

Abstract
Highly selective mTOR inhibitors have been discovered through the exploration of the heteroaromatic ring engaging the binding affinity region in mTOR kinase. Compound 11 showed predicted BBB permeability in a MDCK-MDR1 permeability in vitro assay, being the first pyrimido-pyrrolo-oxazine with potential application in the treatment of neurological disorders.
Here we present the first pyrimido-pyrrolo-oxazine-based mTOR kinase inhibitor (11) predicted to penetrate the blood brain barrier (BBB). Thus, 11 has a potential in treatments of neurological disorders.
Introduction
The mechanistic target of rapamycin (mTOR) operates downstream of phosphoinositide 3-kinase (PI3K). The mTOR protein kinase is part of two functionally distinct multiprotein mTOR complexes, mTORC1 and mTORC2.1,2 mTOR promotes cell growth and proliferation and is dysregulated in cancer and central nervous system (CNS) disorders,3 such as Parkinson's, Alzheimer's, Huntington's disease, and epilepsy.4 Tuberous sclerosis complex (TSC) is a negative regulator of TORC1. TSC loss-of-function causes hyperactivation of TORC1 signalling leading to the development of tuberous sclerosis, which causes epilepsy, behavioural changes and cognitive impairment in >90% of patients. Rapamycin is an allosteric inhibitor of TORC1, and is the prototype of first generation mTOR inhibitors. Everolimus, a rapalog, showed efficacy in decreasing the frequency and severity of epileptic seizures and has been approved for the treatment of TSC patients in 2018.5 Rapalogs have, however, significant immunosuppressive effects, which are pronounced due to their limited ability to cross the blood brain barrier (BBB).6 Therefore, the identification of highly selective mTOR inhibitors with optimized brain permeability is an ongoing challenge for the treatment of CNS disorders. Lead optimization strategies on different scaffolds led to the discovery of ATP-competitive mTOR kinase inhibitors (TORKi) targeting both mTORC1 and mTORC2, including CC-223,7 INK128,8 AZD2014,9 PQR620,10 PQR626 (ref. 11) and a thiazolopyrimidine derivative.12 Recently, we have disclosed a conformational restriction strategy to identify highly selective TORKi.13 A systematic structure–activity relationship (SAR) study pinpointed the pyrazine derivative 12b as potent inhibitor of mTOR kinase (Ki = 8.0 nM), showing a 212-fold selectivity over the structurally related PI3Kα (see Table 1 for chemical structure).14 However, 12b displayed a limited ability to cross the BBB, preventing its application in the treatment of CNS disorders such as epilepsy. Given the unexplored activity of the tricyclic pyrimido-pyrrolo-oxazine compounds in CNS indications, we selected this privileged scaffold as starting point for the development of novel lead compounds with potential brain permeability.
Heteroaromatic rings investigation.
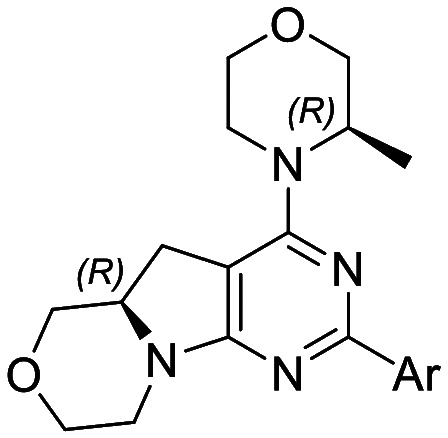
|
Cellular assays IC50a [nM] | In vitro binding assays Kib [nM] | SIc | clog Pd | PSAd | |||
|---|---|---|---|---|---|---|---|---|
| Name | Ar | pPKB S473 | pS6 S235/236 | PI3Kα | mTOR | |||
| 12b e |
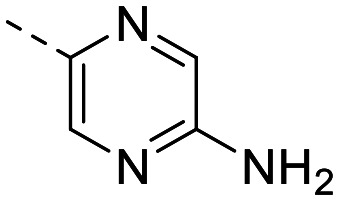
|
94 ± 2.5 | 60 ± 13 | 1695 ± 277 | 8.0 ± 0.92 | 212 | 1.74 | 102.5 |
| 1 e |
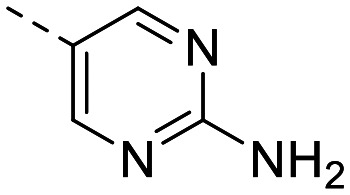
|
77 ± 6.8 | 55 ± 5.5 | 536 ± 22 | 5.1 ± 0.4 | 105 | 1.85 | 102.5 |
| 2 |
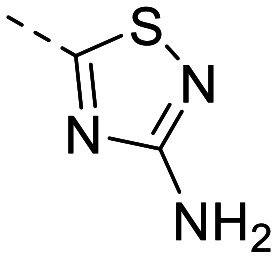
|
512 ± 59 | 261 ± 7.1 | 806 ± 20 | 58 ± 5.8 | 14 | 2.38 | 102.5 |
| 3 |
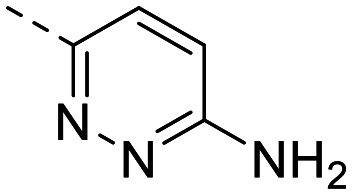
|
162 ± 3.1 | 84 ± 20 | 847 ± 216 | 11 ± 2.4 | 77 | 1.84 | 102.5 |
| 4 |
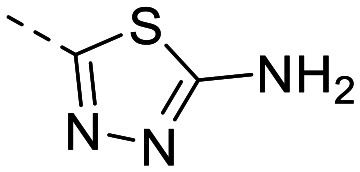
|
135 ± 14 | 80 ± 7.8 | 1547 ± 278 | 8.2 ± 0.6 | 189 | 1.80 | 102.5 |
| 5 e |
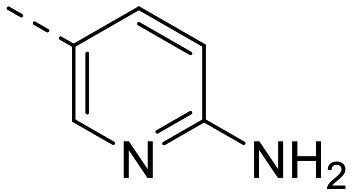
|
133 ± 16 | 61 ± 12 | 2147 ± 400 | 10 ± 1.6 | 215 | 2.59 | 89.6 |
| 6 |
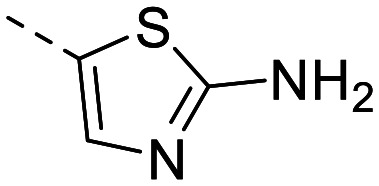
|
157 ± 5.4 | 102 ± 4.2 | 2485 ± 251 | 15 ± 2.8 | 166 | 2.37 | 89.6 |
| 7 |
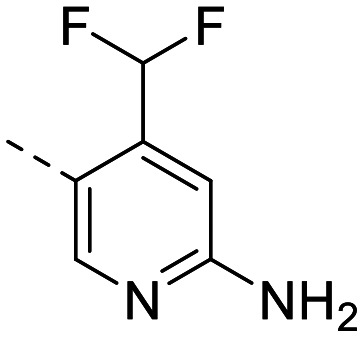
|
253 ± 10 | 67 ± 4.8 | 232 ± 33 | 12 ± 0.7 | 19 | 2.82 | 89.6 |
| 8 |
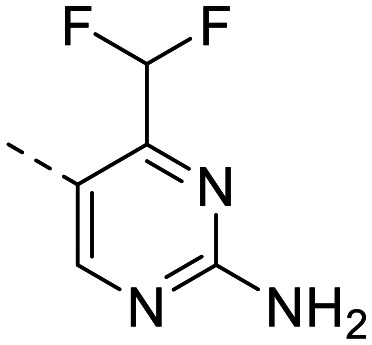
|
116 ± 7.5 | 119 ± 9.9 | 17 ± 0.30 | 34 ± 9.9 | 0.5 | 2.33 | 102.5 |
| 9 |
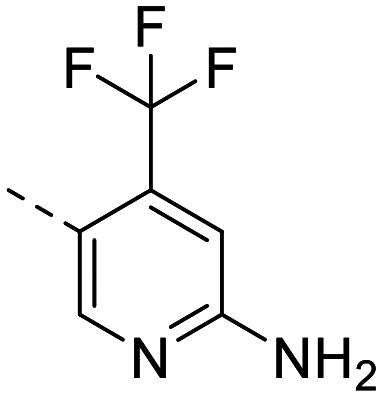
|
1089 ± 34 | 776 ± 138 | 272 ± 10 | 118 ± 9.6 | 2.3 | 3.46 | 89.6 |
| 10 |
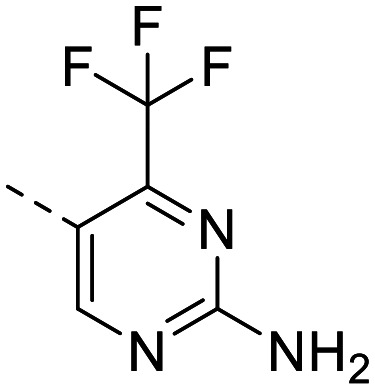
|
446 ± 45 | 694 ± 131 | 55 ± 0.93 | 249 ± 39 | 0.22 | 3.11 | 102.5 |
| 11 |
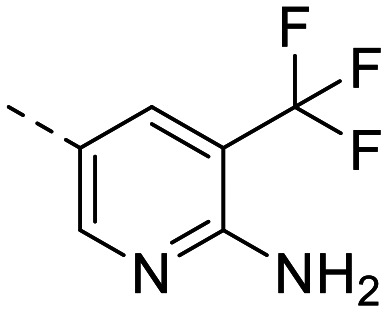
|
549 ± 56 | 348 ± 44 | >20 000 | 33 ± 1.3 | >600 | 3.46 | 89.6 |
IC50s were measured using a 7-point 1 : 2 serial dilution and each concentration was measured in triplicate.
Compounds were tested for the in vitro binding to the ATP-binding site of PI3Kα and mTOR using a commercially available LanthaScreen time-resolved FRET (TR-FRET) displacement assay. IC50s were measured using a 10-point 1 : 4 serial dilution. Each concentration was performed in duplicate, IC50 to Ki conversion factor = 9.84 (p110α), 2.05 (mTOR). Data are reported as mean ± SD.
Selectivity index calculated as Ki(mTOR)/Ki(p110α).
Calculation of log P and PSA was performed with ChemAxon (https://www.chemaxon.com).
Previously reported in ref. 14.
Study design and synthesis
Herein, we investigated the effect of the heteroaryl moiety, engaging the binding affinity region in the mTOR kinase, on ligand potency and brain penetration. In several compound classes, isosterism between 6-membered diazaheterocycles and thiadiazole derivatives has been exploited to improve potency, enhance selectivity, alter physical properties, modify and reduce metabolism.15 The introduction of fluorine into molecular scaffolds16 is known to modulate lipophilicity, and plays an important role in BBB permeability.
We explored different bioisosters, and difluoro/trifluoro-substituted heteroaromatic rings on the tricyclic scaffold. The synthetic procedures for library preparation are reported in Schemes 1 and 2. Sulfamidate 22 was prepared starting from a chiral amino alcohol – (S)-serine – in several steps (ESI† for details).17,18 The tricyclic pyrimido-pyrrolo-oxazine scaffold was generated by sequential alkylation and a nucleophilic aromatic substitution reaction of dichloropyrimidine 12 with (S)-sulfamidate 22 using n-BuLi and copper(i) iodide (cat.) followed by acidic hydrolysis of the intermediary sulfaminic acid and base-mediated ring closure.14 To introduce a 1,2,4-thiadiazole and a pyrazine, a tributyltin group was appended on 13, followed by Stille coupling between 14 and halides (Scheme 1a). The Stille reaction of 3-bromo-5-chloro-1,2,4-thiadiazole was regioselective for C-5 coupling to give bromo derivative 15. In contrast, no site-selective cross-coupling between the tin derivative 14 and 3-chloro-6-iodopyridazine was observed, and a mixture of chloro and iodo derivatives was obtained (16, 50% chloro, 44% iodo; see ESI†). The heteroaryl halide 16 was aminated by treatment with aqueous ammonia at 100 °C to yield the desired pyridazine-substituted compound 3 in moderate yield. The conversion of the bromide 15 into an amine by using ammonia was not successful. tert-Butyl carbamate in presence of caesium carbonate was employed for the Pd-catalysed synthesis of the N-Boc-protected derivative of 15. In a one-pot procedure, treatment with HCl in dioxane generated the final 1,2,4-thiadiazole derivative 2. To prepare the 1,3,4-thiadiazole-substituted compound 4, the chloropyrimidine 13 was converted into the nitrile 17 using tetraethylammonium cyanide in presence of DABCO. Coupling of nitrile 17 and thiosemicarbazide followed by cyclization at 65 °C in presence of TFA yielded compound 4 (Scheme 1c) Subsequently, thiazole and difluoromethyl- and trifluoromethyl-substituted pyridines/pyrimidines were installed on intermediate 13 using a Suzuki cross-coupling reaction with boronic acid pinacol esters to generate the final compounds 6–11 (Schemes 1b and 2).
Scheme 1. Synthesis of compounds 2–4, 6. Reagents and conditions: (a) (i) Pd(amphos)Cl2, bis(tributyltin), dioxane, reflux, 3 h; (ii) halide derivative, Pd(PPh3)4, dioxane, 90 °C, o/n; (iii) 1) Pd2(bda)3/xantphos, tert-butyl carbamate, Cs2CO3, dioxane, 90 °C, o/n, 2) HCl in dioxane (for 2); (iv) NH3 aq., DMSO, 100 °C, o/n (for 3). (b) (v) 1) boronic ester, XPhosPdG2 (cat.), K3PO4, dioxane/H2O, 95 °C, 2–16 h; 2) HCl, dioxane/H2O, 60 °C, 3–16 h; (c) (vi) tetraethylammonium cyanide, DABCO, DMSO, 140 °C, 1.5 h. (vii) TFA, 65 °C, o/n. See ESI† for details.

Scheme 2. Synthesis of compounds 7–11. Reagents and conditions: (viii) morpholine derivative, DIPEA, DCM, 0 °C → rt, o/n; (ix) 1) n-BuLi, CuI, −78 °C → r.t., o/n; 2) HCl conc., MeOH, 45 °C, 4–6 h; 3) NaOH, H2O, r.t., 1–16 h; (x) 1) boronic ester (for 7, 9–11) or boronic ester generated in situ (for 8), XPhosPdG2 (cat.), K3PO4, dioxane/H2O, 95 °C, 2–16 h; 2) HCl, dioxane/H2O, 60 °C, 3–16 h (for 7–9, 11). See ESI† for details.

Results and discussion
The library was tested for in vitro binding to recombinant mTOR and PI3Kα (Ki), and for PI3K/mTOR signalling in A2058 melanoma cells (IC50 for phosphorylated S6 ribosomal protein [pS6, Ser235/236] and phosphorylated protein kinase B [PKB/Akt, Ser473] to detect mTORC1 and mTORC2 activity). The in vitro and cellular profiling of the novel set of conformationally restricted compounds is reported in Table 1. Pyrimidine (1), pyrazine (3) and pyridine (5) have been compared with their bioisosters involving the replacement of –CH CH– by sulphur. Replacement of the pyrimidine with a 1,2,4-thiadiazole led to a >10-fold decrease in mTOR affinity, reducing compound selectivity [Ki(mTOR) 1 = 5.1 nM, 2 = 58 nM; Ki(PI3Kα)/Ki(mTOR) 1 = 105, 2 = 14]. On the contrary, comparable activity and selectivity were observed for the bioisosteric pairs 3–4 and 5–6 (Table 1). The presence of a difluoro or trifluoromethyl group in para-position of 2-amino pyridines (7 and 9) caused a selectivity drop compared to 12b [Ki(PI3Kα)/Ki(mTOR) <20]. The pyrimidine derivatives 8 and 10 were more potent on PI3Kα than on mTOR kinase [Ki(PI3Kα)/Ki(mTOR) <1]. A gain in mTOR potency and selectivity over PI3K was achieved moving the CF3-group from para (compound 9) to meta position (11) on the pyridine scaffold. 11 showed the highest selectivity [Ki(PI3Kα)/Ki(mTOR) >600] among these tricyclic derivatives, and outperformed the selectivity of 12b. Compound 11 displayed a moderate cellular activity (IC50 for phosphorylated PKB/Akt and for phosphorylated S6 <550 nM).
While difluoromethyl- and trifluoromethyl-substituted pyridines/pyrimidines have been explored in pre-clinical and clinical candidates targeting the PI3K–mTOR pathway,10,19–21 to the best of our knowledge no mTOR inhibitor bearing thiazole or 1,3,4-thiadiazole moieties has been disclosed yet. With no prior hints to stability information, we characterised compounds 4 and 6 with respect to their CYP related metabolism, after incubation with individual human recombinant CYP isoenzymes CYP1A1 and CYP1A2. These isoenzymes have been selected since they are known to be implicated in the metabolism of structurally similar molecules.194 was highly stable with both CYP1A1 and 1A2 (>70% remaining compound after 60 minutes of incubation, Fig. 1). 6 displayed moderate to high stability with CYP1A1 and CYP1A2 (48% and 87% remaining compound, respectively), and comparable to that of the corresponding bioisosteric compound 5 (Fig. 1). Moreover, the (R)-3-methylmorpholine derivative 5 presented a higher stability with 1A1 isoenzyme with respect to the corresponding unsubstituted-morpholine 27 (see ESI†). Due to the highly variable expression of CYP1A1 in liver and extrahepatic tissues, the high stability towards degradation by CYP1A1 isoenzyme is an asset in drug development and facilitates the establishment of a therapeutic dose.
Fig. 1. Pie chart showing the percentage of remaining compounds 4–6 and 27 (blue) after 60 min of incubation with the indicated human recombinant CYP isoenzymes.

The observed SAR was rationalized using computational modelling studies. We investigated the putative interactions of 1–6 and 11 with ATP-binding sites of mTOR and PI3Kα. After energy minimization, the mTOR–ligand complexes maintained pivotal interactions including H-bonds (i) between Asp2195 and the NH2-group of the aminopyridine, and (ii) between the oxygen atom of the (R)-3-methylmorpholine and the hinge region backbone amide of Val2240. The (R)-3-methylmorpholine has been previously reported as a crucial feature to achieve selectivity for mTOR kinase over PI3Ks.12,14 The orientation of the amino group on the 1,3,4-thiadiazole in 4 and on thiazole in 6 resembles that of the 2-amino substituted pyrimidine (1), pyrazine (3) and pyridine (5, Fig. 2b for 3–4), leading to the establishment of an H-bond network with Asp2195 and Glu2190. On the contrary, the NH2-group on the 1,2,4-thiadiazole (2) is 2.8 Å away from the 2-amino group of 1. The different orientation prevents an H-bond interaction with Glu2190 (6 Å distance), which plays a pivotal role in the stabilization of inhibitor binding (Fig. 2a). This explains the reduced affinity for mTOR kinase of 2 with respect to compounds 1, 3–6. Docking studies of compound 11 into PI3Kα and mTOR revealed that the 3-trifluoromethyl group is well accommodated in mTOR (Fig. 2c), while it induces steric clashes with Lys802 and Asp933 in PI3Kα (Fig. 2d). This is due to the different orientation of Lys2187 (mTOR) and Lys802 (PI3Kα), which leads to a broader binding affinity pocket in mTOR compared to that of PI3Kα. The orientation of Lys2187 (mTOR)/Lys802 (PI3Kα) is conserved among the different X-ray crystallographic structures (Fig. S1†).
Fig. 2. (a and b) Superimposition of (a) mTOR–1 and mTOR–2 complexes and (b) mTOR–3 and mTOR–4 complexes. mTOR, grey; 1, green; 2, brown; 3, yellow; 4, cyan. The distances between the 2-amino group and Glu2190/Asp2195 are depicted as dashed black lines. Distances values are coloured as the inhibitors. (c and d) Docking of compound 11 (magenta) to (a) mTOR (grey) starting from PDB 4JT6,22 and (b) PI3Kα (wheat) starting from PDB 6OAC.21 No steric clashes are present with Asp2357 and Lys2187 in mTOR (c). Steric clashes with Asp933 and Lys802 in PI3Kα are shown (d).

To assess the potential for BBB penetration of 4, 6 and 11, the apparent permeability (Papp) and the efflux ratio (ER = Papp,basolateral-apical/Papp,apical-basolateral) across cell monolayers were assessed using an in vitro TransWell assay. CRISPR-Cas9 generated MDCK cells expressing human MDR1, but no endogenous canine MDR1 (MDCK-hMDR1cMDR1-ko) were employed,23 and the results compared with a control cell line expressing neither MDR1 ortholog (MDCKcMDR1-ko). Compounds 4 and 6, as well as 12b included as negative control, were identified as hMDR1 substrates (ER > 20) with low permeability (Papp < 60 × 10−6 cm s−1), predicting poor BBB permeability in vivo. On the contrary, compound 11 was no hMDR1 substrate (ER ∼ 1) and showed a Papp comparable to PQR30919 and PQR620,10 which are known brain penetrant PI3K and/or mTOR inhibitors (Fig. 3).
Fig. 3. (a) Efflux ratio (ER) in MDCK cells with and without hMDR1 grown on TransWell filters. An ER of 1 is indicative of the compound not being a hMDR1 substrate. (b) Apparent permeability (Papp) from apical to basolateral site in MDCK-hMDR1cMDR1ko cells. Numerical values are reported in Tables S1 and S2.† *p < 0.05, unpaired t-test, GraphPad Prism 8.0.2.

Conclusion
In summary, we have identified 3-CF3-substituted pyridine, thiazole and 1,3,4-thiadiazole as unexplored heteroaromatic rings for mTOR inhibitors providing an exquisite mTOR selectivity. We have reported the first tricyclic pyrimido-pyrrolo-oxazine derivative showing MDCK-based BBB permeability. Altogether, the good in vitro/cellular potency, and the predicted brain penetration qualify 11 as second-generation tricyclic pyrimido-pyrrolo-oxazine with potential application in the treatment of CNS disorders.
Conflicts of interest
PH is a past employee of PIQUR Therapeutics AG, Basel; and PH and MPW are shareholders of PIQUR Therapeutics AG.
Supplementary Material
Acknowledgments
This work was supported by the Innosuisse grant 37213.1 IP-LS, EU Horizon 2020, ITN 675392 – Phd; the Novartis Foundation for medical-biological Research grant 14B095; the Swiss Commission for Technology and Innovation (CTI) by PFLS-LS grant 17241.1; the Stiftung für Krebsbekämpfung grant 341, the Swiss National Science Foundation grant 310030_189065 to MPW. We thank D. Rageot, F. Beaufils, J. B. Langlois, A. Dall'Asen and E. Teillet for advice, discussions and contributions to synthetic efforts. A. T. and M. H. thank P. Artursson's lab (Uppsala University, Sweden) for providing the MDCK cells.
Electronic supplementary information (ESI) available: Experimental procedures and characterisation for all new compounds. Experimental details and raw data for biological assays, 1H, 13C NMR, HRMS spectra and HPLC chromatograms for all novel compounds. See DOI: 10.1039/d0md00408a
References
- Wymann M. P. Schneiter R. Nat. Rev. Mol. Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- Sarbassov D. D. Guertin D. A. Ali S. M. Sabatini D. M. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Wymann M. P. Marone R. Curr. Opin. Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Chong Z. Z. Shang Y. C. Wang S. Maiese K. Future Neurol. 2012;7:733–748. doi: 10.2217/fnl.12.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechuga L. Franz D. N. Expert Rev. Neurother. 2019;19:913–925. doi: 10.1080/14737175.2019.1635457. [DOI] [PubMed] [Google Scholar]
- Brandt C. Hillmann P. Noack A. Römermann K. Öhler L. A. Rageot D. Beaufils F. Melone A. Sele A. M. Wymann M. P. Fabbro D. Löscher W. Neuropharmacology. 2018;140:107–120. doi: 10.1016/j.neuropharm.2018.08.002. [DOI] [PubMed] [Google Scholar]
- Jin Z. Niu H. Wang X. Zhang L. Wang Q. Yang A. Oncotarget. 2017;8:58469–58479. doi: 10.18632/oncotarget.17753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin E. K. Patwardhan P. P. Vasudeva S. D. de Stanchina E. Tap W. D. Schwartz G. K. Mol. Cancer Ther. 2015;14:395–406. doi: 10.1158/1535-7163.MCT-14-0711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike K. G. Malagu K. Hummersone M. G. Menear K. A. Duggan H. M. Gomez S. Martin N. M. Ruston L. Pass S. L. Pass M. Bioorg. Med. Chem. Lett. 2013;23:1212–1216. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Rageot D. Bohnacker T. Melone A. Langlois J. B. Borsari C. Hillmann P. Sele A. M. Beaufils F. Zvelebil M. Hebeisen P. Loscher W. Burke J. Fabbro D. Wymann M. P. J. Med. Chem. 2018;61:10084–10105. doi: 10.1021/acs.jmedchem.8b01262. [DOI] [PubMed] [Google Scholar]
- Borsari C. Keles E. Rageot D. Treyer A. Bohnacker T. Bissegger L. De Pascale M. Melone A. Sriramaratnam R. Beaufils F. Hamburger M. Hebeisen P. Löscher W. Fabbro D. Hillmann P. Wymann M. P. J. Med. Chem. 2020;63(22):13595–13617. doi: 10.1021/acs.jmedchem.0c00620. [DOI] [PubMed] [Google Scholar]
- Bonazzi S. Goold C. P. Gray A. Thomsen N. M. Nunez J. Karki R. G. Gorde A. Biag J. D. Malik H. A. Sun Y. Liang G. Lubicka D. Salas S. Labbe-Giguere N. Keaney E. P. McTighe S. Liu S. Deng L. Piizzi G. Lombardo F. Burdette D. Dodart J.-C. Wilson C. J. Peukert S. Curtis D. Hamann L. G. Murphy L. O. J. Med. Chem. 2020;63:1068–1083. doi: 10.1021/acs.jmedchem.9b01398. [DOI] [PubMed] [Google Scholar]
- Cmiljanovic V., Hebeisen P., Jackson E., Beaufils F., Bohnacker T. and Wymann M. P., WO2015049369, 2015
- Borsari C. Rageot D. Dall'Asen A. Bohnacker T. Melone A. Sele A. M. Jackson E. Langlois J.-B. Beaufils F. Hebeisen P. Fabbro D. Hillmann P. Wymann M. P. J. Med. Chem. 2019;62:8609–8630. doi: 10.1021/acs.jmedchem.9b00972. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Danon J. J. Reekie T. A. Kassiou M. Trends Chem. 2019;1:612–624. doi: 10.1016/j.trechm.2019.04.009. [DOI] [Google Scholar]
- Rousseau J. F. Chekroun I. Ferey V. Labrosse J. R. Org. Process Res. Dev. 2015;19:506–513. doi: 10.1021/op500264v. [DOI] [Google Scholar]
- Hebeisen P. Alker A. Buerkler M. Heterocycles. 2012;85:65–72. doi: 10.3987/COM-11-12360. [DOI] [Google Scholar]
- Beaufils F. Cmiljanovic N. Cmiljanovic V. Bohnacker T. Melone A. Marone R. Jackson E. Zhang X. Sele A. Borsari C. Mestan J. Hebeisen P. Hillmann P. Giese B. Zvelebil M. Fabbro D. Williams R. L. Rageot D. Wymann M. P. J. Med. Chem. 2017;60:7524–7538. doi: 10.1021/acs.jmedchem.7b00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsari C. Rageot D. Beaufils F. Bohnacker T. Keles E. Buslov I. Melone A. Sele A. M. Hebeisen P. Fabbro D. Hillmann P. Wymann M. P. ACS Med. Chem. Lett. 2019;10:1473–1479. doi: 10.1021/acsmedchemlett.9b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rageot D. Bohnacker T. Keles E. McPhail J. A. Hoffmann R. M. Melone A. Borsari C. Sriramaratnam R. Sele A. M. Beaufils F. Hebeisen P. Fabbro D. Hillmann P. Burke J. E. Wymann M. P. J. Med. Chem. 2019;62:6241–6261. doi: 10.1021/acs.jmedchem.9b00525. [DOI] [PubMed] [Google Scholar]
- Yang H. Rudge D. G. Koos J. D. Vaidialingam B. Yang H. J. Pavletich N. P. Nature. 2013;497:217–223. doi: 10.1038/nature12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlgren M. Simoff I. Backlund M. Wegler C. Keiser M. Handin N. Müller J. Lundquist P. Jareborg A. C. Oswald S. Artursson P. J. Pharm. Sci. 2017;106:2909–2913. doi: 10.1016/j.xphs.2017.04.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.