

Abstract
Transcription networks and epigenetic mechanisms including DNA methylation, histone modifications, and noncoding RNAs control lineage commitment of multi-potent mesenchymal progenitor cells. Proteins that read, write, and erase histone tail modifications curate and interpret the highly intricate histone code. Epigenetic reader proteins that recognize and bind histone marks provide a crucial link between histone modifications and their downstream biological effects. Here, we investigate the role of bromodomain-containing (BRD) proteins, which recognize acetylated histones, during osteogenic differentiation. Using RNA-sequencing (RNA-seq) analysis, we screened for BRD proteins (n = 40) that are robustly expressed in MC3T3 osteoblasts. We focused functional follow-up studies on Brd2 and Brd4 which are highly expressed in MC3T3 preosteoblasts and represent “bromodomain and extra terminal domain” (BET) proteins that are sensitive to pharmacological agents (BET inhibitors). We show that small interfering RNA depletion of Brd4 has stronger inhibitory effects on osteoblast differentiation than Brd2 loss as measured by osteoblast-related gene expression, extracellular matrix deposition, and alkaline phosphatase activity. Similar effects on osteoblast differentiation are seen with the BET inhibitor +JQ1, and this effect is reversible upon its removal indicating that this small molecule has no lasting effects on the differentiation capacity of MC3T3 cells. Mechanistically, we find that Brd4 binds at known Runx2 binding sites in promoters of bone-related genes. Collectively, these findings suggest that Brd4 is recruited to osteoblast-specific genes and may cooperate with bone-related transcription factors to promote osteoblast lineage commitment and maturation.
Keywords: bone, Brd4, bromodomain, epigenetics, osteoblast
1 |. INTRODUCTION
Skeletal development in vertebrates is highly complex and involves the concerted interaction of many signaling pathways and cell types (Kobayashi & Kronenberg, 2014). New bone tissue forms by either endochondral ossification in which osseous tissue replaces previously established cartilaginous centers or intramembranous ossification in which osseous tissue is generated within the mesenchyme de novo (Berendsen & Olsen, 2015). Central to both these processes is the recruitment of osteoblast progenitors and their successful differentiation to mature osteoblasts capable of generating a calcified matrix (Maes et al., 2010; Stein, Lian, Stein, Van Wijnen, & Montecino, 1996). As such, proper osteoblast maturation and function is important for development of the entire vertebrate skeleton.
Osteoblasts are mesenchymal in origin and their differentiation to mature bone forming cells is directed by tightly regulated changes in gene expression (Huang, Yang, Shao, & Li, 2007; Lian et al., 2006). Alterations in gene expression are controlled by epigenetic mechanisms such as noncoding RNAs, DNA methylation, and posttranslational modifications to histone tails (Allis & Jenuwein, 2016; Gibney & Nolan, 2010). Histone tail modifications are dynamic and can be altered by epigenetic regulators, proteins that read, write, and erase these marks. The resulting epigenomic landscape is complex and has been show to modulate osteogenic gene expression and bone formation (Dudakovic & van Wijnen, 2017; Gordon, Stein, Westendorf, & van Wijnen, 2015; Mortada & Mortada, 2018; van Wijnen & Westendorf, 2019).
Histone tail acetylation has been of particular interest as this mark induces euchromatin formation and is indicative of active transcription (Grunstein, 1997; Sabari, Zhang, Allis, & Zhao, 2017). The levels of histone acetylation are controlled by the opposing actions of histone lysine acetyl transferases that generate acetylated histone marks and histone deacetylases (HDACs) that remove acetyl groups from histone tails. Early studies have demonstrated that histone acetylation on H3 and H4 accompanies bone-related gene activation during osteoblast differentiation (Shen et al., 2002; Shen et al., 2003). Furthermore, the genetic and mechanistic contributions of HDACs to bone formation have been well-studied in skeletal development (Bradley, Carpio, van Wijnen, McGee-Lawrence, & Westendorf, 2015; Cantley, Zannettino, Bartold, Fairlie, & Haynes, 2017; McGee-Lawrence & Westendorf, 2011). Several studies have demonstrated that HDAC loss or inhibition promotes hyperacetylation of histones and accelerates osteoblast differentiation (Di Bernardo et al., 2009; Dudakovic et al., 2013; Iwami & Moriyama, 1993; Jensen, Schroeder, Bailey, Gopalakrishnan, & Westendorf, 2008; H. W. Lee et al., 2006; S. Lee et al., 2009; Schroeder, Kahler, Li, & Westendorf, 2004; Xu et al., 2013). However, it must be noted that these pro-osteogenic effects are context dependent as other studies have reported inhibitory effects on osteoblast differentiation and bone formation upon HDAC loss and inhibition (Dudakovic, Camilleri, Lewallen, et al., 2015; Dudakovic et al., 2017; McGee-Lawrence et al., 2011; Nissen-Meyer et al., 2007; Pratap et al., 2010; Razidlo et al., 2010; Senn et al., 2010). Considering the biological complexities of these previous results, it is appreciated that hyperacetylation of bone-related loci does not suffice to alter gene expression levels and that multiple factors contribute to epigenetic control of the osteogenic gene expression program. Rather, other downstream factors (e.g., epigenetic readers) that depend on histone acetylation may functionally modulate gene expression by bridging the molecular gap between histone marks and transcriptional activation.
Previously, we have shown that inhibition of the epigenetic transcriptional repressor and histone methyltransferase Ezh2 results in activation of bone-specific genes in different biological contexts (Dudakovic & van Wijnen, 2017; Dudakovic et al., 2016; Dudakovic, Camilleri, Xu, et al., 2015). This activation could be supported by acquisition of activating histone lysine methylation marks (e.g., H3K4me3) or histone acetylation marks (e.g., H3 and H4 acetylation). Interestingly, it has been proposed that histone acetylation may prevail over H3K4 methylation in bone specific gene activation (Wu et al., 2014; Wu et al., 2017). Therefore, we focused our present studies on bromodomain-containing (BRD) proteins, which are readers of acetylated lysine residues and provide a link between histone modifications and transcriptional activation (Sanchez & Zhou, 2009).
Within the BRD class of epigenetic readers is a subfamily of bromodomain and extra terminal domain (BET) proteins. These proteins are not only able to bind acetylated lysine residues, but can interact with additional transcription factors and epigenetic regulators via the extra terminal domain (Taniguchi, 2016). Of particular interest among the members of the BET protein family is bromodomain-containing 4 (Brd4) as it is linked to transcriptional initiation and super-enhancer formation (Devaiah, Gegonne, & Singer, 2016; Loven et al., 2013). This family of epigenetic reader proteins provides a plausible link between histone modifications and subsequent activation of the osteoblastic transcriptional network. Indeed, prior studies have suggested that Brd4 may contribute to biological regulation of osteogenesis (Baud’huin et al., 2017; Najafova et al., 2017). In the current study, we evaluated the expression levels of a large panel of BRD proteins (n = 40), including multiple BET proteins (n = 7) in MC3T3 osteoblasts and assessed the impact of Brd2 and Brd4 loss of function by small interfering RNA (siRNA)-mediated knock-down and small molecule inhibition. Our results indicate that Brd4 is required for proper differentiation of MC3T3 osteoblasts and functions as a molecular interpreter of the histone acetylome during osteogenesis.
2 |. METHODS
2.1 |. MC3T3 cell culture
MC3T3-E1 sc4 cells were used for all experiments. Cells were maintained and expanded in MEM alpha media with no ascorbic acid (Gibco) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals), 100 U/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco). +JQ1 (Cayman Chemicals) was dissolved in DMSO and added to culture media at indicated time points. Vehicle control groups were supplemented with equivalent concentration of DMSO when appropriate.
2.2 |. Osteogenic differentiation
For osteogenic differentiation experiments, MC3T3 cells were plated at 10,000 cells/cm2 on tissue culture plastic (Gibco) in standard MEM alpha media (Gibco) supplemented with 10% FBS (Atlanta Biologicals), 100 U/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco). One day after seeding, media was removed and replaced with osteogenic media. Osteogenic media consisted of standard MEM alpha plus 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin supplemented with osteogenic cocktail (50 μg/ml ascorbic acid [Sigma] and 10 mM β-glycerol phosphate [Sigma]). Media was changed every 3 days and osteogenic cocktail was replenished.
2.3 |. Messenger RNA (mRNA) isolation
At indicated time points, cells were lysed using TRI-Reagent (Zymo Research) and RNA was isolated using the Direct-zol RNA isolation kit (Zymo Research). Purified RNA was quantified and quality tested using a NanoDrop 2000 spectrophotometer (Thermo Fischer Scientific).
2.4 |. RNA sequencing
RNA libraries were prepared according to the manufacturer’s instructions for the TruSeq RNA Sample Prep Kit v2 (Illumina). Briefly, poly-A mRNA was purified from total RNA using oligo dT magnetic beads. Purified mRNA was fragmented at 95°C for 8 min, eluted from the beads and primed for first strand complementary DNA (cDNA) synthesis. RNA fragments were copied into first strand cDNA using SuperScript III reverse transcriptase and random primers (Invitrogen). Next, second strand cDNA synthesis was performed using DNA polymerase I and RNase H. The double-stranded cDNA was purified using a single AMPure XP bead (Agencourt) clean-up step. The cDNA ends were repaired and phosphorylated using Klenow, T4 polymerase, and T4 polynucleotide kinase followed by a single AMPure XP bead clean-up. Blunt-ended cDNAs were modified to include a single 3′ adenylate (A) residue using Klenow exo- (3′ to 5′ exo minus). Paired-end DNA adaptors (Illumina) with a single “T” base overhang at the 3′ end were immediately ligated to the “A tailed” cDNA population. Unique indexes, included in the standard TruSeq Kits (12-Set A and 12-Set B) were incorporated at the adaptor ligation step for multiplex sample loading on the flow cells. The resulting constructs were purified by two consecutive AMPure XP bead clean-up steps. The adapter-modified DNA fragments were enriched by 12 cycles of polymerase chain reaction (PCR) using primers included in the Illumina Sample Prep Kit. The concentration and size distribution of the libraries was determined on an Agilent Bioanalyzer DNA 1000 chip. A final quantification, using Qubit fluorometry (Invitrogen), confirmed sample concentrations.
Libraries were loaded onto flow cells at concentrations of 8–10 p.m. to generate cluster densities of 700,000/mm2 following the standard protocol for the Illumina cBot and cBot Paired end cluster kit version 3. Flow cells were sequenced as 51 × 2 paired end reads on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 and HCS v2.0.12 data collection software. Base-calling was performed using Illumina’s RTA version 1.17.21.3. The RNA-Seq data were analyzed using the standard RNA-Seq workflow by Mayo Bioinformatics Core called MAPRSeq v.1.2.1 (Kalari et al., 2014), which includes alignment with TopHat 2.0.6 (Kim et al., 2013) and quantification of gene expression using the HTSeq software (Anders, Pyl, & Huber, 2015). Normalized gene counts were also obtained from MAPRSeq where expression values for each gene were normalized to 1 million reads and corrected for gene length (reads per kilobase pair per million mapped reads [RPKM]). Complete data sets generated from RNA-sequencing are archived in the Gene Expression Omnibus (GEO) database (MC3T3 A and B Accession #GSE83506, MC3T3 C Accession #GSE135984).
2.5 |. siRNA-mediated knockdown
MC3T3s were plated at 10,000 cells/cm2 in MEM alpha media with no ascorbic acid (Gibco) supplemented with 10% FBS (Atlanta Biologicals), 100U/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco). The next day, cells were transfected with siRNAs using Lipofectamine RNAiMAX (Thermo Fisher Scientific) transfection reagent in standard MEM alpha media. Dharmacon smart-pool siRNAs (GE Healthcare) targeting Brd2 (Cat #L-043404–00) and Brd4 (Cat #L-041493–00) as well as nontargeting control (Cat #D-001810-10-20) were used at a final concentration of 20 nM for these studies. Osteogenic differentiation cocktail (50 μg/ml ascorbic acid [Sigma] and 10 mM β-glycerol phosphate [Sigma]) was added 1 day after transfection. Media was changed 3 days post-transfection with subsequent media changes occurring every 3 days.
2.6 |. Western blot analysis
Western blot analysis was performed according to standard protocol as previously described (Dudakovic, Camilleri, Xu, et al., 2015). Cells were lysed using RIPA buffer at indicated time points. Proteins were visualized using SuperSignal West Femto Reagent (Thermo Fisher Scientific) and imaged using the ChemiDoc Imaging System (BioRad). Antibodies and concentrations used for western blot analysis were as follows: Brd2 (Cell Signaling; Cat #D89B4 1:1000), Brd4 (Bethyl Labs; Cat #A301–985A; 1:2,000), Runx2 (in house; 1:3,000), Gapdh (Cell Signaling; Cat #51745; 1:5,000), H3K27Me3 (Cell Signaling; Cat #9733S, 1:1,000), H3K27Ac (Millipore; Cat #07–360; 1:1,000), Histone 3 (Millipore; Cat #05–982; 1:10,000).
2.7 |. Quantitative reverse transcription PCR (RT-qPCR)
RNA was isolated as described above and reverse transcribed into cDNA using the Promega Reverse Transcription kit and protocol. Gene expression was quantified using real-time quantitative PCR with QuantiTect SYBR Green PCR Kit (Qiagen) and the CFX384 Real-Time System (BioRad). Transcript levels were quantified using the method and normalized to the housekeeping gene Gapdh (set at 100). Primer pairs were utilized at a final concentration of 0.8 μM (Table S1).
2.8 |. Picrosirius red staining
Media was removed and cells were washed with phosphate buffered saline (PBS) before fixing in 10% neutral buffered formalin (NBF). After 1 hour, NBF was removed and cells were washed with H2O. Cultures were then stained with Picrosirius Red F3BA Kit (Poly-sciences Inc.) according to the manufacturer’s protocol.
2.9 |. MTS activity assay
MTS assay (Promega) was conducted according to the manufacturer’s protocol at indicated time points. Enzymatic activity was calculated from absorbance at 490 nm determined using a SpectraMAX Plus spectrophotometer (Molecular Devices).
2.10 |. Hoechst staining
Media was removed and cells were washed with PBS. After washing, Tris–EDTA buffer (×0.1) was added to the wells to completely cover the cells. The plate was then stored at −80°C for at least 2 hours and then thawed at room temperature. An equal volume of Hoechst 33258 staining solution (0.5 μg Hoechst 33258 [Sigma 94403] per 1 ml of buffer solution [0.3 M NaCl, 5 mM Tris, pH 8.0 in H2O]) was added to each well. The plate was incubated in the dark at room temperature for 20 min. Fluorescence intensity was measured at 340 nm excitation wavelength and 460 nm emission wavelength using an F200 Infinite Pro (Tecan) plate reader. Measurements were fit to a standard DNA curve to determine relative DNA content.
2.11 |. Alkaline phosphatase activity assay
To the same plate from the Hoechst staining described above, 500 μl of para-nitrophenylphosphate solution (2.5 mg 4-nitrophenylphosphate disodium salt hexahydrate [Sigma] per 1 ml of buffer [0.1 M diethanolamine, 150 mM NaCl, 2 mM MgCl2]) was added to each well. The plate was incubated for 30 min (time may vary) at room temperature before measuring absorbance at 405 nm using the SpectraMAX Plus spectrophotometer. Values were fit to a standard curve prepared using reconstituted alkaline phosphatase enzyme (Roche) to determine relative enzymatic activity.
2.12 |. Alizarin red staining
At indicated time points, media was removed and cells were washed with PBS. Cells were then fixed in 10% NBF. After 1 hr, NBF was removed, cells were washed with PBS, and stained with 2% alizarin red (Thermo Fisher Scientific) for 10 min. Stain was removed and wells were washed five times with H2O. Images were taken of the wells and staining was quantified using the ImageJ software (Schneider, Rasband, & Eliceiri, 2012).
2.13 |. Immunoprecipitation
MC3T3 cells were plated at 10,000 cells/cm2 in maintenance medium and allowed to proliferate overnight. The next day, media was removed and replaced with osteogenic differentiation media (MEM alpha plus 10% FBS, 100U/ml penicillin, and 100 μg/ml streptomycin supplemented with ascorbic acid [Sigma; final concentration 50 μg/ml] and β-glycerol phosphate [Sigma; final concentration 10 mM]). Three days later, cells were lysed using RIPA buffer and quantified using a BSA standard curve. Ten micrograms of antibody against Runx2 (Santa Cruz; Cat #M-70) or Brd4 (Bethyl Labs; Cat #A301–985A) was incubated with 500 μg of protein lysate. Normal Rabbit IgG (Cell Signaling; Cat #27295) was used as a nontargeting control. Protein G Dynabeads (Thermo Fisher Scientific) were used to isolate antibody bound proteins from whole lysate. Western blot analysis was conducted on resulting pull-down as described previously.
2.14 |. Chromatin immunoprecipitation (ChIP)
MC3T3 cells were plated at 10,000 cells/cm2 in maintenance media and allowed to proliferate for 2 days (~95% confluence). Then, media was removed and replaced with osteogenic differentiation media. Cells were fixed 3 days later using 16% formaldehyde (Methanol-free; Pierce; Cat #28906) and ChIP procedure was carried out as previously described (Dudakovic et al., 2013, 2016). Cell pellets were sonicated using 10 cycles of 30”ON/30”OFF in a Bioruptor Pico (Diagenode) sonicator. 10 μg of anti-Brd4 antibody (Bethyl Labs; Cat #A301–985A) was used for pull-downs. Normal Rabbit IgG (Cell Signaling; Cat #27295) was used as a nontargeting control. Real-time quantitative PCR was performed on the resulting DNA as previously described using indicated primers (Table S1).
3 |. RESULTS
3.1 |. Expression of BRD proteins in MC3T3 cells
We evaluated the mRNA expression levels of a large panel of BRD proteins (n = 40) in undifferentiated MC3T3-E1 cells (Wang et al., 1999) (Figure 1a). We note robust expression of most BRD proteins in undifferentiated MC3T3s. All BRD proteins have the capacity to bind acetylated lysine residues; however, proteins with a bromodomain and an extra terminal domain (BET) can also interact with other transcription factors and epigenetic regulators (Devaiah et al., 2016). Therefore, we assessed expression levels in this subfamily of BET proteins as they are the most functionally relevant to our study (Figure 1a, blue). Interestingly, we find Brd2 and Brd4 among the top ten highest expressed BRD proteins as well as the highest expressed BET proteins suggesting a potential role in osteoblasts. To further investigate this observation, we evaluated the expression levels of Brd2 and Brd4 over the course of osteogenic differentiation in MC3T3s (Figure 1b). We note a reduction in both Brd2 and Brd4 expression over time during early stages of osteogenic differentiation. Together, these findings suggest Brd2 and Brd4 may play a role in early osteogenic lineage commitment and contribute to activation of the early osteogenic transcriptional network.
FIGURE 1.
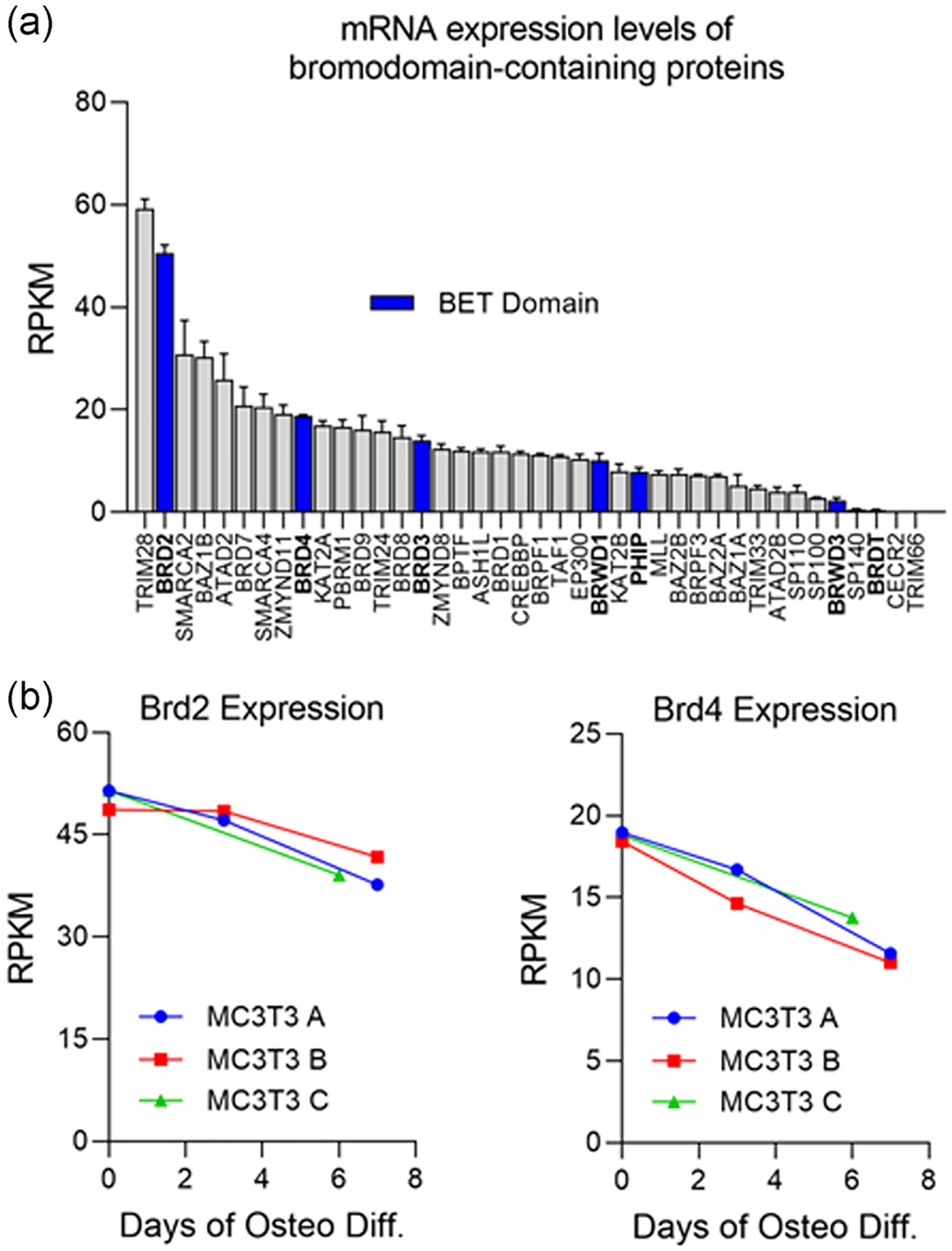
Evaluation of messenger RNA (mRNA) expression levels of bromodomain-containing proteins in MC3T3 cells. (a) mRNA expression levels (RPKM) for all bromodomain-containing proteins determined by RNA-sequencing in MC3T3 preosteoblasts (n = 3, mean ± STD) before induction of osteogenic differentiation. Bromodomain and extra terminal domain-containing proteins (BET) are highlighted in blue. (b) Expression levels of Brd2 and Brd4 throughout osteogenic differentiation time course in MC3T3s
3.2 |. Knockdown of Brd2 and Brd4 inhibits MC3T3 osteogenesis
We conducted siRNA knockdown experiments targeting Brd2 and/or Brd4 in MC3T3s. Cells were transfected 1 day before addition of osteogenic differentiation cocktail to ensure sufficient knockdown of each protein before lineage commitment. Successful knockdown of both Brd2 and Brd4 was confirmed by western blot analysis 2 days post-transfection (Figure 2a) and via RT-qPCR 4 days posttransfection (Figure 2b). After 7 days of osteogenic differentiation, collagen deposition was significantly reduced in the Brd4 knockdown groups when compared to nontargeting control (Figure 2c). Brd2 knockdown did not impair collagen deposition. Alkaline phosphatase (Alpl) enzymatic activity was significantly reduced in both Brd2 and Brd4 knockdown groups at Day 14 of osteogenic differentiation (Figure 2d). Dual loss of Brd2 and Brd4 was more detrimental to Alpl enzymatic activity when compared to single knockdowns. Additionally, we observe significant reduction in early expression of key osteoblastic genes on day three of differentiation (Figure 2e). Interestingly, knockdown of Brd2 alone appears to have only a mild impact on osteogenic differentiation. However, the impact of Brd4 knockdown is more severe in all cases, and in combination with Brd2 knockdown is detrimental to lineage commitment. These data demonstrate that Brd2 and Brd4 may have slightly redundant roles, but Brd4 is the main driver of osteogenic lineage commitment. In total, these data demonstrate that Brd4 loss reduces the expression of osteoblastic genes and results in impaired matrix deposition and a reduction in Alpl enzymatic activity.
FIGURE 2.
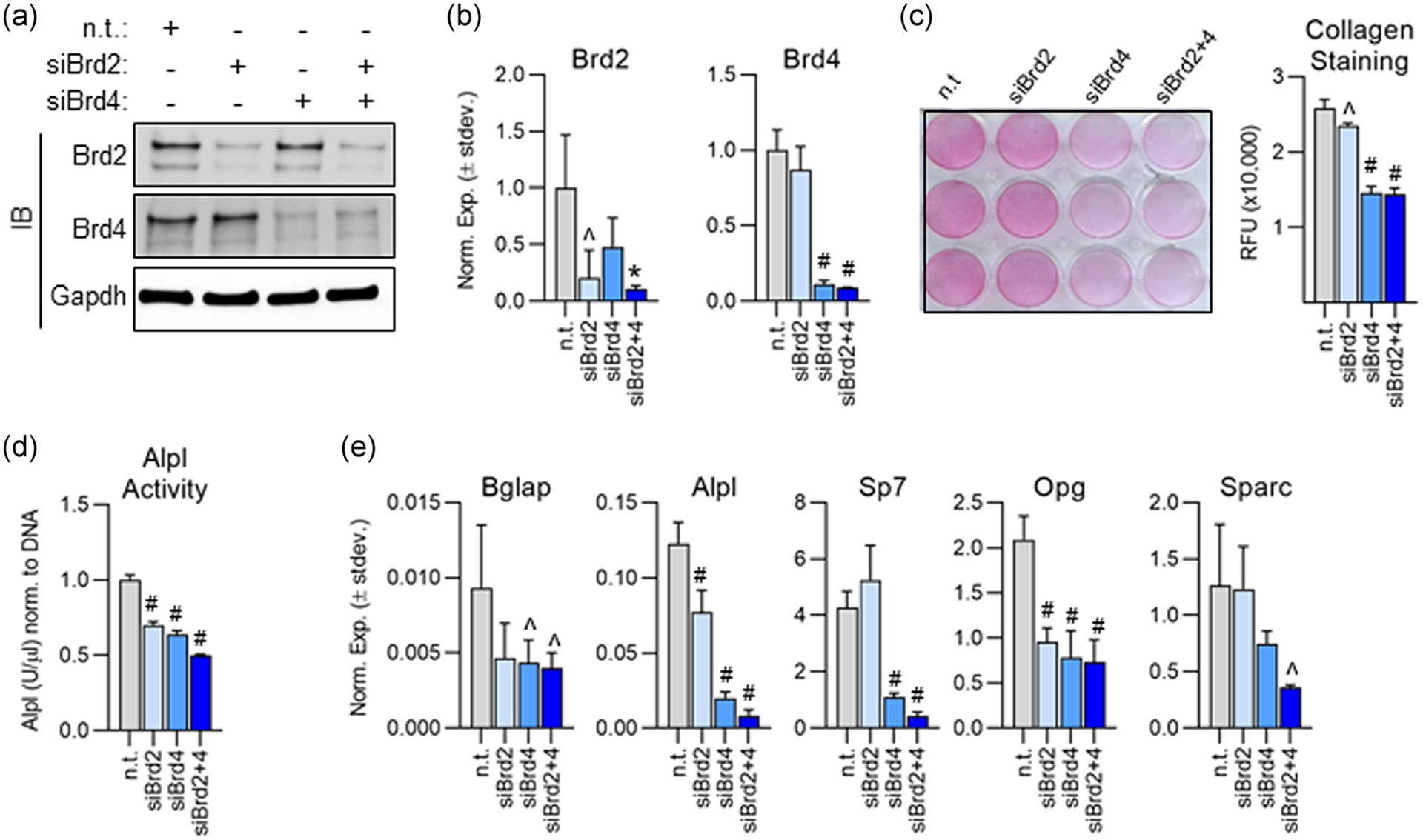
Knockdown of Brd2 and/or Brd4 impairs osteogenic differentiation of MC3T3 cells. (a) Western blot analysis of protein lysates collected 2 days after small interfering RNA (siRNA) transfection. (b) Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis performed on messenger RNA (mRNA) isolated 4 days post-transfection (n = 3, mean ± STD). (c) Pico Sirius Red staining and quantification for collagen deposition performed 7 days after induction of osteogenic differentiation (n = 3, mean ± STD). (d) Alkaline phosphatase (Alpl) enzymatic activity measured on Day 14 of osteogenic differentiation (n = 3, mean ± STD). (e) RT-qPCR conducted on mRNA isolated 3 days after induction of osteogenic differentiation (n = 3, mean ± STD). All error bars represent ± STD of three biological replicates. Statistical significance to nontargeting control group indicated as follows: ^p < .05, *p < .01, #p < .001
3.3 |. Toxicity profile of +JQ1 in MC3T3 cells
To further investigate the involvement of Brd4 on osteogenesis, we utilized a small molecule inhibitor, +JQ1, which binds with high specificity to both Brd4 bromodomains to prevent recognition of acetylated lysine residues (Filippakopoulos et al., 2010). We first conducted a concentration-response assay in which MC3T3 cells were cultured in the presence of +JQ1 for 24 hr and determined the median lethal dose (LD50) to be 161 nM (Figure 3a). +JQ1′s previously established Brd4 IC50 of 78 nM provides a reasonable therapeutic window for MC3T3 experiments (Filippakopoulos et al., 2010). In light of these results, we proceeded with a concentration of 100 nM to ensure sufficient inhibition of Brd4 binding while preserving cell viability. Subconfluent MC3T3s were treated with 100 nM +JQ1 for 3 days. After 3 days, media containing +JQ1 was replaced with maintenance media. Modest cytotoxicity is observed 24 hr after treatment, but cell viability is restored to vehicle levels 3 days after the treatment as measured by MTS assay (Figure 3b). Similarly, there is a minor reduction in cell number 24 hr after +JQ1 treatment but no appreciable difference is observed at day seven post-treatment (Figure 3c). In addition to the 3 day +JQ1 treatment, we also replenished the drug during the day three media change to assess cytotoxicity after prolonged exposure to the compound. We observed no change in cell viability after 6 days of continuous treatment with 100 nM +JQ1 (Figure 3d). In light of these experiments, 100 nM concentration of +JQ1 was selected for all subsequent MC3T3 studies.
FIGURE 3.
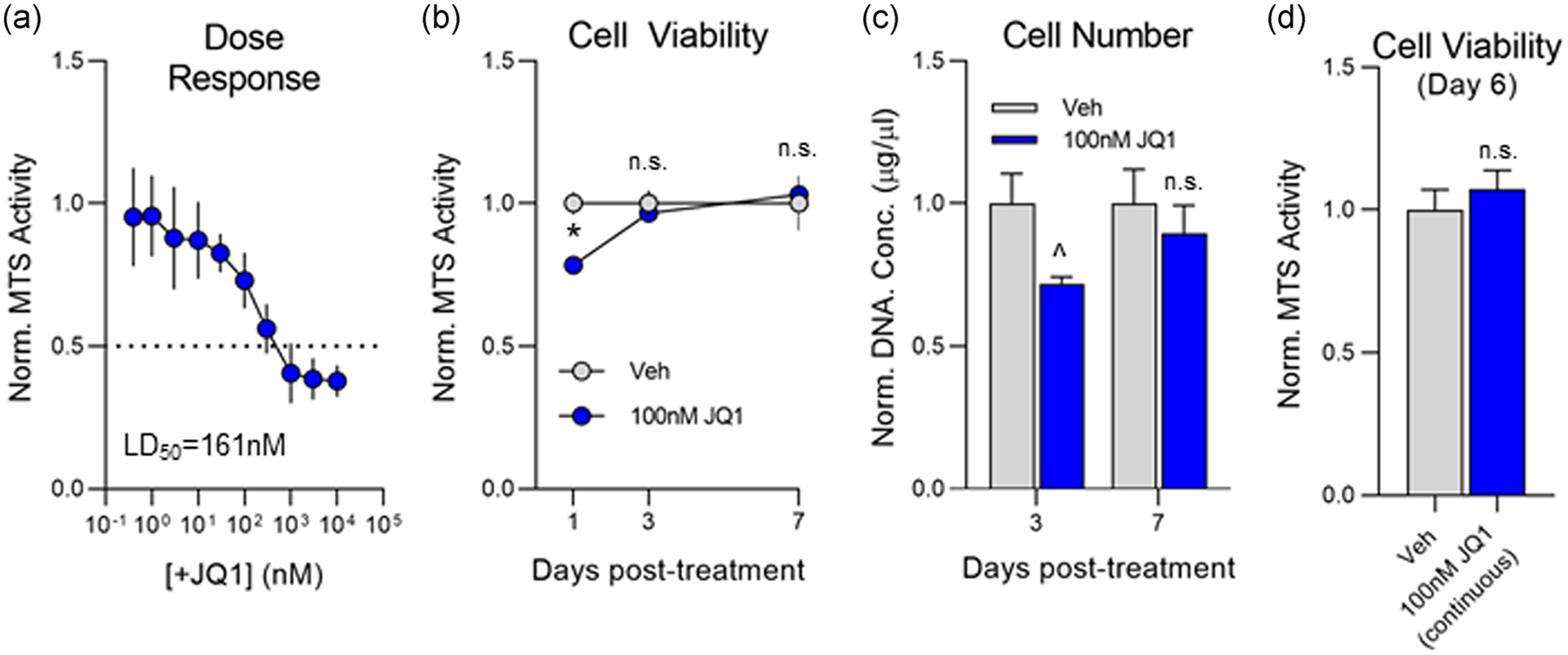
Assessment of +JQ1 toxicity in MC3T3 cells. (a) Subconfluent MC3T3 cells were treated with increasing concentrations of +JQ1. Cell viability was measured 24 hr posttreatment via MTS assay (n = 3, mean ± STD). (b) Subconfluent MC3T3 cells were treated with 100 nM +JQ1 for 3 days. Cell viability was measured 24 hr, 3 days, and 7 days post-treatment via MTS assay (n = 3, mean ± STD) and normalized to dimethyl sulfoxide (DMSO)-treated control. (c) Subconfluent MC3T3 cells were treated with 100 nM +JQ1 for 3 days. Cell number was measured by relative fluorescence of Hoechst staining (DNA) and normalized to DMSO-treated control (n = 3, mean ± STD). (d) Subconfluent MC3T3 cells were treated with 100 nM +JQ1 for 6 continuous days. Cell viability was measured on Day 6 via MTS assay (n = 3, mean ± STD) and normalized to DMSO-treated control. All error bars represent ± STD of 3 biological replicates. Statistical significance to vehicle (DMSO) control group indicated as follows: ^p < .05, *p < .01, n.s.p > .05
3.4 |. Brd4 inhibition suppresses osteogenic gene expression
To assess the impact of Brd4 inhibition on osteogenesis, MC3T3s were treated with 100 nM +JQ1 for the first 3 days of osteogenic differentiation. A significant reduction in mRNA expression of key osteogenic markers (Alpl, Bglap, and Ibsp) at day three (Figure 4a) as well as a reduction in alkaline phosphatase enzymatic activity at day six (Figure 4b) is observed in the +JQ1 treated group. We next investigated whether these initial short-term effects persisted throughout the remainder of the osteogenic differentiation time course. Interestingly, we note that the initial effect of +JQ1 at day three is reversible as gene expression returns to baseline levels (vehicle control) 1–2 weeks after treatment (Figure 4c). Consequently, there is no long-term impact on matrix mineralization as demonstrated by alizarin red staining of the cultures (Figure 4d). These findings demonstrate that Brd4 inhibition prevents osteogenic initiation, but is reversible and does not confer lasting effects on osteogenic differentiation.
FIGURE 4.
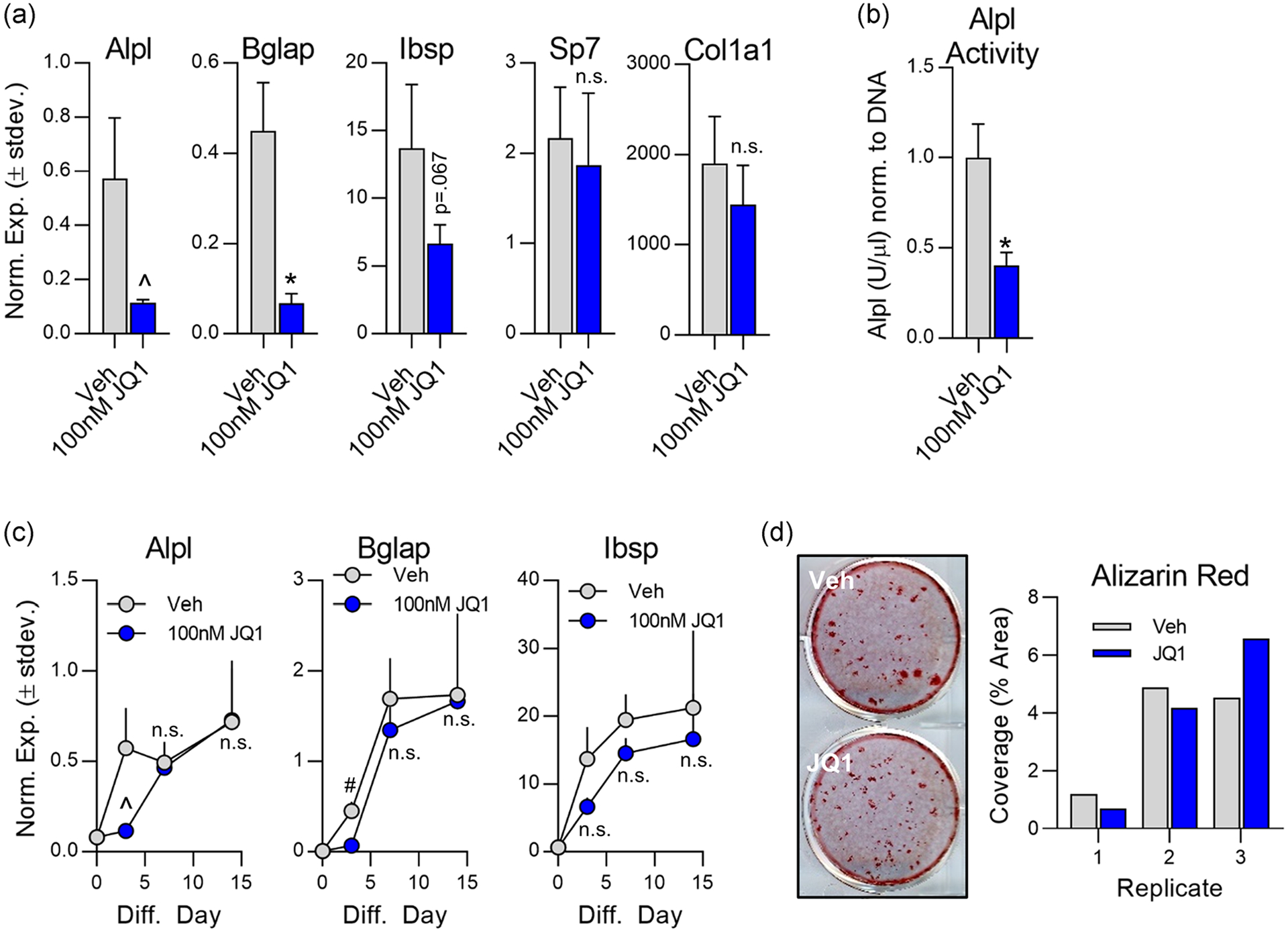
Inhibition of osteogenic differentiation by +JQ1 is reversible in MC3T3 cells. Subconfluent MC3T3 cells were treated from Day 0 to Day 3 with 100 nM +JQ1. Osteogenic differentiation supplements were added on Day 0 and replenished throughout the entire culture period. (a) Quantitative reverse transcription polymerase chain reaction (RT-qPCR) performed on messenger RNA (mRNA) isolated on Day 3 (n = 3, mean ± STD). (b) Alkaline phosphatase (Alpl) enzymatic activity measured on Day 6 (n = 3, mean ± STD). (c) mRNA expression levels assessed via RT-qPCR for indicated genes throughout the osteogenic differentiation time course (n = 3, mean ± STD). (d) Representative image (left) and ImageJ quantification (right) of alizarin red staining for hydroxyapatite deposition. All error bars represent ± STD of three biological replicates. Statistical significance to vehicle (DMSO) control group indicated as follows: ^p < .05, *p < .01, #p < .001, n.s.p > .05
3.5 |. Persistent Brd4 inhibition prevents osteogenic differentiation of MC3T3 cells
To address the observation that MC3T3s can recover after a short-term 3 day treatment with +JQ1, we conducted a long-term treatment in which the inhibitor was present throughout the entire duration of the osteogenic differentiation time course (Figure 5a). Again, short-term administration of the inhibitor (one time addition of drug for 3 days) results in impaired expression of osteogenic genes on day three, but osteogenic gene expression recovers to vehicle control levels after the removal of the inhibitor (Figure 5b). However, when +JQ1 was kept in culture continuously, mRNA expression levels of key osteoblast genes (Bglap and Alpl) remained significantly lower in the treatment group and did not recover to the vehicle control levels (Figure 5b) resulting in a significant reduction in matrix mineralization at Day 24 of differentiation (Figure 5c).
FIGURE 5.
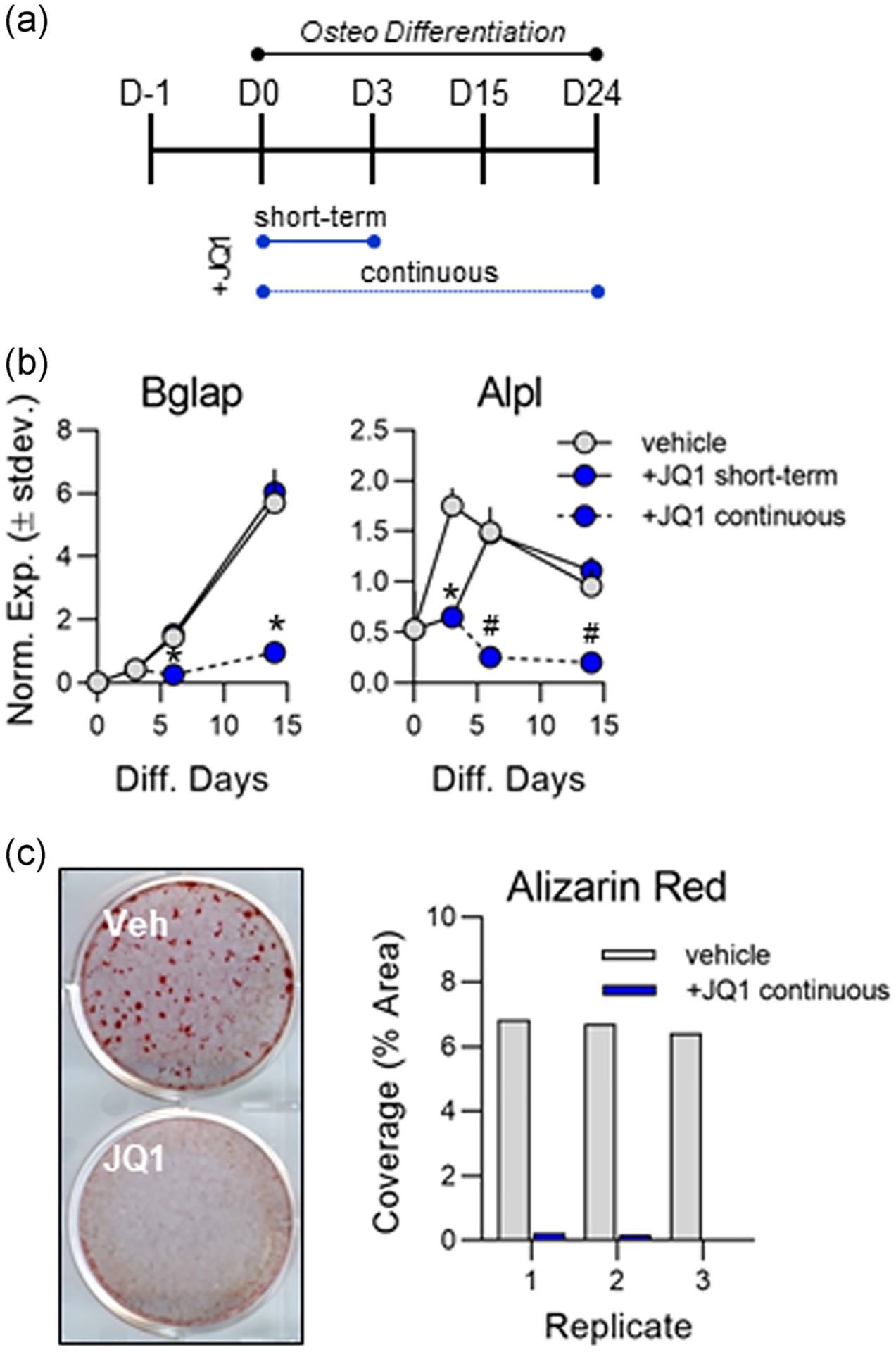
Continuous +JQ1 treatment prevents osteogenesis of MC3T3 cells. (a) Experimental outline indicating short-term (3 days) and continuous 100 nm +JQ1 treatment in differentiating MC3T3 cells. (b) Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis performed on messenger RNA (mRNA) collected throughout the osteogenic differentiation time course (n = 3, mean ± STD). (c) Representative image (left) and ImageJ quantification (right) of alizarin red staining for hydroxyapatite deposition on Day 24 of osteogenic differentiation. All error bars represent ± STD of three biological replicates. Statistical significance to vehicle (dimethyl sulfoxide [DMSO]) control group indicated as follows: *p < .01, #p < .001
3.6 |. Pro-osteogenic effects of Ezh2 inhibition are Brd4 dependent
Our previous studies have shown that inhibition of Ezh2, a histone 3 lysine 27 (H3K27) methyltransferase, accelerates osteogenic differentiation of MC3T3 cells (Dudakovic et al., 2016; Dudakovic, Camilleri, Xu, et al., 2015). Those studies demonstrated that the pro-osteogenic effect of Ezh2 inhibition was conveyed by a reduction in H3K27 tri-methylation (H3K27Me3) which promoted expression of genes that support osteoblastogenesis. We hypothesized that Brd4 recognition of acetylated histones is required for the pro-osteogenic effects of Ezh2 inhibition. Thus, differentiating MC3T3 cells were exposed to 5 μM GSK126, a selective Ezh2 inhibitor, and/or 100 nM +JQ1. We note a reduction in H3K27Me3 and an increase in H3K27Ac in the presence of GSK126 (Figure 6a). +JQ1 treatment resulted in a slight reduction in H3K27Me3, but did not alter H3K27Ac levels (Figure 6a). Enhanced osteogenic differentiation by GSK126 treatment is supported by enhanced alizarin red staining at Day 22 (Figure 6b) and in the form of elevated mRNA expression levels of osteogenic genes throughout the time course (Figure 6c, green). Strikingly, the stimulatory effects of GSK126 on osteoblastogenesis are almost completely negated by the addition of +JQ1 as measured by matrix mineralization (Figure 6b, column 4) and mRNA expression (Figure 6c, orange). Together, these data demonstrate that Brd4 not only supports basal osteogenesis, but its function is also required during hyperacetylated states which have been shown to enhance osteogenic differentiation.
FIGURE 6.
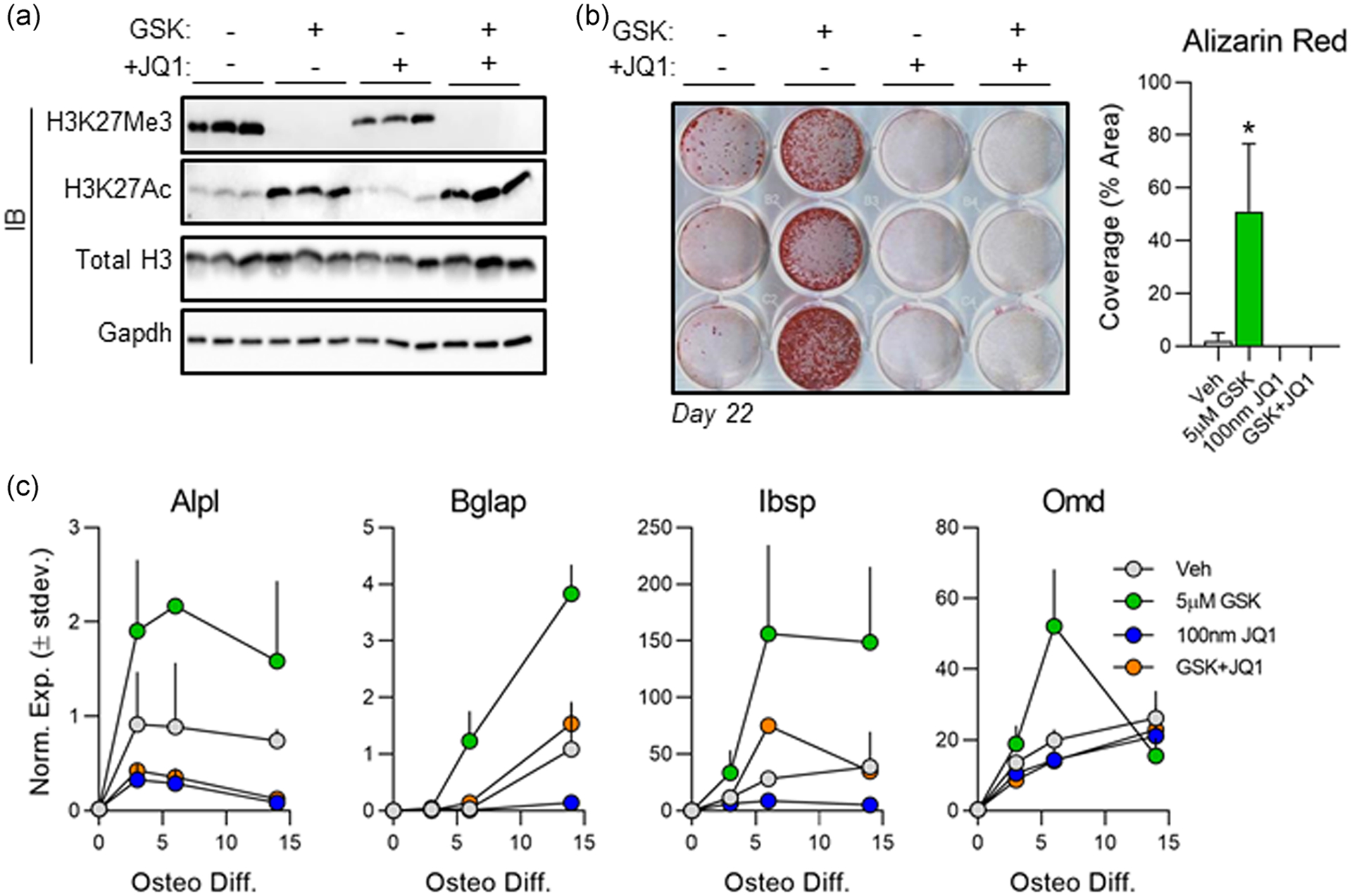
Brd4 inhibition negates the pro-osteogenic effects of Ezh2 inhibition. Subconfluent MC3T3s were treated with 5 μM GSK126 and/or 100 nM +JQ1 for the first 3 days of osteogenic differentiation. (a) Western blot analysis of protein lysates collected on day three. (b) Alizarin red staining (left) and quantification (right) conducted on Day 22 of osteogenic differentiation (n = 3, mean ± STD). (c) Quantitative reverse transcription polymerase chain reaction (RT-qPCR) analysis performed on messenger RNA (mRNA) collected throughout the osteogenic differentiation time course for each treatment group (n = 3, mean ± STD). All error bars represent ± STD of three biological replicates. Statistical significance to vehicle (DMSO) control group indicated as follows: *p < .01
3.7 |. Brd4 binds to established Runx2 binding sites
To understand the molecular mechanism by which Brd4 may be regulating osteoblast differentiation, immunoprecipitation studies were conducted to probe for interactions with Runx2, a known master-regulator of osteoblast differentiation (Komori et al., 1997; Schroeder, Jensen, & Westendorf, 2005; Stein et al., 2004). We did not observe evidence for a direct Runx2-Brd4 interaction after Runx2 (Figure 7a) or Brd4 (Figure 7b) immunoprecipitation. Interestingly, ChIP for Brd4 followed by RT-qPCR revealed that Brd4 is localized to regions of the genome that have previously been established as Runx2 binding sites (Meyer, Benkusky, & Pike, 2014) (Figure 7c). Additionally, Brd4 occupancy of these sites increases from day zero to day three of osteogenic differentiation. Together, these findings indicate that although Brd4 and Runx2 may not interact directly, these proteins may be recruited to the same regions of the genome to enhance osteogenic differentiation.
FIGURE 7.
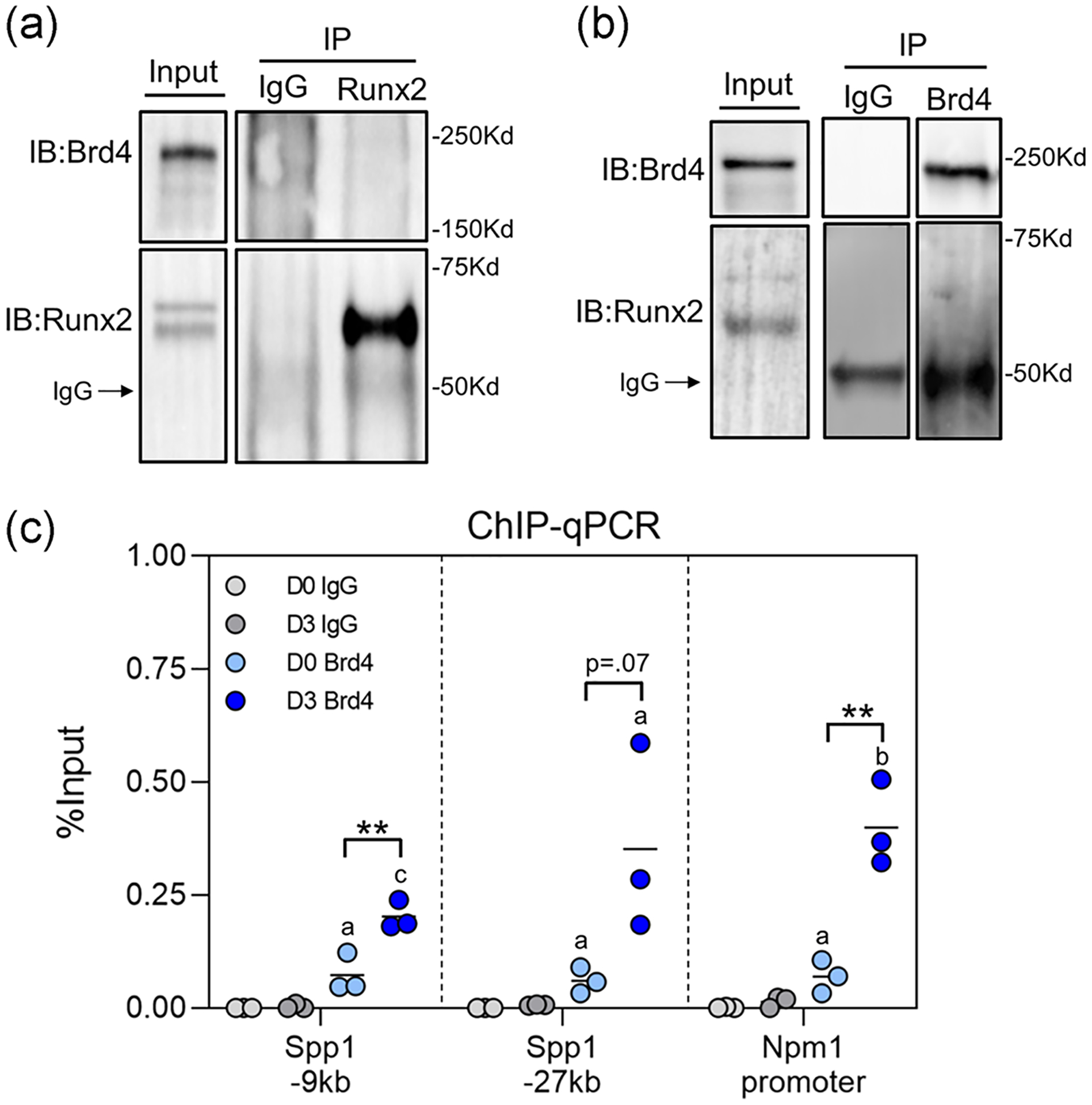
Brd4 occupies Runx2 binding sites on genomic DNA. (a) Immunoprecipitation of Runx2 from MC3T3 protein lysates collected on Day 3 of osteogenic differentiation followed by western blot analysis. (b) Immunoprecipitation of Brd4 from MC3T3 protein lysates collected on Day 3 of osteogenic differentiation followed by western blot analysis. (c) ChIP with anti-Brd4 antibody followed by RT-PCR using primers that flank known Runx2 binding sites (n = 3). ChIP, chromatin immunoprecipitation; IgG, immunoglobulin G; IP, immunoprecipitation; qPCR, quantitative polymerase chain reaction; RT-PCR, reverse transcription polymerase chain reaction. Statistical significance indicated as follows: ap < .05 compared to reverse transcription polymerase chain reaction (IgG) control on corresponding day, bp < .01 compared to IgG control on corresponding day, cp < .001 compared to IgG control on corresponding day. **p < .01 for indicated groups
4 |. DISCUSSION
In the present study, we demonstrate that Brd2 and Brd4, which belong to the BET family of bromodomain-containing proteins, are robustly expressed in MC3T3 preosteoblasts. We note similar expression levels compared to those observed for other tissues in the Genotype-Tissue Expression (GTEx) Project database (GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2 on 09/06/2018]) and the robust Musculoskeletal RNA-seq Atlas we developed at Mayo Clinic (unpublished data). The ubiquitous expression of Brd2 and Brd4 across a wide array of tissue types suggests an important role for these epigenetic readers in all contexts of cellular differentiation and tissue homeostasis. Additionally, we show that Brd2 and Brd4 expression levels are highest early in the osteogenic differentiation time course. This pattern suggests that these epigenetic reader proteins are crucial during early osteoblast lineage commitment which is similar to findings in other cell types (J. E. Lee et al., 2017; Sakurai et al., 2017).
Our studies show that knock-down of Brd4 has a more severe impact on osteoblast differentiation than depletion of Brd2. These findings were validated with the small molecule inhibitor +JQ1 at a low concentration (100 nM) that effectively prevents Brd4 binding, but remains below the IC50 for other bromodomain proteins (Filippakopoulos et al., 2010). Administration of +JQ1 reduces osteoblast-specific gene expression and alkaline phosphatase activity suggesting that Brd4 function is required for proper osteoblast lineage progression. These findings support previous studies which demonstrate that early tissue-specific loss of Brd4 prevents lineage commitment of myoblasts and adipocytes (J. E. Lee et al., 2017). Additionally, Brd4 inhibition has been shown to prevent differentiation of human fetal osteoblasts (Najafova et al., 2017) and slow progression of osteosarcoma cells (Lamoureux et al., 2014). Our findings corroborate these previous observations in MC3T3 osteoblasts. Of note, we find that the early inhibitory effects of +JQ1 in MC3T3s is reversible upon removal from culture media with no long-term effect on matrix mineralization. This finding is remarkable as other studies on small molecule inhibitors targeting epigenetic regulators have shown lasting effects after a short pulse early in the differentiation time course (Dudakovic et al., 2013, 2016; Dudakovic, Camilleri, Xu, et al., 2015; Schroeder & Westendorf, 2005).
Mechanistically, our study revealed that Brd4 binding in MC3T3 osteoblasts occurs at or near Runx2 binding sites that were previously established in promoter regions of osteoblast-related genes (Spp1 and Npm1) (Meyer et al., 2014). Importantly, we note an increase in Brd4 occupancy at these sites during early stages of osteogenic differentiation. This observation may reflect shifts in Brd4 binding from a broad and nonspecific distribution throughout the genome to highly localized interactions at active enhancers after induction of differentiation (J. E. Lee et al., 2017). Our finding that Brd4 binds close to Runx2 binding sites in murine MC3T3 osteoblasts corroborates previous studies with human fetal osteoblasts (Najafova et al., 2017) and is consistent with the broader concept that Brd4 supports cellular differentiation by cooperating with lineage-specific transcription factors to activate transcriptional networks (Gilmour et al., 2018; Najafova et al., 2017; Sakurai et al., 2017).
Our current study builds on previous investigation into the role of Ezh2 and H3K27me3 during osteogenic differentiaiton. Pharmacological inhibitors of Ezh2 block formation of H3K27me3 marks, and we show that reduction in H3K27me3 promotes hyperacetylation (H3K27Ac) in this study. Remarkably, even though a hyper-acetylated state was achieved upon Ezh2 inhibition, there was no subsequent increase in gene expression when Brd4 was unable to bind acetylated cites (+JQ1 treatment). Previous studies have shown that treatment with +JQ1 removes Brd4 from promoter regions of genes with the highest level of Brd4 binding. As a result, these genes exhibited the greatest reduction in expression after administration of the inhibitor (Najafova et al., 2017). The same may occur in MC3T3 osteoblasts in which administration of +JQ1 may reduce expression of osteoblastic genes by removing Brd4 from their promoters. The unique ability of Brd4 to interact with RNA-Pol II via CDK9 and the p-TEFb complex renders it a key component of transcriptional activation (Sakurai et al., 2017). These findings affirm the role of Brd4 as the molecular bridge between hyper-acetylated histone tails and the RNA transcription machinery.
In conclusion, our studies show that Brd4 is a key regulator of osteoblast differentiation in MC3T3 cells and that administration of the BET inhibitor +JQ1 is detrimental to proper osteoblast maturation and function. BET protein inhibitors have recently become promising therapeutic agents in the field of cancer as well as orthopedics (Baud’huin et al., 2017; Doroshow, Eder, & LoRusso, 2017). Before their widespread adoption, it is important to consider the impact these pharmacological agents may have on bone tissues within the developing skeleton and on bone homeostasis.
Supplementary Material
ACKNOWLEDGMENTS
We thank our Mayo Clinic colleagues and members of the Orthopedic Research Laboratories, including Drs. Jennifer Westendorf and David Deyle for stimulating discussions and sharing reagents. We also thank Oksana Pichurin for technical support and Marina Ganshina for administrative support. The Genotype-Tissue Expression (GTEx) Project was utilized in our study. We would like to thank the Common Fund of the Office of the Director of the National Institutes of Health, as well as the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS for funding and curating this valuable resource. This publication was made possible through an intramural award from the Mayo Clinic (Career Development Award in Orthopedics Research to AD) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR069049 to AvW). We also thank William and Karen Eby for generous philanthropic support.
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
DATA AVAILABILITY STATEMENT
Data sets utilized in this study are archived in the Gene Expression Omnibus (GEO) database (MC3T3 A and B Accession #GSE83506, MC3T3 C Accession #GSE135984).
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- Allis CD, & Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nature Reviews Genetics, 17(8), 487–500. 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, & Huber W (2015). HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud’Huin M, Lamoureux F, Jacques C, Rodriguez Calleja L, Quillard T, Charrier C, … Ory B (2017). Inhibition of BET proteins and epigenetic signaling as a potential treatment for osteoporosis. Bone, 94, 10–21. 10.1016/j.bone.2016.09.020 [DOI] [PubMed] [Google Scholar]
- Berendsen AD, & Olsen BR (2015). Bone development. Bone, 80, 14–18. 10.1016/j.bone.2015.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bernardo G, Squillaro T, Dell’Aversana C, Miceli M, Cipollaro M, Cascino A, … Galderisi U (2009). Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem Cells and Development, 18(4), 573–581. 10.1089/scd.2008.0172 [DOI] [PubMed] [Google Scholar]
- Bradley EW, Carpio LR, van Wijnen AJ, McGee-Lawrence ME, & Westendorf JJ (2015). Histone deacetylases in bone development and skeletal disorders. Physiological Reviews, 95(4), 1359–1381. 10.1152/physrev.00004.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley MD, Zannettino ACW, Bartold PM, Fairlie DP, & Haynes DR (2017). Histone deacetylases (HDAC) in physiological and pathological bone remodelling. Bone, 95, 162–174. 10.1016/j.bone.2016.11.028 [DOI] [PubMed] [Google Scholar]
- Devaiah BN, Gegonne A, & Singer DS (2016). Bromodomain 4: A cellular Swiss army knife. Journal of Leukocyte Biology, 100(4), 679–686. 10.1189/jlb.2RI0616-250R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroshow DB, Eder JP, & LoRusso PM (2017). BET inhibitors: A novel epigenetic approach. Annals of Oncology, 28(8), 1776–1787. 10.1093/annonc/mdx157 [DOI] [PubMed] [Google Scholar]
- Dudakovic A, Camilleri ET, Lewallen EA, McGee-Lawrence ME, Riester SM, Kakar S, … van Wijnen AJ (2015). Histone deacetylase inhibition destabilizes the multi-potent state of uncommitted adipose-derived mesenchymal stromal cells. Journal of Cellular Physiology, 230(1), 52–62. 10.1002/jcp.24680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic A, Camilleri ET, Riester SM, Paradise CR, Gluscevic M, O’Toole TM, … van Wijnen AJ (2016). Enhancer of Zeste homolog 2 inhibition stimulates bone formation and mitigates bone loss caused by ovariectomy in skeletally mature mice. Journal of Biological Chemistry, 291(47), 24594–24606. 10.1074/jbc.M116.740571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic A, Camilleri ET, Xu F, Riester SM, McGee-Lawrence ME, Bradley EW, … van Wijnen AJ (2015). Epigenetic control of skeletal development by the histone methyltransferase Ezh2. Journal of Biological Chemistry, 290(46), 27604–27617. 10.1074/jbc.M115.672345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic A, Evans JM, Li Y, Middha S, McGee-Lawrence ME, van Wijnen AJ, & Westendorf JJ (2013). Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. Journal of Biological Chemistry, 288(40), 28783–28791. 10.1074/jbc.M113.489732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic A, Gluscevic M, Paradise CR, Dudakovic H, Khani F, Thaler R, … van Wijnen AJ (2017). Profiling of human epigenetic regulators using a semi-automated real-time qPCR platform validated by next generation sequencing. Gene, 609, 28–37. 10.1016/j.gene.2017.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudakovic A, & van Wijnen AJ (2017). Epigenetic control of osteoblast differentiation by enhancer of Zeste homolog 2 (EZH2). Current Molecular Biology Reports, 3(2), 94–106. 10.1007/s40610-017-0064-8 [DOI] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, … Bradner JE (2010). Selective inhibition of BET bromodomains. Nature, 468(7327), 1067–1073. 10.1038/nature09504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney ER, & Nolan CM (2010). Epigenetics and gene expression. Heredity, 105(1), 4–13. 10.1038/hdy.2010.54 [DOI] [PubMed] [Google Scholar]
- Gilmour J, Assi SA, Noailles L, Lichtinger M, Obier N, & Bonifer C (2018). The co-operation of RUNX1 with LDB1, CDK9 and BRD4 drives transcription factor complex relocation during haematopoietic specification. Scientific Reports, 8(1), 10410. 10.1038/s41598-018-28506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JAR, Stein JL, Westendorf JJ, & van Wijnen AJ (2015). Chromatin modifiers and histone modifications in bone formation, regeneration, and therapeutic intervention for bone-related disease. Bone, 81, 739–745. 10.1016/j.bone.2015.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M (1997). Histone acetylation in chromatin structure and transcription. Nature, 389(6649), 349–352. 10.1038/38664 [DOI] [PubMed] [Google Scholar]
- Huang W, Yang S, Shao J, & Li YP (2007). Signaling and transcriptional regulation in osteoblast commitment and differentiation. Frontiers in Bioscience, 12, 3068–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwami K, & Moriyama T (1993). Effects of short chain fatty acid, sodium butyrate, on osteoblastic cells and osteoclastic cells. The International Journal of Biochemistry, 25(11), 1631–1635. http://www.ncbi.nlm.nih.gov/pubmed/8288032 [DOI] [PubMed] [Google Scholar]
- Jensen ED, Schroeder TM, Bailey J, Gopalakrishnan R, & Westendorf JJ (2008). Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. Journal of Bone and Mineral Research, 23(3), 361–372. 10.1359/jbmr.071104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, … Kocher JP (2014). MAP-RSeq: Mayo analysis pipeline for RNA sequencing. BMC Bioinformatics, 15, 224. 10.1186/1471-2105-15-224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, & Salzberg SL (2013). TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14(4), R36. 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, & Kronenberg HM (2014). Overview of skeletal development. Methods in Molecular Biology, 1130, 3–12. 10.1007/978-1-62703-989-5_1 [DOI] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, … Kishimoto T (1997). Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 89(5), 755–764. 10.1016/s0092-8674(00)80258-5 [DOI] [PubMed] [Google Scholar]
- Lamoureux F, Baud’huin M, Rodriguez Calleja L, Jacques C, Berreur M, Redini F, … Ory B (2014). Selective inhibition of BET bromodomain epigenetic signalling interferes with the bone-associated tumour vicious cycle. Nature Communications, 5, 3511. 10.1038/ncomms4511 [DOI] [PubMed] [Google Scholar]
- Lee HW, Suh JH, Kim AY, Lee YS, Park SY, & Kim JB (2006). Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Molecular Endocrinology, 20(10), 2432–2443. 10.1210/me.2006-0061 [DOI] [PubMed] [Google Scholar]
- Lee JE, Park YK, Park S, Jang Y, Waring N, Dey A, … Ge K (2017). Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nature Communications, 8(1), 2217. 10.1038/s41467-017-02403-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Park JR, Seo MS, Roh KH, Park SB, Hwang JW, … Kang KS (2009). Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Proliferation, 42(6), 711–720. 10.1111/j.1365-2184.2009.00633.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, … Young DW (2006). Networks and hubs for the transcriptional control of osteoblastogenesis. Reviews in Endocrine & Metabolic Disorders, 7(1–2), 1–16. 10.1007/s11154-006-9001-5 [DOI] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, … Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell, 153(2), 320–334. 10.1016/j.cell.2013.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, … Kronenberg HM (2010). Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Developmental Cell, 19(2), 329–344. 10.1016/j.devcel.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee-Lawrence ME, McCleary-Wheeler AL, Secreto FJ, Razidlo DF, Zhang M, Stensgard BA, … Westendorf JJ (2011). Suberoylanilide hydroxamic acid (SAHA; vorinostat) causes bone loss by inhibiting immature osteoblasts. Bone, 48(5), 1117–1126. 10.1016/j.bone.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee-Lawrence ME, & Westendorf JJ (2011). Histone deacetylases in skeletal development and bone mass maintenance. Gene, 474(1–2), 1–11. 10.1016/j.gene.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MB, Benkusky NA, & Pike JW (2014). The RUNX2 cistrome in osteoblasts: Characterization, down-regulation following differentiation, and relationship to gene expression. Journal of Biological Chemistry, 289(23), 16016–16031. 10.1074/jbc.M114.552216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortada I, & Mortada R (2018). Epigenetic changes in mesenchymal stem cells differentiation. European Journal of Medical Genetics, 61(2), 114–118. 10.1016/j.ejmg.2017.10.015 [DOI] [PubMed] [Google Scholar]
- Najafova Z, Tirado-Magallanes R, Subramaniam M, Hossan T, Schmidt G, Nagarajan S, … Johnsen SA (2017). BRD4 localization to lineage-specific enhancers is associated with a distinct transcription factor repertoire. Nucleic Acids Research, 45(1), 127–141. 10.1093/nar/gkw826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen-Meyer LS, Svalheim S, Tauboll E, Reppe S, Lekva T, Solberg LB, … Jemtland R (2007). Levetiracetam, phenytoin, and valproate act differently on rat bone mass, structure, and metabolism. Epilepsia, 48(10), 1850–1860. 10.1111/j.1528-1167.2007.01176.x [DOI] [PubMed] [Google Scholar]
- Pratap J, Akech J, Wixted JJ, Szabo G, Hussain S, McGee-Lawrence ME, … Lian JB (2010). The histone deacetylase inhibitor, vorinostat, reduces tumor growth at the metastatic bone site and associated osteolysis, but promotes normal bone loss. Molecular Cancer Therapeutics, 9(12), 3210–3220. 10.1158/1535-7163.MCT-10-0572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, … Westendorf JJ (2010). Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLOS One, 5(7), 0011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Zhang D, Allis CD, & Zhao Y (2017). Metabolic regulation of gene expression through histone acylations. Nature Reviews Molecular Cell Biology, 18(2), 90–101. 10.1038/nrm.2016.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai N, Inamochi Y, Inoue T, Hariya N, Kawamura M, Yamada M, … Mochizuki K (2017). BRD4 regulates adiponectin gene induction by recruiting the P-TEFb complex to the transcribed region of the gene. Scientific Reports, 7(1), 11962. 10.1038/s41598-017-12342-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez R, & Zhou MM (2009). The role of human bromodomains in chromatin biology and gene transcription. Current Opinion in Drug Discovery and Development, 12(5), 659–665. [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, & Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods, 9(7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder TM, Jensen ED, & Westendorf JJ (2005). Runx2: A master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Research. Part C, Embryo Today: Reviews, 75(3), 213–225. 10.1002/bdrc.20043 [DOI] [PubMed] [Google Scholar]
- Schroeder TM, Kahler RA, Li X, & Westendorf JJ (2004). Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. The Journal of Biological Chemistry, 279(40), 41998–42007. 10.1074/jbc.M403702200 [DOI] [PubMed] [Google Scholar]
- Schroeder TM, & Westendorf JJ (2005). Histone deacetylase inhibitors promote osteoblast maturation. Journal of Bone and Mineral Research, 20(12), 2254–2263. 10.1359/jbmr.050813 [DOI] [PubMed] [Google Scholar]
- Senn SM, Kantor S, Poulton IJ, Morris MJ, Sims NA, O’Brien TJ, & Wark JD (2010). Adverse effects of valproate on bone: Defining a model to investigate the pathophysiology. Epilepsia, 51(6), 984–993. 10.1111/j.1528-1167.2009.02516.x [DOI] [PubMed] [Google Scholar]
- Shen J, Hovhannisyan H, Lian JB, Montecino MA, Stein GS, Stein JL, & Van Wijnen AJ (2003). Transcriptional induction of the osteocalcin gene during osteoblast differentiation involves acetylation of histones h3 and h4. Molecular Endocrinology, 17(4), 743–756. 10.1210/me.2002-0122 [DOI] [PubMed] [Google Scholar]
- Shen J, Montecino M, Lian JB, Stein GS, Van Wijnen AJ, & Stein JL (2002). Histone acetylation in vivo at the osteocalcin locus is functionally linked to vitamin D-dependent, bone tissue-specific transcription. Journal of Biological Chemistry, 277(23), 20284–20292. 10.1074/jbc.M112440200 [DOI] [PubMed] [Google Scholar]
- Stein GS, Lian JB, Stein JL, Van Wijnen AJ, & Montecino M (1996). Transcriptional control of osteoblast growth and differentiation. Physiological Reviews, 76(2), 593–629. 10.1152/physrev.1996.76.2.593 [DOI] [PubMed] [Google Scholar]
- Stein GS, Lian JB, van Wijnen AJ, Stein JL, Montecino M, Javed A, … Pockwinse SM (2004). Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene, 23(24), 4315–4329. 10.1038/sj.onc.1207676 [DOI] [PubMed] [Google Scholar]
- Taniguchi Y (2016). The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. International Journal of Molecular Sciences, 17(11), 1849. 10.3390/ijms17111849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Christensen K, Chawla K, Xiao G, Krebsbach PH, & Franceschi RT (1999). Isolation and characterization of MC3T3-E1 preosteoblast subclones with distinct in vitro and in vivo differentiation/mineralization potential. Journal of Bone and Mineral Research, 14(6), 893–903. 10.1359/jbmr.1999.14.6.893 [DOI] [PubMed] [Google Scholar]
- van Wijnen AJ, & Westendorf JJ (2019). Epigenetics as a new frontier in orthopedic regenerative medicine and oncology. Journal of Orthopaedic Research, 37(7), 1465–1474. 10.1002/jor.24305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Gordon JA, Whitfield TW, Tai PW, van Wijnen AJ, Stein JL, … Lian JB (2017). Chromatin dynamics regulate mesenchymal stem cell lineage specification and differentiation to osteogenesis. Biochimica et Biophysica Acta, Gene Regulatory Mechanisms, 1860(4), 438–449. 10.1016/j.bbagrm.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Whitfield TW, Gordon JA, Dobson JR, Tai PW, van Wijnen AJ, … Lian JB (2014). Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biology, 15(3), R52. 10.1186/gb-2014-15-3-r52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, De Veirman K, Evans H, Santini GC, Vande Broek I, Leleu X, … Van Riet I (2013). Effect of the HDAC inhibitor vorinostat on the osteogenic differentiation of mesenchymal stem cells in vitro and bone formation in vivo. Acta Pharmacologica Sinica, 34(5), 699–709. 10.1038/aps.2012.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.