interacting with an anionic site or participating in H-bonding within a hydrophobic pocket of the channel. Furthermore, the terminal hydroxy group was necessary for M-channel opening activity, as compounds lacking a terminal OH group were either M-channel inactive (103, 104) or antagonistic (105, 106).314
The ester analog 101 enhanced the Kv7.2/7.3 current in a concentration dependent fashion with an EC50 value of 22 μM in CHO cells, and it also significantly slowed down the deactivation kinetics of Kv7.2/7.3 channels. In sensory dorsal root ganglion and hippocampal rat neurons, 101 robustly and reversibly depressed the number of evoked action potentials. It also decreased hippocampal glutamate and GABA release by reducing the frequency of spontaneous excitatory and inhibitory post-synaptic currents. The compound showed anti-convulsant activity in a MES seizure model with an ED50 value of 12 mg kg−1. In contrast, the amide derivative of diclofenac, NH29, dose-dependently activated Kv7.2/7.3 channels with an EC50 of 14 μM. Most importantly, NH29 was selective for M-channels (Kv7.2/7.3) versus other Kv channels, including the cardiac Kv11.1 (hERG or IKr) and Kv7.1/KCNE1 (IKs), Kv7.3, Kv7.4 channels. In addition, NH29 also did not interact with AMPA/NMDA and GABAA channels, and significantly increased the current amplitude of the RTG-sensitive mutant W236L, suggesting that the target binding site of NH29 was different from that of RTG.315,316
Mutagenesis studies on Kv7.2 showed that two mutant residues, L197G and R198A, and two epilepsy mutations, R207W and R214W, in the S4 domain exhibited significantly weaker stimulation by NH29 compared to wild type Kv7.2, which suggested that NH29 interacted with the VSD. Docking studies using a homology model of human Kv7.2 indicated that NH29 bound at an external groove formed by the interfaces of helices S1, S2 and S4 in a pocket formed by residues K120 in loop S1–S2, Y127, E130 in helix S2, and L200 and R207 in helix S4. The data also suggested that NH29 stabilized an electrostatic interaction between the charged residues E130 and R207, thereby favoring the activated VSD conformation.315
5.5. Other Kv7.2–Kv7.5 agonists
Recently, a series of benzoimidazol-1,2-yl amides 107 was reported as positive modulators of Kv7 channels (Scheme 17). The lead compound, benzimidazole KB-3061, was a potent activator of Kv7.2/7.3 channels with an EC50 = 440 nM in a fluorescence-based assay. It produced a V1/2 shift of −16 ± 1.1 mV using an automated patch clamp technique on QPatch HTX instrumentation. KB-3061 had little to no effect in the human α1β3γ2 GABA receptor PAM assay at 10 μM. It also did not have significant activity against GPCR, ion channel, transporter, and kinase targets at 10 μM. In a photostability assay, KB-3061 was stable exposed to visible light for up to 75 h, whereas RTG was completely degraded. In Kv7.1/mink and hERG cardiac channels, KB-3061 didn't produce significant inhibition up to 30 μM (23% and 9% inhibition, respectively). In a rat MES seizure model, KB-3061 showed in vivo anticonvulsion activity and a wide therapeutic index compared to RTG. KB-3061 provided 100% protection against seizures starting at 3 mg kg−1 with no change in neurological scoring (NS) up to 30 mg kg−1, whereas RTG provided 100% protection against seizures at 75 mg kg−1 with changes in NS starting at 45 mg kg−1. Overall, compared to RTG, KB-3061 was superior in terms of potency, tolerability, stability towards photo-oxidation, and lack of off-target effects, which supported its further development toward a new treatment for Kv7.2-NEE patients.
Scheme 17. General synthesis of benzoimidazol-1,2-yl amides 107.

A general synthesis of benzoimidazol-1,2-yl amides 107 commenced with a nucleophilic substitution of appropriately substituted 1-fluoro-2-nitrobenzene 108a with primary amines to provide 1-amino-2-nitrobenzenes 109. Alternatively, coupling of 1-chloro-2-nitrobenzene 108b with primary amines under Pd-catalysis afforded 109. Reduction of the nitro group in 109 with a variety of well-established methods gave the corresponding 1,2-diaminobenzenes 110. Reaction with cyanogen bromide then formed the 2-amino-benzimidazoles 111. Amide coupling of the 2-amino group in 111 with appropriate carboxylic acids or acid chlorides provided the target benzoimidazol-1,2-yl amides 107 (Scheme 17).317
5.5.1. Celecoxib
The NSAID celecoxib (Celebrex®), a commonly used COX-2 inhibitor, also modulates Kv7 channels, featuring a mechanism of action similar to RTG's (Chart 16).318 Celecoxib significantly enhanced the homomeric Kv7.2, Kv7.4 and heteromeric Kv7.3/7.5 currents in HEK293 cells at −20 mV in a concentration range from 1 μM to a maximum effect at 10–30 μM. The concentration-response relationship in HEK293 cells indicated EC50 values of in Kv7.2, Kv7.4 and Kv7.3/7.5 of 3.09 ± 0.27 μM, 3.37 ± 0.26 μM and 2.27 ± 0.4 μM, respectively. The effect on homomeric Kv7.3 channels was not significant at −20 mV, but at a more negative potential (−60 mV), celecoxib concentration-dependently increased Kv7.3 currents. However, the compound inhibited Kv7.1 (IC50 4.00 ± 0.13 μM) and Kv7.1/KCNE1 currents to a larger extent than RTG. Similar to RTG but less pronounced, celecoxib shifted the V1/2 of Kv7.2–7.4 and Kv7.3/7.5 currents to more negative potentials. In HEK293 cells, celecoxib increased Kv7.2/7.3 currents in a concentration-dependent manner with an EC50 of 4.95 ± 1.16 μM. It also dose-dependently enhanced M-current (Kv7.2/7.3) on rat SCG neurons and slowed their deactivation. It is likely that celecoxib forms a complex with Kv7.2 in the open channel conformation, involving residues near Trp236 at S5 and the gating hinge (Gly301) of S6. Importantly, the modulating effect of celecoxib was not depended on its COX-2 inhibition, as DM-celecoxib, a dimethyl analog of celecoxib that did not inhibit COX-2, had similar outcomes as celecoxib on currents in both wild-type and mutant Kv7.2 channels.129
Chart 16. Structures of other Kv7.2–Kv7.5 agonists.

5.5.2. Zinc pyrithione (ZnPy)
Zinc pyrithione (ZnPy) is a 1 : 2 complex of Zn2+ and pyrithione that was found to display unusual potency in elevating Kv7 currents as determined by the nonradioactive rubidium efflux assay (Chart 16).248 Considering the open channel probability (Po) in CHO cells, the potentiation by ZnPy followed the order Kv7.5 > Kv7.4 > Kv7.2 > Kv7.1. However, ZnPy did not show any significant effect on Kv7.3 potentiation. The activating effect of this complex was dose-dependent, with EC50 values of 1.5 ± 0.3 μM for Kv7.2 and 2.4 ± 0.4 for Kv7.2/7.3 in CHO cells. ZnPy positively modulated native M-current in hippocampal neurons in a similar fashion to recombinant Kv7.2/7.3 channels. The compound was selective for Kv7 channels and did not potentiate other ion channels, e.g. hERG, Kv2.1 delay rectifier voltage gated potassium channel, Kv4.2 A-type potassium channel or N-type calcium channel. Experiments to determine which components of ZnPy, Zn2+ or pyrithione, were responsible for the potentiation, suggested that only the 1 : 2 complex caused the observed channel current increase.
The sensitivity of the RTG-insensitive mutant Kv7.2 W236L to ZnPy suggested its binding interaction deviated from the RTG site. Furthermore, ZnPy potentiated Kv7.1 but did not have an effect on Kv7.3; whereas RTG was most active on Kv7.3 but did not affect Kv7.1. ZnPy most likely bound at an extracellular site between the S5 and S6 domains, close to many key residues of the gating machinery. Important residues for ZnPy effects included Leu249, Leu275, and Arg306. A Kv7.2 homology model was used to predict that Leu249 and Leu275, situated on two separate helices and facing each other with a linear distance of 10.6 Å, were close to the binding site of ZnPy.248
5.5.3. Pyrazolo[1,5-a]pyrimidin-7(4H)-ones (PPOs)
The novel pyrazolo[1,5-a]pyrimidin-7(4H)-ones 112 and 113 were identified as lead structures in an atomic absorption Rb+ efflux assay (Chart 16).319 Subsequent SAR studies indicated that a trifluoromethyl group at the 2-position was necessary for activity; the presence of a phenyl or naphthyl group at the 3-position afforded optimal activity, and electron-withdrawing substituents on the aromatic ring at the 5-position were also important. Synthetic optimizations led to the discovery of the lead compound QO-58, with a potent EC50 = 0.06 ± 0.01 μM for activation of Kv7.2/7.3 channels in CHO cells.320
The synthesis of QO-58 utilized the condensation of phenyl acetonitrile with trifluoroethylacetate in the presence of freshly prepared sodium ethoxide to form the sodium enolate 114, which, after reaction with hydrazine sulfate in dimethyl carbonate (DMC) at 80 °C, formed 5-amino pyrazole 115. Cyclo-condensation of 115 with a substituted pyridyl β-keto ester 116 provided QO-58 in good overall yield (Scheme 18).
Scheme 18. Synthesis of QO-58.

QO-58 demonstrated a high subtype selectivity for Kv7.2/7.3 over Kv7.1 channels in CHO cells. In HEK cells, QO-58 significantly potentiated Kv7.1, Kv7.2, Kv7.4 and Kv7.3/7.5, but only slightly increased Kv7.3 currents.320
In contrast, Kv7.5 did not produce significant currents in the presence of QO-58. In HEK cells, QO-58 was more potent in potentiating Kv7.2 and Kv7.4 than other channels, with EC50's of 1.3 ± 1.0 μM and 0.6 ± 0.1 μM, respectively. At 10 μM, QO-58 produced a significant leftward shift of the V1/2 of Kv7.1, Kv7.2, Kv7.4 and Kv7.3/7.5 currents. In addition, 10 μM QO-58 considerably slowed the activation kinetics of Kv7.4 and Kv7.3/7.5 currents, and also slowed the deactivation kinetics of Kv7 currents. QO-58 significantly increased the mutant Kv7.2 W236L current, suggesting a mechanism of action different from RTG. A mutagenesis study indicated that Val224, Val225, and Tyr226 in Kv7.2 were involved in activation by QO-58.320 A docking model further identified an interaction with residues Ala306 and Leu275 in Kv7.2 channels.
QO-58 also potentiated native M-type K+ currents in rat DRG neurons and demonstrated anti-seizure and anti-nociceptive effects in a model of sciatic nerve chronic constriction injury.320 However, the physical properties of QO-58 resulted in low bioavailability and irregular absorption after oral administration.321 In order to increase aqueous solubility, a salt form, QO58-lysine, was prepared, and the pharmacokinetics of QO58-lysine were investigated in rat plasma, which indicated that the salt was more readily absorbed than QO-58.322,323
QO58-lysine displayed anti-nociceptive effects during the second phase of pain induced by formalin. The maximum anti-nociceptive effect was observed after 4 h, consistent with a Tmax of ∼3 h. The anti-nociceptive effect of QO58-lysine was tested on chronic inflammatory pain induced by Complete Freund's Adjuvant (CFA). QO58-lysine increased the paw withdrawal threshold in rats in both a time- and dose-dependent manner. Overall, QO58-lysine alleviated inflammatory pain in rodent models without apparent toxic effects.323
5.5.4. NS1643
LQT2 syndrome and BFNC are associated with a mutation of hERG and KCNQ genes, respectively. Due to overwhelming evidence that LQT2 and epilepsy may coexist in some patients, a fundamental approach to investigate and treat the two diseases might be more effective. In this regard, a previously known hERG activator, NS1643324,325 was found to also activate Kv7 channels (Chart 16).326
The known Kv7 agonist ztz240 (Chart 14) is structurally related to NS1643, featuring two aryl groups connected by a functionalized linker. In CHO-K1 cells, NS1643 potentiated all Kv7.2/7.3, Kv7.4, and Kv7.2 channels, whereas Kv7.3 channels were not sensitive and the cardiac Kv7.1 channels were inhibited. For Kv7.2 channels, the EC50 value of 2.44 ± 0.25 μM was calculated from a dose–response curve of current amplitude. Similar to ztz240, which bound in the VSD, the hydrophobic residue F137 was also critical for NS1643 activity on Kv7.2 channels; upon mutation of F137 to Trp (F137W), NS1643 was unable to potentiate Kv7.2 channels.326
To understand the inhibitory effects of NS1643 on Kv7.2 F137W, the compound was docked to a closed-state structure of the mutant Kv7.2 F137W channel in a pocket near residue W137, and the docking data were considered as consistent with the notion that NS1643 may bind to the closed state of Kv7.2 F137W, thus resulting in inhibition. Overall, NS1643 showed a strong binding potency to the VSD region of Kv7.2 channels. In CHO-K1 cells, NS1643 exhibited stronger potentiation effects on the four tested BFNC mutants, e.g. R207W, M208V, R214W, and Y284C, which indicated that it could rescue BFNC mutants. Accordingly, the capability of NS1643 for restoring LQT2 and BFNC mutants suggested that the two seemingly different disease phenotypes might be treated with a single pharmacological agent.326
5.5.5. PF-05020182
In order to improve some of the pharmacological drawbacks of RTG, e.g. subtype selectivity and the aniline structural alert,327 a new series of dimethoxypyrimidines characterized by PF-05020182 was developed by Pfizer (Chart 16).328 In these compounds, the aniline was replaced by an 4,6-dimethoxypyrimidine, with an amide substitution at the 5-position. Among the 18 reported analogs, PF-05020182 emerged as a promising candidate surpassing the target product profile EC50 <500 nM, LipE >3,329 and adequate in vitro ADME and safety properties.
An SNAr reaction of 2-chloro-4,6-dimethoxy-5-nitropyrimidine (117) with 4-methoxypiperidine provided the 2-(4-methoxypiperidinyl)pyrimidine 118, which, after nitro group reduction followed by acylation with 3,3-dimethylbutanoyl chloride, afforded PF-05020182 in high yield (Scheme 19).
Scheme 19. Synthesis of the Kv7 agonist PF-05020182.

PF-05020182 potentiated Kv7.2/7.3 currents preferentially over Kv7.4, and better selectivity was observed for neuronal channels (Kv7.2/7.3, Kv7.4 and Kv7.3/7.5) over the cardiac Kv7.1/KCNE1 channels. Unlike RTG, PF-05020182 did not exhibit significant activity on GABAA channels. PF-05020182 was also able to cross the blood brain barrier readily, and in an in vivo MES seizure model it displayed promising activity, as it dose-dependently reduced the number of rats exhibiting full tonic extension convulsions in response to corneal stimulation.328
5.5.6. Benzbromarone, ZG1732 and ZG2083
Benzbromarone (BBR),330,331 an inhibitor of urate transporters,332,333 is widely used for the treatment of gout (Chart 16). BBR was found to also act as a Kv7 activator, primarily on peripheral channels.334
In CHO cells, BBR potentiated recombinant Kv7.1, Kv7.2/7.3, Kv7.4, Kv7.5 but not Kv7.3 channels at 10 μM concentration. Based on the concentration-dependence of current amplitude, the EC50 value on Kv7.2 channels was 4.32 ± 1.40 μM. BBR potentiated the native M-current in cultured rat DRG and hippocampal neurons, and at 10 μM it also suppressed the generation of action potential firing evoked by injection of a +400 pA current. Furthermore BBR dampened neuronal overexcitability induced by inflammatory mediators such as OXO-M and BK.335–337
BBR dose-dependently suppressed a formalin-induced inflammatory pain phenotype in rats and mice. The compound also attenuated monosodium urate (MSU)-induced acute inflammatory pain in rats, which suggested that potentiation of Kv7 channels was beneficial for relief of gout pain.334
A mutagenesis study revealed that the R198 residue at the external S4 chain terminus in Kv7.2 channels might be critical for BBR binding, since the R198A mutation significantly reduced potentiation. In addition to R198A, C106A, V111A, S121A, L126A, F137A, G186A, T194A, and L200W mutations, as well as 2 pairs of double mutants, C106AR207W and C106AN190A, also significantly reduced potentiation of outward currents.334 Overall, these results indicated an essential role of VSD in the potentiation effect of BBR on Kv7.2 channels.
An improved high-throughput screen of a collection of 80 000 compounds using a conventional patch clamp technique identified two additional lead compounds, ZG1732 and ZG2083, as novel Kv7.2 channel activators, with EC50 values of 1.04 ± 0.18 μM and 1.37 ± 0.06 μM, respectively.338 Further details regarding their subtype selectivity and mechanism of action have not yet been reported.
5.5.7. Substituted piperidines and N-phenylbutanamides
Based on the structures of RTG and PF-05020182, and using a hybridization design strategy, a series of piperidines were designed as Kv7 openers.339 The target compounds were prepared in three different strategies (Scheme 20). The key intermediate 120 was formed by amidation of 4-bromo-2,6-dimethylaniline (121) with 3,3-dimethyl butanoyl chloride. Pd-Catalyzed cross-coupling of 120 with substituted piperidines provided 122–124. In a second approach, the bromoaniline 120 was converted to the corresponding hydroxymethyl derivative 125 in two steps; first, the corresponding benzaldehyde was formed with n-BuLi–DMF, followed by reduction with NaBH4. Mesylation of alcohol 125 and nucleophilic substitution with piperidine or tetrahydroquinoline led to 126 and 127, respectively. In a third route, the 4-aryl piperidine intermediate 128 was prepared by a three-step sequence of Suzuki coupling-reduction, catalytic hydrogenation, and deprotection of the Boc-group from the commercially available boronic acid 129. SNAr reaction of 128 with 4-fluoro-2,6-dimethylnitrobenzene followed by nitro group reduction and amide coupling provided the target compounds 132–134 in good yields.339
Scheme 20. Synthesis of substituted piperidines as Kv7 agonists.

The Kv7.2 potentiation activities of these compounds were initially determined at 10 μM by a fluorescence-based thallium influx assay and described as activation ratios (Table 12). When the R group was an unsubstituted piperidine, the resulting analog 122 was more active than RTG (69.8% vs. 40.2%), and upon substitution of the piperidine ring with 4-dimethyl (123) and carbonyl (124) groups, the activity further increased (100.0% and 110.3%). Insertion of a methylene group between the piperidine and the core scaffold in 126 led to a dramatic decrease in activity (10.3%). Interestingly, introducing a fused benzene at the 2,3-position of the piperidine in 127 increased the activity 3.5-fold (138.8%) over RTG. Compounds with 3-(EWG)-substituted phenyl groups at the 4-position of the piperidine, e.g.132 (3-F), 133 (3-CF3) and 134 (3-Cl), also displayed significantly better activity than RTG (3.7–4.2 fold).
Activation profile of piperidine derivatives on Kv7.2 channels.
| |||||
|---|---|---|---|---|---|
| Compound | R substituent | Kv7.2 activation ratio (%, at 10 μM) | Compound | R substituent | Kv7.2 activation ratio (%, at 10 μM) |
| 122 |
|
69.8 | 127 |
|
138.8 |
| 123 |
|
100.0 | 132 |
|
151.9 |
| 124 |
|
110.3 | 133 |
|
149.1 |
| 126 |
|
10.3 | 134 |
|
168.2 |
| RTG | 40.2 | ||||
Compounds 122, 123, 126 and 132 were selected for a confirmatory atomic absorption Rb+-ion flux assay, a faster and more reliable analytical measurement.340 Analog 122 showed a similar potency to RTG, whereas 123, 127 and 132 had slightly lower efficacy and potency.339
In vivo PK studies in rats revealed a high exposure (8.36 h μg mL−1) and peak concentration (9.11 μg mL−1) at a dose of 1 mg kg−1 i.v. for compound 132, which also had moderate clearance (0.12 L h−1 kg−1) and a good half-life (1.38 h). Overall, 132 had a satisfactory metabolic profile and progressed to further development.339
Recently, N-phenylbutanamide derivatives were also reported as Kv7 openers.341 Various fused heterocycles were linked at the N-phenyl moiety to generate 135–146 using a Pd-catalyzed Buchwald coupling of bromide 120 with heterocyclic amines (Scheme 21). A Rb+ flow test was first used to determine Kv7 opening activity in vitro, and later a thallium assay and a patch-clamp test were used to verify activity on Kv7.2 channels.
Scheme 21. Synthesis of N-phenyl butanamides as Kv7 agonists.

Thieno[3,2-c]pyridine 135 was found to have a 17-fold higher potency (EC50 0.02 μM) than RTG (EC50 = 0.35 μM) in a Rb+ flow test. Introduction of electron-withdrawing groups, e.g. CF3 (136) or CN (137), at the ortho-position significantly decreased activity, while a methyl group at the 3-position (138) had an equivalent potency (EC50 = 0.3 μM) to RTG. Inverting the thiophene in thieno[2,3-c]pyridines 139–141 provided an analog 139 with an equivalent potency (EC50 = 0.02 μM) to 135, whereas the fluorinated 140 was 150-fold less potent.
While the electron-donating methyl group in 141 slightly increased its potency (EC50 = 0.3 μM) compared to 139, pyrrolo[1,2-a]pyrazine 142 had almost 2× decreased potency (EC50 = 0.034 μM) vs. its bioisostere 135. Replacement of the piperidine with a 1-benzazepine ring produced 143–146 (Scheme 21). Except for the 7-bromo substituted 145, fluoro- or chloro-substituted analogs 143 and 144 showed higher potency than RTG, and the methyl-substituted 146 was significantly more potent (EC50 = 0.04 μM).341 Among the most potent analogs, 135, 139 and 142, were further evaluated in the thallium and patch clamp tests to confirm Kv7 opening activity. Based on these in vitro results, 135 was advanced to an in vivo study. The compound displayed significant anti-convulsant activity in the MES test with a calculated EC50 of 1.83 mg kg−1 while no adverse effects were observed during a rotarod test and PK experiments in rats. Thiophene 135 was able to diffuse rapidly into the brain and establish an equilibrium distribution between brain and plasma within 2 h in mice. After i.v. injection in rats, 135 showed a favorable half-life (T1/2 = 5.49 h) and moderate plasma exposure (AUClast = 687.21 h ng mL−1) and clearance (CLobs = 1416.50 mL −1 kg−1). Based on these in vitro and in vivo data, 135 appears to be a potential lead compound for the development of antiepileptic drugs.341
5.5.8. Substituted benzamides and cyclohexamides
An HTS screen of the GSK compound library using heterodimeric Kv7.2/7.3 channels identified two structurally distinct families, a benzamide series and a cyclohexamide series, as pan-Kv7 activators (Schemes 22 and 23).342
Scheme 22. Synthesis of benzamides 150 and 151.

Scheme 23. Synthesis of cyclohexamide 152.

In the benzamide series, phenyl ethers delivered the highest level of Kv7.2/7.3 activation. The methyl sulfone at the 3-position of the phenyl ring provided both solubility and high Kv7.2/7.3 activity (Scheme 22). An SNAr reaction of o-bromo benzoic acid derivatives 147 with phenol provided aryl ethers 148 which upon amide coupling with chiral benzyl amines led to benzamides 150 and 151. For the cyclohexamide series, the presence of an (R)-hydroxymethyl group α to the amide significantly improved solubility, and a gem-difluorosubstitution on the cyclohexyl ring also increased metabolic stability. In addition, lipophilic groups at the meta-position of the benzyl group increased activity without decreasing solubility. The synthesis of cyclohexamide 152 used a base-mediated α-functionalization of 4,4′-difluorocyclohexanone with dimethyl carbonate to provide β-ketoester 154 which was converted to the corresponding enol-triflate 155 using Comins' reagent. Suzuki coupling with phenyl boronic acid provided the aryl-substituted ester 156. Hydrogenation followed by saponification under basic conditions produced racemic trans-cyclohexyl carboxylic acid (±)-157. Amide coupling of (±)-157 with substituted (R)-benzyl amine provided a diastereomeric mixture of amides which were separated by chiral HPLC to provide the Kv7.2/7.3 activator 152.342
Ultimately, compounds 150, 151 and 152 were selected for further studies.342 These compounds were very potent in Kv7.2–Kv7.5 channel assays (EC50 = 0.06–0.36 μM using a SyncroPatch platform),343 except for 150 which had reduced activity in Kv7.4 (EC50 > 10 μM). In primary cultured rat neurons, 151 and 152 inhibited spontaneous neuronal firing. Both compounds showed good blood–brain-barrier permeability and demonstrated anti-seizure effects in a dose-dependent manner in MES, a PTZ-induced seizure model. This activity was also proportional to the concentration of non-protein bound compound in the brain. Lead structures passed the rotarod CNS tolerability model.342 From the study of point mutations (Kv7.2 W236L and Kv7.3 W265L), it was found that both compounds interacted with the Kv7.2/7.3 pore domain. Finally, in order to understand how individual Kv7.2–Kv7.5 isoforms contributed to bladder contractions, 150, an analog with significantly reduced Kv7.4 activity, was tested ex vivo on expressed human detrusor tissue. Interestingly, 150 did not reduce bladder contraction, suggesting that homomeric Kv7.4 may not play a critical role in this process.342
Recently, the neurotransmitter GABA was found to activate voltage-gated Kv7.3, Kv7.5 and Kv7.2/7.3 channels at subthreshold potential.344 The tryptophan residue W265 in the Kv7.3 channel was responsible for GABA's affinity to heteromeric Kv7.2/7.3 channels. Similar to GABA, the synthetic antiepileptic and antinociceptive gabapentin was also a potent activator of heteromeric Kv7.2/7.3 and homomeric Kv7.3 and Kv7.5 channels (Chart 16).345 In contrast, the structurally related gabapentinoid pregabaline did not activate Kv7.2/7.3 channels, and, at high concentration (≥10 μM), demonstrated inhibitory effects.345
Similar to RTG and other Kv7 activators, GABA and gabapentine exhibited a negative electrostatic surface potential near their carbonyl oxygen, which was thought to be responsible for their affinity to Kv7.2/7.3 channels.346
Using Xenopus oocyte expression and two-electrode voltage-clamp electrophysiology, gabapentine was found to dose-dependently activate Kv7.2/7.3 channels with an EC50 of 4.2 ± 0.13 nM, ca. 1000 times more potent than RTG. However, gabapentine did not activate homomeric Kv7.2 and Kv7.4 channels. In contrast, Kv7.5 channels exhibited even higher sensitivity (EC50 = 1.9 nM) but lower efficacy towards gabapentine. While gabapentin and GABA exhibited similar efficacy, gabapentin was 200-fold more potent. Analogous to GABA, the mutant channel Kv7.2/7.3 W265L was insensitive to gabapentine, which indicated that both compounds shared a common binding site, i.e. near the conserved tryptophan W265, at the S5 helix of Kv7.3.345
5.5.9. Pyrazolopyrimidines
An HTS campaign using a FLIPR-based thallium (Tl+) influx assay and a HEK293 cell line identified a pyrazolopyrimidine series as novel Kv7.2/7.3 openers (Chart 17).347
Chart 17. Pyrazolopyrimidines as Kv7.2/7.3 modulators. a % efficacy relative to 10 μM RTG.

Multiple compounds of the structural type 158 were screened, identifying 159 as an excellent lead molecule with a Kv7.2/7.3 EC50 of 0.91 μM and an efficacy of 149% relative to 10 μM RTG. SAR modifications of the substitution pattern around the pyrazolopyrimidine core of 159 led to 2,3-substitued 160.347
Further SAR studies on 160 revealed that a 4-CF3/4-fluoro-substituted phenyl group at the 3-position of the pyrazolopyrimidine provided the most active analogs (Chart 17). In addition, alkyl or cycloalkyl groups were optimal R2 substituents, whereas substituents larger than a methyl group were not tolerated at the R3 position.347
A general synthesis of analogs of 160 used a Pd-catalyzed coupling of aryl iodides 165 with malononitrile to provide the corresponding phenyl malononitriles 166. Reaction with hydrazine hydrate gave pyrazole-3,5-diamines 167 (Scheme 24). Diamines 167 were condensed with enones 168 to give pyrazolopyrimidine amines 169, which, upon reaction with alkyl chloroformates, afforded the target compounds 160.
Scheme 24. Synthesis of pyrazolopyrimidines 160.

Among analogs with an unsubstituted pyrimidine ring (R3 = H), SAR studies suggested that cyclopentane 162 had a superior potency (EC50 = 0.05 μM) amongst the initial group of analogs studied at the Hit-to-Lead stage. A further investigation to improve the overall potency and efficacy by substitution of methyl groups at the pyrimidine rings led to two more potent compounds, 163 and 164 (Chart 17).347
Most importantly, 164 displayed a Kv7.2/7.3 EC50 of 0.07 μM and an efficacy of 205% compared to 10 μM RTG. However, ultimately analog 161, in spite of more modest potency and efficacy (EC50 = 0.41 μM and 143%, respectively), was selected for further in vivo studies due to its favorable PK properties. This compound had a good plasma clearance of 0.8 L h−1 kg−1, favorable oral bioavailability of 56%, and a high brain to plasma ratio of 3.8. It also showed good efficacy in the in vivo capsaicin-induced secondary mechanical hyperalgesia (SMH) inflammatory pain model.348
5.5.10. BMS-204352
The unfortunate failure of the phase III clinical candidate BMS-204352 was attributed to a lack of efficacy compared to patients treated with placebo. This result was difficult to associate with a PD issue since the drug showed potent in vitro and in vivo activity as well as a good PK profile. After single or multiple i.v. dosing in humans, BMS-204352 was found to have an excellent half-life (t1/2 = 20 h) as compared to animal studies (t1/2 = 1.6 h in rats), with a tendency to accumulate in brain (brain/plasma ratio of 9.6, brain half-life = 1.9 h in rats). No significant side effects were observed in rats and dogs.122
A gram-scale synthesis of BMS-204352 started with the chlorination of commercially available 2-methoxyphenylacetic acid (170) with sulfuryl chloride followed by carboxy-protection using dimethylsulfate to form ester 171 (Scheme 25).
Scheme 25. Synthesis of BMS-204352.

An SNAr reaction of 4-fluoro-3-nitrobenzotrifluoride (172) with the lithium enolate generated from ester 171 using excess LiHMDS generated intermediate 173 which upon in situ electrophilic fluorination using NFSi provided fluoroester 174. Saponification of 174 formed the racemic acid 175 which was resolved with (S)-α-methylbenzylamine to provide acid (S)-175 in 99.3% enantiomeric purity. Reduction of the nitro group by sodium dithionite followed by treatment with methanolic HCl furnished BMS-204352 in 23% overall yield in seven steps and an ee of 94.9% for the (S)-isomer.349
5.5.11. Synthetic approaches to the Kv7.1-selective agonists ML277 and R-L3
ML277 was the result of an intense SAR study performed on amide, piperidine, and sulfonamide moieties, leading to the observation that only the (R)-isomer showed Kv7.1 activity. In addition to a peculiar PD profile, ML277 demonstrated in vitro PK deficiencies, especially a metabolic instability in rat and human hepatic microsomes resulting in the NADPH-dependent oxidation of the tolyl moiety and oxidative demethylation of the methoxy group.112
The synthesis of ML277 commenced with the commercially available (R)-N-Boc-2-piperidine carboxylic acid (176), which was coupled with 4-(4-methoxyphenyl)thiazol-2-amine in presence of HATU and DIEA to provide the corresponding amide 177 (Scheme 26). TFA-mediated Boc-deprotection of 177 followed by sulfonamide formation with p-toluenesulfonyl chloride yielded ML277 (Chart 2).112
Scheme 26. Synthesis of ML277.

Since the R-L3 enantiomers showed opposite effects in modulating the IKs current (d-enantiomer was an activator while l-enantiomer was an inhibitor), a stereoselective synthesis of R-L3 was performed. Reaction of 2-amino-2′-fluorobenzophenone (178) with 179 formed the Trp derivative 180 which underwent intramolecular cyclization in base to form benzodiazepine 181 (Scheme 27). N-Methylation of the amide group in 181 using NaH and MeI provided R-L3 (Chart 2).252,350
Scheme 27. Synthesis of R-L3.

6. Syntheses and biological properties of Kv7 negative modulators
Kv7 blockers promote a reduction of outward K+ current, and, consequently, shift the channels to more positive membrane potentials. In Chart 18, chemical structures of both selective and unselective Kv7 inhibitors are shown.
Chart 18. Structures of Kv7 antagonists.

Linopirdine was found to inhibit all Kv7 subtypes with a non-negligible affinity for other targets (Tables 13 and 14),351 and a cryo-EM structure revealed its binding site in a cytosolic pocket underneath the inner gate in Kv7.4.116 This indolinone was evaluated as an anti-Alzheimer's agent since it was able to release acetylcholine in rat hippocampus, cortex and caudate nucleus slices, in addition to causing increased dopamine, glutamate, aspartate, GABA, and serotonin levels.352,353In vivo studies confirmed that neurotransmitter release provided improvements in cognitive and mnemonic functions.353,354 Nevertheless, linopirdine failed in phase III clinical trials in Alzheimer's patients due to undesired pro-epileptic side effects.355
Potency of Kv7 blockers based on current inhibition in Kv7 subtypes.
| Linopirdine | XE991 | (3R,4S)-Chromanol 293B | HMR 1556 | |
|---|---|---|---|---|
| Kv7.1 IC50 (μM) | Xenopus oocytes: 8.9 ± 0.6 (ref. 362) | Xenopus oocytes: 0.75 ± 0.05 (ref. 362) | Xenopus oocytes: 16.4 ± 0.7 (ref. 363) | Xenopus oocytes: 0.42 ± 0.02 (ref. 363) |
| Xenopus oocytes: 65.4 ± 1.7 (ref. 364) | ||||
| Xenopus oocytes: 26.9 ± 0.8 (ref. 365) | ||||
| Kv7.1/KCNE1 IC50 (μM) | CHO cells: 2.08 ± 0.78 (ref. 366) | CHO cells: 1.60 ± 0.17 (ref. 366) | Xenopus oocytes: ∼6.5 μM (ref. 367) | |
| Xenopus oocytes: 15.1 ± 3.3 (ref. 364) | ||||
| Xenopus oocytes: 6.9 ± 0.5 (ref. 365) | ||||
| Kv7.1/KCNE3 IC50 (μM) | Xenopus oocytes: 0.54 ± 0.18 (ref. 364) | |||
| Kv7.2 IC50 (μM) | Xenopus oocytes: 4.8 ± 0.6 (ref. 362) | Xenopus oocytes: 0.71 ± 0.07 (ref. 362) | ||
| HEK293 cells: 3.4 ± 2.6 (ref. 105) | ||||
| HEK293 cells: 1.5 ± 0.4 (ref. 105) | ||||
| Kv7.2/7.3 IC50 (μM) | HEK293 cells: 2.7 ± 0.9 (ref. 105) | HEK293 cells: 0.9 ± 0.3 (ref. 105) | ||
| Xenopus oocytes: 10 ± 1 (ref. 363) | ||||
| Kv7.3/7.4 IC50 (μM) | HEK293 cells: 1.4 ± 0.4 (ref. 105) | HEK293 cells: 1.4 ± 0.5 (ref. 105) | ||
| Kv7.4 IC50 (μM) | HEK293 cells: 1.7 ± 0.7 (ref. 105) | HEK293 cells: 1.2 ± 0.1 (ref. 105) | ||
| Kv7.5/7.3 IC50 (μM) | CHO cells: 7.7 ± 1.1 (ref. 107) | |||
| Xenopus oocytes: 15 ± 2 (ref. 363) |
Inhibition of non-Kv7 channels (IC50) by Kv7 antagonists and their effects on non-IKs currents.
| Linopirdine | Xenopus oocytes: eag1 = 31 ± 3 μM; erg1 = 53 ± 4 μM; erg3 = 85 ± 5 μM; elk1 = 37 ± 4 μM; Kv1.2 = 68 ± 6 μM; Kv4.3 = 86 ± 14 μM; inhibition of Kv2.1 (ref. 362 and 371) |
| XE991 | Xenopus oocytes: eag1 = 49 ± 6 μM; erg1 = >100 μM; erg3 = >100 μM; elk1 = >100 μM; Kv1.2 = >100 μM; Kv4.3 = 43 ± 7 μM; inhibition of Kv2.1 (ref. 362 and 371) |
| Racemic Chromanol 293B | Xenopus oocytes: Kv1.1, Kir2.1, ICa–L, and hERG = no inhibition |
| Xenopus oocytes: ICFTR = 19 μM; KATP channel: between 50 and 100 μM | |
| Guinea pig ventricles: IKr = >30 μM | |
| Guinea pig myocytes: IK1, INa, ICa–L, IKr = no inhibition | |
| Human myocytes: INa, ICa–L = no inhibition | |
| Human atrial myocytes: Ito1 = 31.2 μM; IKur = 30.9 μM | |
| Human ventricles: Ito = 24.0 μM (ref. 372–377) | |
| (3R,4S)-Chromanol 293B | CHO cells: hERG = 224 ± 6.3 μM; Kv4.3 = 54 ± 2.5 μM; Kv1.5 = 27 ± 1.6 μM (ref. 373) |
| HMR 1556 | Canine myocytes: Ito = 33.9 μM; ICa,L= 27.5 μM; IKr = 12.6 μM; no effect on IK1 |
| Xenopus oocytes: no effects on Kv1.3, Kir2.1, and HCN2. Less than 15% inhibition at 10 μM against hERG and Kv1.5 (ref. 378 and 379) | |
| DMP 543 | α1AKi = 1.1–3.1 μM; σ1R: Ki = 3.9 μM (ref. 368) |
A possible therapeutic application of Kv7 channel blockers for the treatment of hypotension was supported by the reduction of systolic blood pressure and heart rate observed after prolonged use of the Kv7 activator flupirtine in humans. Due to the expression of Kv7.1, Kv7.3, Kv7.4, Kv7.5 in vascular smooth muscle cells, linopirdine (0.01–3 mg kg−1 i.v.) demonstrated a dose-dependent increase in both mesenteric vascular resistance and systolic blood pressure in rats. Although further investigations of the Kv7 subtype in vascular tone are needed, the results achieved by linopirdine are consistent with a possible use of Kv7 antagonists in hypotension and haemorrhagic shock treatments.356,357
XE991 is a more potent pan-Kv7 antagonist than linopirdine (Table 13).358 Several studies evaluated a possible neuroprotective effect of XE991 in Parkinson's disease (PD), following the detection of Kv7.2 and Kv7.4 in nigral dopaminergic neurons. Both in vitro and in vivo studies demonstrated an increase in dopamine levels in substantia nigra compacta as well as in the spontaneous firing rate of dopaminergic neurons.359,360
XE991 microinjection (1 μM, 0.5 μL) into the substantia nigra compacta in a rat model for PD partially reverted the rigidity and akinesia induced by administration of the D2 receptor antagonist haloperidol.360 In order to confirm these results, rats with 6-hydroxydopamine-induced neurodegeneration were used to mimic late-stage PD and treated with XE991 (10 μmol L−1 s.c.). Subsequently, balance and motor coordination improved and an attenuation in muscle rigidity was observed.361
The absence of seizures in an animal model after XE991 and linopirdine treatment and their neuroprotective properties could be explained by their slow binding kinetics to the active-conformation of Kv7 channels, mainly exerting their inhibition in hyperexcited neurons.71
Interestingly, the antagonist DMP 543 was claimed to exhibit anti-AD properties as a consequence of effects in a phencyclidine-induced schizophrenia mouse model of drug addiction.368,369 Moreover, DMP 543 and XE991 showed an improved ACh release from rat brain slices (490 nM and 700 nM, respectively) compared to linopirdine (4.2 μM). This effect was reproduced transferred in a rat model of AD, leading to increases in hippocampal extracellular ACh levels of about 100% (XE991 dose = 5 mg kg−1 and DMP 543 dose = 1 mg kg−1).370
Studies of selective Kv7 channel blockers have intensified following the therapeutic applications of pan-Kv7 antagonists and the realization of the large range of biological processes in which Kv7 involvement has been demonstrated.366,380–382
The (−)-(3R,4S)-chromanol 293B is a blocker of the delayed rectifier K+ current (IKs) with a high affinity for Kv7.1/KCNE1 and Kv7.1/KCNE3 over Kv7.1 (Tables 13 and 14).372,373 Kv7.1 inhibition by 293B arises from its binding to a cavity in the pore region, blocking the channel in the AO conformation without affecting the C-terminus. In particular, 293B stops ion permeation by engaging in hydrophobic interactions with the S6 transmembrane domain and exhibiting electrostatic interactions within the lower selectivity filter. Kv7.1/Kv7.2 chimera studies and a Kv7.1 point mutation approach revealed Ile337, Phe340 (in S6) and Thr312 (in the selectivity filter) as crucial residues for 293B interactions. Further information was obtained by docking 293B into a homology model based on KcsA and Kv1.2 X-ray structures of the open Kv7.1 pore, demonstrating the formation of a tertiary complex between 293B, the lowest K+ ion in the selectivity filter and residues in the pore cavity. The resulting complex was stabilized by electrostatic effects with the oxygen atoms of the sulfonamide moiety and the alkoxide group of 293B.365
Kv7.1 channels have also been detected in islet of Langerhans cells in rodent pancreas and in GLP-1-positive L-cells in the intestinal crypt.383 Since its in vitro inhibition (initial infusion of 8 mg kg−1 h−1 followed by 4 mg kg−1 h−1 as a maintenance dose) led to insulin secretion,384,385 chromanol 293B was investigated in a mouse model of type 2 diabetes, where it was found to promote an increased glucose-stimulated insulin secretion compared to control.383 Moreover, IKs blockers have been evaluated as potential antiarrhythmic agents with the purpose to overcome the limitation of class III antiarrhythmics based on their greater efficacy at slow rate rather than tachycardic rate, leading to proarrhythmic side effects.386 Chromanol 293B and HMR1556 (30 mg kg−1 d−1; p.o.), a more potent 293B analog,387 showed an inverted rate-dependent profile of action potential duration compared to class III antiarrhythmics,388,389 restoring sinus rhythm and preventing heart failure in persistent atrial fibrillation-induced pigs.390
To evaluate the neuroprotection effects of linopirdine during clinical trials studies, a limited gram-scale synthesis was adapted to an industrial manufacturing process (Scheme 28).391,392 In the scaled-up approach, the oxindole scaffold 183 was replaced with the isatin 184 in order to avoid the need of large amounts of aluminum chloride during the highly exothermic Stollé synthesis of 1-phenyloxindole 183. Accordingly, 1-phenylisatin 184 was obtained by reacting diphenylamine with oxalyl chloride to give amide 185 at gentle reflux. Moreover, the use of a catalyst for the ring closure of 185 was thereby made unnecessary, contrary to the synthesis of 183.
Scheme 28. Optimized synthetic approach for linopirdine on an industrial manufacturing scale, compared with a previous gram-scale synthesis.

The 4-pyridinylmethyl group was introduced in the isatin scaffold 183 by exploiting the ease of the methyl substituent of 4-picoline to undergo an aldol-like condensation, affording intermediate 186. In this way, the amount of expensive 4-picolyl chloride (two equivalents were used in the final step of the original synthetic approach) was reduced. Moreover, LDA was no longer needed. Intermediate 186 was then dehydrated to alkene 187, which was the first isolated intermediate and was purified by crystallization. The reduction of the double bond of 187 with NaBH4 in alkaline solution followed by in situ alkylation of 188 gave linopirdine. This seven-step synthesis of linopirdine was conducted in a single reaction vessel, isolating only the intermediate 187, with an overall yield of 45–55% from diphenylamine.391,392
Despite its in vitro potency, linopirdine did not ameliorate the cognitive state in humans affected by Alzheimer's disease. Possible reasons for this setback might be its low blood–brain barrier (BBB) penetration and short half-life (t1/2 = 0.4–3.2 hours).393 In fact, linopirdine was shown to be metabolized by cytochrome P-450, leading to inactive mono- and bis-pyridine N-oxides, i.e.189 and 190 (Scheme 29).394
Scheme 29. Chemical structures of linopirdine metabolites.

To address these drawbacks, a library of linopirdine analogues was synthesized. The oxindole in the linopirdine scaffold was replaced with anthrone or 4-azafluorene cores, enhancing the lipophilic character and fostering BBB penetration. Moreover, a fluorine atom was inserted in a position adjacent to the pyridine nitrogen, thus reducing the basicity of the heterocycle and preventing metabolism to the N-oxide. As a result, DMP 543 and XE991, discovered at DuPont Pharmaceuticals, showed an improved inhibitory activity (DMP 543 IC50 = 48 nM, XE991 IC50 = 55 nM in HEK-293 cells expressing Kv7.2 channels), brain-to-plasma ratio (linopirdine = 1 : 6; DMP 543 = 1 : 1) and about 4-fold greater half-life (for DMP 543 only).
DMP 543 and XE991 were prepared in a single-step synthesis, alkylating anthrone with 2-fluoro-4-(iodomethyl)pyridine using NaH as a base, and treatment with 4-picolyl chloride hydrochloride under phase-transfer catalysis conditions, respectively (Scheme 30).395
Scheme 30. Syntheses of DMP 543 and XE991.

Optimization of the DMP 543 synthesis generated 2-fluoro-4-(iodomethyl)pyridine in situ from 2-fluoro-4-mesylpyridine and NaI, and directly used it for the anthrone dialkylation in the presence of lithium tert-butoxide instead of NaH, raising the yield, simplifying the purification and avoiding anthrone O-alkylation.396 The vivid interest in the development of Kv7 antagonists as neuroprotective agents was based on additional evidence of their efficacy in relevant neurodegenerative disease models. The chemical exploration and SAR studies of anthrone-based derivatives highlighted the importance of a rigid scaffold surrounded by two hydrogen bond acceptors to maintain the Kv7 antagonist property.397 In spite of the considerable Kv7 inhibitory potency of DMP 543, its PK properties needed to be optimized. Mainly, its acid instability represented an obstacle for oral administration. Under acidic conditions, the pyridine nitrogen was protonated, allowing fluorine elimination by an SNAr reaction with water and resulting in the inactive derivative 191 (Scheme 31).398
Scheme 31. DMP 543 conversion into inactive derivative by acid hydrolysis.

To avoid this undesired transformation, the 2-fluoropyridines were replaced with 2-trifluoromethyl or 3,5-difluoropyridine groups in compounds 192 and 193, which also prevented the formation of pyridine N-oxide metabolites.368
The low water solubility of DMP 543 (6 μM) was another aspect considered and partially resolved by introducing a pyridazine moiety as shown in 194, attenuating the lipophilicity of the analog.
Since DMP 543 and XE991 inhibited CYP P-450, probably due to the coordination of the pyridine nitrogen to the heme iron, a combination of weak and strong hydrogen bond accepting groups was introduced in pyridazine JDP-107. The PK properties of JDP-107 satisfied a lead compound profile in terms of acid stability, water solubility, and CYP inhibition as well as strong Kv7 inhibitory activity (IC50 = 0.16 μM In HEK-293 cells expressing Kv7.2 channels), and yet further optimizations were warranted. In Schemes 32 and 33, the synthetic approaches used for symmetric and asymmetric anthrone analogs, respectively, are summarized.
Scheme 32. Synthesis of symmetrical DMP 543 analogs 192, 193, and 194.

Scheme 33. Synthesis of racemic JDP-107.

Interestingly, in order to prepare differentially substituted anthrone derivatives, a replacement of NaH with LiOt-Bu was instrumental to achieve the mono-substituted intermediate 195, because only disubstituted product was obtained when NaH was used.368
From an off-target point of view, XE991, DMP 543, and JDP-107 showed hERG inhibition at greater than or equal to 32 μM based on a thallium flux assay using HEK 293 cells expressing hERG channels.368
In some cases, opposite enantiomers of Kv7 inhibitors showed different PK and PD properties.121,366 In this context, an enantioselective synthesis of chromanols led to the identification of about one order of magnitude more potent (3R,4S)-293B and (3R,4S)-HMR1556 stereoisomers with superior properties over (3S,4R)-293B and (3S,4R)-S5557, respectively (Scheme 34). Both chromanols were prepared by a regioselective epoxide-ring opening of 196 and 197, using (ethylsulfonyl)(methyl)amide. For the synthesis of HMR1556, the benzyl group was deprotected to give 198, followed by alkylation with 4,4,4-trifluorobutyl iodide.373,399
Scheme 34. Stereocontrolled synthetic approaches for HMR1556 and chromanol 293B.

In addition to the different Kv7 modulator properties exhibited by enantiomers, there are few examples in the literature reporting the discovery of Kv7 inhibitors (or Kv7 activators) by apparently small changes in the chemical structure of Kv7 agonists (or Kv7 antagonists), demonstrating the subtlety of the mechanism for modulating these channels.
The sub-micromolar Kv7.2 inhibitor HN38 represented the result of a hit-to-lead campaign following the discovery of a Kv7 antagonist during the screening of RTG analogs modified at N-2 and N-3 positions. The synthesis of HN38 started with the reductive amination of 2-nitrobenzene-1,4-diamine and 4-fluorobenzaldehyde to afford 200 (Scheme 35).
Scheme 35. Synthesis of HN38.

The nitro group of 200 was then reduced to 201 followed by a Boc-protection step of the aniline to give 202. The free secondary amine of 202 was alkylated with methylallyl bromide, and the resulting intermediate 203 was deprotected and acylated to yield the final target HN38.400
Another example of how a relatively simple structural change resulted in a switch from a Kv7 agonist to a Kv7 antagonist and vice versa was the serendipitous discovery of a potent Kv7.2 activator derived from an SAR study of novel Kv7 inhibitors. As a result of a high-throughput screen of compounds from the Molecular Library Small Molecule Repository (MLSMR), the Kv7.2 inhibitor 204 was identified as a hit compound for investigation as an Alzheimer's disease treatment. The evaluation of analogs showed that the removal of the ethyl group (205) or its replacement with a fluorine atom (206) led to an inhibitor/activator switch (Chart 19). Encouraged by this discovery, a program for Kv7 activators was initiated, and the potent, Kv7.2 selective, BBB permeable inhibitor ML252 was identified in addition to the sub-micromolar Kv7.2 activator 207.
Chart 19. Identification of Kv7.2 antagonist ML252 and Kv7.2 agonist 207 from 204.

As with chromanols 293B and HMR1556,363 the two enantiomers of ML252 showed different inhibitory activities, with ML252 designated as the more potent (S)-enantiomer. This compound was prepared in a one-step synthesis by coupling 2-(1-pyrrolidinyl)aniline (208) and (S)-2-phenylbutyric acid (209) with HATU (Scheme 36).366
Scheme 36. Synthesis of ML252.

Despite ML252's interesting pharmacodynamic profile and attractive brain-to-plasma ratio of 1.9 : 1, further developments are required to address remaining deficiencies such as metabolic instability.366
The modulation of the afterhyperpolarization of electrically excitable cells can also potentially be exploited to repristinate altered excitatory conditions which underlie neuronal disorders such as memory and learning impairment. However, the afterhyperpolarization phase is not fully understood yet, the complexity arising from a sum of different currents acting in different time frames and varying from cell to cell, making it difficult to distinguish the outcomes based on afterhyperpolarization subtypes (slow-, medium-, and fast-afterhyperpolarizations).22,401,402
Since the M-current plays a role in both slow and medium afterhyperpolarizations, the development of UCL-2077 raised considerable interest (Chart 18). The compound was able to suppress slow afterhyperpolarization (sAHP) at low micromolar concentration, without affecting medium afterhyperpolarization.382 Subsequently, UCL-2077 was used to alter the electrophysiology of endocrine cells by modulating currents which can play a role in the sAHP. An understanding of this compound's ability to modulate the sAHP is still under investigation. However, this property is probably derived from a combination of UCL-2077's inhibitory effects on a wide variety of channels. Among these, selective Kv7.1 and Kv7.2 inhibition was observed as well as hERG current suppression (IC50 = 4.7 μM), inhibition of intermediate-conductance Ca2+-activated K+ (IKCa) channels, and a weak effect against the large-conductance Ca2+-activated K+ (BKCa) channels (Table 15).366,403
Kv7 channel selectivity of UCL-2077.
| Kv7.1 | Kv7.1/KCNE1 | Kv7.2 | Kv7.2/7.3 | Kv7.4 | |
|---|---|---|---|---|---|
| IC50 (μM) | 0.06 ± 0.02 | 0.13 ± 0.12 | 9.42 ± 1.12 | >30 | 19.29 ± 11.3 |
In vivo assay data for UCL-2077 are expected to be forthcoming. Due to its unusual mechanism of action, UCL-2077 might become useful as a probe molecule to improve our understanding of the etiology of endocrine and neuroendocrine disfunctions, as well as neuronal diseases.403–405
7. Conclusions
Ion channels have long served as attractive targets for chemical probe and drug development, in part due to their involvement in the regulation of an exceptionally wide spectrum of physiological processes. Voltage-gated potassium channels (Kv) are encoded by 40 different genes, with further division into 12 subfamilies. The Kv7 protein subfamily, encoded by the KCNQ genes, plays a critical role in membrane excitability. Kv7 channel family members are voltage-gated, non-inactivating ion channels that show negative voltage sensitivity compared to other Kv channels. Consequently, this ion channel family contributes to the maintenance of the resting membrane potential in many cell types. Activation of Kv7 channels by cell depolarization or pharmacological events exerts a strong negative control over hyperexcitability. Each Kv7 isoform has a specific distribution within the body, and mutations within the gene family have been implicated in several human diseases.
Kv7.1 is widely expressed in cardiac myocytes and plays a critical role in the repolarization of the cardiac potential. Kv7.1 is also expressed in the inner ear, thyroid gland, lung, ovaries, proximal and distal tubule of the nephron, pancreas, intestinal system, and vascular smooth muscles.
The Kv7.2 channel is located both in the central and peripheral nervous system, with a high density in neocortex and hippocampus. High current amplitude Kv7.2/7.3 heteromers are the most abundant Kv7.2 channel assembly, and particular attention has been given to Kv7.2/7.3 channels for their importance to M-current contributions in neuronal tissues. Non-specific and specific Kv7.2/7.3 modulators include a large number of electron-rich N-acylated anilines and aminopyridines, such as RTG and flupirtine. The control of metabolic and spontaneous oxidation of these scaffolds, as well as channel subtype selectivity and tissue distribution continue to challenge the design ingenuity of medicinal chemists interested to bring new analogs of these clinically validated lead structures to market.
The availability of cryo-EM structures in 2.5–3.3 Å resolutions of human Kv7.2 and Kv7.4 in the apo state as well as in complex with activators and blockers with different binding modes represents a major advancement in our ability to rationally design and optimize current and future modulator chemotypes.115,116 For example, the Kv7.2 cryo-EM ligand analysis illustrates a hydrogen bonding network with Ser303 and Phe305 at the NH2-substituent in RTG as well as the positioning of the fluorine substituent of the benzylamine moiety in a hydrophobic cavity near residue Leu272 (Fig. 7, panels A and B). An increase in the hydrophobic interactions at the latter site might, in part, be responsible for the significantly higher potency of analog RL648-81 (Fig. 7, panel C).270
Fig. 7. Cryo-EM based hydrophobic surface structures of human Kv7 (pdb 7cr2) showing RTG bound to the pore region in overall (panel A) and close-up views (panel B); docking model of RL648-81 derived from this structure (panel C).

The Kv7.4 channel variant is predominantly expressed in homomeric forms in dopaminergic neurons, auditory, and vestibular systems, and as Kv7.4/7.5 heteromers in smooth muscle cells. In contrast, the Kv7.5 channel is expressed in various regions of the brain, both in homomeric forms as well as Kv7.3/7.5 heteromers, and in skeletal muscles as Kv7.4/7.5 heteromers. Recently, N-propargylated RTG was shown to have selectivity for Kv7.4/7.5 subtypes, suggesting further opportunities to explore this chemotype as for Kv7.4 channelopathies as a therapeutic or pharmacological tool compound.
Beyond isoform specificity and physicochemical features, critical challenges in the chemical modulation of Kv7 function lie in the complex regulatory mechanisms of this channel family. Members integrate voltage and secondary messaging of Ca2+ through calmodulin (CaM) and phosphatidylinositol-4,5-bisphosphate (PIP2), both of which are downstream of neuro-transmitter sensitive G protein-coupled receptors (GPCRs). To date, only two drugs have been approved by regulatory agencies and marketed with Kv7 agonism as the main mechanism of action for the treatment of partial on-set seizures in adults: RTG (USA & EU) and the structurally related flupirtine (EU). RTG is a relatively unselective Kv7.2–5 channel opener, and its ability to reduce neuronal hyperexcitability originally led to a rapid transition to market. Despite its efficacy, however, RTG was withdrawn in 2017 due to off-target effects such as blue skin discoloration and eye abnormalities. In 2018, the European Medicines Agency also recommended discontinuing flupirtine because of the risk for serious liver injury.
There is now a substantial void in the treatment of drug-resistant epilepsies and other hyperexcitability disorders despite the therapeutic potential of Kv7 agonists. No available current treatment engages Kv7 channels as a main mechanism of action, despite the extremely broad biomedical relevance of these channels in epilepsy, pain, tinnitus, cancer, cardiovascular disease, and neurodegeneration. Furthermore, many inherited diseases have been attributed to mutations in KCNQ genes, encoding Kv7 proteins with a higher or lower ability to hyperpolarize the cell membrane. Significantly, the range of chemical structure diversity and target sites that has been summarized in this review remains still limited, and there is much room left to explore in ongoing and future medicinal chemistry programs. There is also a clear need to develop new lead structure families based on innovative new synthetic methods.
Conflicts of interest
One of the authors (P. W.) is a co-inventor of patents on the composition of matter and use of RL-81 and related compounds, filed and assigned to the University of Pittsburgh. M. B., P. M. and R. L. have no conflicts to declare.
Acknowledgments
This work was supported in part by DOD Award W81XWH1810623.
Biographies
Biography
Matteo Borgini.

Matteo Borgini graduated from the University of Siena, Italy, in 2015 and received his PhD in Chemical and Pharmaceutical Sciences under the supervision of Professor Maurizio Botta. During his PhD studies he worked on the synthesis of epigenetic modulators as anticancer agents, host protein inhibitors as antiviral compounds and antifungal drugs. He then pursued his postdoctoral studies with Professor Peter Wipf at the University of Pittsburgh, USA. His current work focuses on the synthesis of neuroprotective PARP inhibitors.
Biography
Pravat Mondal.

Pravat Mondal received his M.Sc. in Chemistry from IIT Guwahati, India, in 2009, and his Ph.D. under the guidance of Dr. Narshinha P. Argade from CSIR-National Chemical Laboratory, Pune. in 2016, working on the total synthesis of bioactive indole alkaloids and analogs. He was a research scientist in Syngenta Biosciences, Goa (2016–2017), before he moved to Technion-Israel Institute of Technology as a postdoctoral fellow. There, he worked in supramolecular organic chemistry of hetero-bambusurils in the group of Professor Ofer Reany and Professor Ehud Keinan (2017–2019). Currently, he is a postdoctoral associate with Professor Peter Wipf at University of Pittsburgh where he is working on the synthesis of small molecule modulators for medicinal chemistry targets. His research interests includes synthetic methodology development and the synthesis of bioactive natural and unnatural compounds.
Biography
Ruiting Liu.

Ruiting Liu obtained her B. S. degree in Chemistry at Lanzhou University, China, in 2011. Currently, she is a staff member as well as pursuing her graduate studies at the University of Pittsburgh, working under the direction of Professor Peter Wipf. Her research interests include natural product synthesis, organometallic chemistry and the synthesis of bioactive compounds.
Biography
Peter Wipf.

After receiving a Dipl. Chem. in 1984, Peter Wipf graduated with a Ph.D. from the University of Zürich in Switzerland in 1987 under the direction of Professor Heinz Heimgartner. He then joined the laboratory of Professor Robert E. Ireland at the University of Virginia as a Swiss NSF postdoctoral fellow, and, in 1990, the University of Pittsburgh as an Assistant Professor. Since 2004, he is the Distinguished University Professor of Chemistry at Pittsburgh. His research interests include the total synthesis of natural products, organometallic, heterocyclic and medicinal chemistry.
Notes and references
- Delmas P. Brown D. A. Nat. Rev. Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Barrese V. Stott J. B. Greenwood I. A. Annu. Rev. Pharmacol. Toxicol. 2018;58:625–648. doi: 10.1146/annurev-pharmtox-010617-052912. [DOI] [PubMed] [Google Scholar]
- Dorange I. Swahn B. Annu. Rep. Med. Chem. 2011;46:53–65. [Google Scholar]
- Stott J. B. Jepps T. A. Greenwood I. A. Drug Discovery Today. 2014;19:413–424. doi: 10.1016/j.drudis.2013.12.003. [DOI] [PubMed] [Google Scholar]
- Bagal S. K. Brown A. D. Cox P. J. Omoto K. Owen R. M. Pryde D. C. Sidders B. Skerratt S. E. Stevens E. B. Storer R. I. Swain N. A. J. Med. Chem. 2013;56:593–624. doi: 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- Miceli F. Soldovieri M. V. Ambrosino P. Manocchio L. Mosca I. Taglialatela M. Curr. Med. Chem. 2018;25:2637–2660. doi: 10.2174/0929867324666171012122852. [DOI] [PubMed] [Google Scholar]
- Miceli F. Cilio M. R. Taglialatela M. Bezanilla F. Channels. 2009;3:274–283. doi: 10.4161/chan.3.4.9477. [DOI] [PubMed] [Google Scholar]
- Battefeld A. Tran B. T. Gavrilis J. Cooper E. C. Kole M. H. J. Neurosci. 2014;34:3719–3732. doi: 10.1523/JNEUROSCI.4206-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H. Castle N. A. Pardo L. A. Nat. Rev. Drug Discovery. 2009;8:982–1001. doi: 10.1038/nrd2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. Pharmacol. Ther. 2001;90:1–19. doi: 10.1016/S0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Brueggemann L. I. Haick J. M. Cribbs L. L. Byron K. L. Mol. Pharmacol. 2014;86:330–341. doi: 10.1124/mol.114.093799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder B. C. Hechenberger M. Weinreich F. Kubisch C. Jentsch T. J. J. Biol. Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Lerche C. Scherer C. R. Seebohm G. Derst C. Wei A. D. Busch A. E. Steinmeyer K. J. Biol. Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Hu H. Vervaeke K. Storm J. F. J. Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O. Lara L. S. Gamper N. Hilgemann D. W. Jaffe D. B. Shapiro M. S. J. Physiol. 2006;575:49–67. doi: 10.1113/jphysiol.2006.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas J. Harper A. A. Trequattrini C. Adams D. J. J. Neurophysiol. 1997;78:1890–1902. doi: 10.1152/jn.1997.78.4.1890. [DOI] [PubMed] [Google Scholar]
- Brown D. A. Passmore G. M. Br. J. Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. Vo T. Rotstein H. G. McCarthy M. M. Kopell N. J. Math. Neurosci. 2018;8:13. doi: 10.1186/s13408-018-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doan T. N. Kunze D. L. J. Physiol. 1999;514:125–138. doi: 10.1111/j.1469-7793.1999.125af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupica C. R. Bell J. A. Hoffman A. F. Watson P. L. J. Neurophysiol. 2001;86:261–268. doi: 10.1152/jn.2001.86.1.261. [DOI] [PubMed] [Google Scholar]
- Aponte Y. Lien C. C. Reisinger E. Jonas P. J. Physiol. 2006;574:229–243. doi: 10.1113/jphysiol.2005.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis A. V. Heidenreich M. Kharkovets T. Spitzmaul G. Jensen H. S. Nicoll R. A. Jentsch T. J. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S. Appel S. B. J. Neurophysiol. 2006;96:535–543. doi: 10.1152/jn.00574.2005. [DOI] [PubMed] [Google Scholar]
- Gu N. Vervaeke K. Hu H. Storm J. F. J. Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters H. C. Hu H. Pongs O. Storm J. F. Isbrandt D. Nat. Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Schwarz J. R. Glassmeier G. Cooper E. C. Kao T. C. Nodera H. Tabuena D. Kaji R. Bostock H. J. Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M. M. Migliore M. Valencia I. Cooper E. C. Brown D. A. Proc. Natl. Acad. Sci. U. S. A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire M. Castaldo P. D'Amico M. Preziosi P. Annunziato L. Taglialatela M. J. Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H. Vervaeke K. Storm J. F. J. Physiol. 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon B. Yarom Y. Trends Neurosci. 2000;23:216–222. doi: 10.1016/S0166-2236(00)01547-2. [DOI] [PubMed] [Google Scholar]
- Marcelin B. Chauvière L. Becker A. Migliore M. Esclapez M. Bernard C. Neurobiol. Dis. 2009;33:436–447. doi: 10.1016/j.nbd.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Vera J. Pezzoli M. Pereira U. Bacigalupo J. Sanhueza M. PLoS One. 2014;9:e85826. doi: 10.1371/journal.pone.0085826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhikevich E. M. Desai N. S. Walcott E. C. Hoppensteadt F. C. Trends Neurosci. 2003;26:161–167. doi: 10.1016/S0166-2236(03)00034-1. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos M. A. Rigas P. Tawara-Hirata Y. J. Physiol. 2007;578:173–191. doi: 10.1113/jphysiol.2006.119016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L. Liu X. D. Acta Pharmacol. Sin. 2016;37:67–74. doi: 10.1038/aps.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M. Jentsch T. J. Friedrich T. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R. J. Clark K. A. Holton J. M. Minor Jr. D. L. Neuron. 2007;53:633–765. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R. Haitin Y. Shamgar L. Fernández-Alonso M. C. Martos A. Chomsky-Hecht O. Rivas G. Attali B. Hirsch J. A. J. Biol. Chem. 2008;283:5815–5830. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- Jentsch T. J. Nat. Rev. Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Hughes S. Marsh S. J. Tinker A. Brown D. A. Pflugers Arch. 2007;455:115–124. doi: 10.1007/s00424-007-0259-6. [DOI] [PubMed] [Google Scholar]
- Haitin Y. Yisharel I. Malka E. Shamgar L. Schottelndreier H. Peretz A. Paas Y. Attali B. PLoS One. 2008;3:e1935. doi: 10.1371/journal.pone.0001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldovieri M. V. Miceli F. Taglialatela M. Physiology. 2011;26:365–376. doi: 10.1152/physiol.00009.2011. [DOI] [PubMed] [Google Scholar]
- Sanguinetti M. C. Curran M. E. Zou A. Shen J. Spector P. S. Atkinson D. L. Keating M. T. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Bett G. C. Morales M. J. Beahm D. L. Duffey M. E. Rasmusson R. L. J. Physiol. 2006;576:755–767. doi: 10.1113/jphysiol.2006.116012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen A. P. Steffensen A. B. Grunnet M. Olesen S. P. Biophys. J. 2011;101:818–827. doi: 10.1016/j.bpj.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng F. L. Davis A. J. Jepps T. A. Harhun M. I. Yeung S. Y. Wan A. Reddy M. Melville D. Nardi A. Khong T. K. Greenwood I. A. Br. J. Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-de-Souza J. L. Bezanilla F. J. Gen. Physiol. 2018;150:307–321. doi: 10.1085/jgp.201711882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P. Eldstrom J. Shi J. Zhong L. McFarland K. Gao Y. Fedida D. Cui J. Nat. Commun. 2017;8:1730. doi: 10.1038/s41467-017-01911-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman M. A. Kasimova M. A. McFarland K. Beller Z. Hou P. Kinser H. E. Liang H. Zhang G. Shi J. Tarek M. Cui J. eLife. 2014;3:e03606. doi: 10.7554/eLife.03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. Pugsley M. K. Fermini B. Curtis M. J. Koerner J. Accardi M. Authier S. J. Pharmacol. Toxicol. Methods. 2017;87:11–23. doi: 10.1016/j.vascn.2017.04.002. [DOI] [PubMed] [Google Scholar]
- Authier S. Pugsley M. K. Koerner J. E. Fermini B. Redfern W. S. Valentin J. P. Vargas H. M. Leishman D. J. Correll K. Curtis M. J. J. Pharmacol. Toxicol. Methods. 2017;86:34–43. doi: 10.1016/j.vascn.2017.02.021. [DOI] [PubMed] [Google Scholar]
- Diao Y. Tian Y. Han S. Zhang N. Li J. Yin Y. Neuropsychiatry. 2017;7:26–31. doi: 10.4172/Neuropsychiatry.1000175. [DOI] [Google Scholar]
- Bal M. Zhang J. Zaika O. Hernandez C. C. Shapiro M. S. J. Biol. Chem. 2008;45:30668–30676. doi: 10.1074/jbc.M805216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills T. A. Greenwood S. L. Devlin G. Shweikh Y. Robinson M. Cowley E. Hayward C. E. Cottrell E. C. Tropea T. Brereton M. F. Dalby-Brown W. Wareing M. Placenta. 2015;36:638–644. doi: 10.1016/j.placenta.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Brueggemann L. I. Kakad P. P. Love R. B. Solway J. Dowell M. L. Cribbs L. L. Byron K. L. Am. J. Physiol. 2012;302:120–132. doi: 10.1152/ajplung.00194.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipavec V. Martire M. Barrese V. Taglialatela M. Currò D. Pharmacol. Res. 2011;64:397–409. doi: 10.1016/j.phrs.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps T. A. Greenwood I. A. Moffatt J. D. Sanders K. M. Ohya S. Am. J. Physiol. 2009;297:107–115. doi: 10.1152/ajpgi.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum L. A. Greenwood I. A. Tribe R. M. Pflugers Arch. 2009;457:1111–1120. doi: 10.1007/s00424-008-0567-5. [DOI] [PubMed] [Google Scholar]
- McCallum L. A. Pierce S. L. England S. K. Greenwood I. A. Tribe R. M. J. Cell. Mol. Med. 2011;15:577–586. doi: 10.1111/j.1582-4934.2010.01021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svalø J. Sheykhzade M. Nordling J. Matras C. Bouchelouche P. PLoS One. 2015;10:e0117350. doi: 10.1371/journal.pone.0117350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evseev I. A. Semenov I. Archer C. R. Medina J. L. Dube P. H. Shapiro M. S. Brenner R. Front. Physiol. 2013;4:277. doi: 10.3389/fphys.2013.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepps T. A. Olesen S. P. Greenwood I. A. Dalsgaard T. Br. J. Pharmacol. 2016;173:1478–1490. doi: 10.1111/bph.13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyroud N. Tesson F. Denjoy I. Leibovici M. Donger C. Barhanin J. Fauré S. Gary F. Coumel P. Petit C. Schwartz K. Guicheney P. Nat. Genet. 1997;15:186–189. doi: 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Kosenko A. Kang S. Smith I. M. Greene D. L. Langeberg L. K. Scott J. D. Hoshi N. EMBO J. 2012;31:3147–3156. doi: 10.1038/emboj.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Gamper N. Hilgemann D. W. Shapiro M. S. J. Neurosci. 2005;25:9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberdi A. Gomis-Perez C. Bernardo-Seisdedos G. Alaimo A. Malo C. Aldaregia J. Lopez-Robles C. Areso P. Butz E. Wahl-Schott C. Villarroel A. J. Cell Sci. 2015;128:4014–4023. doi: 10.1242/jcs.176420. [DOI] [PubMed] [Google Scholar]
- Hoshi N. Zhang J. S. Omaki M. Takeuchi T. Yokoyama S. Wanaverbecq N. Langeberg L. K. Yoneda Y. Scott J. D. Brown D. A. Higashida H. Nat. Neurosci. 2003;6:564–571. doi: 10.1038/nn1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z. Kao T. Horvath Z. Lemos J. Sul J. Y. Cranstoun S. D. Bennett V. Scherer S. S. Cooper E. C. J. Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C. C. Zaika O. Shapiro M. S. J. Gen. Physiol. 2008;132:361–381. doi: 10.1085/jgp.200810007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. S. Duignan K. M. Hawryluk J. M. Soh H. Tzingounis A. V. Biophys. J. 2016;110:1089–1098. doi: 10.1016/j.bpj.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene D. L. Hoshi N. Cell. Mol. Life Sci. 2017;74:495–508. doi: 10.1007/s00018-016-2359-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winks J. S. Hughes S. Filippov A. K. Tatulian L. Abogadie F. C. Brown D. A. Marsh S. J. J. Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B. C. Inoue T. Meyer T. Hille B. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion N. V. Annu. Rev. Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- Gamper N. Shapiro M. S. J. Gen. Physiol. 2003;122:17–31. doi: 10.1085/jgp.200208783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N. Li Y. Shapiro M. S. Mol. Biol. Cell. 2005;16:3538–3551. doi: 10.1091/mbc.e04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroz D. Rodriguez N. Choveau F. Baró I. Mérot J. Loussouarn G. J. Physiol. 2008;586:1785–1789. doi: 10.1113/jphysiol.2007.148254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. A. J. Physiol. 2008;586:1781–1783. doi: 10.1113/jphysiol.2008.153007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman A. Thouta S. Mancini G. M. S. Naidu S. van Slegtenhorst M. McWalter K. Person R. Mwenifumbo J. Salvarinova R. Guella I. McKenzie M. B. Datta A. Connolly M. B. Kalkhoran S. M. Poburko D. Friedman J. M. Farrer M. J. Demos M. Desai S. Claydon T. Am. J. Hum. Genet. 2017;101:65–74. doi: 10.1016/j.ajhg.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maljevic S. Wuttke T. V. Seebohm G. Lerche H. Pflugers Arch. 2010;460:277–288. doi: 10.1007/s00424-010-0831-3. [DOI] [PubMed] [Google Scholar]
- Maljevic S. Lerche H. Prog. Brain Res. 2014;213:17–53. doi: 10.1016/B978-0-444-63326-2.00002-8. [DOI] [PubMed] [Google Scholar]
- Gilling M. Rasmussen H. B. Calloe K. Sequeira A. F. Baretto M. Oliveira G. Almeida J. Lauritsen M. B. Ullmann R. Boonen S. E. Brondum-Nielsen K. Kalscheuer V. M. Tümer Z. Vicente A. M. Schmitt N. Tommerup N. Front. Genet. 2013;4:54. doi: 10.3389/fgene.2013.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C. Link A. Future Med. Chem. 2019;11:337–355. doi: 10.4155/fmc-2018-0350. [DOI] [PubMed] [Google Scholar]
- Szelenyi I. Inflammation Res. 2013;62:251–258. doi: 10.1007/s00011-013-0592-5. [DOI] [PubMed] [Google Scholar]
- Kornhuber J. Bleich S. Wiltfang J. Maler M. Parsons C. G. J. Neural Transm. 1999;106:857–867. doi: 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- Bock C. Surur A. S. Beirow K. Kindermann M. K. Schulig L. Bodtke A. Bednarski P. J. Link A. ChemMedChem. 2019;14:952–964. doi: 10.1002/cmdc.201900112. [DOI] [PubMed] [Google Scholar]
- Kim R. Y. Yau M. C. Galpin J. D. Seebohm G. Ahern C. A. Pless S. A. Kurata H. T. Nat. Commun. 2015;6:8116. doi: 10.1038/ncomms9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann W. M. Kern U. Aigner M. Postgrad. Med. J. 1987;63:87–103. [PubMed] [Google Scholar]
- Kaplan S. Ehlken B. Hamann X. Curr. Med. Res. Opin. 2019;35:33–40. doi: 10.1080/03007995.2018.1499507. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency, Withdrawal of pain medicine flupirtine endorsed, www.ema.europa.eu/docs/en GB/document library/Press release/2018/03/WC500246353.pdf, (accessed May 2020)
- Surur A. S. Bock C. Beirow K. Wurm K. Schulig L. Kindermann M. K. Siegmund W. Bednarski P. J. Link A. Org. Biomol. Chem. 2019;17:4512–4522. doi: 10.1039/C9OB00511K. [DOI] [PubMed] [Google Scholar]
- Surur A. S. Beirow K. Bock C. Schulig L. Kindermann M. K. Bodtke A. Siegmund W. Bednarski P. J. Link A. ChemistryOpen. 2019;8:41–44. doi: 10.1002/open.201800244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom C. E. Carmant L. Cold Spring Harbor Perspect. Med. 2015;5:a022426. doi: 10.1101/cshperspect.a022426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee for medicinal products for human use, Assessment report Trobalt, www.ema.europa.eu/docs/en GB/document library/EPAR - Public assessment report/human/001245/WC500104839.pdf, (accessed May 2020)
- Tatulian L. Delmas P. Abogadie F. C. Brown D. A. J. Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe M. J. Large C. H. Sankar R. Epilepsia. 2012;53:412–424. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- Barrese V. Miceli F. Soldovieri M. V. Ambrosino P. Iannotti F. A. Cilio M. R. Taglialatela M. Clin. Pharmacol. 2010;2:225–236. doi: 10.2147/CPAA.S15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen B. H. Schmitt N. Calloe K. Dalby-Brown W. Grunnet M. Olesen S. P. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Wuttke T. V. Seebohm G. Bail S. Maljevic S. Lerche H. Mol. Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Shi S. Li J. Sun F. Chen Y. Pang C. Geng Y. Qi J. Guo S. Wang X. Zhang H. Zhan Y. An H. J. Membr. Biol. 2020;253:167–181. doi: 10.1007/s00232-020-00113-6. [DOI] [PubMed] [Google Scholar]
- Syeda R. Santos J. S. Montal M. J. Biol. Chem. 2016;291:2931–2937. doi: 10.1074/jbc.M115.683185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob R. Krieglstein J. Br. J. Pharmacol. 1997;122:1333–1338. doi: 10.1038/sj.bjp.0701519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger F. Bajric M. Salzer I. Dorostkar M. M. Khan D. Pollak D. D. Kubista H. Boehm S. Koenig X. Br. J. Pharmacol. 2015;172:4946–4958. doi: 10.1111/bph.13262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi I. Nickel B. Borbe H. O. Brune K. Br. J. Pharmacol. 1989;97:835–842. doi: 10.1111/j.1476-5381.1989.tb12023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrøder R. L. Jespersen T. Christophersen P. Strøbaek D. Jensen B. S. Olesen S. P. Neuropharmacology. 2001;40:888–898. doi: 10.1016/S0028-3908(01)00029-6. [DOI] [PubMed] [Google Scholar]
- Dupuis D. S. Schrøder R. L. Jespersen T. Christensen J. K. Christophersen P. Jensen B. S. Olesen S. P. Eur. J. Pharmacol. 2002;437:129–137. doi: 10.1016/S0014-2999(02)01287-6. [DOI] [PubMed] [Google Scholar]
- Wickenden A. D. Zou A. Wagoner P. K. Jegla T. Br. J. Pharmacol. 2001;132:381–384. doi: 10.1038/sj.bjp.0703861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stas J. I. Bocksteins E. Jensen C. S. Schmitt N. Snyders D. J. Sci. Rep. 2016;6:35080. doi: 10.1038/srep35080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanovic I. M. Yonekawa W. D. Kupferberg H. J. Epilepsy Res. 1995;22:167–173. doi: 10.1016/0920-1211(95)00050-X. [DOI] [PubMed] [Google Scholar]
- Treven M. Koenig X. Assadpour E. Gantumur E. Meyer C. Hilber K. Boehm S. Kubista H. Epilepsia. 2015;56:647–657. doi: 10.1111/epi.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribkoff V. K. Starrett J. E. Dworetzky S. I. Hewawasam P. Boissard C. G. Cook D. A. Frantz S. W. Heman K. Hibbard J. R. Huston K. Johnson G. Krishnan B. S. Kinney G. G. Lombardo L. A. Meanwell N. A. Molinoff P. B. Myers R. A. Moon S. L. Ortiz A. Pajor L. Pieschl R. L. Post-Munson D. J. Signor L. J. Srinivas N. Taber M. T. Thalody G. Trojnacki J. T. Wiener H. Yeleswaram K. Yeola S. W. Nat. Med. 2001;7:471–477. doi: 10.1038/86546. [DOI] [PubMed] [Google Scholar]
- Mattmann M. E. Yu H. Lin Z. Xu K. Huang X. Long S. Wu M. McManus O. B. Engers D. W. Le U. M. Li M. Lindsley C. W. Hopkins C. R. Bioorg. Med. Chem. Lett. 2012;22:5936–5941. doi: 10.1016/j.bmcl.2012.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Lin Z., Xu K., Huang X., Long S., Wu M., McManus O. B., Le Engers J., Mattmann M. E., Engers D. W., Le U. M., Lindsley C. W., Hopkins C. R. and Li M., Probe Reports from the NIH Molecular Libraries Program [Internet], 2013
- Yu H. Wu M. Townsend S. D. Zou B. Long S. Daniels J. S. McManus O. B. Li M. Lindsley C. W. Hopkins C. R. ACS Chem. Neurosci. 2011;2:572–577. doi: 10.1021/cn200065b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Zhang Q. Guo P. Fu J. Mei L. Lv D. Wang J. Lai D. Ye S. Yang H. Guo J. Cell Res. 2021;31:52–61. doi: 10.1038/s41422-020-00410-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T. Wu K. Yue Z. Wang Y. Zhang F. Shen H. Mol. Cell. 2021;81:25–37. doi: 10.1016/j.molcel.2020.10.037. [DOI] [PubMed] [Google Scholar]
- Clark S. Antell A. Kaufman K. Ther. Adv. Drug Saf. 2015;6:15–19. doi: 10.1177/2042098614560736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beacher N. G. Brodie M. J. Goodall C. BMC Oral Health. 2015;15:122. doi: 10.1186/s12903-015-0102-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Medicines Agency, Trobalt: procedual steps taken and scientific information after the authorizaition, www.ema.europa.eu/docs/en GB/document library/EPAR - Procedural steps taken and scientific information after authorisation/human/001245/WC500120129.pdf, (accessed May 2020)
- Brickel N. Hewett K. Rayner K. McDonald S. De'Ath J. Daniluk J. Joshi K. Boll M. C. Tiamkao S. Vorobyeva O. Cooper J. Epilepsy Behav. 2020;102:106580. doi: 10.1016/j.yebeh.2019.106580. [DOI] [PubMed] [Google Scholar]
- Korsgaard M. P. Hartz B. P. Brown W. D. Ahring P. K. Strøbaek D. Mirza N. R. J. Pharmacol. Exp. Ther. 2005;314:282–292. doi: 10.1124/jpet.105.083923. [DOI] [PubMed] [Google Scholar]
- Jensen B. S. CNS Drug Rev. 2002;8:353–360. doi: 10.1111/j.1527-3458.2002.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://www.clinicaltrials.gov/ct2/show/NCT03887325, (accessed May 2020)
- Wickenden A. D. Krajewski J. L. London B. Wagoner P. K. Wilson W. A. Clark S. Roeloffs R. McNaughton-Smith G. Rigdon G. C. Mol. Pharmacol. 2008;73:977–986. doi: 10.1124/mol.107.043216. [DOI] [PubMed] [Google Scholar]
- Blom S. M. Schmitt N. Jensen H. S. Pharmacology. 2010;86:174–181. doi: 10.1159/000317525. [DOI] [PubMed] [Google Scholar]
- Padilla K. Wickenden A. D. Gerlach A. C. McCormack K. Neurosci. Lett. 2009;465:138–142. doi: 10.1016/j.neulet.2009.08.071. [DOI] [PubMed] [Google Scholar]
- Roeloffs R. Wickenden A. D. Crean C. Werness S. McNaughton-Smith G. Stables J. McNamara J. O. Ghodadra N. Rigdon G. C. J. Pharmacol. Exp. Ther. 2008;326:818–828. doi: 10.1124/jpet.108.137794. [DOI] [PubMed] [Google Scholar]
- Dalby-Brown W. Jessen C. Hougaard C. Jensen M. L. Jacobsen T. A. Nielsen K. S. Erichsen H. K. Grunnet M. Ahring P. K. Christophersen P. Strøbæk D. Jørgensen S. Eur. J. Pharmacol. 2013;709:52–63. doi: 10.1016/j.ejphar.2013.03.039. [DOI] [PubMed] [Google Scholar]
- Kardosh A. Wang W. Uddin J. Petasis N. A. Hofman F. M. Chen T. C. Schonthal A. H. Cancer Biol. Ther. 2005;4:571–582. doi: 10.4161/cbt.4.5.1699. [DOI] [PubMed] [Google Scholar]
- Wickenden A. D. Yu W. Zou A. Jegla T. Wagoner P. K. Mol. Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- St John Smith E. J. Neurol. 2018;265:231–238. doi: 10.1007/s00415-017-8641-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilliox L. A. Continuum. 2017;23:512–532. doi: 10.1212/CON.0000000000000462. [DOI] [PubMed] [Google Scholar]
- Cohen S. P. Mao J. BMJ. 2014;348:f7656. doi: 10.1136/bmj.f7656. [DOI] [PubMed] [Google Scholar]
- Dworkin R. H. Clin. J. Pain. 2002;18:343–349. doi: 10.1097/00002508-200211000-00001. [DOI] [PubMed] [Google Scholar]
- Wieseler-Frank J. Maier S. F. Watkins L. R. Neurosignals. 2005;14:166–174. doi: 10.1159/000087655. [DOI] [PubMed] [Google Scholar]
- Colloca L. Ludman T. Bouhassira D. Baron R. Dickenson A. H. Yarnitsky D. Freeman R. Truini A. Attal N. Finnerup N. B. Eccleston C. Kalso E. Bennett D. L. Dworkin R. H. Raja S. N. Nat. Rev. Dis. Primers. 2017;3:17002. doi: 10.1038/nrdp.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti F. Truini A. Cruccu G. J. Headache Pain. 2006;7:61–69. doi: 10.1007/s10194-006-0287-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J. Erol F. Viatchenko-Karpinski V. Kanda H. Gu J. G. Mol. Pain. 2017;13:1–10. doi: 10.1177/1744806917724715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd-Elsayed A. Jackson M. Gu S. L. Fiala K. Gu J. Mol. Pain. 2019;15:1744806919864256. doi: 10.1177/1744806919864256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd-Elsayed A. A. Ikeda R. Jia Z. Ling J. Zuo X. Li M. Gu J. G. Mol. Pain. 2015;11:45. doi: 10.1186/s12990-015-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn-Munro G. Jensen B. S. Eur. J. Pharmacol. 2003;460:109–116. doi: 10.1016/S0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Lawson K. Expert Opin. Invest. Drugs. 2002;11:1437–1445. doi: 10.1517/13543784.11.10.1437. [DOI] [PubMed] [Google Scholar]
- Stoll A. L. Psychosomatics. 2000;41:371–372. doi: 10.1176/appi.psy.41.4.371. [DOI] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT00612105, (accessed May 2020)
- Zhang F. Liu S. Jin L. Tang L. Zhao X. Yang T. Wang Y. Huo B. Liu R. Li H. Pharmacology. 2020;105:471–476. doi: 10.1159/000505934. [DOI] [PubMed] [Google Scholar]
- Yu T. Li L. Liu H. Li H. Liu Z. Li Z. Mol. Pain. 2018;14:1744806918793229. doi: 10.1177/1744806918793229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seretny M. Currie G. L. Sena E. S. Ramnarine S. Grant R. MacLeod M. R. Colvin L. A. Fallon M. Pain. 2014;155:2461–2470. doi: 10.1016/j.pain.2014.09.020. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith P. Curr. Opin. Support Palliat. Care. 2011;5:1–7. doi: 10.1097/SPC.0b013e328342f9cc. [DOI] [PubMed] [Google Scholar]
- Li L. Li J. Zuo Y. Dang D. Frost J. A. Yang Q. J. Pain. 2019;20:528–539. doi: 10.1016/j.jpain.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore A. Neufeld-Cohen A. Egoz I. Baranes S. Gez R. Grauer E. Chapman S. Lazar S. Toxicol. Appl. Pharmacol. 2020;395:114963. doi: 10.1016/j.taap.2020.114963. [DOI] [PubMed] [Google Scholar]
- Haut S. R. Lipton R. B. Epilepsy Behav. 2013;28:241–242. doi: 10.1016/j.yebeh.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Zhang X. Jakubowski M. Buettner C. Kainz V. Gold M. Burstein R. Epilepsy Behav. 2013;28:243–248. doi: 10.1016/j.yebeh.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston G. N. Cereb. Cortex. 2003;13:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Peng H. Bian X. L. Ma F. C. Wang K. W. Acta Pharmacol. Sin. 2017;38:1248–1256. doi: 10.1038/aps.2017.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A. K. Juarez B. Ku S. M. Zhang H. Calizo R. C. Walsh J. J. Chaudhury D. Zhang S. Hawkins A. Dietz D. M. Murrough J. W. Ribadeneira M. Wong E. H. Neve R. L. Han M. H. Nat. Commun. 2016;7:11671. doi: 10.1038/ncomms11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A. Costi S. Morris L. S. Van Dam N. T. Kautz M. Whitton A. E. Friedman A. K. Collins K. A. Ahle G. Chadha N. Do B. Pizzagalli D. A. Iosifescu D. V. Nestler E. J. Han M. H. Murrough J. W. Mol. Psychiatry. 2020;25:1323–1333. doi: 10.1038/s41380-018-0283-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H. Stellwagen D. J. Neurosci. 2013;33:15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J. Martel J. Lin C. S. Chang C. J. Wu T. R. Lu C. C. Ko Y. F. Lai H. C. Ojcius D. M. Young J. D. Brain, Behav., Immun. 2018;69:1–8. doi: 10.1016/j.bbi.2017.08.026. [DOI] [PubMed] [Google Scholar]
- Feng M. Crowley N. A. Patel A. Guo Y. Bugni S. E. Luscher B. Neuroscience. 2019;406:109–125. doi: 10.1016/j.neuroscience.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeberlein B. S. Harris T. Clin. Pharmacol. Ther. 2015;98:492–501. doi: 10.1002/cpt.195. [DOI] [PubMed] [Google Scholar]
- Solanki I. Parihar P. Parihar M. S. Neurochem. Int. 2016;95:100–108. doi: 10.1016/j.neuint.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Bian G. Liu J. Guo Y. Yang Y. Li L. Qiao H. Li W. Xu T. Zhang Q. Neuropharmacology. 2020;168:108012. doi: 10.1016/j.neuropharm.2020.108012. [DOI] [PubMed] [Google Scholar]
- Du C. X. Liu J. Guo Y. Zhang L. Zhang Q. J. Neurosci. Lett. 2018;662:162–166. doi: 10.1016/j.neulet.2017.10.027. [DOI] [PubMed] [Google Scholar]
- Sander S. E. Lemm C. Lange N. Hamann M. Richter A. Neuropharmacology. 2012;62:1052–1061. doi: 10.1016/j.neuropharm.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Sander S. E. Lambrecht C. Richter A. Neurosci. Lett. 2013;545:59–63. doi: 10.1016/j.neulet.2013.04.017. [DOI] [PubMed] [Google Scholar]
- Cooper E. C. Harrington E. Jan Y. N. Jan L. Y. J. Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper J. M. Bolam J. P. Curr. Opin. Neurobiol. 2004;14:685–692. doi: 10.1016/j.conb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Kreir M. De Bondt A. Van den Wyngaert I. Teuns G. Lu H. R. Gallacher D. J. Eur. J. Pharmacol. 2019;858:172474. doi: 10.1016/j.ejphar.2019.172474. [DOI] [PubMed] [Google Scholar]
- Wainger B. J. Kiskinis E. Mellin C. Wiskow O. Han S. S. Sandoe J. Perez N. P. Williams L. A. Lee S. Boulting G. Berry J. D. Brown R. H. Cudkowicz M. E. Bean B. P. Eggan K. Woolf C. J. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk M. O. Heuberger J. A. A. C. Sleutjes B. T. H. M. Ziagkos D. van den Berg L. H. Ferguson T. A. Franssen H. Groeneveld G. J. Clin. Pharmacol. Ther. 2018;104:1136–1145. doi: 10.1002/cpt.1096. [DOI] [PubMed] [Google Scholar]
- Okano H. Yasuda D. Fujimori K. Morimoto S. Takahashi S. Trends Pharmacol. Sci. 2020;41:99–109. doi: 10.1016/j.tips.2019.12.002. [DOI] [PubMed] [Google Scholar]
- Brown R. H. Al-Chalabi A. N. Engl. J. Med. 2017;377:162–172. doi: 10.1056/NEJMra1603471. [DOI] [PubMed] [Google Scholar]
- Morimoto S. Takahashi S. Fukushima K. Saya H. Suzuki N. Aoki M. Okano H. Nakahara J. Regen. Ther. 2019;11:143–166. doi: 10.1016/j.reth.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr J. Wernecke K. D. Würfel J. Bellmann-Strobl J. Siffrin V. Sättler M. B. Simons M. Linsa A. Tumani H. Paul F. Front. Neurol. 2018;9:842. doi: 10.3389/fneur.2018.00842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C. Cummings D. M. André V. M. Holley S. M. Levine M. S. ASN Neuro. 2010;2:e00033. doi: 10.1042/AN20090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenstock S. Dudanova I. Front. Neurosci. 2020;14:82. doi: 10.3389/fnins.2020.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L. Sathasivam K. Seller M. Cozens B. Harper A. Hetherington C. Lawton M. Trottier Y. Lehrach H. Davies S. W. Bates G. P. Cell. 1996;87:493–506. doi: 10.1016/S0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Cao Y. Bartolomé-Martín D. Rotem N. Rozas C. Dellal S. S. Chacon M. A. Kadriu B. Gulinello M. Khodakhah K. Faber D. S. Proc. Natl. Acad. Sci. U. S. A. 2015;112:2239–2244. doi: 10.1073/pnas.1405748112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley R. W. Medel R. Kassell N. F. Dumont A. S. Drug Discovery Today. 2008;13:254–260. doi: 10.1016/j.drudis.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Mani B. K. O'Dowd J. Kumar L. Brueggemann L. I. Ross M. Byron K. L. J. Cardiovasc. Pharmacol. 2013;61:51–62. doi: 10.1097/FJC.0b013e3182771708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L. L. Diaz-Arrastia R. Vespa P. M. Neurosurg. Clin. N. Am. 2016;27:499–508. doi: 10.1016/j.nec.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Vespa P. Tubi M. Claassen J. Buitrago-Blanco M. McArthur D. Velazquez A. G. Tu B. Prins M. Nuwer M. Ann. Neurol. 2016;79:579–590. doi: 10.1002/ana.24606. [DOI] [PubMed] [Google Scholar]
- Vigil F. A. Carver C. M. Shapiro M. S. Front. Physiol. 2020;11:688. doi: 10.3389/fphys.2020.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P. Kumar D. Jha S. K. Jha N. K. Ambasta R. K. Adv. Protein Chem. Struct. Biol. 2016;103:97–136. doi: 10.1016/bs.apcsb.2015.10.006. [DOI] [PubMed] [Google Scholar]
- Shah N. H. Aizenman E. Transl. Stroke Res. 2014;5:38–58. doi: 10.1007/s12975-013-0297-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. Mucke L. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durán-González J. Michi E. D. Elorza B. Perez-Córdova M. G. Pacheco-Otalora L. F. Touhami A. Paulson P. Perry G. Murray I. V. Colom L. V. Neurobiol. Aging. 2013;34:2071–2076. doi: 10.1016/j.neurobiolaging.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão R. N. Colom L. V. Borgius L. Kiehn O. Fisahn A. Neurobiol. Aging. 2012;33:2046–2061. doi: 10.1016/j.neurobiolaging.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Chang X. Wang J. Jiang H. Shi L. Xie J. Front. Mol. Neurosci. 2019;12:141. doi: 10.3389/fnmol.2019.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan-Barmatz V. Nahon-Crystal E. Shteinfer-Kuzmine A. Gupta R. Pharmacol. Res. 2018;131:87–101. doi: 10.1016/j.phrs.2018.03.010. [DOI] [PubMed] [Google Scholar]
- Atri A. Goldfarb D. Sheard S. Shaughnessy L. J. Clin. Psychiatry. 2019;80:MS18002AH3C. doi: 10.4088/JCP.MS18002AH3C. [DOI] [PubMed] [Google Scholar]
- Briggs R. Kennelly S. P. Clin. Med. 2016;16:247–253. doi: 10.7861/clinmedicine.16-3-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Xue B. Wang J. Liu H. Shi L. Xie J. Neurosci. Bull. 2018;34:341–348. doi: 10.1007/s12264-017-0177-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge S. I. Smith P. J. Stewart P. E. Bever C. T. Recent Pat. CNS Drug Discovery. 2007;2:200–228. doi: 10.2174/157488907782411765. [DOI] [PubMed] [Google Scholar]
- Raza C. Anjum R. Shakeel N. U. A. Life Sci. 2019;226:77–90. doi: 10.1016/j.lfs.2019.03.057. [DOI] [PubMed] [Google Scholar]
- Cacabelos R. Expert Opin. Drug Discovery. 2018;13:523–538. doi: 10.1080/17460441.2018.1457645. [DOI] [PubMed] [Google Scholar]
- Carrillo-Reid L. Day M. Xie Z. Melendez A. E. Kondapalli J. Plotkin J. L. Wokosin D. L. Chen Y. Kress G. J. Kaplitt M. Ilijic E. Guzman J. N. Chan C. S. Surmeier D. J. eLife. 2019;8:e40818. doi: 10.7554/eLife.40818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garret M. Du Z. Chazalon M. Cho Y. H. Baufreton J. CNS Neurosci. Ther. 2018;24:292–300. doi: 10.1111/cns.12826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Hernández A. Ávila G. Rueda A. Eur. J. Pharmacol. 2014;739:26–38. doi: 10.1016/j.ejphar.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Bachoud-Lévi A. C. Ferreira J. Massart R. Youssov K. Rosser A. Busse M. Craufurd D. Reilmann R. De Michele G. Rae D. Squitieri F. Seppi K. Perrine C. Scherer-Gagou C. Audrey O. Verny C. Burgunder J. M. Front. Neurol. 2019;10:710. doi: 10.3389/fneur.2019.00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S. M. Walterfang M. Velakoulis D. Looi J. C. Australas. Psychiatry. 2018;26:376–380. doi: 10.1177/1039856218766120. [DOI] [PubMed] [Google Scholar]
- Dickey A. S. La Spada A. R. Am. J. Med. Genet., Part A. 2018;176:842–861. doi: 10.1002/ajmg.a.38494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friese M. A. Schattling B. Fugger L. Nat. Rev. Neurol. 2014;10:225–238. doi: 10.1038/nrneurol.2014.37. [DOI] [PubMed] [Google Scholar]
- Vergo S. Craner M. J. Etzensperger R. Attfield K. Friese M. A. Newcombe J. Esiri M. Fugger L. Brain. 2011;134:571–584. doi: 10.1093/brain/awq337. [DOI] [PubMed] [Google Scholar]
- Mercado F. Almanza A. Rubio N. Soto E. Neurosci. Lett. 2018;677:88–93. doi: 10.1016/j.neulet.2018.04.052. [DOI] [PubMed] [Google Scholar]
- Repovic P. Continuum. 2019;25:655–669. doi: 10.1212/CON.0000000000000739. [DOI] [PubMed] [Google Scholar]
- Cree B. A. C. Mares J. Hartung H. P. Curr. Opin. Neurol. 2019;32:365–377. doi: 10.1097/WCO.0000000000000700. [DOI] [PubMed] [Google Scholar]
- Howells J. Matamala J. M. Park S. B. Garg N. Vucic S. Bostock H. Burke D. Kiernan M. C. J. Physiol. 2018;596:5379–5396. doi: 10.1113/JP276624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do-Ha D. Buskila Y. Ooi L. Mol. Neurobiol. 2018;55:1410–1418. doi: 10.1007/s12035-017-0392-y. [DOI] [PubMed] [Google Scholar]
- Jaiswal M. K. Med. Res. Rev. 2019;39:733–748. doi: 10.1002/med.21528. [DOI] [PubMed] [Google Scholar]
- Scott K. Shannon R. Roche-Green A. Searcy R. Woolyhand G. Meeks G. Schuh M. J. Palliative Med. 2016;19:335–336. doi: 10.1089/jpm.2015.0362. [DOI] [PubMed] [Google Scholar]
- Boscia F. Annunziato L. Taglialatela M. Neuropharmacology. 2006;51:283–294. doi: 10.1016/j.neuropharm.2006.03.024. [DOI] [PubMed] [Google Scholar]
- Bierbower S. M. Choveau F. S. Lechleiter J. D. Shapiro M. S. J. Neurosci. 2015;35:2101–2111. doi: 10.1523/JNEUROSCI.3805-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil F. A. Bozdemir E. Bugay V. Chun S. H. Hobbs M. Sanchez I. Hastings S. D. Veraza R. J. Holstein D. M. Sprague S. M. Carver C. Cavazos J. E. Brenner R. Lechleiter J. D. Shapiro M. S. J. Cereb. Blood Flow Metab. 2020;40:1256–1273. doi: 10.1177/0271678X19857818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y. Yan W. Sun W. Luo Y. Li J. Yin Y. J. Cell. Physiol. 2019;234:12714–12726. doi: 10.1002/jcp.27889. [DOI] [PubMed] [Google Scholar]
- Sampath D. Lam P. M. Laoprasert M. Diaz M. J. Busquet N. White A. M. Gonzalez M. I. Raol Y. H. Pediatr. Res. 2020;88:202–208. doi: 10.1038/s41390-019-0734-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redrobe J. P. Nielsen A. N. Behav. Brain Res. 2009;198:481–485. doi: 10.1016/j.bbr.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Kristensen L. V. Sandager-Nielsen K. Hansen H. H. J. Neurochem. 2012;121:373–382. doi: 10.1111/j.1471-4159.2012.07704.x. [DOI] [PubMed] [Google Scholar]
- Al-Mousawi A. H. Evans N. Ebmeier K. P. Roeda D. Chaloner F. Ashcroft G. W. Br. J. Psychiatry. 1996;169:509–516. doi: 10.1192/bjp.169.4.509. [DOI] [PubMed] [Google Scholar]
- Brooks J. O. Hoblyn J. C. Ketter T. A. Psychiatry Res. 2010;181:136–140. doi: 10.1016/j.pscychresns.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Blumberg H. P. Stern E. Martinez D. Ricketts S. de Asis J. White T. Epstein J. McBride P. A. Eidelberg D. Kocsis J. H. Silbersweig D. A. Biol. Psychiatry. 2000;48:1045–1052. doi: 10.1016/S0006-3223(00)00962-8. [DOI] [PubMed] [Google Scholar]
- Luykx J. J. Boks M. P. Terwindt A. P. Bakker S. Kahn R. S. Ophoff R. A. Eur. Neuropsychopharmacol. 2010;20:357–368. doi: 10.1016/j.euroneuro.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Henry J. A. Roberts L. E. Caspary D. M. Theodoroff S. M. Salvi R. J. J. Am. Acad. Audiol. 2014;25:5–22. doi: 10.3766/jaaa.25.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimitsuki T. Ohashi M. Umeno Y. Yoshida T. Komune N. Noda T. Komune S. Neurosci. Lett. 2011;504:28–31. doi: 10.1016/j.neulet.2011.08.050. [DOI] [PubMed] [Google Scholar]
- Sheppard A. Hayes S. H. Chen G. D. Ralli M. Salvi R. Acta Otorhinolaryngol. Ital. 2014;34:79–93. [PMC free article] [PubMed] [Google Scholar]
- Sheppard A. M. Chen G. D. Salvi R. Hear. Res. 2015;327:1–8. doi: 10.1016/j.heares.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Lobarinas E. Dalby-Brown W. Stolzberg D. Mirza N. R. Allman B. L. Salvi R. Physiol. Behav. 2011;104:873–879. doi: 10.1016/j.physbeh.2011.05.022. [DOI] [PubMed] [Google Scholar]
- Jung J. Choi H. B. Koh Y. I. Rim J. H. Choi H. J. Kim S. H. Lee J. H. An J. Kim A. Lee J. S. Joo S. Y. Yu S. Choi J. Y. Kang T. M. Gee H. Y. Sci. Rep. 2018;8:16659. doi: 10.1038/s41598-018-34876-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. Zhang Q. Jia Y. Shu Y. Yang J. Yang H. Yan Z. Hear. Res. 2020;388:107884. doi: 10.1016/j.heares.2020.107884. [DOI] [PubMed] [Google Scholar]
- Leitner M. G. Feuer A. Ebers O. Schreiber D. N. Halaszovich C. R. Oliver D. Br. J. Pharmacol. 2012;165:2244–2259. doi: 10.1111/j.1476-5381.2011.01697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D. H. Jung J. Koh Y. I. Rim J. H. Lee J. S. Choi H. J. Joo S. Y. Yu S. Cha D. H. Lee S. Y. Lee J. H. Lee M. G. Choi J. Y. Gee H. Y. Hum. Mutat. 2019;40:335–346. doi: 10.1002/humu.23698. [DOI] [PubMed] [Google Scholar]
- Jepps T. A. Bentzen B. H. Stott J. B. Povstyan O. V. Sivaloganathan K. Dalby-Brown W. Greenwood I. A. Br. J. Pharmacol. 2014;171:4413–4424. doi: 10.1111/bph.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode F. Svalø J. Sheykhzade M. Rønn L. C. Eur. J. Pharmacol. 2010;638:121–127. doi: 10.1016/j.ejphar.2010.03.050. [DOI] [PubMed] [Google Scholar]
- Svalø J. Bille M. Parameswaran Theepakaran N. Sheykhzade M. Nordling J. Bouchelouche P. Eur. J. Pharmacol. 2013;715:312–320. doi: 10.1016/j.ejphar.2013.05.005. [DOI] [PubMed] [Google Scholar]
- Provence A. Malysz J. Petkov G. V. J. Pharmacol. Exp. Ther. 2015;354:290–301. doi: 10.1124/jpet.115.225268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afeli S. A. Malysz J. Petkov G. V. PLoS One. 2013;8:e75875. doi: 10.1371/journal.pone.0075875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provence A. Angoli D. Petkov G. V. J. Pharmacol. Exp. Ther. 2018;364:131–144. doi: 10.1124/jpet.117.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioakeimidis N. Kostis J. B. J. Cardiovasc. Pharmacol. Ther. 2014;19:53–64. doi: 10.1177/1074248413504034. [DOI] [PubMed] [Google Scholar]
- Gratzke C. Christ G. J. Stief C. G. Andersson K. E. Hedlund P. Eur. Urol. 2010;57:342–349. doi: 10.1016/j.eururo.2008.12.024. [DOI] [PubMed] [Google Scholar]
- Reffelmann T. Kloner R. A. Expert Opin. Drug Saf. 2005;4:531–540. doi: 10.1517/14740338.4.3.531. [DOI] [PubMed] [Google Scholar]
- Lee J. H. Chae M. R. Kang S. J. Sung H. H. Han D. H. So I. Park J. K. Lee S. W. Pflugers Arch. 2020;472:89–102. doi: 10.1007/s00424-019-02343-7. [DOI] [PubMed] [Google Scholar]
- Cannon S. C. Compr. Physiol. 2015;5:761–790. doi: 10.1002/cphy.c140062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. R. Zei W. S. Su C. C. Hsiao G. Lin M. J. Evid. Based Complement. Alternat. Med. 2012;2012:803082. doi: 10.1155/2012/803082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont C. Denman K. S. Hawash A. A. Voss A. A. Rich M. M. Exp. Neurol. 2019;315:52–59. doi: 10.1016/j.expneurol.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. Lin A. H. Ru F. Patil M. J. Meeker S. Lee L. Y. Undem B. J. JCI Insight. 2019;4:124467. doi: 10.1172/jci.insight.124467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abitbol I. Peretz A. Lerche C. Busch A. E. Attali B. EMBO J. 1999;18:4137–4148. doi: 10.1093/emboj/18.15.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. Xiong Q. Sun H. Li M. J. Biol. Chem. 2008;283:22649–22658. doi: 10.1074/jbc.M802426200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Q. Sun H. Li M. Nat. Chem. Biol. 2007;3:287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Zhu X. Zhou P. Lan X. Xu H. Li M. Gao Z. PLoS One. 2012;7:e51820. doi: 10.1371/journal.pone.0051820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G. Pusch M. Chen J. Sanguinetti M. C. Circ. Res. 2003;93:941–947. doi: 10.1161/01.RES.0000102866.67863.2B. [DOI] [PubMed] [Google Scholar]
- Gao H. Boillat A. Huang D. Liang C. Peers C. Gamper N. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E6410–E6419. doi: 10.1073/pnas.1620598114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salata J. J. Jurkiewicz N. K. Wang J. Evans B. E. Orme H. T. Sanguinetti M. C. Mol. Pharmacol. 1998;54:220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- Mruk K. Kobertz W. R. PLoS One. 2009;4:e4236. doi: 10.1371/journal.pone.0004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. Wang Y. Zhang M. Jiang M. Rosenhouse-Dantsker A. Wassenaar T. Tseng G. N. Biophys. J. 2015;108:62–75. doi: 10.1016/j.bpj.2014.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D. Wei H. Lu J. Huang D. Liu Z. Loh L. J. Islam O. Liew R. Shim W. Cook S. A. Stem Cell Res. Ther. 2015;6:39. doi: 10.1186/s13287-015-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P. Shi J. White K. M. Gao Y. Cui J. eLife. 2019;8:e48576. doi: 10.7554/eLife.48576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaporis G. Kalik Z. M. Blatter L. A. J. Physiol. 2019;597:723–740. doi: 10.1113/JP277188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinitz J. S. Vaishnava P. Narayan R. L. Fuster V. Circulation. 2013;127:408–416. doi: 10.1161/CIRCULATIONAHA.112.120758. [DOI] [PubMed] [Google Scholar]
- Morillo C. A. Europace. 2014;16:785–786. doi: 10.1093/europace/eut433. [DOI] [PubMed] [Google Scholar]
- Xu X. Salata J. J. Wang J. Wu Y. Yan G. X. Liu T. Marinchak R. A. Kowey P. R. Am. J. Physiol. 2002;283:664–670. doi: 10.1152/ajpheart.00076.2002. [DOI] [PubMed] [Google Scholar]
- Magyar J. Horváth B. Bányász T. Szentandrássy N. Birinyi P. Varró A. Szakonyi Z. Fülöp F. Nánási P. P. Naunyn-Schmiedeberg's Arch. Pharmacol. 2006;373:85–89. doi: 10.1007/s00210-006-0047-4. [DOI] [PubMed] [Google Scholar]
- Corici C. Kohajda Z. Kristóf A. Horváth A. Virág L. Szél T. Nagy N. Szakonyi Z. Fülöp F. Muntean D. M. Varró A. Jost N. Can. J. Physiol. Pharmacol. 2013;91:586–592. doi: 10.1139/cjpp-2012-0407. [DOI] [PubMed] [Google Scholar]
- Seebohm G. Biophys. J. 2013;105:2437–2438. doi: 10.1016/j.bpj.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen J. D. Thomsen M. B. Bentzen B. H. Diness J. G. Diness T. G. Jespersen T. Grunnet M. J. Cardiovasc. Pharmacol. 2012;59:142–150. doi: 10.1097/FJC.0b013e318238727a. [DOI] [PubMed] [Google Scholar]
- Seydel J. K. Schaper K. J. Coats E. A. Cordes H. P. Emig P. Engel J. Kutscher B. Polymeropoulos E. E. J. Med. Chem. 1994;37:3016–3022. doi: 10.1021/jm00045a005. [DOI] [PubMed] [Google Scholar]
- Kinarivala N. Patel R. Boustany R.-M. Al-Ahmad A. Trippier P. C. J. Med. Chem. 2017;60:9739–9756. doi: 10.1021/acs.jmedchem.7b01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methling K. Reszka P. Lalk M. Vrana O. Scheuch E. Siegmund W. Terhaag B. Bednarski P. J. Drug Metab. Dispos. 2009;37:479–493. doi: 10.1124/dmd.108.024364. [DOI] [PubMed] [Google Scholar]
- Groseclose M. R. Castellino S. Chem. Res. Toxicol. 2019;32:294–303. doi: 10.1021/acs.chemrestox.8b00313. [DOI] [PubMed] [Google Scholar]
- Dousa M. Srbek J. Radl S. Cerny J. Klecan O. Havlicek J. Tkadlecova M. Pekarek T. Gibala P. Novakova L. J. Pharm. Biomed. Anal. 2014;94:71–76. doi: 10.1016/j.jpba.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Liu R. Tzounopoulos T. Wipf P. ACS Med. Chem. Lett. 2019;10:929–935. doi: 10.1021/acsmedchemlett.9b00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalappa B. I. Soh H. Duignan K. M. Furuya T. Edwards S. Tzingounis A. V. Tzounopoulos T. J. Neurosci. 2015;35:8829–8842. doi: 10.1523/JNEUROSCI.5176-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan F., Li M., Gao Z., Chen F., Zhang Y., Zhou P., Hu H., Xu H. and Liu S., People's Republic of China Pat., WO2013060097, 2012
- Zhou P. Zhang Y. Xu H. Chen F. Chen X. Li X. Pi X. Wang L. Zhan L. Nan F. Gao Z. Mol. Pharmacol. 2015;87:31–38. doi: 10.1124/mol.114.095190. [DOI] [PubMed] [Google Scholar]
- Dalby-Brown W., Jessen C. and Strøbaek D., PCT Int. Appl., WO2010060955, 2010
- Dalby-Brown W. Jessen C. Hougaard C. Jensen M. L. Jacobsen T. A. Nielsen K. S. Erichsen H. K. Grunnet M. Ahring P. K. Christophersen P. Strøbaek D. Jorgensen S. Eur. J. Pharmacol. 2013;709:52–63. doi: 10.1016/j.ejphar.2013.03.039. [DOI] [PubMed] [Google Scholar]
- Wang L. Q. G.-H. Hu H.-N. Gao Z.-B. Nan F.-J. ACS Med. Chem. Lett. 2019;10:27–33. doi: 10.1021/acsmedchemlett.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmerhirt C. J. Rombach M. Bodtke A. Bednarski P. J. Link A. ChemMedChem. 2015;10:368–379. doi: 10.1002/cmdc.201402442. [DOI] [PubMed] [Google Scholar]
- Bock C. Beirow K. Surur A. S. Schulig L. Bodtke A. Bednarski P. J. Link A. Org. Biomol. Chem. 2018;16:8695–8699. doi: 10.1039/C8OB02530D. [DOI] [PubMed] [Google Scholar]
- Kumar M. Reed N. Liu R. Aizenman E. Wipf P. Tzounopoulos T. Mol. Pharmacol. 2016;89:667–677. doi: 10.1124/mol.115.103200. [DOI] [PubMed] [Google Scholar]
- Grupe M. Bentzen B. H. Benned-Jensen T. Nielsen V. Frederiksen K. Jensen H. S. Jacobsen A.-M. Skibsbye L. Sams A. G. Grunnet M. Rottländer M. Bastlund J. F. Eur. J. Pharmacol. 2020;887:173440. doi: 10.1016/j.ejphar.2020.173440. [DOI] [PubMed] [Google Scholar]
- Vernier J. M., Chen H. and Song J., Valeant Pharmaceuticals (US), WO2009023677A1, 2009
- Ostacolo C. Miceli F. Sarno V. D. Nappi P. Iraci N. Soldovieri M. V. Ciaglia T. Ambrosino P. Vestuto V. Lauritano A. Musella S. Pepe G. Basilicata M. Manfra M. Perinelli D. R. Novellino E. Bertamino A. Gomez-Monterrey I. M. Campiglia P. Taglialatela M. J. Med. Chem. 2020;63:163–185. doi: 10.1021/acs.jmedchem.9b00796. [DOI] [PubMed] [Google Scholar]
- Boger D. L. Zarrinmayeh H. J. Org. Chem. 1990;55:1379–1390. doi: 10.1021/jo00291a059. [DOI] [Google Scholar]
- Fried J. Sabo E. F. J. Am. Chem. Soc. 1954;76:1455–1456. doi: 10.1021/ja01634a101. [DOI] [Google Scholar]
- Purser S. Moore P. R. Swallow S. Gouverneur V. Chem. Soc. Rev. 2008;37:320–330. doi: 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. J. Med. Chem. 2018;61:5822–5880. doi: 10.1021/acs.jmedchem.7b01788. [DOI] [PubMed] [Google Scholar]
- Alverez C. Arkin M. R. Bulfer S. L. Colombo R. Kovaliov M. LaPorte M. G. Lim C. Liang M. Moore W. J. Neitz R. J. Yan Y. Yue Z. Huryn D. M. Wipf P. ACS Med. Chem. Lett. 2015;6:1225–1230. doi: 10.1021/acsmedchemlett.5b00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipf P. Mo T. Geib S. J. Caridha D. Dow G. S. Gerena L. Roncal N. Milner E. E. Org. Biomol. Chem. 2009;7:4163–4165. doi: 10.1039/B911483A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilenkin B. Burris K. D. Eastwood B. J. Sher E. Williams A. C. Priest B. T. Assay Drug Dev. Technol. 2019;17:310–321. doi: 10.1089/adt.2019.942. [DOI] [PubMed] [Google Scholar]
- Wu Y. J. Boissard C. G. Greco C. Gribkoff V. K. Harden D. G. He H. L'Heureux A. Kang S. H. Kinney G. G. Knox R. J. Natale J. Newton A. E. Lehtinen-Oboma S. Sinz M. W. Sivarao D. V. Starrett J. E. Sun L. Q. Tertyshnikova S. Thompson M. W. Weaver D. Wong H. S. Zhang L. Dworetzky S. I. J. Med. Chem. 2003;46:3197–3200. doi: 10.1021/jm034073f. [DOI] [PubMed] [Google Scholar]
- Barhanin J. Lesage F. Guillemare E. Fink M. Lazdunski M. Romey G. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Wu Y. J. He H. Sun L. Q. L'Heureux A. Chen J. Dextraze P. Starrett J. E. Boissard C. G. Gribkoff V. K. Natale J. Dworetzky S. I. J. Med. Chem. 2004;47:2887–2896. doi: 10.1021/jm0305826. [DOI] [PubMed] [Google Scholar]
- Wu Y.-J. Davis C. D. Dworetzky S. Fitzpatrick W. C. Harden D. He H. Knox R. J. Newton A. E. Philip T. Polson C. Sivarao D. V. Sun L.-Q. Tertyshnikova S. Weaver D. Yeola S. Zoeckler M. Sinz M. W. J. Med. Chem. 2003;46:3778–3781. doi: 10.1021/jm034111v. [DOI] [PubMed] [Google Scholar]
- Wu Y. J. Boissard C. G. Chen J. Fitzpatrick W. Gao Q. Gribkoff V. K. Harden D. G. He H. Knox R. J. Natale J. Pieschl R. L. Starrett J. E. Sun L. Q. Thompson M. Weaver D. Wu D. Dworetzky S. I. Bioorg. Med. Chem. Lett. 2004;14:1991–1995. doi: 10.1016/j.bmcl.2004.01.069. [DOI] [PubMed] [Google Scholar]
- Wu Y. J. Sun L. Q. He H. Chen J. Starrett J. E. Dextraze P. Daris J. P. Boissard C. G. Pieschl R. L. Gribkoff V. K. Natale J. Knox R. J. Harden D. G. Thompson M. W. Fitzpatrick W. Weaver D. Wu D. Gao Q. Dworetzky S. I. Bioorg. Med. Chem. Lett. 2004;14:4533–4537. doi: 10.1016/j.bmcl.2004.06.035. [DOI] [PubMed] [Google Scholar]
- Wu Y. J. Conway C. M. Sun L. Q. Machet F. Chen J. Chen P. He H. Bourin C. Calandra V. Polino J. L. Davis C. D. Heman K. Gribkoff V. K. Boissard C. G. Knox R. J. Thompson M. W. Fitzpatrick W. Weaver D. Harden D. G. Natale J. Dworetzky S. I. Starrett J. E. Bioorg. Med. Chem. Lett. 2013;23:6188–6191. doi: 10.1016/j.bmcl.2013.08.092. [DOI] [PubMed] [Google Scholar]
- Blom S. M. Schmitt N. Jensen H. S. PLoS One. 2009;4:e8251. doi: 10.1371/journal.pone.0008251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen H. S. Grunnet M. Olesen S. P. Biophys. J. 2007;92:2747–2756. doi: 10.1529/biophysj.106.101287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H. Chung J. M. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Tesch G. H. Allen T. J. Nephrology. 2007;12:261–266. doi: 10.1111/j.1440-1797.2007.00796.x. [DOI] [PubMed] [Google Scholar]
- Blom S. M. Rottlander M. Kehler J. Bundgaard C. Schmitt N. Jensen H. S. PLoS One. 2014;9:e100209. doi: 10.1371/journal.pone.0100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton-Smith G. A., Gross M. F. and Wickenden A. D., U.S. Pat., US2002091122, 2001
- Amato G. Roeloffs R. Rigdon G. C. Antonio B. Mersch T. McNaughton-Smith G. Wickenden A. D. Fritch P. Suto M. J. ACS Med. Chem. Lett. 2011;2:481–484. doi: 10.1021/ml200053x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlen A. Schwake M. Dost R. Kunert A. Fidzinski P. Heinemann U. Gebhardt C. Br. J. Pharmacol. 2013;168:1182–1200. doi: 10.1111/bph.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z. Zhang T. Wu M. Xiong Q. Sun H. Zhang Y. Zu L. Wang W. Li M. J. Biol. Chem. 2010;285:28322–28332. doi: 10.1074/jbc.M110.116392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. Chen Z. Xu H. Sun H. Li H. Liu H. Yang H. Gao Z. Jiang H. Li M. Cell Res. 2013;23:1106–1118. doi: 10.1038/cr.2013.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Y. Banerjee A. MacKinnon R. PLoS Biol. 2009;7:e1000047. doi: 10.1371/journal.pbio.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V. Baker D. Catterall W. A. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas E. Bezanilla F. Roux B. Neuron. 2011;72:713–720. doi: 10.1016/j.neuron.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleo M. D. Aubrecht J. Bonin P. D. Burt D. A. Colangelo J. Luo L. Schomaker S. Swiss R. Kirby S. Rigdon G. C. Dua P. Pharmacol. Res. Perspect. 2019;7:e00467. doi: 10.1002/prp2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst D. E. and Munster T., in Nonsteroidal ant-inflammatory drugs, disease modifying antirheumatic drugs, nonopioid analgesics and drugs used in gout, ed. B. G. Katzung, McGraw-Hill Book Company, New York, 2001, pp. 596–623 [Google Scholar]
- Peretz A. Degani N. Nachman R. Uziyel Y. Gibor G. Shabat D. Attali B. Mol. Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Brueggemann L. I. Mackie A. R. Martin J. L. Cribbs L. L. Byron K. L. Mol. Pharmacol. 2011;79:10–23. doi: 10.1124/mol.110.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A. Degani-Katzav N. Talmon M. Danieli E. Gopin A. Malka E. Nachman R. Raz A. Shabat D. Attali B. PLoS One. 2007;2:e1332. doi: 10.1371/journal.pone.0001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A. Pell L. Gofman Y. Haitin Y. Shamgar L. Patrich E. Kornilov P. Gourgy-Hacohen O. Ben-Tal N. Attali B. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15637–15642. doi: 10.1073/pnas.0911294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornilov P. Peretz A. Lee Y. Son K. Lee J. H. Refaeli B. Roz N. Rehavi M. Choi S. Attali B. FASEB J. 2014;28:2591–2602. doi: 10.1096/fj.14-250647. [DOI] [PubMed] [Google Scholar]
- Resnick L., Topalov G. T., Belardi J. K., Flentge C. A., Hale J. S. and Mareska D. A., US Pat., 2020/0062714 Al, 2020
- Du X. N. Zhang X. Qi J. L. An H. L. Li J. W. Wan Y. M. Fu Y. Gao H. X. Gao Z. B. Zhan Y. Zhang H. L. Br. J. Pharmacol. 2011;164:1722–1737. doi: 10.1111/j.1476-5381.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi J. Zhang F. Mi Y. Fu Y. Xu W. Zhang D. Wu Y. Du X. Jia Q. Wang K. Zhang H. Eur. J. Med. Chem. 2011;46:934–943. doi: 10.1016/j.ejmech.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Zhang F. Mi Y. Qi J. L. Li J. W. Si M. Guan B. C. Du X. N. An H. L. Zhang H. L. Br. J. Pharmacol. 2013;168:1030–1042. doi: 10.1111/j.1476-5381.2012.02232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. F. Qi J. L. Zhang H. L. Jia Q. Z. Zhongguo Yaolixue Tongbao. 2014;30:574–577. [Google Scholar]
- Ma T. Wang K. J. Chin. Pharm. Sci. 2014;23:153–158. [Google Scholar]
- Teng B. C. Song Y. Zhang F. Ma T. Y. Qi J. L. Zhang H. L. Li G. Wang K. Acta Pharmacol. Sin. 2016;37:1054–1062. doi: 10.1038/aps.2016.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen R. S. Diness T. G. Christ T. Demnitz J. Ravens U. Olesen S. P. Grunnet M. Mol. Pharmacol. 2006;69:266–277. doi: 10.1124/mol.105.015859. [DOI] [PubMed] [Google Scholar]
- Casis O. Olesen S. P. Sanguinetti M. C. Mol. Pharmacol. 2006;69:658–665. doi: 10.1124/mol.105.019943. [DOI] [PubMed] [Google Scholar]
- Li P. Chen X. Zhang Q. Zheng Y. Jiang H. Yang H. Gao Z. J. Pharmacol. Exp. Ther. 2014;351:596–604. doi: 10.1124/jpet.114.217703. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S. Curr. Med. Chem. 2015;22:438–464. doi: 10.2174/0929867321666141112122118. [DOI] [PubMed] [Google Scholar]
- Davoren J. E. Claffey M. M. Snow S. L. Reese M. R. Arora G. Butler C. R. Boscoe B. P. Chenard L. DeNinno S. L. Drozda S. E. Duplantier A. J. Moine L. Rogers B. N. Rong S. Schuyten K. Wright A. S. Zhang L. Serpa K. A. Weber M. L. Stolyar P. Whisman T. L. Baker K. Tse K. Clark A. J. Rong H. Mather R. J. Lowe 3rd J. A. Bioorg. Med. Chem. Lett. 2015;25:4941–4944. doi: 10.1016/j.bmcl.2015.04.074. [DOI] [PubMed] [Google Scholar]
- Ryckmans T. Edwards M. P. Horne V. A. Correia A. M. Owen D. R. Thompson L. R. Tran I. Tutt M. F. Young T. Bioorg. Med. Chem. Lett. 2009;19:4406–4409. doi: 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- Azevedo V. F. Kos I. A. Vargas-Santos A. B. da Rocha Castelar Pinheiro G. Dos Santos Paiva E. Adv. Rheumatol. 2019;59:37. doi: 10.1186/s42358-019-0080-x. [DOI] [PubMed] [Google Scholar]
- Heel R. C. Brogden R. N. Speight T. M. Avery G. S. Drugs. 1977;14:349–366. doi: 10.2165/00003495-197714050-00002. [DOI] [PubMed] [Google Scholar]
- Caulfield M. J. Munroe P. B. O'Neill D. Witkowska K. Charchar F. J. Doblado M. Evans S. Eyheramendy S. Onipinla A. Howard P. Shaw-Hawkins S. Dobson R. J. Wallace C. Newhouse S. J. Brown M. Connell J. M. Dominiczak A. Farrall M. Lathrop G. M. Samani N. J. Kumari M. Marmot M. Brunner E. Chambers J. Elliott P. Kooner J. Laan M. Org E. Veldre G. Viigimaa M. Cappuccio F. P. Ji C. Iacone R. Strazzullo P. Moley K. H. Cheeseman C. PLoS Med. 2008;5:e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A. Kimura H. Chairoungdua A. Shigeta Y. Jutabha P. Cha S. H. Hosoyamada M. Takeda M. Sekine T. Igarashi T. Matsuo H. Kikuchi Y. Oda T. Ichida K. Hosoya T. Shimokata K. Niwa T. Kanai Y. Endou H. Nature. 2002;417:447–452. doi: 10.1038/nature742. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Xu H. Zhan L. Zhou X. Chen X. Gao Z. Pain. 2015;156:1025–1035. doi: 10.1097/j.pain.0000000000000122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linley J. E. Pettinger L. Huang D. Gamper N. J. Physiol. 2012;590:793–807. doi: 10.1113/jphysiol.2011.223404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linley J. E. Rose K. Patil M. Robertson B. Akopian A. N. Gamper N. J. Neurosci. 2008;28:11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B. Linley J. E. Du X. Zhang X. Ooi L. Zhang H. Gamper N. J. Clin. Invest. 2010;120:1240–1252. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J. F. Qiao G. H. Liu N. Nan F. J. Gao Z. B. Acta Pharmacol. Sin. 2016;37:105–110. doi: 10.1038/aps.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S. Lu D. Ouyang P. Bioorg. Med. Chem. Lett. 2018;28:1731–1735. doi: 10.1016/j.bmcl.2018.04.040. [DOI] [PubMed] [Google Scholar]
- Wang K. McIlvain B. Tseng E. Kowal D. Jow F. Shen R. Zhang H. Shan Q. J. He L. Chen D. Lu Q. Dunlop J. Assay Drug Dev. Technol. 2004;2:525–534. doi: 10.1089/adt.2004.2.525. [DOI] [PubMed] [Google Scholar]
- Yang S. Lu D. Ouyang P. Bioorg. Med. Chem. Lett. 2018;28:3004–3008. doi: 10.1016/j.bmcl.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Seefeld M. A. Lin H. Holenz J. Downie D. Donovan B. Fu T. Pasikanti K. Zhen W. Cato M. Chaudhary K. W. Brady P. Bakshi T. Morrow D. Rajagopal S. Samanta S. K. Madhyastha N. Kuppusamy B. M. Dougherty R. W. Bhamidipati R. Mohd Z. Higgins G. A. Chapman M. Rouget C. Lluel P. Matsuoka Y. Bioorg. Med. Chem. Lett. 2018;28:3793–3797. doi: 10.1016/j.bmcl.2018.09.036. [DOI] [PubMed] [Google Scholar]
- Liu C. Li T. Chen J. Curr. Protoc. Pharmacol. 2019;87:e69. doi: 10.1002/cpph.69. [DOI] [PubMed] [Google Scholar]
- Manville R. W. Papanikolaou M. Abbott G. W. Nat. Commun. 2018;9:1847. doi: 10.1038/s41467-018-04266-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manville R. W. Abbott G. W. Mol. Pharmacol. 2018;94:1155–1163. doi: 10.1124/mol.118.112953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R. Y. Yau M. C. Galpin J. D. Seebohm G. Ahern C. A. Pless S. A. Kurata H. T. Nat. Commun. 2015;6:8116. doi: 10.1038/ncomms9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuma A. T. Xu X. Wang Z. Van Camp J. A. Freiberg G. M. Bioorg. Med. Chem. Lett. 2019;29:126603. doi: 10.1016/j.bmcl.2019.08.007. [DOI] [PubMed] [Google Scholar]
- Joshi S. K. Hernandez G. Mikusa J. P. Zhu C. Z. Zhong C. Salyers A. Wismer C. T. Chandran P. Decker M. W. Honore P. Neuroscience. 2006;143:587–596. doi: 10.1016/j.neuroscience.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Pendri Y. R., Martinez E. J., Thottathil J. K. and Hewawasam P., US Pat., 5892106, 1999
- Evans B. E. Rittle K. E. Bock M. G. DiPardo R. M. Freidinger R. M. Whitter W. L. Gould N. P. Lundell G. F. Homnick C. F. Veber D. F. Anderson P. S. Chang R. S. L. Lotti V. J. Cerino D. J. Chen T. B. King P. J. Kunkel K. A. Springer J. P. Hirshfield J. J. Med. Chem. 1987;30:1229–1239. doi: 10.1021/jm00390a019. [DOI] [PubMed] [Google Scholar]
- Aiken S. P. Zaczek R. Brown B. S. Adv. Pharmacol. 1996;35:349–384. doi: 10.1016/s1054-3589(08)60281-1. [DOI] [PubMed] [Google Scholar]
- Zaczek R. Tinker W. J. Tam S. W. Neurosci. Lett. 1993;155:107–111. doi: 10.1016/0304-3940(93)90684-D. [DOI] [PubMed] [Google Scholar]
- Nickolson V. Tam S. Myers M. Cook L. Drug Dev. Res. 1990;19:285–300. doi: 10.1002/ddr.430190307. [DOI] [Google Scholar]
- Tam S. W. Zaczek R. Adv. Exp. Med. Biol. 1995;363:47–56. doi: 10.1007/978-1-4615-1857-0_7. [DOI] [PubMed] [Google Scholar]
- Rockwood K. Beattie B. L. Eastwood M. R. Feldman H. Mohr E. Pryse-Phillips W. Gauthier S. Can. J. Neurol. Sci. 1997;24:140–145. doi: 10.1017/S031716710002148X. [DOI] [PubMed] [Google Scholar]
- Mackie A. R. Byron K. L. Mol. Pharmacol. 2008;74:1171–1179. doi: 10.1124/mol.108.049825. [DOI] [PubMed] [Google Scholar]
- Nassoiy S. P. Babu F. S. LaPorte H. M. Byron K. L. Majetschak M. Clin. Exp. Pharmacol. Physiol. 2018;45:16–26. doi: 10.1111/1440-1681.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. S. Brown B. S. McKinnon D. Cohen I. S. Mol. Pharmacol. 2000;57:1218–1223. doi: 10.1124/mol.57.4.687. [DOI] [PubMed] [Google Scholar]
- Drion G. Bonjean M. Waroux O. Scuvée-Moreau J. Liégeois J. F. Sejnowski T. J. Sepulchre R. Seutin V. Eur. J. Neurosci. 2010;31:827–835. doi: 10.1111/j.1460-9568.2010.07107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L. Bian X. Qu Z. Ma Z. Zhou Y. Wang K. Jiang H. Xie J. Nat. Commun. 2013;4:1435. doi: 10.1038/ncomms2439. [DOI] [PubMed] [Google Scholar]
- Liu H. Jia L. Chen X. Shi L. Xie J. Brain Res. Bull. 2018;137:132–139. doi: 10.1016/j.brainresbull.2017.11.011. [DOI] [PubMed] [Google Scholar]
- Wang H. S. Pan Z. Shi W. Brown B. S. Wymore R. S. Cohen I. S. Dixon J. E. McKinnon D. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Lerche C. Seebohm G. Wagner C. I. Scherer C. R. Dehmelt L. Abitbol I. Gerlach U. Brendel J. Attali B. Busch A. E. Br. J. Pharmacol. 2000;131:1503–1506. doi: 10.1038/sj.bjp.0703734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett G. C. Morales M. J. Beahm D. L. Duffey M. E. Rasmusson R. L. J. Physiol. 2006;576:755–767. doi: 10.1113/jphysiol.2006.116012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C. Bruhova I. Lerche H. Steinmeyer K. Wei A. D. Strutz-Seebohm N. Lang F. Busch A. E. Zhorov B. S. Seebohm G. Mol. Pharmacol. 2007;71:1503–1511. doi: 10.1124/mol.106.031682. [DOI] [PubMed] [Google Scholar]
- Cheung Y. Y. Yu H. Xu K. Zou B. Wu M. McManus O. B. Li M. Lindsley C. W. Hopkins C. R. J. Med. Chem. 2012;55:6975–6979. doi: 10.1021/jm300700v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suessbrich H. Bleich M. Ecke D. Rizzo M. Waldegger S. Lang F. Szabo I. Lang H. J. Kunzelmann K. Greger R. Busch A. E. FEBS Lett. 1996;396:271–275. doi: 10.1016/0014-5793(96)01113-1. [DOI] [PubMed] [Google Scholar]
- Porter J. D. Vivas O. Weaver C. D. Alsafran A. DiMilo E. Arnold L. A. Dickson E. J. Dockendorff C. Bioorg. Med. Chem. Lett. 2019;29:126681. doi: 10.1016/j.bmcl.2019.126681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemzadeh M. B., US Pat., 20100310681, 2010
- Zaczek R. Chorvat R. J. Saye J. A. Pierdomenico M. E. Maciag C. M. Logue A. R. Fisher B. N. Rominger D. H. Earl R. A. J. Pharmacol. Exp. Ther. 1998;285:724–730. [PubMed] [Google Scholar]
- Wladyka C. L. Kunze D. L. J. Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch R. F. Gaspo R. Busch A. E. Lang H. J. Li G. R. Nattel S. Cardiovasc. Res. 1998;38:441–450. doi: 10.1016/S0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- Yang I. C. Scherz M. W. Bahinski A. Bennett P. B. Murray K. T. J. Pharmacol. Exp. Ther. 2000;294:955–962. [PubMed] [Google Scholar]
- Bachmann A. Quast U. Russ U. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:590–596. doi: 10.1007/s002100100410. [DOI] [PubMed] [Google Scholar]
- Fujisawa S. Ono K. Iijima Y. Br. J. Pharmacol. 2000;129:1007–1013. doi: 10.1038/sj.bjp.0703126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z. Q. Thomas G. P. Antzelevitch C. J. Cardiovasc. Electrophysiol. 2001;12:472–478. doi: 10.1046/j.1540-8167.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- Du X. L. Lau C. P. Chiu S. W. Tse H. F. Gerlach U. Li G. R. J. Mol. Cell. Cardiol. 2003;35:293–300. doi: 10.1016/S0022-2828(03)00007-5. [DOI] [PubMed] [Google Scholar]
- Thomas G. P. Gerlach U. Antzelevitch C. J. Cardiovasc. Pharmacol. 2003;41:140–147. doi: 10.1097/00005344-200301000-00018. [DOI] [PubMed] [Google Scholar]
- Gögelein H. Brüggemann A. Gerlach U. Brendel J. Busch A. E. Naunyn-Schmiedeberg's Arch. Pharmacol. 2000;362:480–488. doi: 10.1007/s002100000284. [DOI] [PubMed] [Google Scholar]
- Zeng H. Wang J. Clouse H. Lagrutta A. Sannajust F. JRSM Cardiovasc. Dis. 2019;8:2048004019854919. doi: 10.1177/2048004019854919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel E. Rothenberg I. Krisp C. Hundt F. Fraenzel B. Eckey K. Linders J. T. Gallacher D. J. Towart R. Pott L. Pusch M. Yang T. Roden D. M. Kurata H. T. Schulze-Bahr E. Strutz-Seebohm N. Wolters D. Seebohm G. Nat. Commun. 2016;7:12795. doi: 10.1038/ncomms12795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H. Tzingounis A. V. Mol. Pharmacol. 2010;78:1088–1095. doi: 10.1124/mol.110.066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. Wang F. Lu H. Ren X. Zou J. Islets. 2014;6:e962386. doi: 10.4161/19382014.2014.962386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K. Senokuchi T. Lu M. Takemoto M. Fazlul Karim M. Go C. Sato Y. Hatta M. Yoshizawa T. Araki E. Miyazaki J. Song W. J. Biochem. Biophys. Res. Commun. 2011;407:620–625. doi: 10.1016/j.bbrc.2011.03.083. [DOI] [PubMed] [Google Scholar]
- Ullrich S. Su J. Ranta F. Wittekindt O. H. Ris F. Rösler M. Gerlach U. Heitzmann D. Warth R. Lang F. Pflugers Arch. 2005;451:428–436. doi: 10.1007/s00424-005-1479-2. [DOI] [PubMed] [Google Scholar]
- Nattel S. Zeng F. D. J. Pharmacol. Exp. Ther. 1984;229:283–291. [PubMed] [Google Scholar]
- Gerlach U. Brendel J. Lang H. J. Paulus E. F. Weidmann K. Brüggemann A. Busch A. E. Suessbrich H. Bleich M. Greger R. J. Med. Chem. 2001;44:3831–3837. doi: 10.1021/jm0109255. [DOI] [PubMed] [Google Scholar]
- Bauer A. Becker R. Freigang K. D. Senges J. C. Voss F. Hansen A. Müller M. Lang H. J. Gerlach U. Busch A. Kraft P. Kübler W. Schöls W. Circulation. 1999;100:2184–2190. doi: 10.1161/01.CIR.100.21.2184. [DOI] [PubMed] [Google Scholar]
- Bauer A. Becker R. Freigang K. D. Senges J. C. Voss F. Kraft P. Kuebler W. Schoels W. Basic Res. Cardiol. 2000;95:324–332. doi: 10.1007/s003950070052. [DOI] [PubMed] [Google Scholar]
- Bauer A. Koch M. Kraft P. Becker R. Kelemen K. Voss F. Senges J. C. Gerlach U. Katus H. A. Schoels W. Basic Res. Cardiol. 2005;100:270–278. doi: 10.1007/s00395-005-0522-y. [DOI] [PubMed] [Google Scholar]
- Bryant W. M. Huhn G. F. Jensen J. H. Pierce M. E. Stammbach C. Synth. Commun. 1993;23:1617–1625. doi: 10.1080/00397919308011258. [DOI] [Google Scholar]
- Myers M. J. and Nickolson V. J., US Pat., 4760083, 1988
- Pieniaszek H. J. Fiske W. D. Saxton T. D. Kim Y. S. Garner D. M. Xilinas M. Martz R. J. Clin. Pharmacol. 1995;35:22–30. doi: 10.1002/j.1552-4604.1995.tb04741.x. [DOI] [PubMed] [Google Scholar]
- Rakestraw D. C. Bilski D. A. Lam G. N. J. Pharm. Biomed. Anal. 1994;12:1055–1061. doi: 10.1016/0731-7085(94)E0040-8. [DOI] [PubMed] [Google Scholar]
- Earl R. A. Zaczek R. Teleha C. A. Fisher B. N. Maciag C. M. Marynowski M. E. Logue A. R. Tam S. W. Tinker W. J. Huang S. Chorvat R. J. J. Med. Chem. 1998;41:4615–4622. doi: 10.1021/jm9803424. [DOI] [PubMed] [Google Scholar]
- Pesti J. A. Huhn G. F. Yin J. Xing Y. Fortunak J. M. J. Org. Chem. 2000;65:7718–7722. doi: 10.1021/jo9804339. [DOI] [PubMed] [Google Scholar]
- Earl R. A., Myers M. J. and Nickolson V. J., US Pat., 5173489, 1988
- Chen J. G. Markovitz D. A. Yang A. Y. Rabel S. R. Pang J. Dolinsky O. Wu L. S. Alasandro M. Pharm. Dev. Technol. 2000;5:561–570. doi: 10.1081/PDT-100102039. [DOI] [PubMed] [Google Scholar]
- Joachim B., Uwe G., Jochen L. H. and Klaus W., US Pat., 6008245, 1999
- Hu H. Zhou P. Chen F. Li M. Nan F. Gao Z. Acta Pharmacol. Sin. 2013;34:1359–1366. doi: 10.1038/aps.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari M. N. Mohan S. Biala Y. Yaari Y. Hippocampus. 2018;28:338–357. doi: 10.1002/hipo.22836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari M. N. Mohan S. Biala Y. Yaari Y. J. Neurosci. 2019;39:9914–9926. doi: 10.1523/JNEUROSCI.1603-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H. T. Lo Y. C. Wu S. N. Int. J. Mol. Sci. 2020;21:1441. doi: 10.3390/ijms21041441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojilkovic S. S. Tabak J. Bertram R. Endocr. Rev. 2010;31:845–915. doi: 10.1210/er.2010-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. N. Yang W. H. Yeh C. C. Huang H. C. Arch. Toxicol. 2012;86:713–723. doi: 10.1007/s00204-012-0805-7. [DOI] [PubMed] [Google Scholar]