

Abstract
NIK is a key kinase required for the activation of alternative NF-κB signaling pathways. Overactivation of NIK in patients has been observed and is implicated in the pathogenesis of inflammatory diseases, B-cell malignances, and solid tumors. Over the past decade, inhibition of NIK overactivation with small molecules has been pursued as an attractive strategy for drug discovery, where numerous potent and selective NIK inhibitors with novel pharmacophores have been identified. This review summarizes the structural features and key efficacy studies of the NIK inhibitors reported, which justify the mechanism of action of such inhibitors in animal models driven by NIK overactivation. Given the strong pathological associations between overactivation of NIK and human diseases, human clinical trials of NIK inhibitors as drug candidates are eagerly awaited. Information showcased in this review article might be helpful for the discovery and clinical development of the next generation of NIK inhibitors in the near future.
This review summarizes structural features and key efficacy studies of NIK inhibitors, which justify the mechanism of action of such inhibitors in animal models driven by NIK overactivation.
Introduction
NF-κB-inducing kinase (NIK) is a cytoplasmic serine/threonine kinase first reported in 1997 (ref. 1) and belongs to the SET branch of the human kinome,2 which also includes MAPK/ERK kinase 1/2 (MEK 1/2) and apoptosis signal-regulating kinase 1 (ASK1).3–7 It lacks the conserved arginine residue in the catalytic loop and is considered as a non-RD kinase.8,9 A biallelic mutation in the MAP3K14 gene led to loss of NIK kinase activity in patients who suffered from recurrent bacterial, viral, and Cryptosporidium infections, suggesting that NIK plays an important role in human lymphoid immunity.10 It is now believed that the significance of NIK is far beyond lymphoid immunity in the human biological system.11–15
NIK is an essential component for the processing of p100 to p52 and subsequent activation of the alternative NF-κB signaling pathway (NF-κB2),14–16 which is characterized by nuclear translocation of p52–RelB dimers17,18 (Fig. 1) and is different from the canonical NF-κB signaling pathway (NF-κB1) featuring the nuclear accumulation of p50–RelA and p50–cRel complexes.19–21 The expression level of NIK is very low in various tissues under normal physiological conditions,1 which is because the TNF receptor-associated factor 2 (TRAF2)/TNF receptor-associated factor 3 (TRAF3)/cellular inhibitor of apoptosis protein (cIAP) destruction complex21,22 constantly recruits and promotes degradation of NIK through the proteasome machinery.
Fig. 1. The NIK protein and alternative NF-κB signaling pathway. (A) In unstimulated cells, TRAF3, TRAF2, and cIAP1/2 form a complex that recruits and ubiquitinates NIK, leading to its proteasomal degradation. (B) In the presence of stimulus, the cell surface receptors trimerize and recruit TRAF3, which will be ubiquitinated by cIAP1/2 and degraded through proteasome. As a consequence, the NIK is released and accumulated in cytosol. Auto-phosphorylation of NIK augments its kinase activity, promoting phosphorylation of IKKα on its S176 residue. Both activated kinases promote the phosphorylation of p100 on the S866 and S870 residues, which results in K855 poly-ubiquitination and subsequent limited processing of p100 to p52. Finally, p52 and RelB form a dimer and migrate into the nucleus where they initiate gene expression. (P) denotes phosphorylation and Ub denotes ubiquitination.

It has been shown that TRAF3 directly binds to NIK and TRAF2 directly binds to cIAP1/2,21 while hetero-dimerization of TRAF2 and TRAF3 leads to formation of the destruction complex of NIK,22,23 which promote ubiquitination of NIK meidated by cIAP1 and cIAP2 and its subsequent proteasomal degradation (Fig. 1A).24,25
In the presence of external stimulus, the subset of TNF family receptors, such as B-cell-activating factor receptor (BAFFR), cluster of differentiation 40 (CD40), lymphotoxin β receptor (LTβR), and receptor activator of nuclear factor κB (RANK), form trimeric structures and recruit TRAF3,26–29 which results in c-IAP2-mediated K48-linked polyubiquitination and subsequent degradation of TRAF3 (ref. 22 and 25) (Fig. 1B). In addition, TRAF2 also functions as an E3 ligase and promotes K63-linked polyubiquitination and subsequent degradation of cIAP2.25 Collectively, these biological cascades result in the deconstruction of the TRAF2/TRAF3/cIAP complex; therefore the cellular stability and the concentration of NIK increase, promoting the phosphorylation of downstream kinase IKKα on its S176 residue30,31 and its activation. Together with activated NIK, the phosphorylated IKKα dimer32,33 further promotes the phosphorylation of the S866 and S870 residues of p100.34 Subsequent ubiquitination at K855 of p100 (ref. 35) initiates limited processing of p100 to p52.34 It is worth noting that the limited processing of p100 to p52 is of critical importance for the functional alternative NF-κB signaling pathway. Single point mutation at D865G of p100 resulted in processing failure and p52 was not formed.36 In line with this, a missense mutation of the NFKB2 gene corresponding to a D865G mutation of p100 has been observed from three patients who suffered from severe B-cell deficiency and needed intravenous immunoglobulin replacement therapy.36 In the final step, hetero-dimerization of p52 and RelB forms the NF-κB2 transcription complex, which migrates into the cell nucleus and initiates transcription of target genes. Collectively, it is evident that NIK is at the strategically convergent point of the alternative NF-κB signaling pathway and its tightly regulated activation is important for proper function of the alternative NF-κB signaling pathway.
Inhibition of NIK kinase activities with small-molecule inhibitors (NIK inhibitors) will lead to suppression of the alternative NF-κB signaling pathway, and this emerging area of research has attracted considerable attention over the past decade. In this review, the topics covered on NIK inhibitors are:
(1) A brief introduction on the structural basis of NIK kinases, which is important information for the structure-based drug design and understanding of the interactions between the NIK inhibitors and the kinase;
(2) Overactivation of NIK and its role in human diseases, which explains why NIK inhibitors could be potential treatments for specific human diseases;
(3) Small-molecule NIK inhibitors discovered over the past 10 years, with focuses on drug design strategies and target–ligand interactions at the atomic level
(4) In the fourth section, we discuss several outstanding examples of potent and selective small-molecule NIK inhibitors achieving high systemic exposure, robust NIK inhibition in vivo, and on-target efficacy in animal models of human diseases. These proof-of-concept efficacy studies are valuable for future discovery and clinical development of drug candidates targeting NIK.
(5) Our perspective regarding the future direction of this field of study will be discussed in the last section.
The structural basis of NIK kinases
The NIK protein consists of an N-terminal TRAF-binding domain, a nuclear localization domain, a kinase domain, and a C-terminal IKKα-binding domain.9 The amino acid sequences of human and murine NIK kinase domains are highly conserved with 87% residue similarity.9 The key residues of kinase sub-regions,37,38 such as the gatekeeper and hinge region, the catalytic base region (HGD), the Mg2+ binding site (DFG), the activation loop, the active site Lys region, and the αC helix, are identical with the exception of two differences on the P loop, i.e.406PRV408 for mouse NIK and 404LRL406 for human NIK.
The X-ray crystal structures of human and murine NIK proteins have been solved,9,39 which show that both kinases adopt a “DFG-in” active conformation.8,37 Superimposition of human and murine NIK X-ray crystal structures (PDB: 4G3D and 4G3C) shows the three-dimensional architectures of both proteins aligned very well, despite a part of the activation loop of mouse NIK and the residues around 404LRL406 (the P loop) of human NIK were not well-solved (Fig. 2). It is worth noting that both the unsolved activation loop and the 404LRL406 moiety are distal to the ATP and substrate binding sites and should not be involved in direct ligand interactions in general. In addition, in vitro enzymatic assays showed that the activities of human and murine NIK kinases were approximately equivalent.9 These data suggested that a murine NIK enzymatic assay in vitro could be a good surrogate for the human counterpart.
Fig. 2. Structures of human and murine NIK are superimposed to show their similarity. Human NIK (PDB: 4G3D) is cyan, mouse NIK (PDB: 4G3C) is green, and the Mg2+ is magenta.
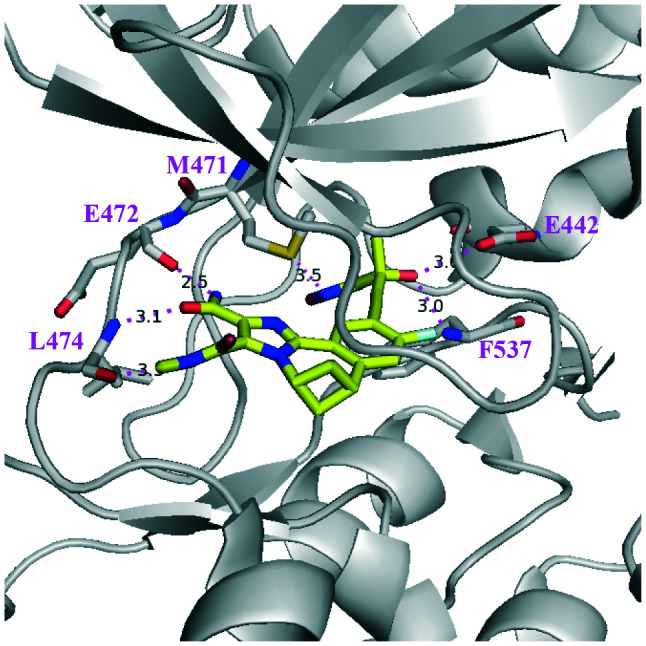
To make it a clear discussion on small molecule/NIK interactions in the following context, a figure of ATP and the substrate binding pocket of human NIK is shown in detail in Fig. 3. Important residues involved in ligand interactions are highlighted in sticks of salmon colour, such as the gatekeeper residue M469, the hinge resides E470/L471/L472, the “DFG” residues D534/F535, the β3 strand K429, the P loop residues L406/R408, and the αD helix residue S476. In addition, the corresponding residue numbers for mouse NIK differ from that of human NIK by 2, for example, M471 for mouse and M469 for human.
Fig. 3. The key amino acid residues of NIK that are essential for inhibitor binding (PDB: 4IDT) where oxygen is red, nitrogen is blue, and sulphur is yellow.

Overactivation of NIK and its role in human diseases
In line with the important role of NIK in the alternative NF-κB signaling pathway, dysregulation of NIK activity has been found to be associated with human diseases. For example, elevated expression of NIK has been found in patient samples and human cell lines of multiple myeloma as a result of genomic alterations or protein stabilization.40,41 Concurrently, overexpression of CD40 and suppression of TRAF3 activity through mutation, deletion, and silencing of the TRAF3 gene have also been observed; all these events are believed to activate NIK/NF-κB2 and contribute to the malignancy of multiple myeloma. In addition, Storz and coworkers have reported elevated expression of NIK found in pancreatic cancer cell lines and patient samples of pancreatic ductal adenocarcinoma cancer, which is attributed to the proteasomal downregulation of TRAF2.42 It has also been reported that high expression of NIK was detected in 51.7% samples of 120 patients diagnosed with NSCLC.43,44
Analysis of liver biopsy samples of patients with alcoholic cirrhosis or primary biliary cirrhosis shows that the levels of both NIK mRNA and p52 protein are higher than that of healthy subjects.45 Mechanistically, overexpression of NIK in liver stimulates the release of numerous chemokines and cytokines that activate marrow-derived macrophages, which further promotes apoptosis of hepatocytes. Furthermore, overexpression of NIK and p52 has been observed in intestinal epithelial cells extracted from patients with uncreative colitis.46 Mechanistic studies suggest that overactivation of intestinal epithelial NIK causes elevated expression of inflammatory cytokines and microfold-cell biomarkers, thus increasing the susceptibility to uncreative colitis. Taken together, these examples show that unbalanced activation of NIK is closely associated with a variety of human diseases.
Overexpression of NIK in animal models has been investigated to elucidate its biological roles in and association with human diseases. The NIK activity in livers of both dietary and genetic obesity mice was tenfold higher than that of normal mice, suggesting an important role of NIK in obesity.47 Interestingly, liver-specific inhibition of NIK improved glucose metabolism and lowered blood glucose concentrations. In a transgenic mouse model, specific overexpression of NIK in islet α-cells resulted in dysfunction of α-cells, death of β-cells, and pancreatitis.48 In addition, specific overexpression of NIK in islet β-cells led to β-cell failure and spontaneous diabetes in male mice at a young age.49
The aforementioned evidence shows that overactivation of NIK plays causative roles in many human diseases. Given that the function of NIK in the alternative NF-κB signaling pathway mainly depends on its kinase activity, inhibition of NIK activity is a reasonable approach to block overactivation of NIK and the alternative NF-κB signaling pathway and thus has been pursued as a potential drug discovery strategy.50,51
A survey of small-molecule inhibitors of NIK reported in the literature
In order to block overactivation of NIK and the alternative NF-κB signaling pathway, there has been a growing interest in discovering small-molecule inhibitors of NIK over the past decade. For example, staurosporine (1),52 a pan kinase inhibitor, was found to inhibit NIK with an IC50 value of 2.3 μM (Scheme 1).53
Scheme 1. Representative examples of NIK inhibitors from high-throughput screening studies.

Early efforts searching for NIK inhibitors involved virtual screening of database and commercially-available compound libraries. Mortier and coworkers performed this approach and identified 224 hits from 67 500 structures.53 Subsequently, 49 hits were selected and tested in a radiometric protein kinase assay, which led to 2 that had a NIK inhibitory IC50 value of 51 μM (Scheme 1). In addition, Pippione and coworkers screened three Prestwick compound libraries comprising 1200 compounds and found that an aminopyrazole-containing compound selectively inhibited IKKβ with an IC50 value of 50.9 μM.54 Medicinal chemistry modifications followed and 3 was synthesized, which inhibited NIK with an IC50 value of 8.4 μM. 3 was a selective NIK inhibitor and had no inhibitory effect against 44 other protein kinases. In order to show the target engagement of 3 in living cells, the EJM cell line with NIK gene amplification was engineered with luciferase reporters to monitor NF-κB activation. Interestingly, 3 at 25 μM suppressed 83.4% of NF-κB reporter activity, indicating that it inhibited cellular NIK activity. In addition, an in-silico screening of the ChemDiv database comprising 1 456 156 compounds for NIK inhibitors furnished 70 hits. Upon subsequent biochemical testing, 4 was found and inhibited NIK with an IC50 value of 71.1 μM.
In 2012, a group of researchers from Genentech reported 5 and 6 (Scheme 2) as potent NIK inhibitors which were discovered from a high-throughput screening followed by additional medicinal chemistry optimization.9 Compounds 5, 6, and 7 have NIK inhibitory IC50 values of 4.4, 3.7, and 4.2 nM, respectively, in an ATP consumption assay.55
Scheme 2. The structures of the first NIK inhibitors reported with NIK co-crystal structures.

In addition, three co-crystal structures of murine NIK complexes of 5, 6, and 7 have been obtained and solved, which provide valuable information about the interactions between ligands and key amino acid residues surrounding the ATP binding site of NIK (Fig. 4). Compounds 5 and 6 share the same core structure and their binding models to NIK are similar. Salt bridges were observed between the NH groups of 5/6 and D536, which is the first residue of the DFG motif that recognizes the ATP-bound Mg2+. The hydroxyl groups of phenols formed H-bonds with K431, E442, and D536 of the N-lobe β-sheet 3. A H-bond was observed between the CN group of 5 and the hinge residue L474, but not for 6. Interestingly, the phenol moieties of 5 and 6 pushed away the gatekeeper residue M471 and partially occupy the back pocket behind M471. On the other hand, three H-bonds were observed between the hinge and gatekeeper residues (M471, E472, and L474) and the 2-aminopyrimidine moiety of 7. The hydroxyl group of 7 formed two H-bonds with E442 and F537, while the oxazole group of 7 formed a H-bond with D536. In particular, the thiazole group of the tertiary alcohol moiety occupied the back pocket behind the gatekeeper residue M471, which is a common feature shared by many potent NIK inhibitors. The triple bond is linear and small in size; therefore it passes the gatekeeper M471 and accesses the back pocket. The NIK binding model of 7 illustrates the key features that are common for many succeeding NIK inhibitors, i.e. a head binds the hinge region, a tail binds the DFG region, and a hydrophilic body links the head and tail. More importantly, this particular model is the source of inspiration for many subsequent drug discovery campaigns that delivered novel NIK inhibitors.
Fig. 4. PyMol figures depict the X-ray co-crystal structures of murine NIK complexes with (A) 5 (PDB: 4G3F), (B) 6 (PDB: 4G3G), and (C) 7 (PDB: 4G3E). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, nitrogen is blue, chlorine is green, fluorine is pale cyan, and sulfur is dark yellow. The magenta dotted lines are H-bonds.

In 2013, Li and coworkers from Amgen reported a high-throughput screening hit 8 that inhibited NIK with an IC50 value of 0.60 μM (Scheme 3).56 In a HT-29 cell-based assay, lymphotoxin (LT) α/β2-induced processing of p100 to p52 was inhibited by 8 with an IC50 of 16 μM, indicating its on-target activity in living cells. Interestingly, 8 structurally resembles a natural indole alkaloid meridianin C;57 therefore the imidazopyridine moiety of 8 was replaced by an indole or other 5–6 fused aromatic heterocycles. For example, 9 was synthesized and had a NIK inhibitory IC50 value of 0.15 μM. Since biaryl compounds such as 9 exist in equilibrium between various conformations, which resulted from the potentially restricted rotation around the highlighted blue C–C bond (Scheme 3), cyclization of two extreme conformations of 9 could provide two cyclic compounds, 10 and 11, that inhibited NIK with IC50 values of 0.10 and 2.6 μM, respectively, which suggested that the conformation of 10 was optimal for NIK binding.
Scheme 3. The representative structures of NIK inhibitors reported by Li and coworkers from Amgen.

The X-ray co-crystal structure of the NIK/10 complex (Fig. 5) has been solved, which not only confirmed the design rationales but also showed that the 2-aminopyrdine moiety of 10 formed two H-bonds with hinge residues E470 and L472 of human NIK.
Fig. 5. PyMol figures depict the X-ray co-crystal structures of human NIK complexes with (A) 10 (PDB: 4IDT) and (B) 13 (PDB: 4IDV). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, nitrogen is blue, and bromine is green. The magenta dotted lines are H bonds.

However, a back pocket behind the gatekeeper residue M469 was not occupied, partially because of restriction from a narrow channel formed by M649 and K429. In order to access the cavity behind the M649 residue, a linear propargylic alcohol was incorporated into 10 and furnished 12, which significantly improved NIK inhibition activity with an IC50 value of 0.041 μM. Subsequently, an X-ray co-crystal structure of an analogous compound 13 and NIK was obtained (Fig. 5), which clearly showed that the back pocket was partially occupied and the hydroxyl group of the tertiary alcohol formed two H-bonds with E440 and F535 in that pocket. In addition, the 2-methoxyl-ethyl group also contributed to NIK binding as it formed two H-bonds with S476 and Q479. The substitution position of the 2-methoxyl-ethyl group was important for optimal NIK activity because a weaker inhibitor was obtained when the substitution position shifted.58
In 2017, Castanedo and coworkers from Genentech reported their discovery and optimization of a series of tricyclic NIK inhibitors (Scheme 4).59 Initially, a high-throughput screening hit 14 was identified with a novel seven-membered tricyclic structure and inhibited NIK with a Ki value of 1.28 μM. The X-ray co-crystal structure of murine NIK/14 had been solved and showed that the CO–NH2 moiety formed two H-bonds with the hinge residues E472 and L474 (Fig. 6). It has been noticed that the back pocket behind gatekeeper M471 was not occupied. In order to reach the back pocket, a tertiary alcohol containing a triple bond was introduced and 15 was synthesized, while the biaryl group of 14 was also changed in order to avoid potential metabolic liability of the thiophene. Compared to 14, 15 has an improved NIK inhibitory Ki value of 0.31 μM and an X-ray co-crystal structure of NIK/15 has also been solved (Fig. 6), which demonstrated that the tertiary alcohol moiety of 15 occupied the back pocket and formed two H-bonds with E442 and F537. Further optimization of the tertiary alcohol moiety provided compound 16, which was a very potent NIK inhibitor with a Ki value of 0.3 nM. However, 16 also inhibited PI3Kδ with a Ki value of 47 nM, partially because the benzoxazepin ring of 16 was also a key feature of taselisib that potently inhibited PI3Kδ.
Scheme 4. The representative structures of NIK inhibitors reported by Castanedo and coworkers.

Fig. 6. PyMol figures depict the X-ray co-crystal structures of murine NIK complexes with (A) 14 (PDB: 5T8P), (B) 15 (PDB: 5T8O), and (C) 17 (PDB: 5T8Q). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, nitrogen is blue, and chlorine is green. The magenta dotted lines are H-bonds.

In order to avoid the potential liability of 16 caused by PI3Kδ inhibition, further optimization was carried out to abolish PI3Kδ activity. Firstly, guided by the X-ray co-crystal structure of NIK/17 (Fig. 6), a series of compounds with substitutions at the 3-position were synthesized. For example, 18 inhibited NIK with a Ki value of 2 nM, while it had no measurable activity against PI3Kδ (Ki >1 μM). Combination of favourable design elements of 16 and 18 provided 20, which inhibited NIK with a Ki value of 0.47 nM but not PI3Kδ inhibition. In addition, replacement of the oxygen atom in the benzoxazepin ring of 16 with a carbon linker also abolished the PI3Kδ activity of 19, yet it was a potent NIK inhibitor with a Ki value of 0.47 nM. Last but not the least, the cellular target of 20 was examined in a cell imaging assay that measured nuclear translocation of the canonical NF-κB p50 versus the alternative NF-κB p52. Satisfactorily, the data showed that 20 inhibited the cellular alternative but not the canonical NF-κB pathway.
Despite 19 selectively inhibiting NIK but not PI3Kδ, it did inhibit PKD1 with a Ki value of 5.9 nM.60 Similar results were observed for 21-R and 21-S (Scheme 5), close analogues of 19, both of which were good inhibitors of NIK and PKD1, given that the R enantiomer was more potent than the S enantiomer in both cases. A co-crystal structure of NIK/22 has been solved (Fig. 7), which showed that the amide group at the 3-position contributed to a H-bond with hinge residue L474, and the 2-position amide formed another two with E472 and L474. In the back pocket behind M471, the tertiary alcohol moiety occupied the cavity and the hydroxyl group and the nitrogen and oxygen atoms of oxazole formed three H-bonds with M471, E442, and F537. The hydroxyl group of 22 was very critical for NIK binding because an analogous compound 23 without an OH group had very weak NIK inhibitory activity (Ki >1250 μM), although it did inhibit PDK1 with sub-nM activity. These studies highlight the importance and structural basis of the tertiary alcohol motif for NIK inhibitors.
Scheme 5. The representative structures of NIK inhibitors reported by Feng and coworkers.

Fig. 7. PyMol figures depict the X-ray co-crystal structures of the murine NIK/22 complex (PDB: 6MYN). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, nitrogen is blue, chlorine is green, and fluorine is pale-cyan. The magenta dotted lines are H-bonds.
In 2018, Blaquiere and coworkers, also from Genentech, reported a series of 3-hydroxypyrrolidin-2-one containing small molecules as NIK inhibitors (Scheme 6).61 The optimization campaign started with 24, which inhibited NIK with a Ki value of 1.5 nM. An X-ray structure of the murine NIK/24 complex has also been solved (Fig. 8), which showed that the amide group formed two H-bonds with hinge residues E472 and L474, whereas the 3-hydroxypyrrolidin-2-one group passed the gatekeeper M471, occupied the back pocket, and formed three H-bonds with E442, D536, and F537. Interestingly, the guanidine of R410 also formed a H-bond with the oxygen atom of tetrahydropyran. Ring opening of the benzoxazepin of 24 and replacement of the imidazole ring with different 5–6 bicyclic heteroaromatic rings furnished a series of NIK inhibitors with improved potency, one example of which was 25 that inhibited NIK with a Ki value of 0.17 nM. Crystallography studies of NIK/25 complexes showed that the amide group formed three H-bonds with R410 and hinge residues E472 and L474, while the H-bonds from the 3-hydroxypyrrolidin-2-one group of 25 were of the same pattern as that of NIK/24 (Fig. 8). Unfortunately, metabolic studies showed that 25 was not stable when incubated with human hepatocytes because the triple bond formed a glutathione (GSH) addition product and the amide group was hydrolysed. In order to improve the stability in human hepatocytes, the pyridine ring of 25 was replaced with a benzene ring, furnishing compound 26. However, these efforts were less successful because 26 was also labile when incubated with human hepatocytes.
Scheme 6. The representative structures of NIK inhibitors reported by Blaquiere and coworkers.

Fig. 8. PyMol figures depict the X-ray co-crystal structures of murine NIK complexes with (A) 24 (PDB: 6G4Y) and (B) 25 (PDB: 6G4Z). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, nitrogen is blue, and fluorine is pale-cyan. The magenta dotted lines are H-bonds.

The metabolic stability problem was eventually resolved when the indazole group of 26 was replaced with a substituted pyridine, which provided 27 that had better human hepatocyte stability compared to 26. More importantly, 27 had very potent biochemical activity (NIK Ki = 0.23 nM) and decent cellular activity (NF-κB reporter IC50 = 34 nM). 27 inhibited nuclear translocation of p52 (RelB) with an IC50 of 70 nM but had no activity against RelA up to 20 μM, which indicated that 27 selectively attenuated the cellular alternative NF-κB signaling pathway via NIK inhibition. Additional mechanism of action studies showed that 27 also inhibited BAFF-stimulated mouse and human B cell survival with IC50 values of 373 and 189 nM, respectively. Good pharmacokinetic properties of 27 have been observed in mice, rats, beagles, and monkeys.
In view of the importance of the tertiary alcohol moiety for optimal NIK inhibition, we designed and synthesized a series of compounds by replacing the 2-aminopyrimidine group of 7 with a 2-amine-5H-pyrrolo[3,2-d]-pyrimidine structure, furnishing 28 (Scheme 7).62
Scheme 7. The representative structures of NIK inhibitors reported by Li and coworkers from the Shanghai Institute of Materia Medica (SIMM).

28 had a NIK inhibitory IC50 value of 303 nM and incorporation of a nitrogen atom into the benzene ring furnished 29 with an improved IC50 value of 22.4 nM. Unfortunately, 29 was not stable in acidic aqueous solution and further optimization based on this core structure was terminated. Alternatively, 30 with the 7-position of 2-amine-5H-pyrrolo[3,2-d]-pyrimidine substituted by the benzene part was found to be a potent NIK inhibitor with an IC50 value of 9.1 nM. 30 was a selective NIK inhibitor against a panel of 98 kinases and had no activities against 97 other kinases up to 500 nM. Mechanistically, 30 dose-dependently inhibited the NIK-mediated proteolysis of p100 to p52 in living cells and NIK-induced expression of inflammatory genes in primary hepatocytes, both of which implied that the cellular target of 30 was NIK proteins.
Recently, Chen and coworkers reported the design and synthesis of a series of 7H-pyrrolo[2,3-d]pyrimidin-4-amine-containing compounds as potent NIK inhibitors (Scheme 8).63 The proparylic alcohol moiety of 7 was adopted in the design and compound 31 was synthesized, which inhibited NIK with a moderate IC50 value of 1639 nM. Interestingly, a morpholine group substitution at the 4-position of the benzene ring significantly improved the NIK inhibitory potency as 32 had an IC50 value of 9.87 nM. In addition, analogous compounds bearing substituted piperazines as replacement of the morpholine group also provided compounds with similar biochemical potencies to that of 32. Compound 33, the R enantiomer of 32, was more potent than the S enantiomer, which is in accordance with the stereochemistry preferences of the aforementioned NIK inhibitors. 32 was a selective NIK inhibitor against a panel of 268 kinases, whereas it only inhibited Aurora B, TRKC, LRRK2 and ACK2 with moderate activity. 32 (1 μM) inhibited 86% NO production of J774 peritoneal macrophages stimulated by lipopolysaccharides (LPSs). In addition, 32 dose-dependently inhibited IL-6 secretion from BEAS-2B cells stimulated by LPS. Both cellular assays indicated that the cellular target of 32 was NIK, while inhibition of cellular NIK attenuated LPS-induced inflammations by 32 as well as the positive control 7. Compound 32 also has favourable pharmacokinetic properties in rats with a good oral bioavailability value of 64%.
Scheme 8. The representative structures of NIK inhibitors reported by Chen and coworkers from China Pharmaceutical University (CPU).

Very recently, two human NIK co-crystal structures containing 34 and 35 (Scheme 9) have been published (PDB codes: 6Z1T and 6Z1Q) with some interesting features (Fig. 9): (1) the stereogenic centres of both compounds are of opposite stereochemistry, i.e. S for 34 and R for 35, while they share very similar core structures; (2) neither compound occupied the back pocket of NIK behind gatekeeper reside M469; (3) the 2-aminopyrimidine moiety formed two H-Bonds with hinge residue L472 in both cases; (4) the 1-nitrogen atoms of 34 indoline and 35 aza-indoline formed salt bridges with D534.
Scheme 9. The representative structures of NIK inhibitors reported in the PDB with code numbers 6Z1T and 6Z1Q.

Fig. 9. PyMol figures depict the X-ray co-crystal structures of human NIK complexes with (A) 34 (PDB: 6Z1T) and (B) 35 (PDB: 6Z1Q). The grey cartoon and lines are NIK protein; the yellow sticks are inhibitors where oxygen is red, and nitrogen is blue. The magenta dotted lines are H-bonds.

In addition, the hydroxyl group of 34 formed an H-bond with R408 and the CN group formed an H-bond with the terminal –NH2 group of K429. On the other hand, the hydroxyl group of 35 formed an H-bond with the carbonyl group of D519 and the 7-nitrogen atom of aza-indoline formed water molecule-bridged H-bonds with K429. These interaction features make 34 and 35 distinct from the aforementioned NIK inhibitors.
Very recently, a cyclopropane-containing compound 37 was reported,64,65 which was designed based on a previously reported compound 36, an analogous compound of 25.61 The bicyclic structure of the 2-azabicyclo[3.1.0]hexanone motif of 37 is a very unique structure and its improved synthesis was recently disclosed.64 Both 36 and 37 potently inhibited NIK with IC50 values of 0.23 nM and 0.88 nM, respectively (Scheme 10).
Scheme 10. Synthesis of the cyclopropane-containing NIK inhibitor.

In order to compare the common protein-binding features of potent NIK inhibitors, we superimposed thirteen X-ray NIK co-crystal structures of 5, 6, 7, 10, 13, 14, 15, 17, 22, 24, 25, 34, and 35 and summarized the key residues that are involved in ligand binding as shown in Fig. 10. On the left edge of the pocket, E470 and L472 are the two hinge residues important for ligand binding. On the right edge, there are three important residues, E440, D534, and F535; the latter two belong to the “DFG” kinase signature motif, while E440 was involved in critical interactions with ligands in 8 of 13 crystal structures.
Fig. 10. Superimposition of the crystal structures of NIK inhibitors. The human NIK structure from PBD 4IDT is shown in grey cartoon, and the NIK residues important for ligand binding are grey sticks. The structures of inhibitors are shown in stacked colorful sticks inside the binding pocket. All residue numbers of murine NIK residues have been corrected to that of human residues.

The upper part of the pocket is the P loop, where L406 and R408 can form H-bonds with ligands; the lower part of the pocket is α-helix D of the C-terminal lobe and the catalytic loop, where S476, Q479, and D517 can also form H-bonds with ligands. The pocket was separated into two sub-pockets by M469. Given its smaller size, it is possible for the tertiary alcohols comprising a triple bond to pass the gatekeeper residue and enter the back pocket. 5 and 6 are outliers because they do not contain a triple bond yet they reached the back pocket behind M471 (Fig. 4). Sufficient interactions with these key residues are essential for inhibitors to have good affinity with NIK.
Efficacy studies of small-molecule NIK inhibitors in animal models of human diseases
Overactivation of NIK has been observed in several human diseases and is believed to play causative roles in disease pathologies. Therefore, inhibition of NIK activities through pharmacological intervention has been studied in several animal models of human diseases, which show the efficacies and potential applications of such a therapy.
Using 7 (B022) and 30 as tool compounds, we and other scientists have shown that NIK inhibition in vivo was efficacious for the treatment of inflammatory and metabolic diseases.49,62,66,67 Both 7 and 30 dose-dependently inhibited the NIK-overexpression-induced processing of p100 to p52 in model hepatic cell lines. As a result, 7 and 30 blocked the expression of NIK-induced inflammatory genes in hepatocytes. In mouse models with liver NIK overexpression induced by either viral transfection or CCl4 exposure, treatment with 7 or 30 significantly reduced liver inflammation and injury.62,66 Mechanistic studies show that 7 inhibited the processing of p100 to p50 in mouse liver tissue and the expression of pro-inflammatory genes. It has also been shown that the liver protective effects of 30 were partially attributed to inhibition of immune cell infiltrations into liver tissues.62
In another study, it has been demonstrated that 7 was efficacious in a mouse model of alcoholic liver disease.67 When wild-type mice were fed with ethanol, the hepatic NIK level increased, which inhibited fatty acid oxidation and promoted alcoholic steatosis through the phosphorylation of peroxisome proliferator-activated receptor alpha (PPARα), the primary regulator of fatty acid oxidation in liver. When the ethanol-fed mice also received the treatment with 7, NIK activation in the liver was inhibited, the phosphorylation of liver PPARα decreased, and the alcohol steatosis was relieved. In a third study, treatment of mice with high doses of streptozotocin, a toxin to islet β-cells, could induce diabetes, while subsequent administration of 7 to diabetic mice ameliorated the streptozotocin-induced diabetes, suggesting that NIK inhibitors have therapeutic potential for the treatment of diabetes associated with islet β-cell impairments.49
The activation of the alternative NF-κB signaling pathway could be induced BAFF, CD40, TWEAK, and RANK signaling pathways,26–29 which are all involved in the pathogenesis of systemic lupus erythematosus, for which the BAFF blockade drug belimumab68 is the only new therapy approved over the past 50 years.69 In a B cell survival study in C57BL/6 mice, the treatment with 27 (Scheme 6) dose-dependently reduced the survival of marginal zone B cells, whose survival was dependent on the activation of the BAFF signaling.61 The inhibition of marginal zone B cell survival was attributed to the blockade of BAFF signaling, indicating that the inhibition of NIK by 27in vivo counteracted the activation of the alternative NF-κB signaling pathway by BAFF. In another study,6927 has also been found to be efficacious in mouse models of systemic lupus erythematosus. Mechanistically, 27 inhibited BAFF-induced, CD40-induced, and OX40-induced activation of the alternative NF-κB signaling pathway in B and T cells; it also suppressed TWEAK-induced activation of the alternative NF-κB signaling pathway in renal proximal tubule-interstitial epithelial cells. In view of its broad cellular effects, 27 was evaluated in an IFNα-accelerated NZB/W-F1 mouse model of systemic lupus erythematosus, in which it suppressed multiple disease-relevant pro-inflammatory biomarkers and improved mouse survival as well as renal functions. These studies proved the effectiveness of small-molecule NIK inhibitors for the treatment of systemic lupus erythematosus in mice, suggesting its therapeutic potential in humans.
In a mouse model of imiquimod cream-induced psoriasis, 32 was administered orally and psoriasis-like dermatitis phenotypes on the back skin such as invasive erythema, roughness, and swelling, were monitored.63 Compared to the vehicle group, the symptoms of the treated groups were significantly relieved. In addition, the skin of the treated group was thicker and the weight of the spleen was lighter than that of the untreated group, which demonstrated the beneficial effects of 32. Pharmacodynamic studies of harvested mouse skin samples showed that the untreated group had a dramatically higher level of p52 compared to that of normal mouse, whereas the processing of p100 to p52 was dose-dependently inhibited upon administration of 32. Consistently, the expression levels of pro-inflammatory genes such as TNFα, IL6, IL10, and IL12 in the 32-treated group were lower than that in the untreated group. Collectively, the in vivo on-target activities and in vivo efficacy of 32 were clearly and compellingly exhibited, offering a new strategy for the treatment of psoriasis.
It has been reported that inhibition of the alternative NF-κB signaling pathway suppresses RANKL-induced osteoclast differentiation from bone marrow cells,70,71 whereas the osteoclast plays an important role in bond resorption.72,73 The bisphosphonate and parathyroid hormone therapy, which function through inhibition of bone resorption, are approved for the treatment of osteoporotic bond loss in elderly patients.74,75 Jimi and coworkers reported that 20 (Scheme 4) suppressed osteoclast differentiation from bone marrow cells, RANKL-induced processing of p100 to p52 in bone marrow macrophages, and bone-resorbing activities of mature osteoclasts.76 In an animal efficacy model, mice were ovariectomized (OVX) followed by administration of 20 or vehicle. Subsequent analysis of bone mineral densities showed that 20 significantly suppressed OVX-induced bond loss in the trabecular bone area, which was attributed to the reduced number of osteoclasts that inhibited bone resorption in the presence of 20. These studies suggested that NIK inhibitors could be developed as potential drugs for the treatment of bond loss arising from excessive bond resorption.
TRC694 is a selective NIK inhibitor with an undisclosed structure, which inhibited processing of p100 to p52 in a multiple myeloma cell line L363.77TRC694 strongly inhibited tumour growth in multiple myeloma cell lines and in a mouse xenograft model of RPMI-8226 and MM.1S cells, both of which harbour mutant TRAF3 and consequently activate NIK and the alternative NF-κB signaling pathway. TRC694 is the first NIK inhibitor that demonstrated antitumor efficacy in mouse tumour models.
Perspective
Several aforementioned studies have clearly demonstrated that small-molecule NIK inhibitors had successfully achieved excellent systemic exposure in animal models. More importantly, robust on-target efficacy has been observed upon oral administration of NIK inhibitors in animal models. Despite extensive studies on and great interest in the therapeutic potential of the NIK-mediated alternative NF-κB signalling pathway, there is no on-going clinical trial of NIK inhibitors, to the best of our knowledge. Given strong pathological associations between overactivation of NIK and human diseases, human clinical trials of NIK-inhibiting drug candidates are eagerly awaited.
Identification of the right clinical indication(s) is the key step for further clinical development of NIK inhibitors, if any. However, there are very limited in vivo proof-of-MOA studies available, partially because of an accessibility issue of drug-like NIK inhibitors to a broader range of biologists and pharmacologists, given the fact that they only had been reported very recently. As more efforts are put in place, extensive pharmacodynamic, efficacy, and toxicity studies of drug-like NIK inhibitors in relevant animal models may lead to a number of research breakthroughs that can drive clinical studies of NIK inhibitors into a new era.
Meanwhile, the chemical structures of most reported NIK inhibitors share a propargylic alcohol moiety, which is required for optimal inhibition of NIK in vitro, could be metabolically labile.61 In multiple species, whether the propargylic alcohol moiety is a potential metabolic liability remains underexplored and warrants further studies. Undoubtedly, NIK inhibitors with more diverse structures will help further validate the target's “druggability” and increase the chance of clinical success by providing an alternative, if not better, choice of clinical candidates. In this respect, medicinal chemists can contribute much more for the identification of future clinical candidates inhibiting NIK.
Conclusions
NIK is the key kinase required for the activation of alternative NF-κB signaling pathways. Overactivation of NIK in patients has been observed and is implicated in the pathogenesis of inflammatory diseases, B-cell malignancies, and solid tumours. Over the past decade, inhibition of NIK overactivation with small molecules has been pursued as an attractive strategy for drug discovery. Numerous potent and selective NIK inhibitors with novel pharmacophores have been identified though high-throughput screening and structure-based drug design. Through extensive evaluations of lead compounds in multiple animal models driven by overactivation of NIK and the alternative NF-κB signaling pathway, it is evident that NIK inhibitors achieved robust inhibition of the processing of p100 to p52 in vivo and demonstrated profound and compelling efficacies in animal models. Information showcased in this review article should be helpful for the discovery of the next generation of NIK inhibitors and hopefully for their clinical development in the near future.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (81872724, 82073682, and 81673295), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (Grant 2018ZX09711002-004-010), the Science and Technology Commission of Shanghai Municipality (18431907100), and the K. C. Wong Education Foundation. We also thank Dr. Xu Ran for careful proofreading.
Notes and references
- Malinin N. L. Boldin M. P. Kovalenko A. V. Wallach D. MAP3K-related kinase involved in NF-kappa B induction by TNF, CD95 and IL-1. Nature. 1997;385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- Manning G. Whyte D. B. Martinez R. Hunter T. Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Lu X. Y. Smaill J. B. Ding K. New promise and opportunities for allosteric kinase inhibitors. Angew. Chem. 2020;59:13764–13776. doi: 10.1002/anie.201914525. [DOI] [PubMed] [Google Scholar]
- Lu X. Smaill J. B. Ding K. Medicinal chemistry strategies for the development of kinase inhibitors targeting point mutations. J. Med. Chem. 2020;63:10726–10741. doi: 10.1021/acs.jmedchem.0c00507. [DOI] [PubMed] [Google Scholar]
- Zhang S. Y. Huang C. Y. Lyu X. L. Wang P. P. Zang Y. Wang Z. T. Wang H. Li J. Zhao Y. J. Discovery of a 2-pyridinyl urea-containing compound YD57 as a potent inhibitor of apoptosis signal-regulating kinase 1 (ASK1) Eur. J. Med. Chem. 2020;195:112277. doi: 10.1016/j.ejmech.2020.112277. [DOI] [PubMed] [Google Scholar]
- Kawarazaki Y. Ichijo H. Naguro I. Apoptosis signal-regulating kinase 1 as a therapeutic target. Expert Opin. Ther. Targets. 2014;18:651–664. doi: 10.1517/14728222.2014.896903. [DOI] [PubMed] [Google Scholar]
- Samatar A. A. Poulikakos P. I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discovery. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- Johnson L. N. Noble M. E. M. Owen D. J. Active and inactive protein kinases: Structural basis for regulation. Cell. 1996;85:149–158. doi: 10.1016/S0092-8674(00)81092-2. [DOI] [PubMed] [Google Scholar]
- de Leon-Boenig G. Bowman K. K. Feng J. W. A. Crawford T. Everett C. Franke Y. Oh A. Stanley M. Staben S. T. Starovasnik M. A. Wallweber H. J. A. Wu J. S. Wu L. C. Johnson A. R. Hymowitz S. G. The crystal structure of the catalytic domain of the NF-kappa B inducing kinase reveals a narrow but flexible active site. Structure. 2012;20:1704–1714. doi: 10.1016/j.str.2012.07.013. [DOI] [PubMed] [Google Scholar]
- Willmann K. L. Klaver S. Dogu F. Santos-Valente E. Garncarz W. Bilic I. Mace E. Salzer E. Conde C. D. Sic H. Majek P. Banerjee P. P. Vladimer G. I. Haskologlu S. Bolkent M. G. Kupesiz A. Condino-Neto A. Colinge J. Superti-Furga G. Pickl W. F. van Zelm M. C. Eibel H. Orange J. S. Ikinciogullari A. Boztug K. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat. Commun. 2014;5:5360. doi: 10.1038/ncomms6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017;17:545–558. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.-C. Controlling the fate of NIK: A central stage in noncanonical NF-κB signaling. Sci. Signaling. 2010;3:pe18. doi: 10.1126/scisignal.3123pe18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cildir G. Low K. C. Tergaonkar V. Noncanonical NF-kappa B signaling in health and disease. Trends Mol. Med. 2016;22:414–429. doi: 10.1016/j.molmed.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Sun S. C. Non-canonical NF-kappa B signaling pathway. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q. Lenardo M. J. Baltimore D. 30 years of NF-kappa B: A blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardam S. Brink R. Non-canonical NF-kappa B signaling initiated by BAFF influences B cell biology at multiple junctures. Front. Immunol. 2014;4:509. doi: 10.3389/fimmu.2013.00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. Q. Zarnegar B. Oganesyan G. Saha S. K. Yamazaki S. Doyle S. E. Dempsey P. W. Cheng G. H. Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J. Exp. Med. 2006;203:2413–2418. doi: 10.1084/jem.20061166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejardin E. Droin N. M. Delhase M. Haas E. Cao Y. X. Makris C. Li Z. W. Karin M. Ware C. F. Green D. R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappa B pathways. Immunity. 2002;17:525–535. doi: 10.1016/S1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Zandi E. Karin M. Bridging the gap: Composition, regulation, and physiological function of the I kappa B kinase complex. Mol. Cell. Biol. 1999;19:4547–4551. doi: 10.1128/MCB.19.7.4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G. Karin M. The two NF-kappa B activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Zarnegar B. J. Wang Y. Y. Mahoney D. J. Dempsey P. W. Cheung H. H. He J. Shiba T. Yang X. L. Yeh W. C. Mak T. W. Korneluk R. G. Cheng G. H. Noncanonical NF-kappa B activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G. X. Zhang M. Y. Harhaj E. W. Sun S. C. Regulation of the NF-kappa B-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J. Biol. Chem. 2004;279:26243–26250. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- He L. S. Grammer A. C. Wu X. L. Lipsky P. E. TRAF3 forms heterotrimers with TRAF2 and modulates its ability to mediate NF-kappa B activation. J. Biol. Chem. 2004;279:55855–55865. doi: 10.1074/jbc.M407284200. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E. Blankenship J. W. Wayson S. M. Fedorova A. V. Kayagaki N. Garg P. Zobel K. Dynek J. N. Elliott L. O. Wallweber H. J. A. Flygare J. A. Fairbrother W. J. Deshayes K. Dixit V. M. Vucic D. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappa B activation, and TNF alpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- Vallabhapurapu S. Matsuzawa A. Zhang W. Z. Tseng P. H. Keats J. J. Wang H. P. Vignali D. A. A. Bergsagel P. L. Karin M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappa B signaling. Nat. Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C. Grusdat M. Letellier E. Brenner D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol. Rev. 2019;99:115–160. doi: 10.1152/physrev.00045.2017. [DOI] [PubMed] [Google Scholar]
- Morrison M. D. Reiley W. Zhang M. Y. Sun S. C. An atypical tumor necrosis factor (TNF) receptor-associated factor-binding motif of B cell-activating factor belonging to the TNF family (BAFF) receptor mediates induction of the noncanonical NF-kappa B signaling pathway. J. Biol. Chem. 2005;280:10018–10024. doi: 10.1074/jbc.M413634200. [DOI] [PubMed] [Google Scholar]
- Cheng G. H. Cleary A. M. Ye Z. S. Hong D. I. Lederman S. Baltimore D. Involvement of Craf1, a relative of Traf, in CD40 signaling. Science. 1995;267:1494–1498. doi: 10.1126/science.7533327. [DOI] [PubMed] [Google Scholar]
- van Kooten C. Banchereau J. CD40-CD40 ligand. J. Leukocyte Biol. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- Ling L. Cao Z. D. Goeddel D. V. NF-kappa B-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3792–3797. doi: 10.1073/pnas.95.7.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senftleben U. Cao Y. Xiao G. Greten F. R. Krähn G. Bonizzi G. Chen Y. Hu Y. Fong A. Sun S.-C. Karin M. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Hayden M. S. Ghosh S. Shared principles in NF-kappa B signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Huynh Q. K. Boddupalli H. Rouw S. A. Koboldt C. M. Hall T. Sommers C. Hauser S. D. Pierce J. L. Combs R. G. Reitz B. A. Diaz-Collier J. A. Weinberg R. A. Hood B. L. Kilpatrick B. F. Tripp C. S. Characterization of the recombinant IKK1/IKK2 heterodimer - Mechanisms regulating kinase activity. J. Biol. Chem. 2000;275:25883–25891. doi: 10.1074/jbc.M000296200. [DOI] [PubMed] [Google Scholar]
- Xiao G. T. Harhaj E. W. Sun S. C. NF-kappa B-inducing kinase regulates the processing of NF-kappa B2 p100. Mol. Cell. 2001;7:401–409. doi: 10.1016/S1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Amir R. E. Haecker H. Karin M. Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF beta-TrCP ubiquitin ligase. Oncogene. 2004;23:2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- Lee C. E. Fulcher D. A. Whittle B. Chand R. Fewings N. Field M. Andrews D. Goodnow C. C. Cook M. C. Autosomal-dominant B-cell deficiency with alopecia due to a mutation in NFKB2 that results in nonprocessable p100. Blood. 2014;124:2964–2972. doi: 10.1182/blood-2014-06-578542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung J. E. Jura N. Structural basis for the non-catalytic functions of protein kinases. Structure. 2016;24:7–24. doi: 10.1016/j.str.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro D. Cowan-Jacob S. W. Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015;172:2675–2700. doi: 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. S. Sudom A. Min X. S. Cao Z. D. Gao X. Ayres M. Lee F. Cao P. Johnstone S. Plotnikova O. Walker N. Chen G. Q. Wang Z. L. Structure of the nuclear factor kappa B-inducing kinase (NIK) kinase domain reveals a constitutively active conformation. J. Biol. Chem. 2012;287:27326–27334. doi: 10.1074/jbc.M112.366658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziata C. M. Davis R. E. Demchenko Y. Bellamy W. Gabrea A. Zhan F. Lenz G. Hanamura I. Wright G. Xiao W. Dave S. Hurt E. M. Tan B. Zhao H. Stephens O. Santra M. Williams D. R. Dang L. Barlogie B. Shaughnessy J. D. Kuehl W. M. Staudt L. M. Frequent engagement of the classical and alternative NF-kappa B pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keats J. J. Fonseca R. Chesi M. Schop R. Baker A. Ching W. J. Van Wier S. Tiedemann R. Shi C. X. Sebag M. Braggio E. Henry T. Zhu Y. X. Fogle H. Price-Troska T. Ahmann G. Mancini C. Brents L. A. Kumar S. Greipp P. Dispenzieri A. Bryant B. Mulligan G. Bruhn L. Barrett M. Valdez R. Trent J. Stewart A. K. Carpten J. Bergsagel P. L. Promiscuous mutations activate the noncanonical NF-kappa B pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doppler H. Liou G. Y. Storz P. Downregulation of TRAF2 mediates NIK-induced pancreatic cancer cell proliferation and tumorigenicity. PLoS One. 2013;8:e53676. doi: 10.1371/journal.pone.0053676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B. X. Wang H. Z. Yang L. T. Zhang Y. W. Wang P. L. Huang G. H. Zheng J. Ren H. Qin S. D. OTUD7B and NIK expression in non-small cell lung cancer: Association with clinicopathological features and prognostic implications. Pathol., Res. Pract. 2016;212:893–898. doi: 10.1016/j.prp.2016.07.011. [DOI] [PubMed] [Google Scholar]
- Saitoh Y. Bruyn V. J. M. Uota S. Hasegawa A. Yamamoto N. Imoto I. Inazawa J. Yamaoka S. Overexpression of NF-kappa B inducing kinase underlies constitutive NF-kappa B activation in lung cancer cells. Lung Cancer. 2010;70:263–270. doi: 10.1016/j.lungcan.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Shen H. Sheng L. Chen Z. Jiang L. Su H. R. Yin L. Omary M. B. Rui L. Y. Mouse Hepatocyte Overexpression of NF-kappa B-Inducing Kinase (NIK) Triggers Fatal Macrophage-Dependent Liver Injury and Fibrosis. Hepatology. 2014;60:2065–2076. doi: 10.1002/hep.27348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan S. K. Zhang H. Ma X. Jung I. Schwartz A. J. Triner D. Devenport S. N. Das N. K. Xue X. Zeng M. Y. Hu Y. Mortensen R. M. Greenson J. K. Cascalho M. Wobus C. E. Colacino J. A. Nunez G. Rui L. Shah Y. M. Intestinal non-canonical NFκB signaling shapes the local and systemic immune response. Nat. Commun. 2019;10:660. doi: 10.1038/s41467-019-08581-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng L. Zhou Y. J. Chen Z. Ren D. C. Cho K. W. Jiang L. Shen H. Sasaki Y. Rui L. Y. NF-kappa B-inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat. Med. 2012;18:943–949. doi: 10.1038/nm.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Z. Jia L. N. Chen X. Y. Dong Y. Ren X. M. Dong Y. F. Chen Y. Xie L. W. Liu M. Shiota C. Gittes G. K. Rui L. Y. Chen Z. Islet alpha-cell inflammation induced by NF-kappa B inducing kinase (NIK) leads to hypoglycemia, pancreatitis, growth retardation, and postnatal death in mice. Theranostics. 2018;8:5960–5971. doi: 10.7150/thno.28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Wu Y. Song Y. Ding N. Lu M. Jia L. Zhao Y. Liu M. Chen Z. Activation of NF-κB-inducing kinase in islet β cells causes β cell failure and diabetes. Mol. Ther. 2020;28:2430–2441. doi: 10.1016/j.ymthe.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valino-Rivas L. Vaquero J. J. Sucunza D. Gutierrez S. Sanz A. B. Fresno M. Ortiz A. Sanchez-Nino M. D. NIK as a druggable mediator of tissue injury. Trends Mol. Med. 2019;25:341–360. doi: 10.1016/j.molmed.2019.02.005. [DOI] [PubMed] [Google Scholar]
- Wong A. H.-H. Shin E. M. Tergaonkar V. Chng W.-J. Targeting NF-κB signaling for multiple myeloma. Cancers. 2020;12:2203. doi: 10.3390/cancers12082203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I. Hunt J. P. Herrgard S. Ciceri P. Wodicka L. M. Pallares G. Hocker M. Treiber D. K. Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Mortier J. Masereel B. Remouchamps C. Ganeff C. Piette J. Frederick R. NF-kappa B inducing kinase (NIK) inhibitors: identification of new scaffolds using virtual screening. Bioorg. Med. Chem. Lett. 2010;20:4515–4520. doi: 10.1016/j.bmcl.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Pippione A. C. Sainas S. Federico A. Lupino E. Piccinini M. Kubbutat M. Contreras J. M. Morice C. Barge A. Ducime A. Boschi D. Al-Karadaghi S. Lolli M. L. N-Acetyl-3-aminopyrazoles block the non-canonical NF-kB cascade by selectively inhibiting NIK. MedChemComm. 2018;9:963–968. doi: 10.1039/C8MD00068A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Cushing T. D., Faulder P., Fisher B., He X., Li K., Li Z., Liu W., McGee L. R., Pattaropong V., Seganish J. and Shin Y., Alkylnyl alcohols as kinase inhibitors, PCT Patent, WO2009158011, 2009
- Li K. X. McGee L. R. Fisher B. Sudom A. Liu J. S. Rubenstein S. M. Anwer M. K. Cushing T. D. Shin Y. Ayres M. Lee F. Eksterowicz J. Faulder P. Waszkowycz B. Plotnikova O. Farrelly E. Xiao S. H. Chen G. Q. Wang Z. L. Inhibiting NF-kappa B-inducing kinase (NIK): Discovery, structure-based design, synthesis, structure-activity relationship, and co-crystal structures. Bioorg. Med. Chem. Lett. 2013;23:1238–1244. doi: 10.1016/j.bmcl.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Gompel M. Leost M. Joffe E. B. D. Puricelli L. Franco L. H. Palermo J. Meijer L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004;14:1703–1707. doi: 10.1016/j.bmcl.2004.01.050. [DOI] [PubMed] [Google Scholar]
- Ye Q. Li Q. Gao A. H. Ying H. Z. Cheng G. Chen J. Che J. X. Li J. Dong X. W. Zhou Y. B. Discovery of novel indoleaminopyrimidine NIK inhibitors based on molecular docking-based support vector regression (SVR) model. Chem. Phys. Lett. 2019;718:38–45. doi: 10.1016/j.cplett.2019.01.031. [DOI] [Google Scholar]
- Castanedo G. M. Blaquiere N. Beresini M. Bravo B. Brightbill H. Chen J. Cui H. F. Eigenbrot C. Everett C. Feng J. W. Godemann R. Gogol E. Hymowitz S. Johnson A. Kayagald N. Kohli P. B. Knuppel K. Kraemer J. Kruger S. Loke P. McEwan P. Montalbetti C. Roberts D. A. Smith M. Steinbacher S. Sujatha-Bhaskar S. Takahashi R. Wane X. L. Wu L. C. Zhang Y. M. Staben S. T. Structure-based design of tricyclic NF-kappa B inducing kinase (NIK) inhibitors that have high selectivity over phosphoinositide-3-kinase (PI3K) J. Med. Chem. 2017;60:627–640. doi: 10.1021/acs.jmedchem.6b01363. [DOI] [PubMed] [Google Scholar]
- Feng J. W. A. Lee P. Alaoui M. H. Barrett K. Castanedo G. Godemann R. McEwan P. Wang X. L. Wu P. Zhang Y. M. Harris S. F. Staben S. T. Structure based design of potent selective inhibitors of protein kinase D1 (PKD1) ACS Med. Chem. Lett. 2019;10:1260–1265. doi: 10.1021/acsmedchemlett.8b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaquiere N. Castanedo G. M. Burch J. D. Berezhkovskiy L. M. Brightbill H. Brown S. Chan C. Chiang P. C. Crawford J. J. Dong T. Fan P. Feng J. W. Ghilardi N. Godemann R. Gogol E. Grabbe A. Hole A. J. Hu B. H. Hymowitz S. G. Ismaili M. H. A. Le H. Lee P. Lee W. Lin X. Y. Liu N. McEwan P. A. McKenzie B. Silvestre H. L. Suto E. Sujatha-Bhaskar S. Wu G. S. Wu L. C. Zhang Y. M. Zhong Z. Staben S. T. Scaffold-hopping approach to discover potent, selective, and efficacious inhibitors of NF-kappa B inducing kinase. J. Med. Chem. 2018;61:6801–6813. doi: 10.1021/acs.jmedchem.8b00678. [DOI] [PubMed] [Google Scholar]
- Li Z. Q. Li X. Z. Su M. B. Gao L. X. Zhou Y. B. Yuan B. C. Lyu X. L. Yan Z. Q. Hu C. J. Zhang H. Luo C. Chen Z. Li J. Zhao Y. J. Discovery of a potent and selective NF-kappa B-inducing kinase (NIK) inhibitor that has anti-inflammatory effects in vitro and in vivo. J. Med. Chem. 2020;63:4388–4407. doi: 10.1021/acs.jmedchem.0c00396. [DOI] [PubMed] [Google Scholar]
- Zhu Y. Q. Ma Y. X. Zu W. D. Song J. N. Wang H. Zhong Y. Li H. M. Zhang Y. M. Gao Q. Q. Kong B. Xu J. Y. Jiang F. Wang X. R. Li S. W. Liu C. H. Liu H. C. Lu T. Chen Y. D. Identification of N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-amine derivatives as novel, potent, and selective NF-kappa B inducing kinase (NIK) inhibitors for the treatment of psoriasis. J. Med. Chem. 2020;63:6748–6773. doi: 10.1021/acs.jmedchem.0c00055. [DOI] [PubMed] [Google Scholar]
- Crawford J. J. Liao D. H. Kolesnikov A. Lee W. Landry M. L. Synthesis of an azabicyclo[3.1.0]hexanone-containing inhibitor of NF-kappa beta inducing kinase via catalytic C-H activation. Synthesis. 2020;52:3420–3426. doi: 10.1055/s-0040-1707279. [DOI] [Google Scholar]
- Crawford J. J., Kolesnikov A. and Feng J. W., 2-Azabicyclo[3.1.0]hexan-3-one derivatives and methods of use, PCT Patent, WO2018037059, 2018
- Ren X. M. Li X. Z. Jia L. N. Chen D. H. Hou H. Rui L. Y. Zhao Y. J. Chen Z. A small-molecule inhibitor of NF-kappa B-inducing kinase (NIK) protects liver from toxin-induced inflammation, oxidative stress, and injury. FASEB J. 2017;31:711–718. doi: 10.1096/fj.201600840R. [DOI] [PubMed] [Google Scholar]
- Li Y. R. Chen M. M. Zhou Y. Tang C. F. Zhang W. Zhong Y. Chen Y. D. Zhou H. Sheng L. NIK links inflammation to hepatic steatosis by suppressing PPAR alpha in alcoholic liver disease. Theranostics. 2020;10:3579–3593. doi: 10.7150/thno.40149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looney R. J. Anolik J. Sanz, I. A perspective on B-cell-targeting therapy for SLE. Mod. Rheumatol. 2010;20:1–10. doi: 10.3109/s10165-009-0213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightbill H. D. Suto E. Blaquiere N. Ramamoorthi N. Sujatha-Bhaskar S. Gogol E. B. Castanedo G. M. Jackson B. T. Kwon Y. C. Haller S. Lesch J. Bents K. Everett C. Kohli P. B. Linge S. Christian L. Barrett K. Jaochico A. Berezhkovskiy L. M. Fan P. W. Modrusan Z. Veliz K. Townsend M. J. DeVoss J. Johnson A. R. Godemann R. Lee W. P. Austin C. D. McKenzie B. S. Hackney J. A. Crawford J. J. Staben S. T. Ismaili M. H. A. Wu L. C. Ghilardi N. NF-kappa B inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun. 2018;9:179. doi: 10.1038/s41467-017-02672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soysa N. S. Alles N. Weih D. Lovas A. Mian A. H. Shimokawa H. Yasuda H. Weih F. Jimi E. Ohya K. Aoki K. The pivotal role of the alternative NF-kappa B pathway in maintenance of basal bone homeostasis and osteoclastogenesis. J. Bone Miner. Res. 2010;25:809–818. doi: 10.1359/jbmr.091030. [DOI] [PubMed] [Google Scholar]
- Maruyama T. Fukushima H. Nakao K. Shin M. Yasuda H. Weih F. T.; Aoki, K.; Alles, N.; Ohya, K.; Hosokawa, R.; Jimi, E. Processing of the NF-kappa B2 Precursor p100 to p52 is critical for RANKL-induced osteoclast differentiation. J. Bone Miner. Res. 2010;25:1058–1067. doi: 10.1359/jbmr.091032. [DOI] [PubMed] [Google Scholar]
- Boyle W. J. Simonet W. S. Lacey D. L. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. L. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Saad F. A. Novel insights into the complex architecture of osteoporosis molecular genetics. Ann. N. Y. Acad. Sci. 2020;1462:37–52. doi: 10.1111/nyas.14231. [DOI] [PubMed] [Google Scholar]
- Tanaka S. Molecular understanding of pharmacological treatment of osteoporosis. EFORT Open Rev. 2019;4:158–164. doi: 10.1302/2058-5241.4.180018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura N. Matsuda M. Khan M. Hiura F. Aoki K. Hirohashi Y. Mori K. Yasuda H. Hirata M. Kitamura C. Jimi E. A novel inhibitor of NF-kappa B-inducing kinase prevents bone loss by inhibiting osteoclastic bone resorption in ovariectomized mice. Bone. 2020;135:115316. doi: 10.1016/j.bone.2020.115316. [DOI] [PubMed] [Google Scholar]
- Versele M. Janssen L. Geerts T. Floren W. Janssens B. Millar H. Jacoby E. Gross G. Ligny Y. Simonnet Y. Amblard N. Querolle O. Csoka I. Poncelet V. Tronel V. Nocquet-Thibault S. Meerpoel L. Edwards J. Salvati M. Balasubramanian S. Lenox L. Theuer C. Attar R. Stansfield I. Inhibition of NF-kB inducing kinase (NIK) selectively abrogates NIK and TRAF3 mutant multiple myeloma tumor growth. Cancer Res. 2017;77:4199. [Google Scholar]