

Abstract
In the search for new antibacterial compounds, we repositioned an antimalarial compound class by derivatising it based on the so-called “eNTRy” rules for enhanced accumulation into Gram-negative bacteria. We designed, synthesised and evaluated a small library of amino acid modified compounds together with the respective Boc-protected analogues, leading to no substantial improvement in antibacterial activity against Escherichia coli wild-type K12, whereas more distinct activity differences were observed in E. coli mutant strains ΔtolC, D22, ΔacrB and BL21(DE3)omp8. A comparison of the activity results of the E. coli mutants with respect to the known rules related to enhanced activity against Gram-negative bacteria revealed that applicability of the rules is not always ensured. Out of the four amino acids used in this study, glycine derivatives showed highest antibacterial activity, although still suffering from efflux issues.
Evaluation of the applicability of Gram-negative activity rules using 16 amino acid modified compounds against a panel of Escherichia coli and its mutant strains.
Introduction
The threat of increasing antimicrobial resistance is alarming, which reinforces the need to continuously nourish the antibiotic pipeline.1 Despite ongoing debates on the “golden set of rules” for antibiotic accumulation into Gram-negative bacteria, the current understanding what makes a molecule a successful antibiotic candidate is still incomplete.2 In another recent review, A. L. Parkes raised the question “what can we design for?” – which still needs to be answered in the antibiotic field.3
The first correlation of physicochemical properties with antibacterial activity dates back to the 1960s, when low non-ionisable lipophilicity, log P, was correlated to enhanced activity against Gram-negative bacteria.4 Later on, low ionisable lipophilicity, log D7.4, strict molecular weight (MW) limit (≤600 Da) and high polar surface area (PSA) were found to be characteristic for marketed antibiotics against Gram-negative infections.5 In 2017, a new direction was given by the so-called “eNTRy” rules. Based on them, a compound needs an ionisable amine (N), low globularity as the factor of three-dimensionality (T) and high rigidity as the measure of rotatable bonds (R) to accumulate into Escherichia coli.6,7 This sparked the research to focus on 3D-properties of the compounds and a scoring function for Gram-negative bacteria was developed based on molecular-dynamics simulations between the outer membrane porins (e.g., E. coli OmpF and OmpC) and the passing molecule. A molecule is more likely to go through the porin with lower size, as the measure of minimal projection area, and with high partial charge determined by dipole moment and charge.8 An alternative scoring profile for Gram-negative bacteria was implemented in the multiparameter software StarDrop in 2018, focusing on physicochemical properties and comparing compounds active against Gram-negative bacteria to other marketed drugs.9 Antibacterial activity requires, however, a delicate balance between permeation and efflux, which can be achieved by focusing on topology, physical properties and atom/bond count of the compounds.10
The eNTRy rules were derived from compound accumulation into E. coli, and some success stories of their applications have already been published, where an introduction of, in particular, an ionisable amine according to the eNTRy rules has increased the antibacterial activity against other Gram-negative bacteria, such as Acinetobacter baumannii and Klebsiella pneumoniae.11–15 In contrast, another recent study argues that there are properties in addition to the ones defined by the eNTRy rules that govern the Gram-negative uptake. Even though the ionisable amine provided the needed activity boost against E. coli, the lower effect on the activity against A. baumannii and Pseudomonas aeruginosa is not yet understood.16 The downside of most rules (Table 1), however, is that they are always representative of only a certain set of compounds or based on a specific bacterium. Sometimes clear boundaries for the applicability of the rules are missing and most of the reported studies are based on known antibacterial compound classes. However, to the authors' knowledge, the general applicability of the rules for a design of a novel series without previous antibacterial activity is questionable. Therefore, it is necessary to evaluate, whether these rules are applicable for repositioning an antimalarial chemical class originating from inhibitors of Plasmodium falciparum (Pf) IspE displaying Pf cell-based activity, but lacking antibacterial activity (unpublished results from this consortium). The purification of PfIspE is reported in the present study for the first time. The kinase IspE is the central and fourth enzyme of the 2-C-methyl-d-erythritol 4-phosphate (MEP) pathway for the biosynthesis of the universal isoprenoid precursors.17 Since most of the reported IspE inhibitors have low-micromolar in vitro activity against Gram-negative homologues but lack activity in cell-based assays, we embarked to address this activity gap by designing a small set of compounds in accordance with the eNTRy rules using l-amino acids.18
Summary of the reported rules for compounds to gain activity against Gram-negative bacteriaa.
| Rules | Governing properties |
|---|---|
| Gram-negative vs. Gram-positive4 | log P ∼ 4 for Gram-negative |
| log P ∼ 6 for Gram-positive | |
| Gram-negative vs. other drugs5 | 4-Fold lower log D7.4 (∼−2.8) |
| Higher MW (∼414, ≤600 Da) | |
| Higher PSA (∼165 Å2) | |
| eNTRy rules6,7 | Ionisable amine (preferably primary) |
| Rotatable bonds (≤5) | |
| Globularity (≤0.25) | |
| Gram-negative scoring function8 | High partial atomic charge (dipole moment and charge) |
| Low size (minimal projection area) | |
| StarDrop scoring profile9 (Gram-negative vs. other drugs) | The most active ones in the range of 0.4–0.6 including TPSA >65.68 Å2, flexibility <0.3656, log S > 0.8232, log D < 1.793, hERG pIC50 <4.938, MW >237.1 Da, BBB category: negative |
| Gram-negative permeation & efflux10 | Escherichia coli: topology, physical properties, atom/bond count |
MW: molecular weight, (T) PSA: (total) polar surface area, log S: log solubility, hERG: the human Ether-à-go-go-related gene, BBB: blood brain barrier.
Results and discussion
Compound 1 originated from a previous publication within the consortium and no antibacterial activity was reported back then for this compound and its close derivatives.19 As it was only used as a reference compound for the E. coli IspE activity assay, the focus was not on its antibacterial activity. Even in the presence of the outer membrane permeabiliser polymyxin B nonapeptide (PMBN) ensuring enhanced uptake, no antibacterial activity (E. coli K12 % inh. = 6 ± 1@100 μM + 1 μg mL−1 PMBN) was observed (Table 2). In the search of new E. coli IspE inhibitors with antibacterial activity, we selected compounds 2 and 3, originating from the antimalarial class featuring moderate E. coli IspE inhibition as suitable starting points to test the applicability of the rules to compounds with weak antibacterial starting activities. The heterocycles, furan and thiophene, on the right-hand side (RHS) could be bioisosterically replaced by amino acids, exploiting amide coupling chemistry. As both compounds 2 and 3 also inhibit undesirably the auxiliary enzymes pyruvate kinase and lactate dehydrogenase (PK/LDH) in the coupled enzyme-activity assay, we focused more on gaining antibacterial activity by modifying them based on the eNTRy rules, while also monitoring activity changes at the enzymatic level to find new scaffolds selectively inhibiting the Pf or E. coli IspE enzymes. Under the cellular assay conditions, compounds 2 and 3 are poorly soluble and percentage (%) growth inhibition could only be measured at 25 and 50 μM, respectively, showing no substantial activity against E. coli K12 wild-type, nor against the ΔtolC efflux-pump mutant (Table 2). Therefore, the compounds 2 and 3 were used to test, whether an application of these eNTRy rules to this series by introducing l-amino acids and glycine will afford the sought-after antibacterial activity. Amino acid modifications were considered a suitable way of evaluating the influence of the free amine vs. the Boc-protected amine on the antibacterial activity. The key interest for the primary amine was influenced by the eNTRy rules and thereby, amino acid modifications opened up a synthetically readily accessible way to introduce them.
Biological-activity results of the reference compoundsa.
| |||
|---|---|---|---|
| 1 | 2 | 3 | |
| PfNF54 IC50 (μM) | n.d. | 5 ± 1 | 6 ± 0 |
| PfIspE IC50 (μM) | >500 | 57 ± 12 | 35 ± 6 |
| EcIspE IC50 (μM) | 1 ± 0 | 91 ± 21 | 68 ± 13 |
| PK/LDH IC50 (μM) | >500 | 65 ± 15 | 56 ± 11 |
| E. coli ΔtolC (% inh.) | n.d. | 30 ± 16@25 μM | 33 ± 3@50 μM |
| E. coli K12 (% inh.) | 6 ± 1@100 μM + 1 μg mL−1 PMBN | 5 ± 6@25 μM | 2 ± 11@50 μM |
Pf: Plasmodium falciparum, E. coli or Ec: Escherichia coli, PK/LDH: pyruvate kinase/lactate dehydrogenase, n.d.: not determined, PMBN: polymyxin B nonapeptide.
For the synthesis, we selected four readily available N-Boc-protected amino acids; l-valine, l-leucine, l-tyrosine and glycine. To obtain derivatives with modifications on the RHS, the 2-aminothiazole building block 6 was accessed via Hantzsch condensation and used for amide couplings followed by Boc-deprotection (Scheme 1).20 In order to evaluate the influence of an increased number of rotatable bonds in accordance with the eNTRy rules, ethyl-pyrrole derivative 7 was used to synthesise the corresponding glycine derivative 12 with one additional rotatable bond.
Scheme 1. Synthetic route for right-hand side modifications. a) Thiourea, EtOH, Δ, 16 h. b) N-Boc-AA-OH, HBTU, TEA, DMF, RT, 18 h. c) DCM, TFA, RT, 30 min. AA: amino acid side chain.

The amide moiety of the amino acids was also considered as a suitable bioisosteric replacement of the pyrrole moiety on the left-hand side (LHS). As 2,4-aminothiazoles are known to be unstable and general concerns about the stability of 2-aminothiazoles emerged from the underlying class, we decided to use building block 18 with a direct phenyl-linker in position 2.21,22 The N-Boc protected compound 19 was accessed via a Curtius rearrangement and used for amide couplings followed by Boc-deprotection to afford a small library of LHS-modified compounds (Scheme 2).23,24
Scheme 2. Synthetic route for left-hand side modifications. a) DPPA, TEA, tBuOH, 80 °C, 18 h. b) 4M HCl/dioxane, RT, 16 h. c) N-Boc-AA-OH, HBTU, TEA, DMF, RT, 18 h. d) DCM, TFA, RT, 30 min. AA: amino acid side chain.

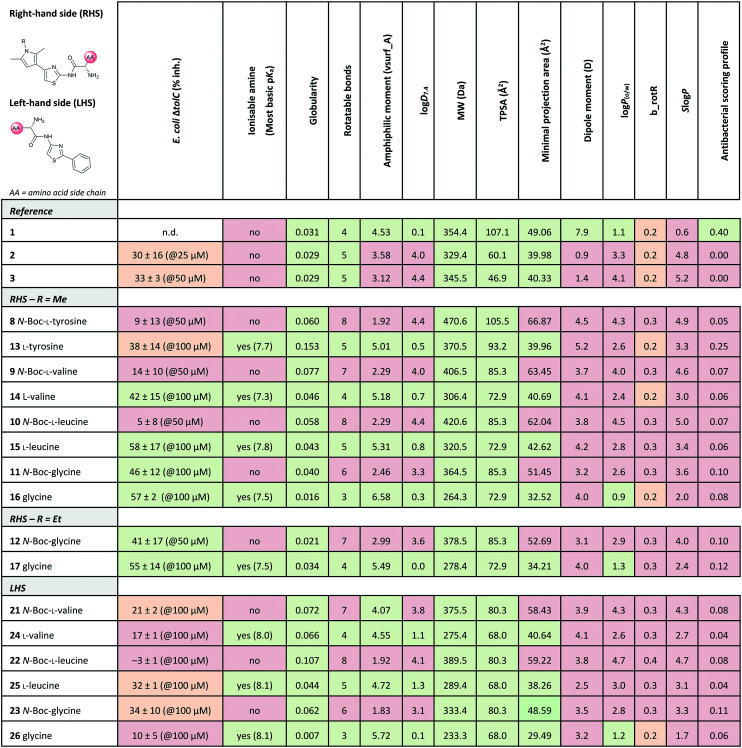
Out of the new compounds, only RHS amine derivative 13 featuring l-tyrosine showed moderate inhibitory activity against E. coli IspE, (IC50 = 200 ± 35 μM), yet selectivity over PK/LDH. In contrast, moderate inhibitory activities selective for PfIspE were observed for the three Boc-derivatives 9, 11 and 12 (Table S2, ESI†) and out of them, both N-Boc-valine derivative 9 (PfIspE IC50 = 61 ± 7 μM and PfNF54 IC50 = 1.9 ± 0.4 μM) and N-Boc-glycine derivative 11 (PfIspE IC50 = 196 ± 44 μM and PfNF54 IC50 = 5.1 ± 0.5 μM) showed low micromolar antimalarial activity. These are the first inhibitors of PfIspE showing also antimalarial activity. The overall weak enzymatic activities suggested minimal E. coli IspE target engagement of the class and we shifted our focus to study how well the reported rules listed in Table 1 apply to this set of compounds. To evaluate antibacterial activity, we screened the compounds primarily against E. coli efflux-pump mutated ΔtolC and wild-type K12 strains. Since most of the compounds only showed antibacterial activity against the efflux-pump mutated E. coli ΔtolC and not against wild-type K12 (Table S3, ESI†), we decided to compare only the ΔtolC results and the respective physicochemical properties to minimise the impact of efflux issues (Table 3). One of the difficulties with the current rules is the lack of clear boundaries of their applicability and therefore, where no reported values were given, we distinguished the physicochemical properties with colour coding (Table 3) to differentiate the N-Boc protected compounds from the free base forms, and not in comparison to other known antibiotics.
Summary of Escherichia coli ΔtolC growth-inhibition results with their respective calculated properties from the different rules related to antibacterial activity in Gram-negative bacteriaa.
|
Colour coding was applied for the reported cut-off limits, where possible, and otherwise, arbitrarily chosen cut-off limits were used to differentiate the N-Boc protected compounds from the free base forms. The colour coding of the results is as follows, (green: obeys the rule, orange: in between and red: disobeys the rule, not statistically determined): E. coli ΔtolC % inh.: red (<20%), orange (20–39%) and green (>40%) at the highest compound solubility, ionisable amine: green (yes) and red (no), globularity: green (≤0.25), rotatable bonds: green (≤5) and red (>5), amphiphilic moment: red (≤4.0) and green (>4.0), log D7.4: green (<1.5), and red (>1.5), MW: green (<600 Da) and red (>600 Da), TPSA: red (<60 Å2) and green (>60 Å2), minimal projection area: green (<50 Å2) and red (>50 Å2), dipole moment: green (>5.5 D) and red (<5.5 D), log P(o/w): green (<1.5) and red (>1.5), b_rotR: green (<0.2), orange (0.2) and red (>0.2), S log P: green (<0.5) and red (>0.5) and antibacterial scoring profile: red (<0.40) and green (≥0.40). n.d.: not determined.
The applicability of the eNTRy rules was tested by comparing the Boc-protected derivatives 8–12 and 21–23 to the amine derivatives 13–17 and 24–26. As a general trend, we observed that most ionisable amine derivatives have improved cellular activity in comparison to the Boc-protected compounds. This trend is more dominant on the RHS-derivative compounds. This is in line with the eNTRy rules as the Boc-protected compounds generally display a higher number of rotatable bonds and lack the ionisable amine. Interestingly, the ethyl-pyrrole derivative featuring glycine 17 showed a similar activity profile to its corresponding methyl-pyrrole derivative 16 despite the increased number of rotatable bonds, but still being within the eNTRy limits. All tested compounds respect the limits of low globularity, although the Boc-protected compounds have higher globularity than the corresponding amine derivatives. Another parameter of rigidity was described by S. J. Cooper et al. as b_rotB (fraction of rotatable bonds), stating that b_rotB should be below 0.2 to achieve higher activities in E. coli in the absence of efflux.10 Using this parameter of rigidity, all of the compounds would have been predicted not to accumulate well. The Boc-protected compounds also show lower solubility in the bacterial assays, which is supported by the calculated log D7.4, log P(o/w) and S log P values that are higher than for the amine derivatives (Table 3). However, the Boc-derivatives have a higher TPSA than the corresponding amine derivatives. In contrast to the antibacterial activity results, the Boc-protected compounds 8–12 are more active in the enzyme assay than the corresponding free amines (Table S2, ESI†). This brings across one of the key challenges in antibiotic research, how to achieve cellular activity and target engagement in a balanced way. For example, it would be expected that the best reference EcIspE inhibitor 1 would show antibacterial activity based on the overall profile of the calculated properties. Additionally, in contradiction to the experimental results of compound 1 (E. coli K12 % inh. with 1 μg mL−1 PMBN = 6 ± 1%@100 μM), the antibacterial scoring profile implemented in StarDrop also predicts high antibacterial activity (score = 0.4), but rates the new amino acid derivatives poorly (Tables 2 and 3). On the other hand, the other reference compounds 2 and 3 do not exhibit any antibacterial activity that is supported by the predicted low antibacterial scoring profile (score = 0.0 for both).
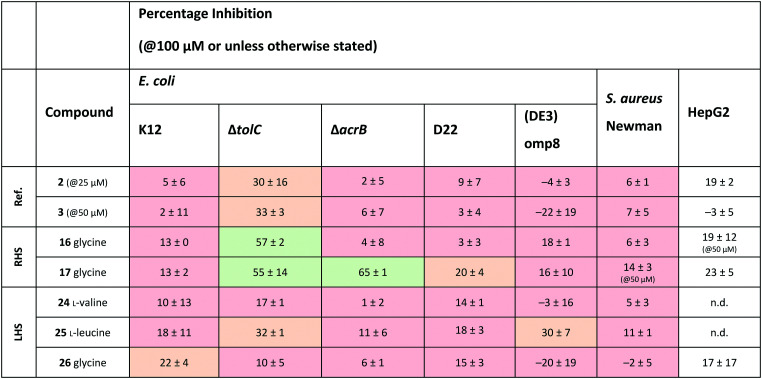
Since the observed antibacterial activities against E. coli ΔtolC did not correlate with those against wild-type E. coli K12, the most active compounds against E. coli ΔtolC were also tested against efflux-pump mutated E. coli ΔacrB, lipopolysaccharide-mutated E. coli D22 and porin-knockdown mutant (BL21(DE3)omp8) to evaluate potential permeation or efflux issues (Tables 4 and S3, ESI†). Reference compounds 2 and 3, however, do not display any antibacterial activity in any of the mutant strains (Table 4). Accumulation into Gram-negative bacteria can occur via active or passive transport. Recent studies show that molecular uptake through porins is governed by other properties than by a previously-defined, simple MW cut-off (≤600 Da).25,26 Most of the rules given in Table 1 are based on the dominating E. coli OmpF. The eNTRy rules demonstrate that the ionisable amine is needed for a key salt–salt interaction within OmpF and the scoring function by S. Acosta-Gutiérrez et al. relies on compound passage through outer membrane porins including E. coli OmpF and OmpC.6,8 Therefore, we used the porin-knockdown mutant (BL21(DE3)omp8) to evaluate, whether the primary amine is needed for permeability via porins and in fact, none of the tested compounds showed antibacterial activity against the omp8 strain lacking the major E. coli porins OmpF, OmpA and OmpC (Table 4).27 With respect to the eNTRy rules, the glycine derivative 17 showed no inhibition against the omp8 strain, suggesting that the amine could indeed play a role in the uptake via the porins. On the other hand, its Boc-derivative 12 also showed no inhibition, thus disagreeing with the necessity of the ionisable amine proposed by the eNTRy rules, leaving the question of the real uptake mechanism of the series (Table S3, ESI†). With respect to the other defining properties for porin passage, the reference compounds 2 and 3 lack ionisable amines and have clearly lower dipole moments than compound 1, but they have smaller minimal projection areas. All amine derivatives have higher dipole moments than the corresponding Boc-protected derivatives, except the LHS derivatives l-leucine 25 and glycine 26. The dipole moments of the amine derivatives are, however, lower than the threshold (5.5 D), which is shown for P. aeruginosa for good outer membrane permeability.10 The minimal projection areas of the amines are also lower compared to their corresponding Boc-derivatives, supporting the general trend seen with the slightly increased antibacterial activities in the efflux-suppressed mutants.
Summary of the activities of the most active compounds against a panel of Escherichia coli strains, Staphylococcus aureus and HepG2 cellsa.
|
The colour coding of the results is as follows: bacterial % inh.: red (0–19%), orange (20–39%), and green (>40%) at the highest compound solubility. RHS: right-hand side, LHS: left-hand side, n.d.: not determined.
As we observed increased antibacterial activities for glycine derivatives 16 and 17 in the efflux-mutated strains, the lack of activity in the wild-type strain K12 is likely to be linked to efflux issues. The TolC-pump is located in the outer membrane, whereas the AcrB-pump is in the inner membrane of the E. coli transmembrane efflux pump AcrAB–TolC.28 Efflux issues have also been linked to high lipophilicity measured as non-ionisable lipophilicity log P(o/w) (>1.5) and S log P (≥0.5).10 Only the glycine derivatives 16 and 17 are within the limits of log P(o/w) (0.9 and 1.4, respectively) and therefore, should avoid efflux in the wild-type strain K12 (Table 3). Interestingly, the effect of the increased activity in E. coli ΔacrB is only present for the ethyl-pyrrole derivative 17 and not for the methyl-pyrrole derivative 16, nor for their corresponding Boc-derivatives 11 and 12, (Table S3, ESI†), nor for the LHS free amine derivatives 24–26. This may also be due to different concentrations of the compounds being present in various compartments of the cell envelope, which may cause distinct parts of the efflux pumps being more engaged than others. The accumulation of the compounds into the different bacterial compartments could be confirmed by MS-based methods.29 However, due to the overall lack of antibacterial activity within the series this path was not followed. Based on the results of the E. coli mutants, it is likely that the ionisable amines engage partly with the outer membrane porins and accumulate in, but are later pumped out, seemingly recognised by the efflux-pump units. No clear inhibitory differences for the Boc-derivatives against the mutants were observed, which contradicts the clear single uptake pathway of the primary amine uptake via porins. Due to the lipophilicity of Boc-derivatives, passive transport through the phospholipid layers could also occur or alternatively, via amino acid specific uptake. Due to the weak target engagement, it is difficult to assess the influence of different amino acids on the antibacterial activities. It is important to remember that amino acids are also important building blocks for bacteria, and specific chemotaxis proteins are linked to different amino acids.30–32 Particularly out of the amino acids used in this study, glycine is reported as an attractant for E. coli growth via Tsr receptor engagement, whereas l-valine, l-leucine and l-tyrosine are reported as repellents.33–36 The most striking difference is that most of the compounds showed no activity against Gram-positive Staphylococcus aureus, except l-leucine derivative 15 (% inh. = 52 ± 15@100 μM). This could be related to different chemotaxis mechanisms or slight target engagement, in particular, as the MEP pathway is absent in most Gram-positive bacteria.37,38 The glycine derivative 17 also weakly inhibited P. aeruginosa (% inh. = 11 ± 7@100 μM) and A. baumannii, (% inh. = 31 ± 17@100 μM), the latter even slightly more than E. coli K12. Further studies are needed to understand if such bacteria-specific amino acid handles could be used to solve uptake issues or in fact, if they are recognised as toxins resulting in efflux. Such amino acid modified compounds could be implemented as “recognition handles” for chemotaxis-enhanced accumulation or for efflux-pump inhibitors into a class with a clear cellular activity and target engagement.
Conclusions
One of the major problems of antibiotic research is to achieve a balance between antibacterial and enzymatic activity, and yet to have a good safety profile. The implementation of l-amino acids in accordance with the eNTRy rules into the old antimalarial class did not result in a highly active antibacterial compound series. Nevertheless, increased antibacterial activity was achieved in comparison to the reference compounds at the expense of efflux issues as demonstrated with the E. coli mutants. Modifications with a non-chiral glycine might be a suitable option to enhance antibacterial activity by introducing an ionisable primary amine, as shown by glycine derivate 17. It might also be worth investigating other amino acids, particularly ones linked to chemotaxis used in Gram-negative bacteria. Eventually, such amino acid-modified compounds could also be used as “recognition handles” for cellular uptake or for efflux-pumps inhibitor. One should, however, avoid designing compound series based on only one set of the current rules available in the literature. We hope that the antibiotic community will continue to investigate the underlying chemical and biological principles of antibiotic accumulation and excretion in a holistic way.
Experimental
Cloning, expression and purification of Plasmodium falciparum IspE
The custom-synthesised DNA fragment coding for IspE of P. falciparum was purchased from GenScript (Piscataway, NJ, USA). The purchased plasmid DNA (based on pET22(+) vector) was used as the template for re-cloning of the DNA fragment coding for IspE with simultaneous re-arrangement of the His6-Tag from N terminus to the C terminus of the protein. The DNA fragment coding for IspE of P. falciparum was amplified by PCR using primers shown in Table S1.† The amplificate was isolated from agarose gel, digested with restriction endonucleases NcoI and HindIII and ligated into the plasmid pNCO113 that had been treated with the same restriction enzymes. The plasmid pNCO113-PFispE-cHis6, containing IspE under control of T5 promoter, was transformed into the E. coli XL1 strain. The transformants were plated on LB medium containing Ap (170 mg L−1). LB medium (50 mL) containing Ap (170 mg L−1) was inoculated with one colony from the plate and incubated for 12 hours at 37 °C in shaking flasks. Preculture (15 mL) was transferred into 1.5 L of terrific broth medium containing Ap (170 mg L−1) and incubated for 3 hours at 37 °C in shaking flasks until the OD600 reached 0.6. The cultures were cooled down to 25 °C, and the incubation was continued for 72 hours in shaking flasks. The cells were harvested by centrifugation (2000 × g, 4 °C, 30 min). The cell pellet was washed once with saline, centrifuged once again and the supernatant was discarded. The cell paste was stored at −20 °C.
All procedures used for cell disruption and protein isolation and purification were performed at 4 °C. Cell paste (10 g) was resuspended in buffer A (50 mM Tris–HCl pH 9.0, 15 mM imidazole 500 mM NaCl, 5% glycerol; 50 mL). Cell disruption was achieved by passing the cell slurry two times through the French press prechilled to 4 °C. Cell debris was centrifuged down (10 000 × g, 4 °C, 30 min). The supernatant was applied onto a chelating Sepharose column (Ni2+-form, 1 cm × 25 cm) equilibrated with buffer A. The column was washed with buffer A (220 mL) and the protein was eluted by a three-step gradient of imidazole concentration (50 mM, 250 mM and 500 mM) in buffer B (50 mM Tris/HCl pH 9.0, 500 mM NaCl, 5% glycerol). Fractions containing IspE were identified with polyacrylamide gel electrophoresis, pooled and applied in 10 mL-aliquots onto a Sephadex G-25 fine cross-linked dextran column (2 cm × 60 cm) equilibrated with buffer C (50 mM Tris/HCl pH 9.0, 100 mM NaCl, 2 mM DTT, 5% glycerol). The column was developed with the same buffer (flow rate, 10 mL min−1). The fractions containing IspE were identified with polyacrylamide gel electrophoresis, concentrated using ultrafiltration cell (Merck, Darmstadt, Germany) equipped with the membrane permeable for proteins with molecular weight up to 10 kDa. The final samples of IspE were stored at −80 °C. The nucleotide sequence of the DNA fragment coding for IspE from P. falciparum is given in Fig. S1, ESI.†
In vitro IspE inhibitory assay
For the IspE assay, CDP-ME (1.0 mM and 0.2 mM for the assaying of IspE from P. falciparum and E. coli, respectively) in 100 mM Tris–HCl, pH 7.6, 0.02% NaN3 (30 μL) were added to a microplate well preloaded either with DMSO or with test compound dissolved in DMSO (3 μL). E. coli IspE was purified as previously described.19,39 The reaction was started by addition of 100 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 60 mM KCl, 10 mM dithiothreitol, 0.02% NaN3, 1 mM NADH, 2 mM phosphoenolpyruvate, 2 mM ATP, pyruvate kinase (1 U mL−1), lactate dehydrogenase (1 U mL−1), and E. coli IspE (0.05 U mL−1) (27 μL per microplate well). For the pyruvate kinase and lactate dehydrogenase (PK/LDH) assay 1 mM ADP in 100 mM Tris–HCl, pH 7.6 (30 μL) was added to a microplate well that had been preloaded with DMSO or with test compound solved in DMSO (3 μL). The reaction was started by addition of 100 mM Tris–HCl, pH 7.6, 10 mM MgCl2, 200 mM KCl, 10 mM dithiothreitol, 0.02% NaN3, 1 mM NADH, 2 mM phosphoenolpyruvate, pyruvate kinase (0.05 U mL−1) and lactate dehydrogenase (0.05 U mL−1) (27 μL per microplate well). The assays were monitored photometrically at room temperature for 30 min. A summary of the results can be found in Table S2, ESI.†
In vitro antibacterial assays
Assays regarding the determination of the minimum inhibitory concentration (MIC) were performed as described recently.40 Our experiments were based on a variety of E. coli strains/mutants (K12, D22, ΔtolC, ΔacrB and BL21(DE3)omp8) as well as S. aureus (Newman strains), P. aeruginosa (PA14) and A. baumannii (DSM30007). In the case no MIC value could be determined due to activity reasons, percentage (%) inhibition at 100 μM (or lower, depending on the solubility of the compounds) was determined. A summary of the results can be found Tables S3 and S4, ESI.†
In vitro cytotoxicity assay
Cytotoxicity assays based on the human hepatocellular carcinoma cell line HepG2 were performed as described previously.41 A summary of the results can be found in Table S3, ESI.†
In vitro antiplasmodial assay
Plasmodium falciparum drug-sensitive NF54 (airport strain from The Netherlands, provided by F. Hoffmann-La Roche Ltd) was cultivated in a variation of the medium consisting of RPMI 1640 supplemented with 0.5% ALBUMAX® II, 25 mM Hepes, 25 mM NaHCO3 (pH 7.3), 0.36 mM hypoxanthine, and 100 μg mL−1 neomycin, as previously described.42,43 Human erythrocytes served as host cells. Cultures were maintained in an atmosphere of 3% O2, 4% CO2, and 93% N2 in humidified modular chambers at 37 °C. Compounds were dissolved in DMSO (10 mM), diluted in hypoxanthine-free culture medium and titrated in duplicate over a 64-fold range in 96 well plates. Infected erythrocytes (1.25% final hematocrit and 0.3% final parasitemia) were added into the wells. After 48 h incubation, 0.25 μCi of [3H]hypoxanthine was added per well and the plates were incubated for an additional 24 h. Parasites were harvested onto glass-fiber filters, and radioactivity was counted using a Betaplate liquid scintillation counter (Wallac, Zurich). The results were recorded and expressed as a percentage of the untreated controls.44 Fifty percent inhibitory concentrations (IC50) were estimated by linear interpolation. A summary of the results can be found in Table S3, ESI.†
Chemistry
All reagents and solvents were of commercial quality and used without further purification. Chemical yields were not optimised. Flash column chromatography (FCC) was performed for compounds packed in ISOLUTE® HM-N (Biotage AB, Uppsala, Sweden) using CombiFlash Rf+ (Teledyne Isco Ltd., Lincoln, NE, USA) equipped with RediSepRf silica columns (Axel Semrau GmbH, Sprockhövel, Germany). Low-resolution mass spectrometry and purity control of final compounds was carried out using an Ultimate 3000-ISQ liquid-chromatography mass spectrometry (LCMS) system (Thermo Fisher Scientific AG, Dreieich, Germany), consisting of a Dionex UltiMate pump, an autosampler, DAD detector and an ESI quadrupole mass spectrometer. Preparative reverse phase-high performance liquid chromatography (RP-HPLC) was performed using an UltiMate 3000 semi-preparative system (Thermo Fisher Scientific AG, Dreieich, Germany) with a Nucleodur® C18 Gravity (250 mm × 10 mm, 5 μm) column. NMR spectra were recorded on a Bruker AV 500 (1H, 500 MHz; 19F, 376 MHz; 13C, 126 MHz) spectrometer. All spectra were measured in CDCl3 or DMSO-d6, and chemical shifts were adjusted based on the residual proton of the internal standard in parts per million (ppm), (CDCl3, δ = 7.27, 77.00 and DMSO-d6, δ = 2.50, 39.51, 1H and 13C respectively). Coupling constants (J) are given in Hertz (Hz) and the following abbreviations were used for multiplicity (s = singlet, d =doublet, t = triplet, m = multiplet, br = broad or combinations of these). High-resolution mass spectrometry (HRMS) was performed on a Thermo Scientific Q Exactive Focus Orbitrap system. The purity of all synthesised compounds used for biological testing was determined by UV tracing at 254 nm in the LCMS platform, being (≥95%) except compound 8 (93%). The reference compound 1 originated from a previous publication within the consortium.19 Reference compounds 2 (Enamine Z26672805/CAS 2094230-26-7) and 3 (Enamine Z26672672/CAS 2391905-54-5) were commercially purchased compounds that were kindly provided by BASF. Synthetic derivatives were synthesised according to Schemes 1 and 2 using general procedures A and B as described in more detail in section S2, ESI.†
Computational methods
StarDrop v. 6.6.4.23412 (Optibrium, Ltd., Cambridge, UK) was used to calculate log D7.4, MW, TPSA, pKa and antibacterial scoring profile and to screen for PAINS showing no hits. The latter two were accessed via the Optibrium community (http://www.optibrium.com/community/). Dipole moment (PM3_dipole), fraction of rotatable bonds (b_rotB), predicted log of the octanol/water partition coefficients (log P(o/w)) and (S log P) and amphiphilic moment (vsurf_A) were calculated with Molecular Operating Environment (MOE) 2018.01 software (Chemical Computing Group ULC, Montreal, Canada). MarvinSketch 20.8, ChemAxon, was used for calculating minimal projection area (https://www.chemaxon.com). The accordance to the eNTRy rules was predicted using eNTRyway, where N: presence of an ionisable primary amine, T: globularity and R: number of rotatable bonds.6,45
Author contributions
H.-K. Ropponen: conceptualisation, data curation, formal analysis, investigation, software, visualisation, writing – original draft, writing – review & editing, E. Diamanti: investigation; supervision, writing – review & editing, A. Siemens: investigation, writing – review & editing, B. Illarionov: data curation, formal analysis, investigation, writing – review & editing, J. Haupenthal: formal analysis, supervision, validation, writing – review & editing, M. Fischer: resources, supervision, writing – review & editing, M. Rottmann: formal analysis, validation and writing – review & editing, M. Witschel: resources and writing – review & editing, A. K. H. Hirsch: conceptualisation, funding acquisition, project administration, resources, supervision and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
H.-K. Ropponen thanks Stiftung Stipendien-Fonds des Verbandes der Chemischen Industrie for the granted Kekulé Mobility Fellowship. A. K. H. Hirsch gratefully acknowledges funding from the Helmholtz Association's Initiative and Networking Fund. The authors thank Jeannine Jung and Dennis-Thomas Jener for excellent technical assistance, Dr. Jennifer Herrmann for kindly providing E. coli ΔacrB and Dr. Mostafa Hamed for critical proofreading of the manuscript (HIPS). The authors are also grateful to Prof. Dr. Jean-Marie Pagés for kindly providing E. coli BL21(DE3)omp8 strain.
Electronic supplementary information (ESI) available: Synthetic protocols and biological data. See DOI: 10.1039/d0md00409j
Notes and references
- Theuretzbacher U. Gottwalt S. Beyer P. Butler M. Czaplewski L. Lienhardt C. Moja L. Paul M. Paulin S. Rex J. H. Silver L. L. Spigelman M. Thwaites G. E. Paccaud J. P. Harbarth S. Lancet Infect. Dis. 2019;19:e40–e50. doi: 10.1016/S1473-3099(18)30513-9. [DOI] [PubMed] [Google Scholar]
- Ropponen H.-K. Richter R. Hirsch A. K. H. Lehr C.-M. Adv. Drug Delivery Rev. 2021 doi: 10.1016/j.addr.2021.02.014. [DOI] [PubMed] [Google Scholar]
- Parkes A. L. Expert Opin. Drug Discovery. 2020;00:1–9. doi: 10.1080/17460441.2020.1767065. [DOI] [PubMed] [Google Scholar]
- Lien E. J. Hansch C. Anderson S. M. J. Med. Chem. 1968;11:430–441. doi: 10.1021/jm00309a004. [DOI] [PubMed] [Google Scholar]
- O'Shea R. Moser H. E. J. Med. Chem. 2008;51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- Richter M. F. Drown B. S. Riley A. P. Garcia A. Shirai T. Svec R. L. Hergenrother P. J. Nature. 2017;545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F. Hergenrother P. J. Ann. N. Y. Acad. Sci. 2019;1435:18–38. doi: 10.1111/nyas.13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Gutiérrez S. Ferrara L. Pathania M. Masi M. Wang J. Bodrenko I. Zahn M. Winterhalter M. Stavenger R. A. Pagè J.-M. Naismith J. H. Van Den Berg B. Page M. G. P. Ceccarelli M. ACS Infect. Dis. 2018;4:1487–1498. doi: 10.1021/acsinfecdis.8b00108. [DOI] [PubMed] [Google Scholar]
- Montefiore B., Klingler F. and Foster N., Poster: A novel scoring profile for the design of antibacterials active against gram-negative bacteria, 2018
- Cooper S. J. Krishnamoorthy G. Wolloscheck D. Walker J. K. Rybenkov V. V. Parks J. M. Zgurskaya H. I. ACS Infect. Dis. 2018;4:1223–1234. doi: 10.1021/acsinfecdis.8b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker E. N. Drown B. S. Geddes E. J. Lee H. Y. Ismail N. Lau G. W. Hergenrother P. J. Nat. Microbiol. 2019;2019(5):1–9. doi: 10.1038/s41564-019-0604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P. A. Koehler M. F. T. Girgis H. S. Yan D. Chen Y. Chen Y. Crawford J. J. Durk M. R. Higuchi R. I. Kang J. Murray J. Paraselli P. Park S. Phung W. Quinn J. G. Roberts T. C. Rougé L. Schwarz J. B. Skippington E. Wai J. Xu M. Yu Z. Zhang H. Tan M. W. Heise C. E. Nature. 2018;561:189–194. doi: 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]
- Liu B. Lee Trout R. E. Chu G.-H. Mcgarry D. Jackson R. W. Hamrick J. C. Daigle D. M. Cusick S. M. Pozzi C. De Luca F. Benvenuti M. Mangani S. Docquier J.-D. Weiss W. J. Pevear D. C. Xerri L. Burns C. J. J. Med. Chem. 2020;63:2789–2801. doi: 10.1021/acs.jmedchem.9b01518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. Shi H. Zhou M. Ren Q. Zhu W. Zhang W. Zhang Z. Zhou C. Liu Y. Ding X. Shen H. C. Frank Yan S. Dey F. Wu W. Zhai G. Zhou Z. Xu Z. Ji Y. Lv H. Jiang T. Wang W. Xu Y. Vercruysse M. Yao X. Mao Y. Yu X. Bradley K. Tan X. J. Med. Chem. 2020;63:9623–9649. doi: 10.1021/acs.jmedchem.0c00768. [DOI] [PubMed] [Google Scholar]
- Motika S. E. Ulrich R. J. Geddes E. J. Lee Y. Lau G. W. Hergenrother P. J. J. Am. Chem. Soc. 2020;142:10856–10862. doi: 10.1021/jacs.0c04427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews L. D. Kane T. R. Dozzo P. Haglund C. M. Hilderbrandt D. J. Linsell M. S. Machajewski T. Mcenroe G. Serio A. W. Wlasichuk K. B. Neau D. B. Pakhomova S. Waldrop G. L. Sharp M. Pogliano J. Cirz R. T. Cohen F. J. Med. Chem. 2019;62:7489–7505. doi: 10.1021/acs.jmedchem.9b00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A. Groll M. Chem. Rev. 2017;117:5675–5703. doi: 10.1021/acs.chemrev.6b00537. [DOI] [PubMed] [Google Scholar]
- Masini T. Hirsch A. K. H. J. Med. Chem. 2014;57:9740–9763. doi: 10.1021/jm5010978. [DOI] [PubMed] [Google Scholar]
- Hirsch A. K. H. Alphey M. S. Lauw S. Seet M. Barandun L. Eisenreich W. Rohdich F. Hunter W. N. Bacher A. Diederich F. Org. Biomol. Chem. 2008;6:2719–2730. doi: 10.1039/b804375b. [DOI] [PubMed] [Google Scholar]
- Mjambili F. Njoroge M. Naran K. De Kock C. Smith P. J. Mizrahi V. Warner D. Chibale K. Bioorg. Med. Chem. Lett. 2014;24:560–564. doi: 10.1016/j.bmcl.2013.12.022. [DOI] [PubMed] [Google Scholar]
- Yokoyama M. Kurauchi M. Imamoto T. Tetrahedron Lett. 1981;22:4–45. [Google Scholar]
- Devine S. M. Mulcair M. D. Debono C. O. Leung E. W. W. Nissink J. W. M. Lim S. S. Chandrashekaran I. R. Vazirani M. Mohanty B. Simpson J. S. Baell J. B. Scammells P. J. Norton R. S. Scanlon M. J. J. Med. Chem. 2015;58:1205–1214. doi: 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- Bolea C., WO2011/86163A1, ADDEX PHARMA S.A., 2011 [Google Scholar]
- Liu B., Liu G., Nelson L., Patel J., Sham H., Xin Z. and Zhao H., US20050014794A1, 2005
- Ruggiu F. Yang S. Simmons R. L. Casarez A. Jones A. K. Li C. Jansen J. M. Moser H. E. Dean C. R. Reck F. Lindvall M. ACS Infect. Dis. 2019;5:1688–1692. doi: 10.1021/acsinfecdis.9b00256. [DOI] [PubMed] [Google Scholar]
- Vergalli J. Bodrenko I. V. Masi M. Moynié L. Acosta-Gutiérrez S. Naismith J. H. Davin-Regli A. Ceccarelli M. van den Berg B. Winterhalter M. Pagès J.-M. Nat. Rev. Microbiol. 2020;18:164–176. doi: 10.1038/s41579-019-0294-2. [DOI] [PubMed] [Google Scholar]
- Prilipov A. Phale P. S. Van Gelder P. Rosenbusch J. P. Koebnik R. FEMS Microbiol. Lett. 1998;163:65–72. doi: 10.1111/j.1574-6968.1998.tb13027.x. [DOI] [PubMed] [Google Scholar]
- Shi X. Chen M. Yu Z. Bell J. M. Wang H. Forrester I. Villarreal H. Jakana J. Du D. Luisi B. F. Ludtke S. J. Wang Z. Nat. Commun. 2019;10:1–6. doi: 10.1038/s41467-019-10512-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnow H. Fetz V. Hotop S. K. García-Rivera M. A. Heumann A. Brönstrup M. Anal. Chem. 2019;91:1863–1872. doi: 10.1021/acs.analchem.8b03586. [DOI] [PubMed] [Google Scholar]
- Adler J., Chemotaxis in bacteria, in Surface Membrane Receptors, NATO Advanced Study Institutes Series (Series A: Life Sciences), Springer, Boston, MA, 1975, p. 11 [Google Scholar]
- Baker M. D. Wolanin P. M. Stock J. B. BioEssays. 2006;28:9–22. doi: 10.1002/bies.20343. [DOI] [PubMed] [Google Scholar]
- Shapiro J. A. Annu. Rev. Microbiol. 1998;52:81–104. doi: 10.1146/annurev.micro.52.1.81. [DOI] [PubMed] [Google Scholar]
- Mesibov R. Adler J. J. Bacteriol. 1972;112:315–326. doi: 10.1128/jb.112.1.315-326.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedblom M. L. Adler J. J. Bacteriol. 1983;155:1463–1466. doi: 10.1128/jb.155.3.1463-1466.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishinuma F. Izaki K. Takahashi H. Agric. Biol. Chem. 1969;33:1577–1586. [Google Scholar]
- Yang Y. Pollard A. M. Höfler C. Poschet G. Wirtz M. Hell R. Sourjik V. Mol. Microbiol. 2015;96:1272–1282. doi: 10.1111/mmi.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuston S. Begley M. Gahan C. G. M. Hill C. Microbiology. 2012;158:1389–1401. doi: 10.1099/mic.0.051599-0. [DOI] [PubMed] [Google Scholar]
- Alreshidi M. M. Dunstan R. H. Macdonald M. M. Gottfries J. Roberts T. K. Front Microbiol. 2020;10:1–12. doi: 10.3389/fmicb.2019.03059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttgen H. Rohdich F. Herz S. Wungsintaweekul J. Hecht S. Schuhr C. A. Fellermeier M. Sagner S. Zenk M. H. Bacher A. Eisenreich W. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1062–1067. doi: 10.1073/pnas.97.3.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgaher W. A. M. Fruth M. Groh M. Haupenthal J. Hartmann R. W. RSC Adv. 2014;4:2177–2194. [Google Scholar]
- Haupenthal J. Baehr C. Zeuzem S. Piiper A. Int. J. Cancer. 2007;121:206–210. doi: 10.1002/ijc.22665. [DOI] [PubMed] [Google Scholar]
- Dorn A. Stoffel R. Matile H. Bubendorf A. Ridley R. G. Nature. 1995;374:269–271. doi: 10.1038/374269a0. [DOI] [PubMed] [Google Scholar]
- Trager W. Jensen J. B. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Huber W. Koella J. C. Acta Trop. 1993;55:257–261. doi: 10.1016/0001-706x(93)90083-n. [DOI] [PubMed] [Google Scholar]
- Entryway, http://www.entry-way.org/, (accessed April 2020)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.