

Abstract
Investigation of novel 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorins, derived from 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin, as PDT agents against melanoma and esophagus cancer is disclosed. Diol and diester fluorinated ring-fused chlorins, including derivatives with 2-(2-hydroxyethoxy)ethanamino groups at the phenyl rings, were obtained via a two-step methodology, combining SNAr and [8π + 2π] cycloaddition reactions. The short-chain PEG groups at the para-position of the phenyl rings together with the diol moiety at the fused pyrazole ring promote a red-shift of the Soret band, a decrease of the fluorescence quantum yield and an increase of the singlet oxygen formation quantum yield, improving the photophysical characteristics required to act as a photosensitizer. Introduction of these hydrophilic groups also improves the incorporation of the sensitizers by the cells reaching cellular uptake values of nearly 50% of the initial dose. The rational design led to a photosensitizer with impressive IC50 values, 13 and 27 nM against human melanoma and esophageal carcinoma cell lines, respectively.
Novel 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorins, derived from 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin, as impressive photosensitizers for photodynamic therapy of melanoma and esophagus cancer have been developed.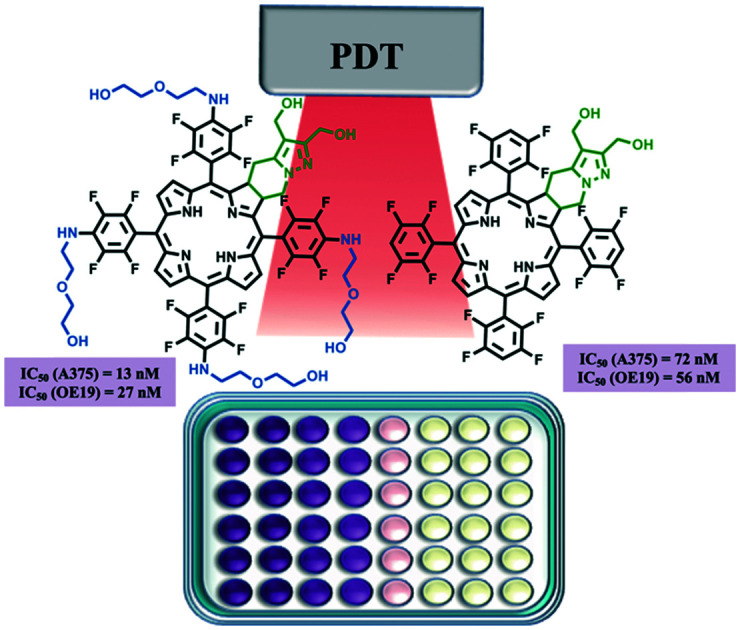
Introduction
The combination of oxygen, light and a photosensitizing agent with the goal of destroying abnormal cells in cancer and other diseases, including pathogenic infections, is the basic principle of photodynamic therapy (PDT).1–3 Briefly, the activation of the photosensitizer (PS) upon light irradiation leads to the formation of so-called reactive oxygen species (ROS) from molecular oxygen via energy and/or electron transfer processes.4 These ROS promote a significant cytotoxic effect, causing damage to cellular organelles, harming associated tissue vasculature, and triggering inflammation and subsequent immune response. As these processes take place within the close environment of the light-absorbing sensitizer, the selective accumulation of the PS in tumor tissues and the utilization of suitable excitation light yields a generalized decrease of off-target damage. Consequently, side-effects are diminished when compared to systemic cancer chemotherapy and radiotherapy treatments.5,6 Therefore, it comes as no surprise that PDT is being increasingly regarded as a fitting therapeutic strategy in the management of cancers, either alone or in combination arrangements, along with radio, chemo and immunotherapies.7–10
Esophageal cancer is the 7th most ordinarily occurring cancer worldwide, with nearly 604 000 new cases and about 537 000 deaths being estimated for 2020, according to the most recent data of the Global Cancer Observatory at the International Agency for Research on Cancer.11,12 Between 40 and 50% of patients with esophageal cancer are deemed surgically unresectable and, of those resected, only around 15 to 20% have a chance for cure or long-time survival.13,14 Therefore, the usual treatment for the majority of cases is simple palliation of symptoms, i.e. the relief of dysphagia. More recently, endoscopic photo-irradiation was found to relieve malignant obstruction of the esophagus, which led to PDT using photofrin and diode laser irradiation as an approved treatment for superficial esophageal cancer.14 While endoscopic submucosal dissection (ESD) is currently a more popular management approach for esophageal cancer, there is plentiful evidence to support PDT as an effective and alternative clinical treatment modality and/or recovery treatment for local failure after chemoradiotherapy.15,16 Another mounting concern in terms of global health is skin cancer. Both melanoma and non-melanoma skin cancers are among the most common malignancies in Caucasians, with increasing incidence over the past few years. There are nearly 302 000 new cases and around 64 000 deaths being projected for 2020 as a result of malignant melanoma.11,12 Albeit being one of the least common forms of skin cancer, it is also one of the deadliest types because of its potential to spread to other parts of the body. Summing up the rising incidence rates of malignant melanoma with its growing resistance to conventional chemotherapy and radiotherapy, an effective and alternative therapeutic strategy, such as photodynamic therapy, can be easily perceived as an urgent need. Although PDT has been successfully applied in the treatment of skin conditions and malignancies,17 its use in malignant melanoma can be compromised due to the natural resistance mechanism of some melanoma tumor cells. Particularly, high melanin levels in pigmented tumors can stimulate an antioxidant effect, due to optical interference via competition with the photosensitizer for the absorption of light.18 Being an aggressive form of skin cancer with an unfavorable prognosis, particularly in advanced stages, the search for new and more proficient PSs able to overcome the resistance of malignant melanoma to photodynamic therapy is highly necessary.19
Tetrapyrrolic macrocycles constitute a class of naturally-occurring heteroaromatic molecules with undeniable biological importance.20 Due to their chemical and photophysical properties, such as effective light absorption and emission within the phototherapeutic window (600–850 nm), great ROS generation capabilities, customarily low toxicity in vivo, and commonly easy structural derivatization, porphyrins and related compounds are broadly recognized as worthy photosensitizers for cancer imaging and therapy.5,6,21,22 A new class of free base ring-fused chlorins (dihydroporphyrins) has been developed by us via a [8π + 2π] cycloaddition methodology,23,24 some of them being successfully evaluated as potent PDT agents against different tumor cell lines.25–27 A similar strategy was also applied in our group to obtain platinum(ii) ring-fused chlorins with improved near-infrared (NIR) luminescence, ratiometric molecular oxygen sensing, and photodynamic action features for cancer theranostic applications.28,29 More recently, the most efficient ring-fused chlorins were studied in vivo, showing an impressive ability as photosensitizers for detection and treatment of malignant tumors.27,29 Although a powerful photodynamic effect has been established for some of these derivatives, both in vitro and in vivo, we decided to widen the range of our ongoing studies in order to encompass ring-fused meso-substituted chlorin derivatives with pentafluorophenyl groups. This would be a strategy to allow further functionalization since it is known that 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin reacts with a variety of nucleophiles via nucleophilic aromatic substitution (SNAr) reactions.30 On the other hand, perfluoroporphyrins are reported to be particularly stable against oxidation (increased photostability)31,32 and efficient singlet oxygen generators.33
Bearing in mind that the inherent features of PSs such as photophysics, hydrophilic/lipophilic nature, shape, size and overall structure are crucial to localize and buildup PSs in several cellular organelles,34,35 the amphiphilic character of photosensitizers was modulated by functional group interconversion and specific peripheral substitutions. The reduction of methyl ester substituents, located at the exocyclic ring, into hydroxymethyl groups was one of the design structural modulations with the purpose of increasing the polarity of the resulting ring-fused chlorins. The second structural modulation was the incorporation of poly(ethylene glycol) (PEG) moieties, a well-known methodology applied in drug design to obtain compounds with the ideal properties to be employed in biological media and improve cell permeability.36 Synthetic details, structural and photophysical characterization, in vitro photocytotoxicity, cell viability and uptake assessment of these novel PEGylated and/or fluorinated 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorin products, in both A375 skin malignant melanoma and OE19 esophageal adenocarcinoma cell lines, are reported herein.
Results and discussion
Synthesis
We have designed and developed a new class of ring-fused free base and Pt(ii) chlorins that were found to be excellent candidates for cancer PDT.23–28 Furthermore, our studies demonstrated that the hydrophilicity of these chlorins is crucial to ensure high photocytotoxicity in tumor cells.25–28 From a chemical standpoint, the 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorin structure bearing meso-substituted pentafluorophenyl moieties can be viewed as a novel and versatile template for further derivatization. Much like its broadly known 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 3 (TPPF20) analogue, a simple nucleophilic aromatic substitution process with a range of suitable nucleophiles provides general access to functionalized derivatives comprising electron-donating substituents at the para positions of the phenyl rings, with this reaction being extremely selective and frequently high yielding.30,37,38 Based on this, we decided to synthesize novel PEGylated derivatives of this type of ring-fused chlorins in order to get compounds with better molecular features to be used as photosensitizers. Two different approaches were carried out to obtain the target ring-fused chlorin 6 (Scheme 1). The first synthetic route (approach A) started with the PEGylation of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 3, adapting a procedure reported by Drain and collaborators30 for the nucleophilic aromatic substitution of the para-fluorine atoms of TPPF20 by primary amines. Thus, the SNAr reaction of porphyrin 3 with 10 equivalents of 2-(2-aminoethoxy)ethanol (amine-PEG) in N-methyl-2-pyrrolidone (NMP), under microwave irradiation (MW) at 200 °C for 2 minutes, afforded the tetrasubstituted porphyrin 5 in only 9% yield after isolation (Table 1, entry 1). A better result was obtained when the reaction time was increased to 5 minutes, giving the desired product in 42% yield (entry 2). Very minor improvements were observed under similar conditions but using a larger excess of amine-PEG (20 equivalents) or by carrying out the reaction at a lower temperature (70 °C) for a longer period of time (18 minutes) (entries 3 and 4, respectively). The best microwave-promoted reaction conditions were achieved by performing the reaction at 200 °C for 10 minutes using 10 equivalents of 2-(2-aminoethoxy)ethanol (entry 5), with porphyrin 5 being isolated in 77% yield. Changing to a conventional heating setting allowed a 10-fold scale-up and after 3 hours at 200 °C and similar work-up, the target porphyrin was obtained in 68% yield (entry 6). The SNAr reaction proved to be very selective occurring, as expected, at the para positions of the meso-pentafluorophenyl groups of porphyrin 3. This can be clearly confirmed by the 19F NMR spectrum of porphyrin 5 showing only two doublets at −143 and −160 ppm, both with a 19.0 Hz coupling constant, corresponding to 8 ortho- and 8 meta-fluorine atoms. On the other hand, peaks characteristic of the para-fluorine atoms at ca. −150 ppm of pentafluorophenyl groups are absent.39 Moreover, in the 1H NMR spectrum, signals corresponding to the short-chained PEG hydrogen atoms (4 NH and 16 CH2) appear at 4.74 ppm and 3.6–3.8 ppm, respectively (see Fig. S1†).
Scheme 1. Schematic routes to PEGylated chlorin 6.

Optimization of the PEGylation of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin with 2-(2-aminoethoxy)ethanol carried out in NMP.
| Entry | Equiv. amine-PEG | Reaction conditions | Yield (%) |
|---|---|---|---|
| 1 | 10 | MW, 200 °C, 2 min | 9 |
| 2 | 10 | MW, 200 °C, 5 min | 42 |
| 3 | 20 | MW, 200 °C, 5 min | 45 |
| 4 | 10 | MW, 70 °C, 18 min | 48 |
| 5 | 10 | MW, 200 °C, 10 min | 77 |
| 6 | 10 | 200 °C, 3 h | 68 |
In order to achieve the desired PEGylated and fluorinated chlorin 6, a [8π + 2π] cycloaddition reaction of porphyrin 5 with diazafulvenium methide 2, generated in situ by thermal extrusion of SO2 from 2,2-dioxo-1H,3H-pyrazolo[1,5-c]thiazole 1,40,41 was carried out as described before for other ring-fused chlorin derivatives (Scheme 1, approach A).23,24 Performing the reaction under microwave irradiation at 250 °C for 20 min, using 1,2,4-trichlorobenzene (1,2,4-TCB) as a solvent, chlorin 6 was obtained in 14% yield (Table 2, entry 1). An excess of porphyrin (2 equivalents) was used in order to prevent the formation of the corresponding bacteriochlorin via bis-cycloaddition of diazafulvenium methide 2 to porphyrin 5. The 19F NMR spectrum of chlorin 6 is a bit more complex than the one of related porphyrin 5, given that the introduction of the exocyclic fused-ring leads to structural asymmetry (see Fig. S3†). Nonetheless, it still presents signals within the ranges −138 to −140 ppm and −158 to −160 ppm, again typical of fluorine atoms at the ortho and meta positions of the meso-pentafluorophenyl groups, respectively, and no signals expected for para-fluorine atoms.39 Switching to conventional heating allowed a 3-fold scale-up of the [8π + 2π] cycloaddition reaction. The reaction of porphyrin 5 with an equimolar and two equivalents of sulfone 1, at 250 °C for 3 hours, afforded chlorin 6 in similar yields as determined by 1H NMR spectroscopy, 47% and 46%, respectively (Table 2, entries 2 and 3). Despite the better yields achieved under conventional heating, a mixture of the target chlorin and starting porphyrin, which we were unable to separate, was obtained. Therefore, the MW-induced reaction was found to be the best synthetic approach to prepare pure chlorin 6. Nevertheless, we found that when derivative 7 was the target (Scheme 2), starting from the mixture proved to be a better option due to the possibility of working on a larger scale. Further details are described below.
Optimization of the [8π + 2π] cycloaddition reaction of PEGylated porphyrin 5 with diazafulvenium methide 2 carried out in 1,2,4-TCB.
| Entry | Equiv. porphyrin 5 | Reaction conditions | Yield (%) |
|---|---|---|---|
| 1 | 2 | MW, 250 °C, 20 min | 14 |
| 2 | 1 | 250 °C, 3 h | 47a |
| 3 | 0.5 | 250 °C, 3 h | 46a |
Yield of chlorin 6 contaminated with the starting porphyrin 5, determined by 1H NMR from the mixture with a chlorin/porphyrin molar ratio of 2/1.
Scheme 2. Synthesis of dihydroxymethyl chlorins 7 and 8.

An alternative strategy to obtain PEGylated compound 6 was pursued, which consisted of the synthesis of chlorin 4 followed by SNAr PEGylation (Scheme 1, approach B). The reaction between TPPF20 and sulfone 1 (molar ratio of 2 : 1), upon microwave irradiation at 250 °C for 20 minutes, provided the new ring-fused 5,10,15,20-tetrakis(pentafluorophenyl)chlorin 4 in 24% yield. Performing the reaction under the same stoichiometric conditions, but under conventional heating for 3 hours at 250 °C, was also tested. Although this procedure led to a slight improvement, with a 28% yield being attained, the MW method was preferred due to the considerably shorter reaction time. Then, we tried to PEGylate chlorin 4 under microwave irradiation under similar conditions used in the synthesis of porphyrin 5 (see Table 1). After several attempts, monitoring the reaction progress by UV-vis spectroscopy, we verified that the formed chlorin 6 partially degrades to its oxidized forms that we were unable to separate. Therefore, we decided to perform the reaction under milder conditions by lowering the temperature. Thus, carrying out the reaction under conventional heating at 50 °C, in 1,4-dioxane, for 192 hours, tetra-PEGylated chlorin 6 was obtained in 12% yield (Scheme 1, approach B). Hence, we have shown that chlorin 6 could be obtained via both synthetic routes. Nevertheless, the overall process through approach A is significantly faster and eco-friendlier and, therefore, more advantageous.
The reduction of the methyl ester substituents of chlorin 4 to their corresponding hydroxymethyl groups was also carried out (Scheme 2). Previously, we have demonstrated that this structural modification led to a considerable improvement in the PDT effectiveness of this type of photosensitizer, which is related to a higher cellular uptake of the hydroxymethyl derivatives.25–29 The reduction was performed by reacting dimethyl ester chlorin 4 with an excess of lithium aluminum hydride (12 equivalents) in tetrahydrofuran (THF), at room temperature and under an inert N2 atmosphere, for 24 hours. After product isolation and 1H NMR analysis, we observed that not only the reduction had occurred, as could be confirmed by the replacement of two singlets corresponding to the methyl esters of chlorin 4 (nearby 3.8 and 4 ppm, see Fig. S2†) by two other singlets assigned to the two methylene groups of the hydroxymethyl substituents of chlorin 7 (around 4.4 and 4.6 ppm, see Fig. S4†) but, somewhat unexpectedly, the complete dehalogenation of the para-fluorine atoms on the aryl groups also took place. The dehalogenation could easily be confirmed by the presence of a multiplet at around 7.5 ppm corresponding to the aromatic protons of the four 2,3,5,6-tetrafluorophenyl groups in the 1H NMR spectrum of chlorin 7, as well as by the significant changes regarding chemical shifts and multiplicities of the 19F NMR signals when compared to the spectroscopic information of the chlorin 4 starting material. These include the disappearance of the above-mentioned typical peaks of the para-fluorine atoms at ca. −150 ppm.39 The signals related to the ortho- and meta-fluorine atoms are observed between −135 and −139 ppm (see Fig. S2 and S4†). The reduction of aryl halides with lithium aluminum hydride is well known42–44 and reductive dehalogenation of aryl fluorides under mild conditions was also demonstrated by Hendrix et al.43 The new dihydroxymethyl chlorin 7 was thus obtained in 42% yield.
The preparation of chlorin 8 followed the same LiAlH4-promoted reduction methodology described above for chlorin 7 (Scheme 2). As referred before, the synthesis of chlorin 6 under conventional heating can be carried out on a larger scale despite not being isolated in a pure form as via the MW-assisted protocol (Table 2). On the other hand, the reduction of the methyl ester substituents of the exocyclic chlorin scaffold to their corresponding hydroxymethyl groups would lead to a much more polar compound than porphyrin 5 contaminant and, hence, easier to separate after suitable chromatographic work-up. In this context, we decided to perform the reduction reaction to achieve 8 starting with the mixture containing chlorin 6 and TPPF20. The reaction with an excess of lithium aluminum hydride in THF, under a saturated N2 atmosphere at 50 °C for 24 hours, led to the target chlorin 8 in 14% yield after isolation. In the 9F NMR spectrum, signals corresponding to the ortho- (between −141 and −145 ppm) and meta-fluorine atoms, from −160 to −162 ppm (see Fig. S5†), clearly demonstrate that there was no unforeseen reductive dehalogenation process.
Photophysical studies
The absorption and fluorescence emission spectra of chlorins 4 and 6–8 in dimethylsulfoxide (DMSO) solution at room temperature are presented in Fig. 1. The spectra show the characteristic absorption features of the chlorin macrocycle, with a strong Soret band with a maximum between 406 and 424 nm and a more pronounced Q band at ∼653 nm, and also typical emission maxima at ∼658 nm. Substitution of the fluorine atoms at the para positions of the meso-aryl rings by short-chained PEG moieties (6 and 8) red-shifts by ∼17 nm the absorption maxima of the Soret band, when compared to the non-PEGylated derivatives 4 and 7 (Table 3). Fluorescence lifetimes (τF) for all chlorin derivatives, collected with excitation at 373 nm in DMSO solution, were found to be well fitted with a single exponential decay law. Similar τF were obtained for the investigated compounds, with values ∼7 ns (Table 3), in agreement with the data reported for a 5,10,15,20-tetrakis(pentafluorophenyl)chlorin bearing a N-benzylisoazolidine ring (τF = 7.3 ns).45
Fig. 1. Room temperature absorption (left hand panel) and fluorescence emission (right hand panel) spectra of chlorins 4 and 6–8 in DMSO solution. The inset in the emission spectra is a magnification of the Soret band, showing the shift with the substitution. See text for more details.

Spectroscopic (absorption, fluorescence and triplet absorption maxima) and photophysical data (including fluorescence (ϕF), internal conversion (ϕIC) and singlet oxygen sensitization (ϕΔ) quantum yields, along with fluorescence lifetimes (τF), triplet lifetimes (τT) and rate constants, kF, kIC and kISC) for chlorins 4 and 6–8 in DMSO solution at 293 K.
| Chlorin | λ Abs max | λ Fluo max |
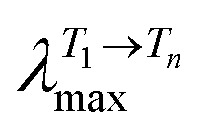
|
ϕ F | ϕ IC a | ϕ Δ | τ F | τ T | k F | k NR b | k ISC a | k IC a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (nm) | (nm) | (nm) | (ns) | (μs) | (ns−1) | (ns−1) | (ns−1) | (ns−1) | ||||
| 4 | 406, 653 | 658, 722 | 460 | 0.39 | 0.21 | 0.40 | 7.0 | 68 | 0.056 | 0.087 | 0.057 | 0.030 |
| 6 | 424, 654 | 659, 725 | 495 | 0.24 | 0.45 | 0.31 | 7.2 | 35 | 0.033 | 0.105 | 0.043 | 0.062 |
| 7 | 408, 653 | 657, 722 | 460 | 0.41 | 0.27 | 0.32 | 7.0 | 46 | 0.059 | 0.085 | 0.046 | 0.039 |
| 8 | 424, 654 | 659, 725 | 495 | 0.28 | 0.27 | 0.45 | 7.3 | 50 | 0.038 | 0.098 | 0.061 | 0.037 |
Assuming ϕT ∼ ϕΔ and ϕIC = 1 − ϕF − ϕT.
k NR = (1 − ϕF)/τF.53
Triplet–triplet transient absorption spectra of deaerated solutions of the chlorin PSs were collected at different times after laser flash photolysis with excitation at 355 nm (see Fig. S6†). Similar transient triplet–triplet absorption spectra and triplet lifetimes (τT = 35–68 μs) were found (Table 3). In general, the triplet–singlet difference absorption spectra present positive broad transient absorption bands in the 300–550 nm range, attributed to the triplet–triplet absorption of the chlorins, together with negative transient absorption bands that are in agreement with the spectroscopic features presented in Fig. 1, which are attributed to the ground state absorption of the chlorins' Soret band. The triplet nature of transient absorption signals is supported by their quenching by oxygen in air-saturated solutions, which follows pseudo first order kinetics. Significant is that, in agreement with the behaviour found in the ground state absorption, the replacement of the para-fluorine atoms at the meso-substituted aryl rings by short-chained PEG groups red-shifts the triplet–triplet absorption maxima by ∼35 nm.
To evaluate the potential of our novel chlorin derivatives as PDT agents, singlet oxygen sensitization quantum yields (ϕΔ) were obtained by direct measurement of the characteristic phosphorescence emission of 1O2, following photolysis of aerated DMSO solutions of the compounds at T = 293 K (Table 3). The ϕΔ values were determined using a comparative method, utilizing meso-tetraphenylporphyrin (TPP) in toluene as a reference photosensitizer, by plotting the initial phosphorescence intensity (at 1270 nm) as a function of the laser dose and comparing the slope with that obtained for the reference compound (Fig. S7†). Singlet oxygen formation quantum yields in the 0.31–0.45 range were obtained for the investigated chlorins (Table 3). Comparison with the parent non-fluorinated diphenylchlorins previously reported26 shows that: (i) the ϕΔ value found for dihydroxymethyl-substituted chlorin 7 (ϕΔ = 0.32) is in agreement with that found for the dihydroxymethyl-bearing diphenylchlorin (ϕΔ = 0.27); (ii) regarding the dimethyl ester-substituted chlorin 4, introduction of the fluorine atoms decreases the singlet oxygen production efficiency (ϕΔ = 0.40) when compared to the non-fluorinated diester diphenylchlorin derivative (ϕΔ = 0.66). Additionally, considering that in general the singlet oxygen quantum yields cannot be higher than the intersystem crossing (triplet formation) quantum yields (ϕΔ ≤ ϕT), and assuming efficient triplet energy transfer from the long-lived triplet state of the fluorinated chlorins to the ground-state triplet molecular oxygen, i.e. SΔ = ϕΔ/ϕT = 1, the obtained ϕΔ values can be considered a good estimate of the triplet formation quantum yields.
Table 3 summarizes the relevant photophysical parameters (quantum yields, lifetimes and rate constants) obtained for the studied chlorin derivatives in DMSO solution. The fluorescence quantum yields values (ϕF) show that the introduction of short-chained PEG moieties decreases the efficiency of the excited-state radiative decay channel (ϕF = 0.39 and 0.41 for chlorins 4 and 7, respectively, vs. ϕF = 0.24 and 0.28 for chlorins 6 and 8). From the overall photophysical parameters in Table 3, we can conclude that, in general, the radiationless deactivation processes are the main excited state deactivation channels, contributing more than 60% (ϕIC + ϕT) to the excited state deactivation of these ring-fused chlorins. However, in contrast with the behaviour found for chlorins 4, 7 and 8 (where the radiationless ISC channel is predominant), for PEGylated chlorin 6 the internal conversion decay channel is now the main radiationless decay process (with ϕIC ∼0.45 vs. 0.21–0.27 for the non-PEGylated chlorins). When the dimethyl ester- and dihydroxymethyl-substituted chlorins and their halogen-free 5,15-diphenylchlorin derivatives26 are compared, a ∼1-fold increase in the fluorescence quantum yield was observed with the introduction of the fluorine atoms. This result is in line with the small or null heavy atom effect observed with the pentafluorophenyl substitution at the porphyrin macrocycle.33,46Table 3 also shows that the excited state deactivation rate constants (kF, kIC and kISC) are all in the same order of magnitude (∼10−11 s−1) thus showing that all the decay processes are competitive with the deactivation of S1.
Cell biology
To evaluate the activity of these new compounds as photosensitizers for photodynamic therapy, the human melanoma A375 and esophageal carcinoma OE19 cell lines were exposed to increasing concentrations of chlorins 4 and 6–8 and then subjected to irradiation with filtered light (cutoff < 560 nm). Control samples to evaluate the cytotoxicity of the PSs and from irradiation alone were also performed. The sigmoid dose–response curves shown in Fig. 2 were adjusted from the experimental results, allowing the IC50 values and the respective 95% confidence intervals to be determined, which are presented in Table 4.
Fig. 2. Dose–response curves of A375 skin malignant melanoma cells (left) and OE19 esophageal adenocarcinoma cells (right). Analysis performed 24 hours after PDT using chlorins 4, 6, 7 and 8 with an energy of 10 J. Data points represent the mean ± SD.

Photocytotoxicity: IC50 and respective CI95 values of chlorins 4 and 6–8 in A375 skin malignant melanoma and OE19 esophageal adenocarcinoma cells. Analysis performed 24 hours after PDT with an energy of 10 J. Cytotoxicity was determined omitting the irradiation step. Values were determined by dose–response sigmoidal fitting (r2 > 0.85).
| Chlorin | Photocytotoxicity (nM) | Cytotoxicity (μM) | ||||
|---|---|---|---|---|---|---|
| A375 | OE19 | A375 | OE19 | |||
| IC50 | CI95 | IC50 | CI95 | IC50 | IC50 | |
| 4 | 6681 | [6179; 7241] | 5606 | [4220; nd] | >10 | >10 |
| 6 | 913 | [749; 1145] | 105 | [91; 122] | >10 | >10 |
| 7 | 72 | [54; 97] | 56 | [49; 68] | >10 | >10 |
| 8 | 13 | [9; 18] | 27 | [13; 37] | >10 | >10 |
Upon light irradiation, chlorins 4 and 6–8 showed dose dependent toxicity against the human melanoma and esophageal carcinoma cells. Not surprisingly, chlorin 4 revealed limited photodynamic activity with IC50 values in the micromolar range in both cell lines. Similar behavior was previously observed with other 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorins bearing diester moieties in the fused ring.25,28,29 The strong hydrophobic character should be the main factor for the low activity.
It has been previously demonstrated that the reduction of diester groups to dihydroxymethyl groups in several 4,5,6,7-tetrahydropyrazolo[1,5-a]pyridine-fused chlorins contributes to a higher photodynamic reaction.25–29 This phototoxicity improvement was also observed, changing from fluorinated chlorin 4 to 7 (Table 4). Unlike chlorin 4, the dihydroxymethyl derivative 7 showed a very interesting activity against both cell lines studied [IC50 (A375) = 72 nM; IC50 (OE19) = 56 nM].
As mentioned above, coupling hydrophobic compounds to PEG moieties is an attractive option for improving their hydrophilicity. This strategy was also explored to achieve a sensitizer with more appropriate features, introduction of PEG moieties in the aryl substituents of chlorin 4 leading to chlorin 6. Undoubtedly, the biological activity of chlorin 6 increased significantly, with greater relevance in the esophageal carcinoma cells with an IC50 value of 105 nM (Table 4).
The two structural modulations, introduction of dihydroxymethyl groups or introduction of PEG moieties, led to new and particularly interesting fluorinated sensitizers, chlorins 6 and 7, with PDT activity in the nanomolar range. These promising results justified the study as a PDT agent of chlorin 8, which incorporates the two referred structural features. Hence, PDT assays were performed in A375 and OE19 cells. Remarkably, chlorin 8 proved to be the most active PS of this series. This compound exhibited impressive IC50 values, values as low as 13 and 27 nM against the human melanoma and esophageal carcinoma cell lines, respectively (Table 4).
Another characteristic of these compounds, which is extremely important considering their application as sensitizers for PDT, is the lack of toxicity in the absence of light. The dark experiments revealed that no effect was seen up to the highest concentration tested of 10 μM, as shown in Table 4.
The differences observed in the PDT activity of the most relevant chlorins 6–8 can be rationalized based on two important properties, the ability to produce ROS, namely singlet oxygen, and the ability to reach intracellular targets. Regarding the production of the cytotoxic oxygen species, chlorins 6 and 7 exhibit similar singlet oxygen sensitization quantum yields in solution (ϕΔ ≈ 0.30), as can be verified in the photophysical data shown in Table 3. Interestingly, chlorin 8 presented the highest singlet oxygen yield (ϕΔ = 0.45). The second crucial property for PDT efficiency is the incorporation of the sensitizers by the cells. Therefore, uptake studies were performed in both cell lines, as represented in Fig. 3. In fact, the highest photodynamic activity of chlorin 8 may be related to the uptake of this compound. In the human melanoma A375 cells, nearly 50% (254 ± 22 nM) of compound 8 entered the cells, significantly higher than chlorin 6 (148 ± 22 nM; p = 0.004) and chlorin 7 (128 ± 15 nM; p = 0.0006). Similarly, in the human esophageal carcinoma OE19 cells, a higher uptake of chlorin 8 was found, with a concentration of 202 ± 32 nM. Hence, these two combined abilities of chlorin 8 lead to the enhancement of intracellular ROS production resulting in its superior PDT efficacy.
Fig. 3. Uptake of chlorins 6, 7 and 8 by A375 skin malignant melanoma and OE19 esophageal adenocarcinoma cells. The cells were incubated for 24 hours with 500 nM of each photosensitizer. Data points represent the mean ± SE.

Conclusions
Fluorinated and PEGylated ring-fused chlorins, bearing methyl ester substituents, have been synthesized by exploring [8π + 2π] cycloaddition of perfluoroporphyrins with the diazafulvenium methide derived from dimethyl 2,2-dioxo-1H,3H-pyrazolo[1,5-c]thiazole-6,7-dicarboxylate, as well as SNAr reactions. The reduction of ring-fused dimethyl 5,10,15,20-tetrakis(pentafluorophenyl)chlorin-dicarboxylate yielded the corresponding tetrakis(tetrafluorophenyl)chlorin with a diol moiety at the exocyclic pyrazole ring, which proved to be an efficient photosensitizer with IC50 values in the nanomolar range, 72 nM and 56 nM against human melanoma and esophageal carcinoma cell lines, respectively. Combining the diol moiety with short-chained PEG moieties in the same fluorinated ring-fused chlorin led to a molecule with improved balance between photophysics and amphiphilicity, with a singlet oxygen formation quantum yield of 0.45 and high cellular uptake (50% of the initial dose), which make it an even more efficient photosensitizer with IC50 values in the nanomolar range, 13 nM and 27 nM, against human melanoma and esophageal carcinoma cell lines, respectively.
Experimental
Chemistry
General
Commercially available high-grade materials and reagents were used as received. Organic solvents were purified by standard procedures prior to utilization.47 Microwave-assisted reactions were carried out with a CEM Discover S-class focused microwave reactor featuring continuous temperature, pressure and microwave power control, under closed vessel conditions. Reaction monitoring was conducted by TLC analysis, on SiO2 60 F254-coated aluminum plates, and via UV-vis absorption spectroscopy, using a PG Instruments T80, Hitachi U-2001 or Shimadzu UV-2100 spectrophotometer. Flash column chromatography was performed using SiO2 60 (35–70 μm) as the stationary phase. Preparative TLC was carried out on SiO2 60 F254-coated glass plates (0.25–0.5 mm, 20 × 20 cm). Melting points were determined with a FALC R132467 electrothermal apparatus, using open glass capillaries, and are uncorrected. NMR spectra were recorded at room temperature with a Bruker Avance III spectrometer, operating at 400 MHz (1H) and 376.5 MHz (19F). Tetramethylsilane (TMS) was used as an internal standard. Chemical shifts (δ) are expressed in parts per million related to TMS and coupling constants (J) are conveyed in hertz. HRMS spectra were obtained with a Waters Micromass VG Autospec M ESI-TOF spectrometer.
Synthesis of compounds
5,10,15,20-Tetrakis(pentafluorophenyl)porphyrin 348,49 was prepared based on Lindsey's method for porphyrin synthesis49–51 and dimethyl 2,2-dioxo-1H,3H-pyrazolo[1,5-c][1,3]thiazole-6,7-dicarboxylate 1 was synthesized as described in the literature.40,41
Porphyrin 5
Microwave heating: a solution of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 3 (50 mg, 0.051 mmol) and 2-(2-aminoethoxy)ethanol (10 equiv., 50 μL, 0.510 mmol) in NMP (2 mL) was stirred and heated under microwave irradiation at 200 °C for 10 min. After cooling to room temperature, the crude product mixture was dissolved in ethyl acetate (50 mL) and washed with a mixture of saturated aqueous NaHCO3 and NaCl (2 × 50 mL). The organic phase was extracted, dried with anhydrous Na2SO4 and evaporated. The resulting residue was purified by flash column chromatography, using ethyl acetate/methanol (9 : 1) as an eluent. After evaporation of the solvents, tetra-PEGylated porphyrin 5 was obtained as a purple solid in 77% yield (52 mg, 0.039 mmol). Conventional heating: a solution of 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 3 (500 mg, 0.51 mmol) and 2-(2-aminoethoxy)ethanol (10 equiv., 500 μL, 5.10 mmol) in NMP (20 mL) was stirred and heated at 200 °C for 3 h. After a similar work-up, porphyrin 5 was isolated in 68% yield (455 mg, 0.346 mmol). MP (°C) >250; 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 9.25 (s, 8H, β-H pyrrolic), 6.43 (s, 4H, NH from PEG), 4.74 (s, 4H, OH from PEG), 3.86–3.72 (m, 16H, CH2 from PEG), 3.67–3.57 (m, 16H, CH2 from PEG), −3.09 (s, 2H, NH); 19F NMR (376.5 MHz, DMSO-d6): δ (ppm) = −142.99 (d, J = 19.0 Hz, 8F, Ar–Fortho), −160.39 (d, J = 19.0 Hz, 8F, Ar–Fmeta); HRMS (ESI): m/z = 1315.3539 (found), 1315.3569 (calculated for C60H51F16N8O8, [M + H]+).
Chlorin 4
Synthesis of fluorinated ring-fused chlorin 4 was carried out following conventional or microwave heating protocols adapted from a previously described approach for other chlorin derivatives of this type.23,24 Conventional heating: a solution of dimethyl 4,6-dihydropyrazolo[1,5-c]thiazole-2,3-dicarboxylate 1 (50 mg, 0.182 mmol) and 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin 3 (2 equiv., 350 mg, 0.359 mmol) in 1,2,4-TCB (3 mL) was bubbled with N2 for 5 min and then stirred and heated at 250 °C for 3 h under an inert atmosphere of N2. After cooling to room temperature, 2–3 drops of triethylamine were added to the crude product mixture, and the resulting residue was purified by flash column chromatography, using first dichloromethane as an eluent to recover the unreacted porphyrin and then dichloromethane/ethyl acetate (95 : 5) to isolate the desired product. After evaporation of the solvents, chlorin 4 was obtained as a purple solid in 28% yield (60 mg, 0.051 mmol). Microwave heating: using the same stoichiometric conditions, the reaction mixture was stirred and heated under microwave irradiation at 250 °C for 20 min. After a similar work-up, chlorin 4 was isolated in 24% yield (51 mg, 0.043 mmol). MP (°C) >250; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.74 (d, J = 5.0 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.72 (d, J = 5.0 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.49 (s, 2H, β-H pyrrolic), 8.45 (d, J = 4.9 Hz, 1H, β-H pyrrolic), 8.35 (d, J = 4.9 Hz, 1H, β-H pyrrolic), 5.64–5.57 (m, 1H, reduced β-H pyrrolic), 5.17–5.10 (m, 1H, reduced β-H pyrrolic), 4.65 (dd, J = 13.4, 8.0 Hz, 1H, CH2 from fused ring), 4.22 (dd, J = 13.4, 10.0 Hz, 1H, CH2 from fused ring), 3.95 (s, 3H, CO2Me), 3.88 (dd, J = 15.5, 6.7 Hz, 1H, CH2 from fused ring), 3.87 (s, 3H, CO2Me), 2.80 (dd, J = 15.5, 10.9 Hz, 1H, CH2 from fused ring), −1.69 (s, 2H, NH); 19F NMR (376.5 MHz, CDCl3): δ (ppm) = −134.61–−134.69 (m, 1F, Ar–Fortho), −134.86–−134.95 (m, 1F, Ar–Fortho), −136.79–−136.99 (m, 4F, Ar–Fortho), −138.11–−138.17 (m, 2F, Ar–Fortho), −148.72 (t, J = 22.6 Hz, 1F, Ar–Fpara), −150.75 (t, J = 22.6 Hz, 1F, Ar–Fpara), −151.35 (t, J = 22.6 Hz, 2F, Ar–Fpara), −158.00–−158.14 (m, 1F, Ar–Fmeta), −158.56–−158.66 (m, 1F, Ar–Fmeta), −159.10–−159.22 (m, 1F, Ar–Fmeta), −159.75–−159.89 (m, 1F, Ar–Fmeta), −161.0–−161.4 (m, 4F, Ar–Fmeta); HRMS (ESI): m/z = 1185.1302 (found), 1185.1299 (calculated for C53H21F20N6O4, [M + H]+).
Chlorin 6
Approach A: a solution of dimethyl 4,6-dihydropyrazolo[1,5-c]thiazole-2,3-dicarboxylate 1 (16 mg, 0.058 mmol) and porphyrin 5 (2 equiv., 150 mg; 0.114 mmol) in 1,2,4-TCB (1 mL) was flushed with N2 for 5 min and then stirred and heated under microwave irradiation at 250 °C for 20 min. After cooling to room temperature, 2–3 drops of triethylamine were added to the crude product mixture, and the resulting residue was purified by flash column chromatography, using ethyl acetate/methanol (9 : 1) as an eluent. After evaporation of the solvents, chlorin 6 was obtained as a purple solid in 14% yield (12 mg, 0.008 mmol). Approach B: a solution of chlorin 4 (50 mg, 0.042 mmol) and 2-(2-aminoethoxy)ethanol (10 equiv., 42 μL, 0.420 mmol) in 1,4-dioxane (1 mL) was stirred and heated at 50 °C for 192 h. The extent of the reaction was followed by TLC. After cooling to room temperature, the crude product mixture was evaporated, and the resulting residue was purified by flash column chromatography, using first ethyl acetate as an eluent to remove undesired by-products and then ethyl acetate/methanol (2–10% v/v) to isolate the desired product. A second purification was carried out via preparative TLC using ethyl acetate/methanol (95 : 5) as an eluent. Chlorin 6 was crystallized from diethyl ether and obtained as a purple solid in 12% yield (8 mg, 0.005 mmol). MP (°C) >250; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.76 (d, J = 5.4 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.74 (d, J = 5.4 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.55 (s, 2H, β-H pyrrolic), 8.46 (d, 1H, J = 4.9 Hz, β-H pyrrolic), 8.39 (d, J = 4.9 Hz, 1H, β-H pyrrolic), 5.69–5.62 (m, 1H, reduced β-H pyrrolic), 5.14–5.07 (m, 1H, reduced β-H pyrrolic), 4.75 (dd, J = 13.3, 8.2 Hz, 1H, CH2 from fused ring), 4.68 (bs, 4H, NH from PEG), 4.19 (dd, J = 13.3, 10.4 Hz, 1H, CH2 from fused ring), 3.95 (s, 3H, CO2Me), 3.91–3.86 (m, 24H, 10xCH2 from PEG, CH2 from fused ring and CO2Me), 3.77–3.72 (m, 12H, CH2 from PEG), 2.69 (dd, J = 15.8, 11.2 Hz, 1H, CH2 from fused ring), −1.67 (s, 2H, NH); 19F NMR (376.5 MHz, CDCl3): δ (ppm) = −138.76 (dd, J = 23.2, 7.7 Hz, 1F, Ar–Fortho), −139.14 (dd, J = 23.2, 7.7 Hz, 1F, Ar–Fortho), −140.38–−140.61 (m, 4F, Ar–Fortho), −141.70 (dd, J = 23.2, 7.7 Hz, 1F, Ar–Fortho), −142.02 (dd, J = 23.2, 7.7 Hz, 1F, Ar–Fortho), −158.17–−159.64 (m, 4F, Ar–Fmeta), −160.26–−161.41 (m, 4F, Ar–Fmeta); HMRS (ESI): m/z = 1525.4164 (found), 1525.4209 (calculated for C69H61F16N10O12, [M + H]+).
Chlorin 7
The reduction of the methyl ester substituents of chlorin 4 to dihydroxymethyl groups was performed based on a procedure previously described by us.26 To a suspension of finely pulverized LiAlH4 (12 equiv., 18 mg, 0.427 mmol) in dry THF (1.5 mL) at 0 °C was dropwise-added a solution of chlorin 4 (42 mg, 0.036 mmol) in dry THF (1.5 mL). The reaction mixture was allowed to warm to room temperature and left stirring under an inert atmosphere of N2 for 24 h. It was then cooled with an ice bath and quenched by the addition of ethyl acetate (0.5 mL), followed by water (0.5 mL) and 10% aqueous HCl (0.5 mL). After vigorous stirring at room temperature for 1 h, the crude product mixture was evaporated and the resulting residue was purified by flash column chromatography, using first dichloromethane/ethyl acetate (95 : 5) as an eluent to remove undesired by-products and then ethyl acetate/methanol (9 : 1) to isolate the desired product. After evaporation of the solvents, chlorin 7 was obtained as a purple solid in 42% yield (16 mg, 0.015 mmol). MP (°C) >250; 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.72 (d, J = 4.9 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.71 (d, J = 4.9 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.49–8.47 (m, 2H, β-H pyrrolic), 8.43–8.42 (m, 1H, β-H pyrrolic), 8.39–8.38 (m, 1H, β-H pyrrolic), 7.61–7.53 (m, 4H, Ar), 5.60–5.53 (m, 1H, reduced β-H pyrrolic), 5.24–5.17 (m, 1H, reduced β-H pyrrolic), 4.59 (s, 2H, CH2OH), 4.46 (dd, J = 13.3, 7.6 Hz, 1H, CH2 from fused ring), 4.37 (s, 2H, CH2OH), 4.20 (dd, J = 13.3, 9.1 Hz, 1H, CH2 from fused ring), 3.23 (dd, J = 15.3, 6.7 Hz, 1H, CH2 from fused ring), 2.82 (dd, J = 15.3, 9.9 Hz, 1H, CH2 from fused ring), −1.66–−1.71 (m, 2H, NH tautomerism); 19F NMR (376.5 MHz, CDCl3): δ (ppm) = −135.23–−135.53 (m, 2F), −135.93–−136.39 (m, 3F), −137.29–−137.71 (m, 5F), −138.53–−138.61 (m, 5F), −139.08–−139.11 (m, 1F); HRMS (ESI): m/z = 1057.1782 (found), 1057.1778 (calculated for C51H25F16N6O2, [M + H]+).
Chlorin 8
The reduction of the methyl ester substituents of chlorin 6 to dihydroxymethyl groups was performed based on a procedure previously described by us.26 To a suspension of finely pulverized LiAlH4 (29 equiv., 40 mg, 1.054 mmol) in dry THF (3 mL) at 0 °C was dropwise-added a solution of a mixture (80 mg) bearing chlorin 6 (0.036 mmol) contaminated with porphyrin 5, in a molar ratio of 2 : 1, respectively, in dry THF (1.5 mL). The reaction mixture was stirred and heated at 50 °C under an inert atmosphere of N2 for 24 h. It was then cooled with an ice bath and quenched by the addition of ethyl acetate (1 mL), followed by water (1 mL) and 10% aqueous HCl (1.3 mL). After vigorous stirring at room temperature for 2 h, the crude product mixture was evaporated and the resulting residue was purified by flash column chromatography, using ethyl acetate/methanol (8 : 2) to isolate the desired product. After evaporation of the solvents, chlorin 8 was obtained as a purple solid in 14% yield (7 mg, 0.005 mmol). MP (°C) >250; 1H NMR (400 MHz, CD4O): δ (ppm) = 8.91 (d, J = 4.2 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.90 (d, J = 4.2 Hz, 1H, overlapping of peaks, β-H pyrrolic), 8.64 (d, J = 4.9 Hz, 1H, β-H pyrrolic), 8.62–8.57 (m, 3H, β-H pyrrolic), 5.61 (dd, J = 16.5, 8.4 Hz, 1H, reduced β-H pyrrolic), 5.41 (dd, J = 16.5, 8.4 Hz, 1H, reduced β-H pyrrolic), 4.61–4.56 (m, 1H, CH2 from fused ring), 4.47–4.40 (m, 2H, CH2OH), 4.37–4.31 (m, 3H, CH2 from fused ring and CH2OH), 3.89–3.84 (m, 16H, CH2 from PEG), 3.81–3.77 (m, 10H, 4xCH2 from PEG and 2xNH from PEG), 3.73–3.70 (m, 10H, 4xCH2 from PEG and 2xNH from PEG), 3.43 (dd, J = 15.2, 7.0 Hz, 1H, CH2 from fused ring), 3.06 (dd, J = 15.2, 8.5 Hz, 1H, CH2 from fused ring); 19F NMR (376.5 MHz, CD4O): δ (ppm) = −141.10–−141.16 (m, 1F, Ar–Fortho), −141.25–−141.31 (m, 1F, Ar–Fortho), −144.00–−144.15 (m, 4F, Ar–Fortho), −145.20–−145.35 (m, 2F, Ar–Fortho), −160.89–−161.04 (m, 2F, Ar–Fmeta), −161.18–−161.24 (m, 1F, Ar–Fmeta), −161.56–−161.62 (m, 1F, Ar–Fmeta), −162.55 (d, J = 9.4 Hz, 4F, Ar–Fmeta); HRMS (ESI): m/z = 735.2183 (found), 735.2192 (calculated for C67H62F16N10O10, [M + H]2+).
Photophysics
Solvents were of spectroscopic grade and used as received. Absorption and fluorescence emission spectra were recorded on a Cary 5000 UV-Vis-NIR and Horiba–Jobin–Yvon Fluoromax 4 spectrometer, respectively. All the fluorescence emission spectra were corrected for the wavelength response of the system. Room temperature fluorescence quantum yields were obtained by the comparative method, using meso-tetraphenylporphyrin (TPP) in toluene as a reference compound (ϕF = 0.11).52 Fluorescence decays were measured with excitation at 373 nm, using a home-built time correlated single photon counting (TCSPC) apparatus described elsewhere.53 Deconvolution of the fluorescence decay curves was performed using the modulating function method, as implemented by G. Striker in the SAND program, as previously reported in the literature.54 The experimental setup used to obtain the triplet–triplet absorption spectra has been described elsewhere.55 The samples were irradiated with the third harmonic (355 nm) of a Nd:YAG Spectra Physics laser (Quanta-Ray model). Low laser energy was used to avoid multiphoton and triplet–triplet annihilation effects. The solutions used to collect the transient singlet–triplet difference absorption spectra were bubbled with nitrogen for at least 20 minutes. In general, the obtained transient absorption signals were assigned to the triplet state of the fluorinated chlorins, since first-order kinetics were found and strong quenching was observed in the presence of oxygen. Singlet oxygen quantum yields were determined by direct measurement of the phosphorescence at 1270 nm, followed by the irradiation of the aerated solution of the samples in DMSO with excitation at 355 nm from a Nd:YAG laser with a setup elsewhere described.53 TPP in toluene was used as a standard (ϕΔ = 0.66).12 For chlorin 8 the singlet oxygen quantum yield was obtained with a Horiba–Jobin–Yvon SPEX Fluorog 3-2.2 using a NIR Hamamatsu R5509-42 photomultiplier, as previously reported.53 The sensitized phosphorescence emission spectra of singlet oxygen from optically matched solutions of the samples and that of the reference compound (TPP) were obtained under identical experimental conditions and the singlet oxygen formation quantum yield was determined by comparing the integrated area under the emission spectra of the sample and that of the reference.53
Cell biology
Cell culture conditions
The A375 human malignant melanoma cell line was purchased from American Type Culture Collection. The OE19 human esophageal carcinoma cell line was purchased from European Collection of Authenticated Cell Cultures. All cell lines were cultured according to standard procedures at 37 °C, in a humidified incubator with 95% air and 5% CO2. The A375 cell line was expanded using Dulbecco's modified Eagle medium (DMEM, Sigma D-5648) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Sigma F7524), 1% penicillin–streptomycin (100 U mL−1 penicillin and 10 mg mL−1 streptomycin, Gibco 15140-122) and 100 mM sodium pyruvate (Gibco Invitrogen Life Technologies; Gibco 1360). The OE19 cell line was propagated using Roswell Park Memorial Institute 1640 medium (RPMI 1640, Sigma R4130) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Sigma F7524), 1% penicillin–streptomycin (100 U mL−1 penicillin and 10 mg mL−1 streptomycin, Gibco 15140-122) and 400 mM sodium pyruvate (Gibco Invitrogen Life Technologies; Gibco 1360). For all studies, cells were detached using a solution of 0.25% trypsin–EDTA (Sigma T4049).
Photodynamic treatment
For each experiment, cells were plated and kept in the incubator overnight, to allow the attachment of the cells. The formulation of the sensitizers consisted of a 1 mg mL−1 solution in DMSO (Fisher Chemical, 200-664-3) and the desired concentrations being achieved by successive dilutions. The sensitizers were administered in several concentrations (from 1 nM to 10 μM) and cells were incubated for 24 h at 37 °C in the dark. Controls were included on every plate, including untreated cell cultures and cultures treated only with the administration vehicle of the sensitizers, DMSO in a concentration of 1%. Cells were washed with phosphate buffered saline (PBS; in mM: 137 NaCl (JMGS), 2.7 KCl (Sigma), 10 Na2HPO4 (Merck), and 1.8 KH2PO4 (Sigma), pH 7.4) and new drug-free medium was added. Each plate was irradiated with a fluence rate of 7.5 mW cm−2 until a total of 10 J was reached, using a light source equipped with a red filter (cut off < 560 nm). Evaluation was performed 24 h after the photodynamic treatment.
Photocytotoxicity and cytotoxicity
The sensitivity of the cell lines to the sensitizers was analyzed using the MTT colorimetric assay (Sigma M2128; Sigma-Aldrich, Inc.) to measure metabolic activity. Cell culture plates were washed with PBS and incubated, for 4 h at 37 °C in the dark, with a solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (0.5 mg mL−1, Sigma M5655) in PBS (pH 7.4). To solubilize formazan crystals, a 0.04 M solution of hydrochloric acid (Merck Millipore 100317) in isopropanol (Sigma 278475) was added. Absorbance was measured using an EnSpire Multimode Plate Reader (PerkinElmer). Cytotoxicity was expressed as the percentage relative to cell cultures treated only with the administration vehicle of the sensitizers. Dose–response curves were obtained using Prism 9.0 and the concentration of sensitizers that inhibits the proliferation of cultures in 50% (IC50) was derived. Dark cytotoxicity studies were performed as described above, but omitting the irradiation step.
Cell uptake
Cells (5 × 105) were incubated with the sensitizers in concentrations of 500 nM for 24 h in the dark. Cells were then washed with PBS and disrupted with DMSO. Cell scrappers were used to ensure full disaggregation. The solutions were collected and centrifuged, with the fluorescence intensity of the supernatants being determined by fluorescence emission spectroscopy with an EnSpire Multimode Plate Reader (Perkin Elmer), using 405 and 420 nm as excitation wavelengths. The intracellular concentration was determined using a calibration curve obtained from the fluorescence intensity in DMSO solutions for each sensitizer.
Conflicts of interest
The authors declare no personal, professional or financial conflicts of interest.
Supplementary Material
Acknowledgments
The authors thank Coimbra Chemistry Centre (CQC), supported by the Portuguese Agency for Scientific Research, “Fundação para a Ciência e a Tecnologia” (FCT) through project UIDB/00313/2020 and UIDP/QUI/00313/2020, co-funded by COMPETE2020-UE. Center for Innovative Biomedicine and Biotechnology (CIBB) is funded by FCT (UID/NEU/04539/2013) and COMPETE-FEDER (POCI-01-0145-FEDER-007440), through the Strategic Project UIDB/04539/2020 and UIDP/04539/2020. Thanks are also due to CIMAGO (Project 06/2019) and FCT, co-funded by the European Regional Development Fund (FEDER) through Portugal 2020/CENTRO 2020 (CENTRO-01-0145-FEDER-000014/MATIS). João Braz thanks FCT/CQC for the PhD scholarship UI/BD/150880/2021. The authors also acknowledge the UC-NMR facility for obtaining the NMR data (www.nmrccc.uc.pt).
Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00433b
References
- Abrahamse H. Hamblin M. R. New photosensitizers for photodynamic therapy. Biochem. J. 2016;473:347–364. doi: 10.1042/BJ20150942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin M. R. Antimicrobial photodynamic inactivation: a bright new technique to kill resistant microbes. Curr. Opin. Microbiol. 2016;33:67–73. doi: 10.1016/j.mib.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmans D. Fukumura D. Jain R. N. K. Photodynamic therapy for cancer. Nat. Rev. Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- Plaetzer K. Krammer B. Berlanda J. Berr F. Kiesslich T. Photophysics and photochemistry of photodynamic therapy: fundamental aspects. Lasers Med. Sci. 2009;24:259–268. doi: 10.1007/s10103-008-0539-1. [DOI] [PubMed] [Google Scholar]
- Brown S. B. Brown E. A. Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497–508. doi: 10.1016/S1470-2045(04)01529-3. [DOI] [PubMed] [Google Scholar]
- Hopper C. Photodynamic therapy: a clinical reality in the treatment of cancer. Lancet Oncol. 2000;1:212–219. doi: 10.1016/s1470-2045(00)00166-2. [DOI] [PubMed] [Google Scholar]
- van Straten D. Mashayekhi V. de Bruijn H. S. Oliveira S. Robinson D. J. Oncologic photodynamic therapy: basic principles, current clinical status and future directions. Cancers. 2017;9:19. doi: 10.3390/cancers9020019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D. Carter K. A. Miranda D. Lovell J. F. Chemophototherapy: an emerging treatment option for solid tumors. Adv. Sci. 2017;4:1600106. doi: 10.1002/advs.201600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski J. M. Arnaut L. G. Photodynamic therapy (PDT) of cancer: from local to systemic treatment. Photochem. Photobiol. Sci. 2015;14:1765–1780. doi: 10.1039/c5pp00132c. [DOI] [PubMed] [Google Scholar]
- Gollnick S. O. Brackett C. M. Enhancement of anti-tumor immunity by photodynamic therapy. Immunol. Res. 2010;46:216–226. doi: 10.1007/s12026-009-8119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F. Ferlay J. Soerjomataram I. Siegel R. L. Torre L. A. Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Pineiro M. Carvalho A. L. Pereira M. M. Gonsalves A. M. d'A. R. Arnaut L. G. Formosinho S. J. Photoacoustic Measurements of Porphyrin Triplet-State Quantum Yields and Singlet-Oxygen Efficiencies. Chem. – Eur. J. 1998;4:2299–2307. [Google Scholar]
- Anand S. Ortel B. J. Pereira S. P. Hasan T. Maytin E. V. Biomodulatory approaches to photodynamic therapy for solid tumors. Cancer Lett. 2012;326:8–16. doi: 10.1016/j.canlet.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila M. L. Photodynamic Therapy. Gastrointest. Endosc. Clin. N. Am. 2011;21:67–79. doi: 10.1016/j.giec.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Allum W. H. Blazeby J. M. Griffin S. M. Cunningham D. Jankowski J. A. Wong R. Guidelines for the management of oesophageal and gastric cancer. Gut. 2011;60:1449–1472. doi: 10.1136/gut.2010.228254. [DOI] [PubMed] [Google Scholar]
- Yano T. Muto M. Yoshimura K. Niimi M. Ezoe Y. Yoda Y. Yamamoto Y. Nishisaki H. Higashino K. Iishi H. Phase I study of photodynamic therapy using talaporfin sodium and diode laser for local failure after chemoradiotherapy for esophageal cancer. Radiat. Oncol. 2012;7:113. doi: 10.1186/1748-717X-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalka K. Merk H. Mukhtar H. Photodynamic therapy in dermatology. J. Am. Acad. Dermatol. 2000;42:389–413. doi: 10.1016/s0190-9622(00)90209-3. [DOI] [PubMed] [Google Scholar]
- Huang Y.-Y. Vecchio D. Avci P. Yin R. Garcia-Diaz M. Hamblin M. R. Melanoma resistance to photodynamic therapy: new insights. Biol. Chem. 2013;394:239–250. doi: 10.1515/hsz-2012-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swavey S. and Tran M., Porphyrin and Phthalocyanine Photosensitizers as PDT Agents: A New Modality for the Treatment of Melanoma, in Recent Advances in the Biology, Therapy and Management of Melanoma, ed. L. Davids, IntechOpen, London, UK, 2013, pp. 253–282 [Google Scholar]
- Milgrom L. R., The colours of life: an introduction to the chemistry of porphyrins and related compounds, Oxford University Press, Oxford, UK, 1997 [Google Scholar]
- Liu T. W., Huynh E., MacDonald T. D. and Zheng G., Porphyrins for imaging, photodynamic therapy, and photothermal therapy, in Cancer Theranostics, ed. X. Chen and S. Wong, Academic Press, Oxford, UK, 2014, pp. 229–254 [Google Scholar]
- Celli J. P. Spring B. Q. Rizvi I. Evans C. L. Samkoe K. S. Verma S. Pogue B. W. Hasan T. Imaging and photodynamic therapy: mechanisms, monitoring and optimization. Chem. Rev. 2010;110:2795–2838. doi: 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira N. A. M. Serra A. C. Pinho e Melo T. M. V. D. Novel approach to chlorins and bacteriochlorins: [8π+2π] cycloaddition of diazafulvenium methides with porphyrins. Eur. J. Org. Chem. 2010:6539–6543. [Google Scholar]
- Pereira N. A. M. Fonseca S. M. Serra A. C. Pinho e Melo T. M. V. D. Burrows H. D. [8π+2π] Cycloaddition of meso-tetra- and 5,15-diarylporphyrins: synthesis and photophysical characterization of stable chlorins and bacteriochlorins. Eur. J. Org. Chem. 2011:3970–3979. [Google Scholar]
- Pereira N. A. M. Laranjo M. Pineiro M. Serra A. C. Santos K. Teixo R. Abrantes A. M. Goncalves A. C. Sarmento Ribeiro A. B. Casalta-Lopes J. Filomena Botelho M. Pinho e Melo T. M. V. D. Novel 4,5,6,7-tetrahydropyrazolo[1,5-a] pyridine fused chlorins as very active photodynamic agents for melanoma cells. Eur. J. Med. Chem. 2015;103:374–380. doi: 10.1016/j.ejmech.2015.08.059. [DOI] [PubMed] [Google Scholar]
- Pereira N. A. M. Laranjo M. Pina J. Oliveira A. S. R. Ferreira J. D. Sanchez-Sanchez C. Casalta-Lopes J. Goncalves A. C. Sarmento-Ribeiro A. B. Pineiro M. Seixas de Melo J. S. Botelho M. F. Pinho e Melo T. M. V. D. Advances on photodynamic therapy of melanoma through novel ring-fused 5,15-diphenylchlorins. Eur. J. Med. Chem. 2018;146:395–408. doi: 10.1016/j.ejmech.2017.12.093. [DOI] [PubMed] [Google Scholar]
- Nascimento B. F. O. Laranjo M. Pereira N. A. M. Dias-Ferreira J. Pineiro M. Botelho M. F. Pinho e Melo T. M. V. D. Ring-Fused Diphenylchlorins as Potent Photosensitizers for Photodynamic Therapy Applications: In Vitro Tumor Cell Biology and in Vivo Chick Embryo Chorioallantoic Membrane Studies. ACS Omega. 2019;4:17244–17250. doi: 10.1021/acsomega.9b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira N. A. M. Laranjo M. Casalta-Lopes J. Serra A. C. Pineiro M. Pina J. Seixas de Melo J. S. Senge M. O. Filomena Botelho M. Martelo L. Burrows H. D. Pinho e Melo T. M. V. D. Platinum(II) ring-fused chlorins as near-infrared emitting oxygen sensors and photodynamic agents. ACS Med. Chem. Lett. 2017;8:310–315. doi: 10.1021/acsmedchemlett.6b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laranjo M. Aguiar M. C. Pereira N. A. M. Brites G. Nascimento B. F. O. Brito A. F. Casalta-Lopes J. Gonçalves A. C. Sarmento-Ribeiro A. B. Pineiro M. Botelho M. F. Pinho e Melo T. M. V. D. Platinum(II) ring-fused chlorins as efficient theranostic agents: Dyes for tumor-imaging and photodynamic therapy of cancer. Eur. J. Med. Chem. 2020;200:112468. doi: 10.1016/j.ejmech.2020.112468. [DOI] [PubMed] [Google Scholar]
- Samaroo D. Vinodu M. Chen X. Drain C. M. meso-Tetra(pentafluorophenyl)porphyrin as an Efficient Platform for Combinatorial Synthesis and the Selection of New Photodynamic Therapeutics using a Cancer Cell Line. J. Comb. Chem. 2007;9:998–1011. doi: 10.1021/cc070067j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya S. Intramolecular Electron Transfer of Diporphyrins Comprised of Electron-Deficient Porphyrin and Electron-Rich Porphyrin with Photocontrolled Isomerization. J. Am. Chem. Soc. 1999;121:48–53. [Google Scholar]
- Tsuchiya S. Seno M. Novel Synthetic Method of Phenol from benzene Catalysed by perfluorinated Hemin. Chem. Lett. 1989:263–266. [Google Scholar]
- Grancho J. C. P. Pereira M. M. Miguel M. G. Rocha Gonsalves A. M. d'A. Burrows H. D. Synthesis, Spectra and Photophysics of some Free Base Tetrafluoroalkyl and Tetrafluoroaryl Porphyrins with Potential Applications in Imaging. Photochem. Photobiol. 2002;75:249–256. doi: 10.1562/0031-8655(2002)075<0249:ssapos>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Benov L. Photodynamic therapy: current status and future directions. Med. Princ. Pract. 2015;24:14–28. doi: 10.1159/000362416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano A. P. Demidova T. N. Hamblin M. R. Mechanisms in photodynamic therapy: Part three - Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagn. Photodyn. Ther. 2005;2:91–106. doi: 10.1016/S1572-1000(05)00060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano M. Brückner C. Modifications of porphyrins and hydroporphyrins for their solubilization in aqueous media. Molecules. 2017;22:980. doi: 10.3390/molecules22060980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa J. I. T. Tomé A. C. Neves M. G. P. M. S. Cavaleiro J. A. S. 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin: a versatile platform to novel porphyrinic materials. J. Porphyrins Phthalocyanines. 2011;15:1116–1133. [Google Scholar]
- Samaroo D. Soll C. E. Todaro L. J. Drain C. M. Efficient microwave-assisted synthesis of amine-substituted tetrakis(pentafluorophenyl)porphyrin. Org. Lett. 2006;8:4985–4988. doi: 10.1021/ol060946z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B. H. Yu B. S. Fluorine-19 NMR spectroscopic studies of phenyl-fluorinated iron tetraarylporphyrin complexes. Bull. Korean Chem. Soc. 2003;24:981–985. [Google Scholar]
- Sutcliffe O. B. Storr R. C. Gilchrist T. L. Rafferty P. Azafulvenium methides: new extended dipolar systems. J. Chem. Soc., Perkin Trans. 1. 2001:1795–1806. [Google Scholar]
- Sutcliffe O. B. Storr R. C. Gilchrist T. L. Rafferty P. Crew A. P. A. Azafulvenium methides: new extended dipolar systems. Chem. Commun. 2000:675–676. [Google Scholar]
- Chung S.-K. Chung F.-f. Reduction of aryl bromides with lithium aluminum hydride: Evidence for a radical mechanism. Tetrahedron Lett. 1979;20:2473–2476. [Google Scholar]
- Hendrix J. A. Stefany D. W. A mild chemospecific reductive dehalogenation of ethylaminobenzamides with lithium aluminum hydride. Tetrahedron Lett. 1999;40:6749–6752. [Google Scholar]
- Karabatsos G. J. Shone R. L. Reaction of lithium aluminum hydride with aromatic halides. J. Org. Chem. 1968;33:619–621. [Google Scholar]
- Silva J. N. Silva A. M. G. Tomé J. P. Ribeiro A. O. Domingues M. R. M. Cavaleiro J. A. S. Silva A. M. S. Neves M. G. P. M. S. Tomé A. C. Serra O. A. Bosca F. Filipe P. Santus R. Morlière P. Photophysical properties of a photocytotoxic fluorinated chlorin conjugated to four β-cyclodextrins. Photochem. Photobiol. Sci. 2008;7:834–843. doi: 10.1039/b800348c. [DOI] [PubMed] [Google Scholar]
- Cavaleiro J. A. S. Görner H. Lacerda P. S. S. MacDonaldc J. G. Mark G. Neves M. G. P. M. S. Nohr R. S. Schuchmann H.-P. von Sonntag C. Tomé A. C. Singlet oxygen formation and photostability of meso-tetraarylporphyrin derivatives and their copper complexes. J. Photochem. Photobiol., A. 2001;144:131–140. [Google Scholar]
- Armarego W. L. F. and Perrin D. D., Purification of Laboratory Chemicals, Butterworth-Heinemann, 4th edn, 1997 [Google Scholar]
- Carvalho de Medeiros M. A. Cosnier S. Deronzier A. Moutet J.-C. Synthesis and Characterization of a New Series of Nickel(II) meso-Tetrakis (polyfluorophenyl)porphyrins Functionalized by Pyrrole Groups and Their Electropolymerized Films. Inorg. Chem. 1996;35:2659–2664. doi: 10.1021/ic9510268. [DOI] [PubMed] [Google Scholar]
- Lindsey J. S. Wagner R. W. Investigation of the synthesis of ortho-substituted tetraphenylporphyrins. J. Org. Chem. 1989;54:828–836. [Google Scholar]
- Lindsey J. S. Hsu H. C. Schreiman I. C. Synthesis of tetraphenylporphyrins under very mild conditions. Tetrahedron Lett. 1986;27:4969–4970. [Google Scholar]
- Lindsey J. S. Schreiman I. C. Hsu H. C. Kearney P. C. Marguerettaz A. M. Rothemund and Adler-Longo reactions revisited: synthesis of tetraphenylporphyrins under equilibrium conditions. J. Org. Chem. 1987;52:827–836. [Google Scholar]
- Montalti M., Credi A., Prodi L. and Gandolfi M. T., Handbook of Photochemistry, CRC Press, 3rd edn, 2006 [Google Scholar]
- Seixas de Melo J. S., Pina J., Dias F. B. and Maçanita A. L., Experimental Techniques for Excited State Characterization, in Applied Photochemistry, ed. R. C. Evans, P. Douglas and H. D. Burrows, Springer, 2013, pp. 533–585 [Google Scholar]
- Striker G. Subramaniam V. Seidel C. A. M. Volkmer A. Photochromicity and Fluorescence Lifetimes of Green Fluorescent Protein. J. Phys. Chem. B. 1999;103:8612–8617. [Google Scholar]
- Pina J. Seixas de Melo J. Burrows H. D. Bilge A. Farrell T. Forster M. Scherf U. Spectral and Photophysical Studies on Cruciform Oligothiophenes in Solution and the Solid State. J. Phys. Chem. B. 2006;110:15100–15106. doi: 10.1021/jp060707t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.