

Long non-coding RNAs (lncRNAs) are a large class of transcripts that do not encode proteins, and while some lncRNAs are known to have important functions in mammalian cells, the function of the large majority of lncRNAs remains to be explored in depth. Thus, large-scale and unbiased approaches to screen for functional lncRNA loci are critical for the advancement of this field. Moreover, as any single approach will likely be impacted by both false positives and false negatives, there is clear value in having a multitude of orthogonal strategies for exploring lncRNA function. Liu et al.1 recently have presented an approach for systematically investigating lncRNA function by targeting the CRISPR/Cas9 nuclease to lncRNA splice acceptor/donor sites. Although the approach is clearly a valuable addition to the arsenal of strategies used to characterize lncRNA function, there are some important caveats as well. Specifically, the authors identified 469 lncRNA loci that confer fitness defects in three cell lines when their splice sites were targeted in this manner. To validate lncRNA hits and their overall approach, they used paired Cas9 targeting to remove lncRNA exons. We find evidence that a substantial fraction of the hits in these screens (at least 30–39% in each cell line) are likely to be false positives due to either nuclease activity in copy number-amplified regions or overlap with protein-coding genes. Furthermore, the validation method chosen by the authors was not sufficiently orthogonal to identify such false positives.
We first analyzed the results from the screen performed by Liu et al.1 in the chronic myeloid leukemia cell line K562. We observed that many of the top hits identified by splice-site targeting were clustered in specific regions of the genome (Fig. 1a), including 22 located in one region of chromosome 22. These hits include BMS1P20, a lncRNA specifically highlighted in their study (see original Figure 4d in Liu et al.1). This region, located between the centromere and the BCR (breakpoint cluster region) gene locus, is a genomic region that has undergone copy number amplification (Fig. 1b)2. An enrichment for false-positive essential protein-coding genes in this region was first noted by Wang et al.3. As this effect results from the DNA damage response triggered by the creation of many double-strand breaks by Cas9 nuclease activity, we did not observe an enrichment of hits in this region in either our protein-coding- or lncRNA locus-targeting screens using CRISPR interference (CRISPRi)4,5, which binds to but does not cleave DNA. ENCODE copy-number data and rolling window medians of the Wang et al.3 K562 screen data, representing regions enriched for negative-growth phenotypes, are plotted for comparison in Figure 1a,b. Applying this analysis systematically, we observed that many of the hits (as defined by Liu et al.1 as “Screen score” ≥ 2) in K562 could be attributed to this copy-number effect on fitness (Fig. 1c,d). Notably, copy-number effect would also impact paired sgRNA targeting of the lncRNA locus, a strategy employed by Liu et al.1 for hit validation. Several hits in the screen performed in HeLa cells also corresponded to amplified regions (Figs 1d and 2a). This analysis may be an underestimate, as we were unable to determine amplification status for ~1200–2500 genes in each cell line due to coverage gaps in copy-number data, and so these genes were considered non-amplified.
Figure 1. LncRNA CRISPR screen hits within copy number amplified regions or overlapping protein-coding genes.
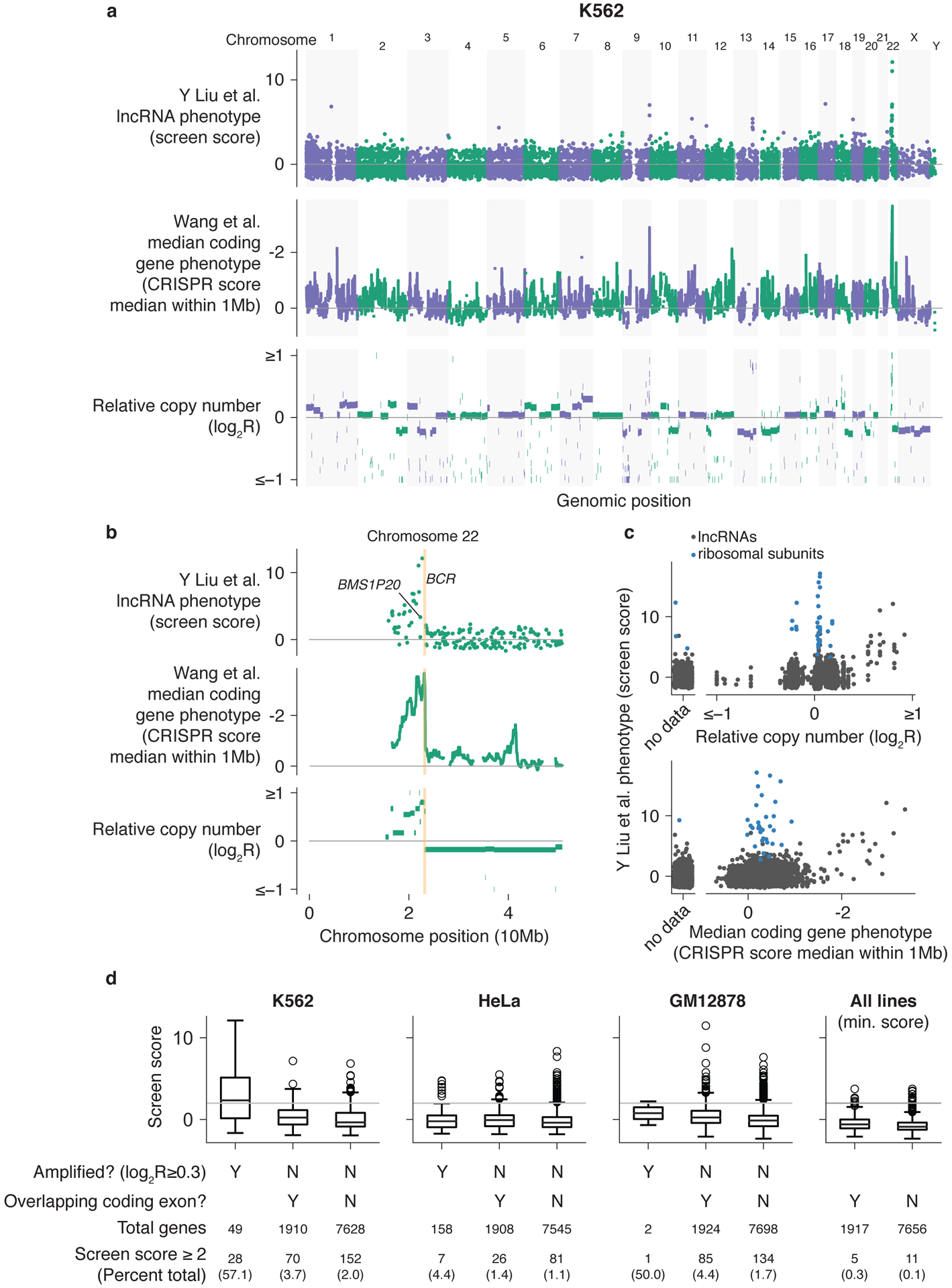
K562 lncRNA screen scores, 1 Mb window median of protein-coding screen scores3, and relative copy number (a) across the genome and (b) on chromosome 22. LncRNA screen scores are more positive for confident hits and protein-coding screen scores are negative for stronger fitness defects. (c) Relationship between relative copy number and window median coding gene phenotype. For each gene, relative copy number was averaged across the gene locus where data were available. Ribosomal subunits included in the screen as positive controls are also plotted for comparison. (d) Boxplots of lncRNA screen scores for each cell line separated by genes located in amplified regions and overlapping coding exons. The minimum score across all three cell lines is also plotted to identify lncRNAs scoring in each cell line. Boxplots show 25th, 50th, and 75th percentiles, with whiskers indicating 1.5× inter-quartile range and outliers individually plotted. Horizontal line represents a screen score of 2 used as the threshold for hits by Liu et al.1
Figure 2. Top lncRNA screen hits in HeLa cells are located in the HPV18 integration locus.
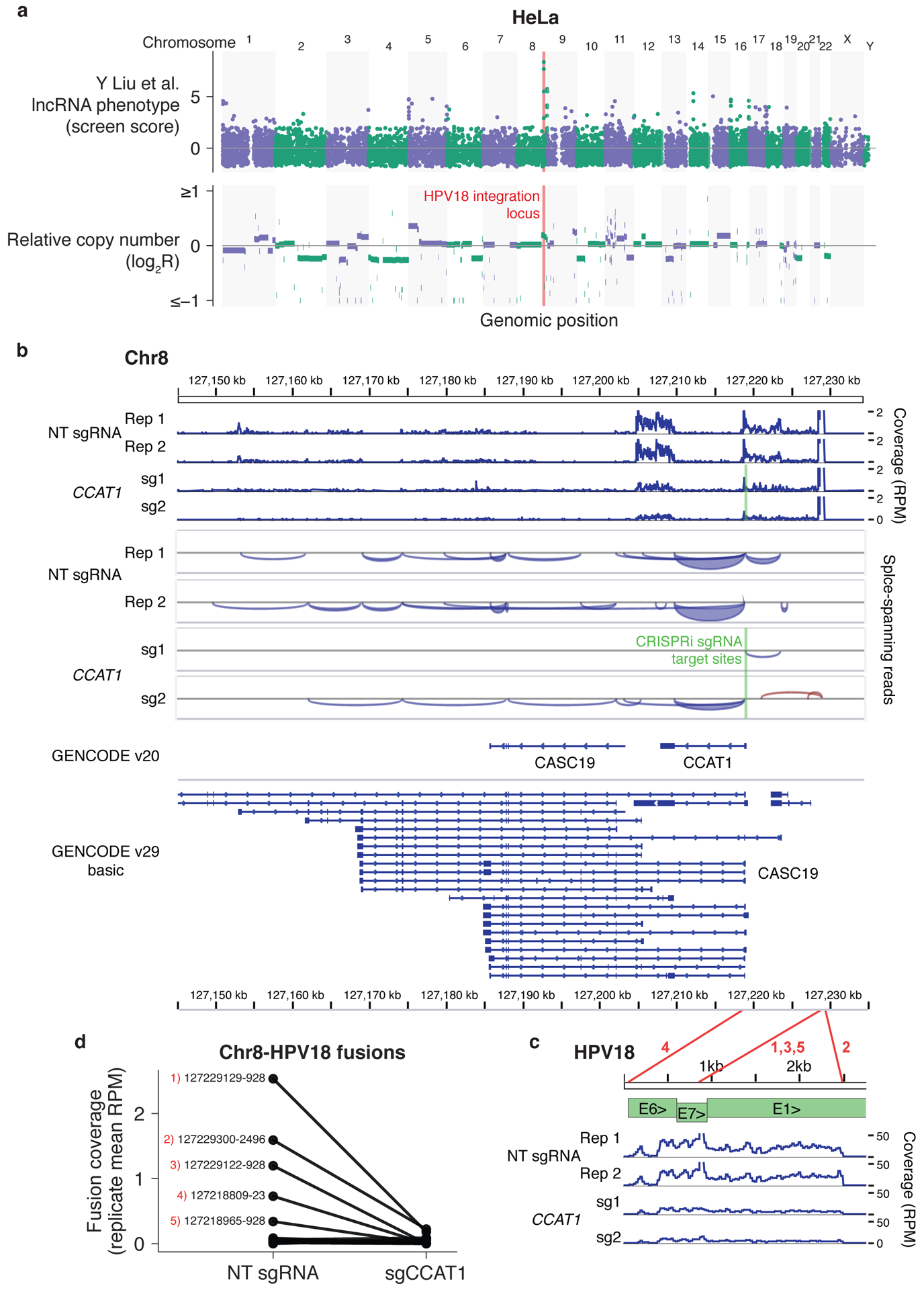
(a) HeLa lncRNA screen scores (top) and relative copy number (bottom) across the genome. (b) RNA-seq coverage across the chromosome 8 locus indicated in (a) with and without CCAT1 knockdown with CRISPRi. (c) RNA-seq coverage across the HPV18 genome using published RNA-seq datasets5 aligned to the hg38 and HPV18 genome sequences with TopHat-Fusion without annotation. Coverage in reads per million (RPM) was calculated with IGVtools count, and coverage, splice spanning reads, and two GENCODE annotations were visualized in IGV. HPV18 gene structure adapted from Adey et al.8 Red lines correspond to fusion sites reported by TopHat-Fusion and numbers correspond to the labeled fusion sites in (d). (d) RNAseq coverage of chromosome 8–HPV18 fusion sites, averaged across two independent sgRNAs. Coverage was determined by the number of fusion-spanning reads reported by TopHat-Fusion normalized to total aligned reads. The top five fusion sites by coverage are labeled with the respective chromosome 8 and HPV18 genome coordinates.
Upon inspection of screen hits located in non-amplified regions, we found that these hits are enriched for lncRNA loci that overlap protein-coding genes. Although such lncRNAs may well have functions that are independent of the activity of the protein-coding gene6, further experiments are required to establish their separable function. By the broadest definition, in which the gene body of the target lncRNA overlapped with a coding gene along any portion of the gene, 46.9% of lncRNAs screened were not intergenic. We applied a less restrictive definition, requiring only that at least one of the sgRNAs targeting a given lncRNA in the screen library also targeted an exon of a protein-coding gene. This identified 20.0% of non-amplified lncRNAs screened and between 24.3–38.8% of hits as non-intergenic after excluding hits in amplified regions (Fig. 1d, Supplementary Fig. 1). Of the hits found in all three cell lines and thus unlikely to exhibit phenotypes solely due to cell line-specific copy number effects, 5 of 16 overlapped protein-coding gene exons (Supplementary Fig. 2).
Finally, we observed that the top two hit genes in the HeLa cell screen, cancer susceptibility 19 (CASC19) and colon cancer associated transcript 1 (CCAT1), a lncRNA that has previously been shown to regulate chromatin looping at the MYC locus in colorectal cancer cells7, neighbored the human papilloma virus 18 (HPV-18) integration site on chromosome 8 in HeLa cells (Fig. 2a)8. This region is not designated as being markedly amplified by ENCODE copy-number data (log2R < 0.3) although higher resolution analysis found areas in the locus with up to 34 repeats8. CCAT1 also modulated HeLa cell growth in our CRISPRi screens as well as in small-interfering RNA (siRNA) and cDNA overexpression experiments by Jia and colleagues9, suggesting the phenotype was not due to a copy-number effect. Instead, gene fusions have been reported between the viral oncogenes E6/E7 and CCAT1 and CASC1910, which may be responsible for the growth defect upon knockdown and nuclease disruption of the genes. CCAT1 and CASC19 may also represent splice isoforms of a single gene, as GENCODE v29 no longer includes CCAT1 as a separate gene (Fig. 2b). Analyzing our previously published RNA-seq datasets, we confirmed that splice junction-spanning reads connect the entire locus. Notably, CRISPRi-mediated inhibition of CCAT1 transcription decreased transcript levels across both CCAT1 and CASC19, the viral oncogenes, and the fusion sites between chromosome 8 and HPV-18 (Fig. 2b–d). As splice-site targeting, siRNA, and CRISPRi all would disrupt the fused viral oncogenes, and cDNA overexpression of the HeLa transcript would also amplify the oncogenes, still further orthogonal methods are required to establish a distinct function for the CCAT1/CASC19 lncRNA locus in HeLa cells.
Every method for perturbing lncRNA function can produce artefacts, and careful filtering is needed to minimize these effects before conclusions are drawn about the method and hits themselves. The potential for false positives due to copy-number amplification has been documented by other groups11,12. Even in euploid regions, CRISPR-mediated double-stranded DNA breaks can cause a measurable growth defect4, especially in P53 wild-type cells12. Although not perfect, algorithmic approaches can help correct this effect (e.g., CERES13 and Crispy14), and library designs that include genome-targeting ‘safe’ controls14 can be employed to preemptively detect phenotypes due solely to nuclease activity. More broadly, these results underscore the importance of validating knock-out results using fully orthogonal methods, such as antisense oligonucleotide targeting of the lncRNA transcripts, as well as confirming that the perturbations indeed affect lncRNA expression or processing6. The comprehensive hit validation performed by Liu et al.1 with paired sgRNAs is also susceptible to the copy-number effect and can also disrupt overlapping coding genes. Thus, the use of paired sgRNAs to delete exons is not sufficiently orthogonal to the primary screening with sgRNAs that target the splice donor/acceptor sites. Where there are bona fide discrepancies in results obtained by different types of perturbations, further analyses combined with a molecular understanding of the methods themselves can in fact reveal novel and important mechanistic insights15.
Supplementary Material
Acknowledgements
The authors were supported by the UCSF Medical Scientist Training Program (M.A.H. and S.J.L.); NIH grants F30NS092319-01 (S.J.L.), 1R01NS0091544 (D.A.L.), and R35CA209919 (H.Y.C.); and the Howard Hughes Medical Institute (M.A.H., H.Y.C., and J.S.W.).
Footnotes
Supplementary Information is available for this paper.
Competing Interests
M.A.H. and J.S.W. have filed a patent application related to CRISPR interference screening (15/326,428).
Data availability
All analysis was performed on previously published datasets. Sources and accession numbers are tabulated in the Methods.
Code availability
All code used for analysis, figure generation, and preparation for visualization in genome browsers is included as a Jupyter Notebook in the Supplementary File. Software dependencies and versions are listed in the Methods.
References
- 1.Liu Y et al. Genome-wide screening for functional long noncoding RNAs in human cells by Cas9 targeting of splice sites. Nat. Biotechnol (2018) doi: 10.1038/nbt.4283. [DOI] [PubMed] [Google Scholar]
- 2.Wu SQ et al. Extensive amplification of bcr/abl fusion genes clustered on three marker chromosomes in human leukemic cell line K-562. Leukemia 9, 858–862 (1995). [PubMed] [Google Scholar]
- 3.Wang T et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horlbeck MA et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu SJ et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassett AR et al. Considerations when investigating lncRNA function in vivo. Elife 3, e03058 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiang J-F et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 24, 513–531 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adey A et al. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500, 207–211 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia L, Zhang Y, Tian F, Chu Z & Xin H Long noncoding RNA colon cancer associated transcript-1 promotes the proliferation, migration and invasion of cervical cancer. Molecular Medicine Reports 16, 5587–5591 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Wu L et al. Full-length single-cell RNA-seq applied to a viral human cancer: applications to HPV expression and splicing analysis in HeLa S3 cells. Gigascience 4, 51 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aguirre AJ et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 6, 914–929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munoz DM et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov 6, 900–913 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Meyers RM et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgens DW et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat Commun 8, 15178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.