

Abstract
The endoplasmic reticulum (ER), responsible for processing approximately one-third of the human proteome including most secreted and membrane proteins, plays a pivotal role in protein homeostasis (proteostasis). Dysregulation of ER proteostasis has been implicated in a number of disease states. As such, continued efforts are directed at elucidating mechanisms of ER protein quality control which are mediated by transient and dynamic protein-protein interactions with molecular chaperones, co-chaperones, protein folding and trafficking factors that take place in and around the ER. Technological advances in mass spectrometry have played a pivotal role in characterizing and understanding these protein-protein interactions that dictate protein quality control mechanisms. Here, we highlight the recent progress from mass spectrometry-based investigation of ER protein quality control in revealing the topological arrangement of the proteostasis network, stress response mechanisms that adjust the ER proteostasis capacity, and disease specific changes in proteostasis network engagement. We close by providing a brief outlook on underexplored areas of ER proteostasis where mass spectrometry is a tool uniquely primed to further expand our understanding of the regulation and coordination of protein quality control processes in diverse diseases.
Introduction
Secretory proteins routed to the endoplasmic reticulum (ER), lysosome, plasma membrane, or extracellular milieu are first folded and processed within the ER [1]. Such proteins account for approximately one third of the proteome. These proteins include prohormones, receptors, and protein transporters amongst others, and are involved in diverse biological processes including signal transduction, protein degradation, cell growth, and metabolism [1]. In the ER, these “clients” encounter sets of chaperones, co-chaperones, folding enzymes, trafficking, and degradation factors that comprise the ER protein homeostasis (proteostasis) network (ER-PN). The ER-PN ensures the trafficking and localization of properly folded clients, while degrading improperly folded, potentially detrimental clients. This process is cumulatively referred to as protein quality control (PQC). PQC requires coordinated and dynamic interactions of client proteins with ER-PN components [1,2]. Polypeptide chains emerging from the ribosome require co-translational engagement with the Sec61 translocon complex to facilitate ER translocation [3,4]. Once the polypeptide chain emerges into the ER, dedicated chaperones such as Hsp70/40s engage with the nascent polypeptide chain until the full protein is translated [5–7]. Post-translational modifications (PTMs), such as glycosylation, later in the folding process can trigger interactions of dedicated chaperones that aid in client-specific processing [8–10]. Proteins found to reach their properly folded conformation are routed by trafficking factors, such as vesicle coat and packaging machinery, to the proper cellular loci [11]. Conversely, destabilized and therefore potentially detrimental proteins are routed toward degradation pathways requiring dedicated ER-associated degradation (ERAD) or autophagy (ER-phagy) components [12–14] [Fig. 1].
Figure 1. Secretory proteostasis and protein quality control is governed by the activity of ER protein folding, trafficking, and degradation pathways.

Proteins routed for secretory environments cotranslationally enter the ER and engage with chaperones and other folding factors or enzymes that aid in client protein folding. Upon reaching a properly folded confirmation, client proteins are packaged and targeted towards secretory environments through engagement with vesicle trafficking machinery. Conversely, client protein unable to reach their proper conformation are routed towards degradation pathways such ERAD or autophagy (ER-phagy). Arrows denoted in the figure are meant to exemplify interactions with different protein folding pathways of the ER. Routing is highly client-specific and a given protein client may not engage with all of the proteostasis factors and pathways.
Insults to the processing of these ER clients or to the ER itself either through genetic, age-related, or environmental perturbations result in imbalances in ER PQC capacity that are linked to a number of disease states including diabetes, neurodegeneration, amyloidosis, and even cancer [15]. Imbalances in ER PQC capacity can lead to the accumulation of non-native protein species that increase protein folding load and result in ER stress. The protein folding load and status of the ER is constantly monitored by the evolutionarily conserved unfolded protein response (UPR). In metazoans, the UPR consists of three signaling branches regulated by the ER transmembrane proteins inositol-required enzyme 1 (IRE1), activating transcription factor 6 (ATF6), and protein kinase R-like ER kinase (PERK) [16]. Yeast, which is also discussed throughout the review, lack the ATF6 pathway and only contain the most evolutionarily conserved IRE1 and PERK branches. When ER stress is encountered, the UPR is activated to adjust protein folding capacity and restore proteostasis [Fig. 2]. ER stress leads to PERK activation through dimerization and autophosphorylation, followed by eIF2α phosphorylation by PERK. Phospho-eIF2α leads to transient attenuation of protein translation, along with activation of transcription factor ATF4, which upregulates UPR gene targets. IRE1 similarly undergoes dimerization/oligomerization and autophosphorylation, promoting the non-canonical RNA splicing activity of the XBP1 mRNA transcript leading to translation of the active XBP1s transcription factor. XBP1s then goes on to upregulate UPR gene targets. ATF6 activation by ER stress leads to ATF6 trafficking to the Golgi apparatus, where ATF6 is proteolytically processed to release an active ATF6 transcription factor that can translocate to the nucleus and upregulate UPR gene targets. These gene targets include ER chaperones, redox enzymes, degradation components and proteins involved in lipid synthesis pathways [17–19]. Additionally, transient translation inhibition by the PERK branch of the UPR decreases protein folding load. Mechanisms of the UPR and its gene targets have been reviewed in depth previously [16,17].
Figure 2. ER protein quality control capacity in metazoans is monitored by the three branches of the unfolded protein response.
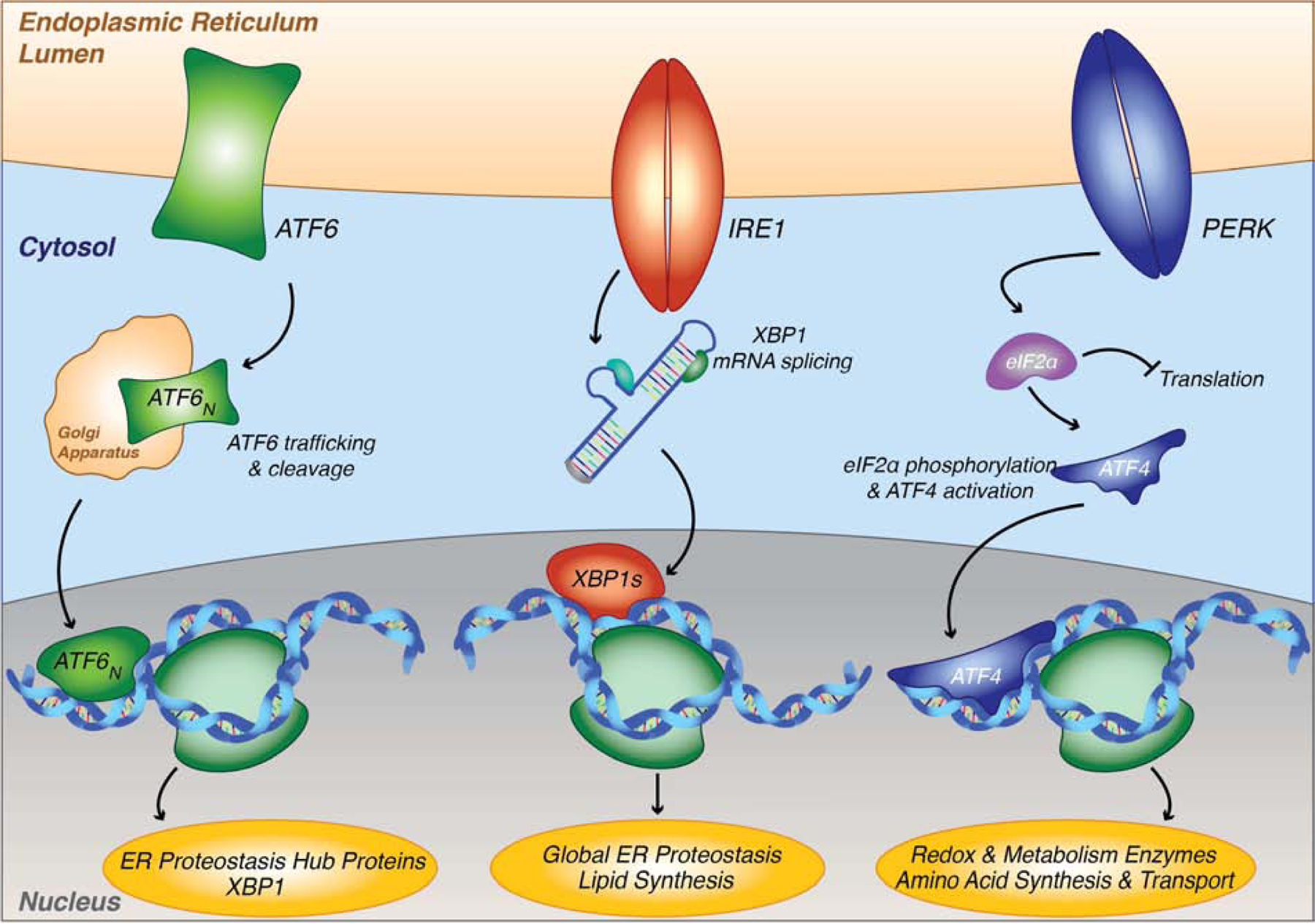
The UPR is regulated by three signaling branches consisting of IRE1, ATF6, and PERK. Yeast lack the ATF6 pathway and only contain the most evolutionarily conserved IRE1 and PERK branches.
Mass spectrometry (MS) has become a key technology to characterize changes in protein expression as a result of ER stress and UPR regulations. At the same time, MS has enabled the identification of protein-protein interactions that facilitate ER PQC, as well as PTMs that play a role in ER PQC [20,21]. In this review, we discuss and highlight a number of investigations and techniques involving MS-based proteomics that have been pivotal in investigating mechanisms of ER stress and PQC.
PQC mechanisms are driven by protein-protein interactions with a diverse set of proteostasis factors motivating efforts to identify the critical components. On the one hand, MS has aided in the global profiling of proteostasis components, as well as changes during disease states and drug treatments. On the other hand, affinity purification – mass spectrometry (AP-MS) has been the most widely used technique to characterize protein-protein interactions enabling interactomics studies [21]. Additionally, liquid chromatography – tandem mass spectrometry (LC-MS/MS) instrumentation and methods have continued to advance as a robust system for investigations across a myriad of sample types, yielding great depth, precision, and throughput in protein identification and quantification [22–24]. In AP-MS, a protein of interest (bait) and interacting proteins (prey) are copurified via affinity bead matrix. After subsequent wash steps to rid the sample of non-specific background, the purified protein and interaction partners are eluted from the bead matrix, digested using one or multiple proteases, and subsequently analyzed via LC-MS/MS [Fig. 3A]. A control AP-MS sample, often using an untagged bait or sample lacking the bait all together, is traditionally used to delineate true interacting proteins from non-specific background bound to the bead matrix during affinity purification. Other methods to investigate interactions take advantage of proximity labeling using a modified biotin ligase (BioID) or ascorbic acid peroxidase (APEX) fused to the bait protein of interest [Fig. 3B] [25,26]. Exogenously supplied biotin substrates are then activated by the enzymes and covalently modify proteins in close proximity to the bait, where the proximity radius is controlled by the lifetime of the activated substrate. This method is useful as biotin enrichment with streptavidin matrix has great specificity and efficiency. AP-MS experiments may utilize protein crosslinkers to covalently link interacting proteins [Fig. 3C]. This is particularly useful in the case of studying protein-protein interactions within the ER-PN, as these interactions are often transient (having fast on-off rates) in nature. Furthermore, the identification of crosslinked peptides can provide structural insight into how these interactions take place [27]. Some crosslinkers, such as DSP, allow interacting proteins to be disassociated under reducing or other chemically controlled conditions prior to protease digestion and LC-MS/MS analysis. DSSO contains an alkyl-sulfoxide linker that is cleavable using collision induced disassociation (CID). This cleavage after CID leaves diagnostic ions in the MS/MS spectra that allow for identification of crosslinked peptides. Cleavage of DSSO via CID allows for MS3-based methods to then sequence the given peptides [28]. Lastly, quantitative methods such as stable isotope labeling with amino acids in cell culture (SILAC) or isobaric labeling such as tandem mass tags (TMT) have aided in quantifying changes in interaction that take place during ER PQC and disease states [Fig. 3D–E] [29–32]. SILAC labeling with “light” or “heavy” amino acids, commonly lysine and arginine, induces a mass shift in peptide m/z that allows the abundance of peptides to be quantified from both samples. TMT compounds consist of an amine reactive group to covalently bind peptides, a mass normalizing linker region to ensure all TMT labels have the same mass prior to analysis, and finally a reporter group that is liberated after CID that provides an indirect quantitative readout of peptide abundance. TMT and TMTpro labeling have allowed up to 11plex and 16plex multiplexing experiments respectively.
Figure 3. Affinity purification – mass spectrometry and tool compounds to elucidate protein-protein interactions implicated in protein quality control.
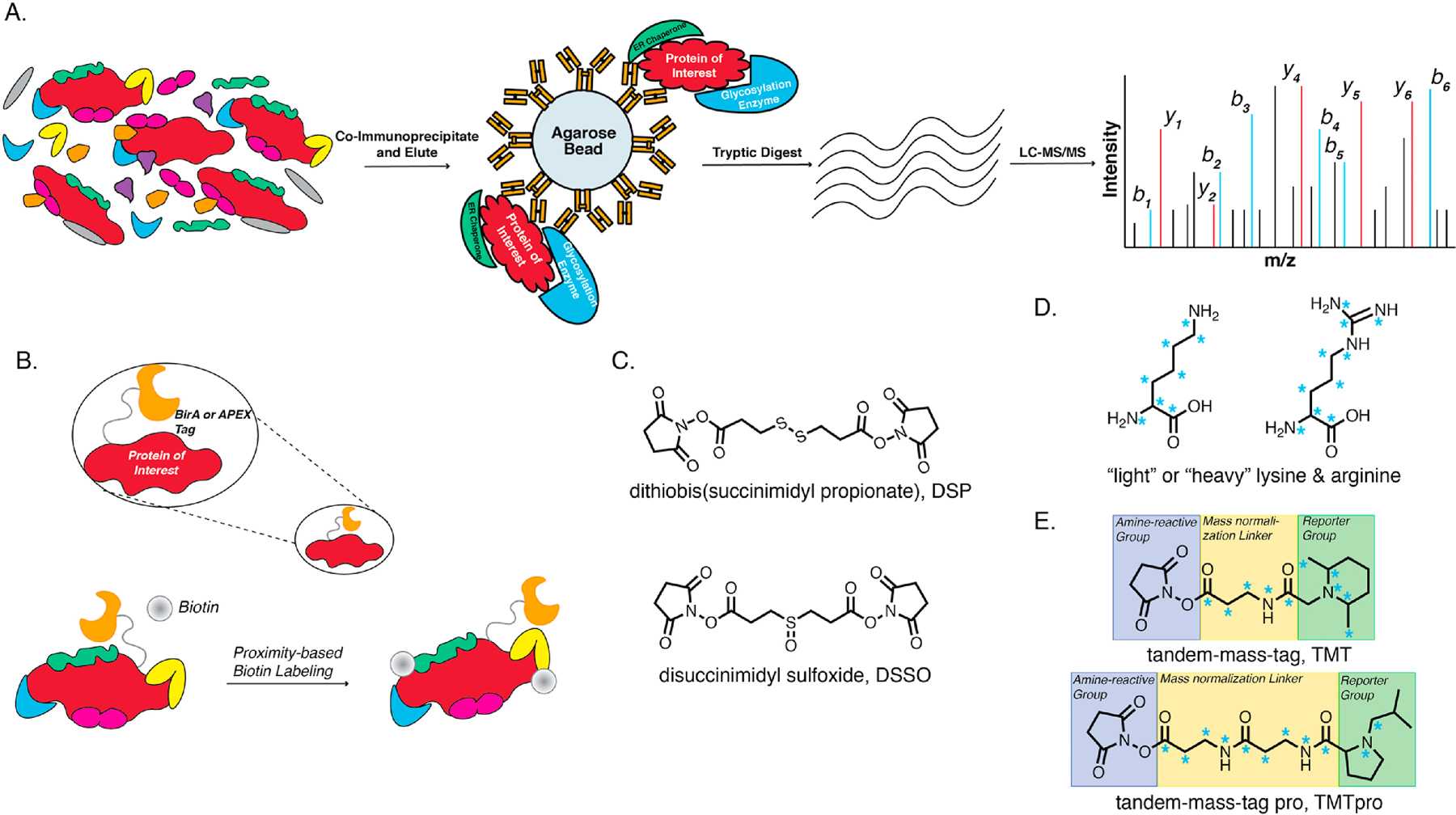
A. Schematic detailing a typical AP-MS workflow. B. Proximity labeling MS often uses a biotin ligase-tagged protein of interest to biotinylate interacting proteins. Biotinylated proteins can then be affinity purified and analyzed via LC-MS/MS to identify interacting partners. C. Protein crosslinkers can be used to aid in affinity purification of interacting proteins by covalently linking them and can subsequently be used to identify crosslinked peptides in some cases. D. Stable isotope labeling with amino acids has aided in quantitative mass spectrometry experiments. Blue asterisks indicate atoms where heavy isotopes can be incorporated. E. Isobaric labeling has additionally aided in quantitative mass spectrometry investigation as well as increased throughput through multiplexing. Blue asterisks indicate points where heavy isotopes can be incorporated.
Of course, these technological advancements do not come without some drawbacks. On the whole, low abundance or proteins lacking peptides that ionize well can still be difficult to identify via LC-MS/MS. AP-MS often requires protein engineering to incorporate epitope tags for affinity purification, or if no such tag is created, an affinity matrix must be manufactured using antibodies that recognize the endogenous protein. Some proteins may not be amenable to epitope tag incorporation, and conversely generating antibody functionalized affinity matrix can be laborious and costly. Proximity labeling methods often suffer from high non-specific background labeling, especially for very abundant proteins, as is the case for many proteostasis components. Careful controls have to be implemented to distinguish true interactions from non-specific background. While isobaric tagging can increase sample throughput compared to SILAC, co-isolated peptides during fragmentation can produce distortions in protein ratio quantification across samples if using LC-MS/MS, but this can be improved with newer instrumentation and eliminated with MS3 quantification [33,34].
Most of the investigations discussed here utilize AP-MS and have largely been separated into two categories: (1) proteostasis network-centric investigations focused on the topological arrangement and functions of ER-PN components with one another, and (2) client-centric investigations focused on the coordination and engagement of ER-PN components with specific client proteins routed through the ER.
Proteostasis Network-Centric Investigations
In this review, we chose to highlight three subsets of proteostasis-centric investigations: chaperone-assisted protein folding, protein degradation, and UPR activation. These categories are not all-encompassing and could easily be broken down into further subcategories.
ER chaperoning complexes
Chaperone/co-chaperone systems and other protein folding complexes are essential to maintaining ER proteostasis. The Hsp70/40 chaperone system and the calreticulin/calnexin cycle are perhaps the most notable examples [6,10]. In the ER, the lone Hsp70 BiP/GRP78 facilitates protein folding and relies on interactions with many Hsp40/J-Domain containing proteins that can directly bind clients to recruit BiP for protein folding, recruit other factors to aid in ERAD, and additionally regulate the ATPase activity of BiP [5,35,36]. In the case of glycoproteins, the ER lectins calreticulin and calnexin coordinate with the protein disulfide isomerase PDIA3/ERp57 and glucosyltransferase UGGT1, along with other glycan modifying enzymes to facilitate glycoprotein folding [10]. AP-MS has aided in the identification of such interactions and complexes.
An early study by Meunier et al. found ER-PN subcomplexes that were hypothesized to be responsible for the processing of ER clients [37]. AP-MS led to the identification of a number of ER components including UGGT1, ERdj3, PPIB, SDF2L1, GRP94, CALR, PDIA, PDIA3, PDIA6, besides BiP, which had been previously identified. Further characterization of preformed ER-PN complexes detailed novel chaperone engagement and rearrangement, for instance showing that BiP, GRP94, ERdj3, and GRP170/HYOU1 could exist as large complexes even in the absence of a client. Additionally, the authors identified a BiP-associated complex including GRP94, GRP170, PDIA6, PPIB, and SDF2L1. Ultimately, this study provided new insight detailing that the formation of ER complexes can concentrate chaperones and folding components onto newly synthesized clients during folding. This was the first investigation to provide evidence for the existence of ER subnetworks from which client proteins are shuttled from one complex to another to facilitate folding.
In subsequent years, a number of studies on global protein-protein interactions in yeast provided further discoveries into ER-PN organization [38,39]. In 2009, Gong et al. published a study of the global landscape of the yeast chaperone network – including all 63 known chaperones [40]. This dataset included a mixture of AP-MS and the Saccharomyces Genome Database (SGD) data. As the authors characterized ER chaperones (human homologs denoted in parentheses throughout the rest of the paper) including, ERj5 (ERdj5/DNAJC10), Hlj1 (DNAJB4), Jem1, Sec63, Scj1 (DNAJC11), Kar2 (BiP), and Lhs1 (HYOU1), revealing key ER-PN components and chaperone organization. All identified ER components were found to interact with various cytoplasmic, nuclear, and mitochondrial components. This organelle crosstalk included interactions across organelle-specific Hsp70/40 systems, along with specific ER Hsp40 to cytosolic Hsp104s and prefoldin proteins. Additionally, the “promiscuity” of given proteostasis components as measured by the number of clients they could bind, revealed ER chaperones to be the least promiscuous (most specific), as compared to PN components found in other organelles. Furthermore, the authors detailed how Scj1/DNAJC11 may play a key role in ER to cytoplasmic and nuclear chaperone communication. On the whole, this work described unseen connectivity between ER chaperone networks and other organelles.
Subsequently, the ER chaperome of mammalian cells was further detailed through a combinatorial approach termed ER membrane yeast two-hybrid system and affinity purification (ER-MAP) [41]. Rather than utilizing antibodies that recognized particular ER-PN components, the authors used a unique method of directly functionalizing sepharose matrix with BiP, PDIA5, PDIA1, ERp29, ERp72, FKBP13, PPIB, PDIA3, or EDEM1–3 for subsequent AP-MS of ER complexes. They identified subnetworks of interactions within the ER across protein folding classes including BiP with peptidyl prolyl isomerases (PPIs) and protein disulfide isomerases (PDIs), further highlighting the role of BiP as a hub for ER multiprotein complexes [35]. Additionally, ER degradation enhancing alpha mannosidase like proteins (EDEMs) were found to interact with PDIs and other oxidative folding components. Particularly, PDI and PPI interactions were specific and formed exclusive pairs. This was especially the case for ERp72 and PPIB. Further work revealed the concerted ability of ERp72 and PPIB interactions to increase the rate of immunoglobulin IgG processing in vitro. These results were particularly revealing as disulfide formation and prolyl isomerization are thought to be the rate-limiting steps of many ER clients [42,43]. Overall the findings uncovered a diverse ER-PN functional landscape and a number of novel interactions between proteostasis components. The concerted effort of these subnetworks can increase protein folding efficiency, further corroborating results and speculations proposed by Meunier et al. [37].
ER membrane complex and transmembrane proteins
With many transmembrane proteins being implicated in protein folding disease states, for instance cystic fibrosis transmembrane conductance regulator (CFTR), potassium voltage-gated channels, GABAA receptors, and Niemann-Pick disease C1 protein (NPC1), elucidating specific ER-PN and mechanisms responsible for folding of such clients has been a key research area [44]. One complex responsible for the processing of transmembrane clients is the ER membrane protein complex (EMC). Since its identification in 2009, multiple investigations have characterized the importance of the EMC [45–48], yet many questions still remained as to whether the EMC played a direct or indirect role in membrane protein biogenesis, what substrates it acted on, and what role the EMC played during client insertion into the membrane. In 2018, a seminal study by Shutleff et al. tackled these questions [32]. Utilizing SILAC and AP-MS for comparative interactome screening of EMC3, an EMC subunit, the authors identified specialized yeast membrane protein chaperones Sop4 (EMC7) and Gsf2 (EMC6), along with other oxidative folding components, Ero1, and general chaperones Ssa1, Ssb1 (both Hsp70 homologs), and Kar2. A series of knock-out experiments of membrane chaperones Ilm1 and Sop4 revealed the EMC could directly bind transmembrane clients to aid in folding. With interactions between transmembrane clients, membrane protein-specific chaperones, general chaperones, and the ribosome itself, the EMC acts as a hub for transmembrane protein folding and processing within the ER. Furthermore, using BioID, the authors found that the EMC can interact cotranslationally with clients, showing a specificity for difficult to fold transmembrane proteins such as transporters and ion channels with charged residues within the lipid bilayer. These findings also held true in mammalian cells, ultimately establishing foundational principles for the action of the EMC. Mass spectrometry experiments in further studies were critical for determining that the EMC is needed for membrane insertion and PQC of diverse transmembrane proteins, for instance connexin 32 involved in Charcot-Marie Tooth disease and sterol homeostasis enzymes [49,50].
ER-associated degradation (ERAD) and ER-phagy
If folding and trafficking of ER clients fails, degradation of misfolded or damaged clients plays an equally important role in maintaining ER proteostasis [12,14,51]. Additionally, turnover of the ER itself plays a key function in ER homeostasis, acting to remove aggregated clients and damaged ER regions, or alter the size and shape of the ER during stress and recovery [13,14,52]. Much work has been dedicated to elucidating PQC mechanisms associated with the two major degradation pathways associated with the ER: ERAD and ER-phagy.
In 2006, Carvalho et al. characterized a new ERAD pathway for integral membrane proteins, termed ERAD-M [53]. Pathways facilitating ERAD of luminal, or membrane clients with misfolded or damaged cytosolic domains, termed ERAD-L and ERAD-C respectively, were previously defined [54,55]. While it was clear that different ERAD substrates employed differing routes of degradation, the organization of ERAD machinery within those pathways and whether clients with misfolded transmembrane domains used the same or a different set of ERAD pathways remained unclear. By characterizing the interactomes of yeast ubiquitin ligases involved in ERAD-C and ERAD-L, Doa10 (MARCHF6), and Hrd1 (HRD1) respectively, the authors defined the complexes implicated in the given ERAD pathways [53]. The Doa10 complex proved to be relatively simple including Ubc7(UBE2G2), Cdc48 (VCP), Npl4 (NPLOC4), and Ubx2 (FAF2). Hrd1, Hrd3 (SEL1L), Der1 (DER1), and a novel complex component, Usa1 (HERP), formed the majority of the Hrd1 complex. Accessory components including Yos9 (OS-9), Ubx2, and Cdc48, highlighted some overlapping complex components between ERAD-C and ERAD-L pathways. Further characterization revealed that the Hrd1 complex was used to facilitate ERAD-M [53]. Additional work by Christianson, Olzman, et al. meticulously detailed these ERAD complexes in mammalian cells to gain further insight into ERAD subnetworks [56]. The authors defined the major ERAD subnetworks, including SEL1L-HRD1, Gp78, the E3 ubiquitin ligase-26S proteasome, and provided the first identification and characterization of the mammalian EMC. Using model ERAD-L, ERAD-C, and ERAD-M substrates, they were able to detail how these subnetworks utilized adaptive mechanisms to facilitate degradation on a client-specific basis. A recent comprehensive interactome study of 25 poorly characterized ER transmembrane E3 ubiquitin ligases and associated complexes revealed new insights into the role of RNF26 in controlling STING levels and associated innate immune signaling pathways [57].
Since it was first characterized, ER-phagy/Reticulophagy (a contraction of the endoplasmic reticulum and macroautophagy) has proven to play a key role in ER protein homeostasis and ER stress [52,58,59]. As such, key regulators and mechanisms of this process have been further studied, and three of the five ER-phagy receptors have been identified and characterized using AP-MS. Grumati et al. performed a comparative interactome screen of reticulon related proteins (RTN1–4) thought to play a role in ER-phagy by recruiting cargo into autophagosome structures [60]. Preliminary work confirmed RTN3 was necessary for ER fragmentation and delivery to the lysosome. The authors found that this class of proteins shared the majority of interacting proteins, yet the full-length isoform of RTN3 was the only one found to act as an autophagy receptor by interacting with GABARAP-L1, an essential protein for autophagosome maturation. Subsequently, Smith et al. used a similar approach and defined the novel interaction of a previously uncharacterized protein, CCPG1, with GABARAP [61]. The authors defined the CCPG1 interactome, leading to the identification of a robust interaction with an additional autophagy component, FIP200. Further characterization showed that CCPG1 expression was regulated by the UPR and drove ER-phagy via distinct interactions with GABARAP and FIP200. In 2019, Chino et al. discovered and characterized the most recently identified ER-phagy receptor, TEX264 [62]. Similarly, using comparative interactome screening of LC3B – another protein involved in autophagosome formation, the authors identified TEX264 as a unique interactor displaying high specificity yet no known role in autophagy. Follow up work characterized the TEX264-LC3B interaction and led to the identification of TEX264 as a major ER-phagy receptor. In two of the three cases presented here, CCPG1 and TEX264, the ER-phagy receptors were either poorly annotated or had no assigned function, highlighting the ability of AP-MS to not only define protein-protein interactions and mechanisms of PQC but discover new or previously unappreciated biological function of those proteins.
Adapting ER proteostasis through the Unfolded Protein Response
As the dysregulation of the UPR is implicated in a number of disease states and has even emerged as a drug target to combat such diseases, characterizing the regulation and consequences of UPR activation has become major topic of ER PQC investigations [17,63,64]. In particular, studies have determined how UPR activation coordinates the upregulation of ER proteostasis factors to influence PQC mechanisms, and furthermore how the modulation of the UPR can be used to combat such protein folding diseases.
Shoulders et al. developed a cell line where chemical genetics tools enabled preferential activation of the ATF6 and IRE1/XBP1s arms of the UPR in isolation or in combination [Fig. 2], independent of ER stress [31]. This allowed for the characterization of changes to the ER-PN composition using SILAC-based quantitative proteomics, revealing distinct, and overlapping gene products of each UPR arm that are used to remodel the ER-PN. Particularly, XBP1s activation provided a more concerted impact on many ER proteostasis pathways. This is in line with IRE1-XBP1s signaling being the single UPR arm conserved from yeast to humans [17,19]. On the contrary, ATF6 specifically upregulated key proteostasis factors in distinct pathways, such as the Hsp70 and Hsp90 chaperones BiP and GRP94, disulfide isomerase PDIA4, as well as several degradation factors. At the same time, several other degradation pathways were cooperatively upregulated through both XBP1s and ATF6 arms.
AP-MS studies on the UPR sensors has produced insights into the mechanisms of regulation during ER stress adaptation. For instance, an IRE1 interactome revealed the E3 ubiquitin ligase CHIP to be a key interactor demonstrating that CHIP-mediated K63-linked ubiquitination plays a role in the UPR regulation [65]. This modification was discovered to play a critical role in controlling the pro-apoptotic activation of the TRAF2/JNK pathway, and is significantly increased under ER stress [66]. As IRE1 undergoes increased phosphorylation during UPR induction, ubiquitination of IRE1 is similarly elevated. Increased signaling of the IRE1/TRAF2/JNK pathway antagonized cellular senescence. These findings revealed another connection between ER stress and UPR activation to aging [67]. Sepulveda et al. conducted a complementary interactome screening of IRE1 under basal and ER stress conditions to investigate regulatory mechanisms of UPR activation [68]. In this study, the collagen-specific chaperone Hsp47/SERPINH1 stood out as an interactor that potentiated the activation of IRE1. Specifically, the authors found that Hsp47 and BiP, often thought of as the major regulator of the UPR, bound to IRE1 with the same affinity [69]. This regulation by Hsp47 held true in D. melanogaster and mouse models of ER stress. The authors proposed a model whereby Hsp47 competes with BiP during UPR activation titrating BiP off from the luminal domain of IRE1 further tuning the IRE1 response of the UPR [68].
Another investigation probed the role ubiquitination plays in the regulation of translational machinery during ER stress and UPR activation [70]. Higgins, Gendron et al. used quantitative diGly proteomics to characterize the ubiquitin modified proteome (ubiquitylome) under conditions of the ER stressors DTT, tunicamycin, and epoxomicin. The authors found that diGly peptides corresponded with proteins enriched in functions related to mRNA translation and were localized to the cytosolic ribosome. Time-course analysis revealed ubiquitylome remodeling of the 40S ribosome as an early UPR event necessary to inhibit protein translation. Furthermore, authors showed that PERK and eIF2α phosphorylation were necessary but not sufficient to mount UPR-induced regulatory ubiquitination of the 40S ribosome. Insults to this process resulted in elevated sensitivity to UPR-induced cell death, with this regulatory mechanism holding true in D. melanogaster and yeast as well.
Client-Centric Investigations
While many investigations have focused on elucidating mechanisms of PQC by investigating subnetworks of the ER-PN, these investigations do not provide an integrated view of how these subnetworks converge to perform the concurrent folding, processing, secretion and degradation of clients routed through the secretory pathway. To address these questions, MS- based studies have also focused on investigating mechanisms of PQC for protein clients, in particular ones involved in protein misfolding and aggregation disease. These client-centric investigations provide a holistic view of client processing by integrating ER-PN subnetworks and have aided in the development of therapeutic treatment strategies for disease states.
Loss-of-function protein misfolding diseases
Cystic fibrosis (CF) is one of the most prevalent protein folding diseases caused by loss of function of the chloride ion channel CFTR, responsible for salt homeostasis in polarized epithelial cells [71]. This results in an inability to clear mucus from the respiratory tract and can lead to chronic infection, respiratory failure, and ultimately death. The most prevalent CFTR mutations in CF patients, deletion of phenylalanine 508 (∆F508), results in improper processing of this ion channel [72]. While ∆F508 CFTR can be functional as an ion channel if trafficked to the plasma membrane, improper folding, and rapid degradation within the secretory pathway result in a loss of expression at the cell surface. To investigate the molecular mechanisms associated with CFTR processing, Pankow et al. detailed the interactomes of WT and ∆F508 CFTR [29,73]. Using a comparative AP-MS based interactome analysis, the authors identified 638 total high confidence interactors with 576 and 430 for ∆F508 and WT CFTR respectively. While some interactions between the two proteins remained the same, the relative abundance of those interactions significantly differed for a number of them. Moreover, 208 and 62 interactors were found to bind exclusively with ∆F508 and WT CFTR respectively, highlighting gain of novel interactions as a hallmark of ∆F508 misprocessing. Furthermore, well-established correction treatment that can restore CFTR biogenesis and trafficking (either by lowering cell culture temperate to 26–30°C, or addition of histone deacetylase inhibitor SAHA) correlated with the abolishment of mutant-specific proteostasis network interactions [74,75]. Many of these interactions were associated with ERAD and heat-shock assisted protein folding, among others. The authors went on to further detail that inhibition of key interactors associated with ER retention, trafficking, and degradation promoted the maturation of ∆F508 CFTR. Ultimately this study resulted in a comprehensive mapping of the WT and ∆F508 CFTR interactomes, provided some of the first insight into the molecular mechanisms of temperature correction for ∆F508 CFTR, and generally detailed disease-specific alterations that take place and may be implicated in other protein misfolding diseases. Follow-up interactomics studies on additional CFTR mutant variants and clinically-approved small molecular correctors and potentiators, VX-809 and VX-770 respectively, have expanded our understanding of the role of proteostasis dysregulation in CF [76]. These studies have established a methodology that can be further expanded to other genetic disorders to implement precision medicine-based approaches for clinical treatment of such diseases.
Collagen proteinopathies such as osteogenesis imperfecta (IO) and Ehlers-Danlos syndrome are caused by divergent missense mutations in collagen that lead to misprocessing and improper protein secretion resulting in weakened connective tissue [77]. AP-MS of collagen I expressed in fibrosarcoma cells was used to define the collagen I PN interactome [78]. Comparative SILAC-based quantitative interactomics identified the non-canonical protein disulfide isomerase ERp29 as a crucial factor promoting collagen retention under ascorbate-limited conditions, a critical cofactor required for post-translational modifications on collagen [79]. A recent follow-up study characterized the PQC defects of the C1163R variant in the C-pro trimerization domain of collagen α2(I) associated with IO resulting in complete loss of secretion [80]. Comparative studies uncovered increased associations with a broad set of proteostasis factors, including BiP, GRP94, co-chaperones, and several PDIs that pointed to a disruption of disulfide network formation and improper targeting of the collagen mutant to ERAD by Hsp70/40 chaperone networks.
Congenital hypothyroidism (CH) can arise from a number of different mutations in genes involved in thyroid hormone biosynthesis. One such gene is thyroglobulin (Tg), the secreted iodoglycoprotein which serves as the precursor for thyroid hormone production. Mutations in the Tg gene can lead to a loss in secretion, ultimately resulting in a decrease or complete loss of thyroid hormone production. Most recently, our lab used a TMT-based multiplexed comparative interactome workflow to elucidate the molecular basis of mutant Tg misprocessing. Common imbalances such as increased chaperoning, oxidative folding, and engagement by ERAD targeting factors were found to be associated with CH-mutant Tg processing. We further characterized mutation specific changes in engagement with the oligosaccharyltransferase (OST) complex that suggest a distinct role for mutant Tg degradation [81].
Protein aggregation diseases
AP-MS studies have also shed light on ER proteostasis defects that promote secretion of gain-of-toxic function protein variants involved in protein aggregation disease, for instance interactions and PQC mechanisms implicated in the processing of amyloidogenic immunoglobulin light chain (LC) protein responsible for light-chain amyloidosis (AL) [30]. Secretion of destabilized LC can result in the formation of toxic extracellular aggregates leading to systemic organ damage in AL, in particular cardiac amyloidosis [82]. Previous studies showed that selective, stress-independent activation of the ATF6 branch of the UPR preferentially reduced the secretion of destabilized LC (ALLC) while leaving a non-amyloidogenic LC and intact IgGs unscathed [83]. Yet, the molecular mechanisms associated with this selective PQC enhancement were not well characterized. By comparing the proteostasis interactomes of ALLC in the presence of ATF6 activation, XBP1s activation, or both, the authors were able to identify key interactions and thus deduce mechanisms responsible for the decreased secretion of the aggregation prone ALLC. XBP1s activation decreased overall ALLC engagement of ER protein folding components, yet increased routing towards degradation, and only modestly reduced secretion. Conversely, ATF6 activation resulted in increased engagement with a subset of ER proteostasis components with ALLC, particularly BiP, GRP94, ERdj3, HYOU1, and PDIA4. The authors further showed that ATF6 activation promoted increased engagement of these ER-PN components with non-amyloidogenic LC, which suggested these increased interactions are independent of client stability. Overexpression of these key ER-PN components showed that the reduction in ALLC secretion is coordinated by ATF6-mediated activation of a cooperative set of proteostasis factors, as overexpression of these individual ER-PN components only partially mimicked the reduction in ALLC secretion found with ATF6 activation [30]. This work highlighted the targeting of proteostasis pathways as a therapeutic intervention for such protein folding and amyloidogenic diseases as it established the mechanism of ATF6-dependent reduction of ALLC secretion [17,83,84].
Viral proteostasis clients
While many client-centric investigations have focused on protein folding diseases, a number of studies investigating viral host-pathogen interactions involving the ER-PN have also emerged. Hafirassou et al. mapped the interactome of the dengue virus (DENV) non-structural protein 1 (NS1) [85]. DENV, like other flaviviruses, uses the ER membrane for replication by forming invaginations to assemble the replication complex (RC) [86]. NS1 plays a variety of roles during DENV infection, including acting as a cofactor during DENV replication. Furthermore, it is the only viral non-structural protein secreted from infected host-cells, and extracellular NS1 disrupts the glycocalyx of endothelial cells increasing vascular leakage, which is the hallmark of Dengue hemorrhagic fever [87]. Yet, the host factors involved in viral replication and secretion of NS1 were not well characterized. The authors mapped 499, 654, and 438 host-NS1 interactions across Daji, HeLa, and HAP1 cells respectively and found 270 host factors to interact across all three cell lines [85]. The NS1 interactome was enriched in ER components involved in ERAD including SEL1L, AUP1, HM13, and the large majority of the OST complex. Further investigation into these host components allowed the authors to delineate host restriction factors found to combat viral production, including ER residents NOMO1, NCLN, NCSTN, PGRMC1, MLEC, and RCN1 and host dependency factors found to promote viral production including Sec63, Sec61A1 and other ER translation and translocation components, STT3A, STT3B, DDOST, and other N-glycosylation components, along with ERAD components previously mentioned. Follow up investigations revealed that glycosylation of NS1 by the OST complex is critical for the folding and stability of the protein, as inhibition of OST complex resulted in decreased DENV infection [85,88].
A different study by Coyaud et al. used two complementary approaches, AP-MS and BioID to map the Zika virus (ZIKV) interactome [89]. Expressing all 10 ZIKV proteins, the authors found that, similar to the DENV interactome, interacting partners were enriched in ER components. Major ER-localized proteins included members of the signal peptidase complex (SPC), EMC, and the BAT3 complex. The authors also found that ER protein folding components prolyl 4-hydroxylase subunit B (P4HB), the lectin calnexin (CANX), along with the ER-phagy receptor FAM134B localized into in these capsid protein-associated structures. Ultimately, these findings revealed that the capsid plays a diverse role in remodeling ER membranes. Additionally, NS3 was found to associate with the ubiquitin proteasome system (UPS), the degradation system responsible for clearing a number of proteins from the cell, including ER clients [14]. Overall, 7 of the 10 expressed ZIKV proteins localized or associated with the ER in some way, highlighting their involvement in forming virus-induced membrane structures used for RC formation.
A tour-de-force comparative AP-MS study mapped the interactomes of DENV and ZIKV flavivirus proteins across human and mosquito cell lines [90]. This was the first study to perform a systematic comparison of flavivirus-host interactions in primate and vector host cells. In line with previous studies, the authors found that viral proteins interacted with host components associated with the ER. Specifically, translocon and calreticulin cycle ER-PN components were found to have distinct interactions with DENV proteins. Additionally, p62, a key accessory protein required for the degradation of a number of substrates and organelles was also identified, potentially highlighting its involvement in DENV protein turnover, or turnover of the ER itself during DENV infection. Investigations into ZIKV in human and mosquito cells allowed the authors to identify key components across flavivirus strains and cell types that may be viable host-centric therapeutic targets. OST subunits STT3A, STT3B and RPN2 as well as HYOU1 and trafficking component interactions were conserved across DENV and ZIKV and the different cell types. The authors identified the Sec61 translocon and signal recognition particle receptor (SRPR) as the most conserved interactors across all flavivirus strains and cell types, and revealed that cotranslational protein translocation inhibitors nearly abolished viral production, even when cells were treated post-infection. Furthermore, inhibiting protein translocation did not affect cell viability. This suggested that inhibition of the Sec61 translocon may serve as a viable anti-viral target. This work highlighted distinct and conserved host-pathogen interactions across multiple flavivirus species and strains and host cell types, furthermore highlighting the ability of AP-MS to not only identify interactions with ER components but identify potential therapeutic targets. Recently, a similar comprehensive AP-MS interactome characterization of 26 SARS-CoV-2 viral proteins revealed important leads for repurposing existing and approved therapeutics that target the identified host cell interactors for COVID-19 treatment [91].
Future Perspectives
Mass spectrometry has clearly proven to be a valuable tool to biological investigations of protein-protein interactions, and as we presented here its impact on elucidating particularly mechanisms of ER stress and PQC. As technology and methodology continues to advance, the scope of mass spectrometry will expand to yield deeper insights into these biological processes., Many underexplored areas of ER stress and PQC still remain, where mass spectrometry would be a well-suited method to make further discoveries. For example, in many of the cases presented here the added complexity of how PTMs on clients and ER-PN components influence mechanisms of PQC are not fully characterized. As a follow up study to the CFTR interactome profiling, Pankow et al. showed that a PTM code on CFTR is altered for ∆F508 CFTR and this alteration correlated with an inability to properly fold and localize to the plasma membrane [92]. The authors further speculated that similar codes may be present in other transmembrane proteins. PTMs have also been implicated in the stability and activity of ER proteostasis components including BiP and PDI. N-terminal arginylation promotes the degradation of BiP via autophagy through p62 binding [93]. Additionally, AMPylation of BiP plays a key role in its chaperoning activity, particularly during ER stress [94]. A number of other BiP modifications, including, phosphorylation, acetylation, ADP-ribosylation, methylation, and more have been mapped and provide insight into BiP activity and subsequent proteostasis mechanisms [95]. A similar regulatory mechanism was recently documented for PDI as it was found that phosphorylation alters PDI oxido-reductase activity to become a “holdase” to attenuate ER stress [96]. How these PTMs on individual proteostasis components regulate the network of proteostasis subcomplexes and their coordination to mediate proper PQC have yet to be realized.
One limitation that current mass spectrometry-based methods have when identifying proteostasis interactions, as well as PTMs, is that interactions in the ER are very dynamic and highly transient in nature and identifying these interactions and modifications using current methods only provides a snapshot of the state of the ER at a given time. Future methods for elucidation of these protein-protein interactions or PTMs in a time-dependent manner would allow for a more complete understanding of ER stress and PQC mechanisms, illuminating previously unseen or poorly understood mechanisms [97].
Acknowledgements
We thank the following funding agencies for support: National Institute of General Medical Sciences (R35 GM133552 – L.P., T32 GM065086 – M.T.W.) and the National Science Foundation Graduate Research Fellowship Program (M.T.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Braakman I, Hebert DN, Protein Folding in the Endoplasmic Reticulum, Cold Spring Harb. Perspect. Biol 5 (2013) a013201–a013201. 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Balchin D, Hayer-Hartl M, Hartl FU, In vivo aspects of protein folding and quality control, Science (80-.) 353 (2016) aac4354. 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- [3].Mandon EC, Trueman SF, Gilmore R, Protein Translocation across the Rough Endoplasmic Reticulum, Cold Spring Harb. Perspect. Biol 5 (2013) a013342–a013342. 10.1101/cshperspect.a013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Voorhees RM, Hegde RS, Structure of the Sec61 channel opened by a signal sequence, Science (80-.) 351 (2016) 88–91. 10.1126/science.aad4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vembar SS, Jonikas MC, Hendershot LM, Weissman JS, Brodsky JL, J domain co-chaperone specificity defines the role of BiP during protein translocation, J. Biol. Chem (2010). 10.1074/jbc.M110.102186. [DOI] [PMC free article] [PubMed]
- [6].Behnke J, Mann MJ, Scruggs F-L, Feige MJ, Hendershot LM, Members of the Hsp70 Family Recognize Distinct Types of Sequences to Execute ER Quality Control, Mol. Cell 63 (2016) 739–752. 10.1016/j.molcel.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feige MJ, Hendershot LM, Quality Control of Integral Membrane Proteins by Assembly-Dependent Membrane Integration, Mol. Cell 51 (2013) 297–309. 10.1016/j.molcel.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ruiz-Canada C, Kelleher DJ, Gilmore R, Cotranslational and Posttranslational N-Glycosylation of Polypeptides by Distinct Mammalian OST Isoforms, Cell (2009). 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed]
- [9].Braunger K, Pfeffer S, Shrimal S, Gilmore R, Berninghausen O, Mandon EC, Becker T, Förster F, Beckmann R, Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum, Science (80-.) 360 (2018) 215–219. 10.1126/science.aar7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lamriben L, Graham JB, Adams BM, Hebert DN, N-Glycan-based ER Molecular Chaperone and Protein Quality Control System: The Calnexin Binding Cycle, Traffic 17 (2016) 308–326. 10.1111/tra.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gomez-Navarro N, Miller E, Protein sorting at the ER–Golgi interface, J. Cell Biol 215 (2016) 769–778. 10.1083/jcb.201610031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Needham PG, Guerriero CJ, Brodsky JL, Chaperoning Endoplasmic Reticulum– Associated Degradation (ERAD) and Protein Conformational Diseases, Cold Spring Harb. Perspect. Biol 11 (2019) a033928. 10.1101/cshperspect.a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schuck S, Gallagher CM, Walter P, ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery, J. Cell Sci 127 (2014) 4078–4088. 10.1242/jcs.154716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pohl C, Dikic I, Cellular quality control by the ubiquitin-proteasome system and autophagy, Science (80-.) 366 (2019) 818–822. 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- [15].Knowles TPJ, Vendruscolo M, Dobson CM, The amyloid state and its association with protein misfolding diseases, Nat. Rev. Mol. Cell Biol 15 (2014) 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- [16].Karagöz GE, Acosta-Alvear D, Walter P, The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum, Cold Spring Harb. Perspect. Biol 11 (2019) a033886. 10.1101/cshperspect.a033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Plate L, Wiseman RL, Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy, Trends Cell Biol 27 (2017) 722–737. 10.1016/j.tcb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Grandjean JMD, Plate L, Morimoto RI, Bollong MJ, Powers ET, Wiseman RL, Deconvoluting Stress-Responsive Proteostasis Signaling Pathways for Pharmacologic Activation Using Targeted RNA Sequencing, ACS Chem. Biol 14 (2019) 784–795. 10.1021/acschembio.9b00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Schneider K, Bertolotti A, Surviving protein quality control catastrophes - from cells to organisms, J. Cell Sci 128 (2015) 3861–3869. 10.1242/jcs.173047. [DOI] [PubMed] [Google Scholar]
- [20].Cravatt BF, Simon GM, Yates JR III, The biological impact of mass-spectrometry-based proteomics, Nature 450 (2007) 991–1000. 10.1038/nature06525. [DOI] [PubMed] [Google Scholar]
- [21].Bludau I, Aebersold R, Proteomic and interactomic insights into the molecular basis of cell functional diversity, Nat. Rev. Mol. Cell Biol (2020). 10.1038/s41580-020-0231-2. [DOI] [PubMed]
- [22].Bian Y, Zheng R, Bayer FP, Wong C, Chang Y-C, Meng C, Zolg DP, Reinecke M, Zecha J, Wiechmann S, Heinzlmeir S, Scherr J, Hemmer B, Baynham M, Gingras A-C, Boychenko O, Kuster B, Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC–MS/MS, Nat. Commun 11 (2020) 157. 10.1038/s41467-019-13973-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rauniyar N, Yates JR, Isobaric Labeling-Based Relative Quantification in Shotgun Proteomics, J. Proteome Res 13 (2014) 5293–5309. 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R, Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system, Nat. Methods 10 (2013) 1246–1253. 10.1038/nmeth.2703. [DOI] [PubMed] [Google Scholar]
- [25].Samavarchi-Tehrani P, Samson R, Gingras A-C, Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches, Mol. Cell. Proteomics 19 (2020) 757–773. 10.1074/mcp.R120.001941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Han S, Li J, Ting AY, Proximity labeling: spatially resolved proteomic mapping for neurobiology, Curr. Opin. Neurobiol 50 (2018) 17–23. 10.1016/j.conb.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Leitner A, Faini M, Stengel F, Aebersold R, Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines, Trends Biochem. Sci 41 (2016) 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- [28].Kao A, Chiu C, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L, Development of a Novel Cross-linking Strategy for Fast and Accurate Identification of Cross-linked Peptides of Protein Complexes, Mol. Cell. Proteomics 10 (2011) M110.002212. 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pankow S, Bamberger C, Calzolari D, Martínez-Bartolomé S, Lavallée-Adam M, Balch WE, Yates JR, ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis, Nature 528 (2015) 510–516. 10.1038/nature15729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Plate L, Rius B, Nguyen B, Genereux JC, Kelly JW, Wiseman RL, Quantitative Interactome Proteomics Reveals a Molecular Basis for ATF6-Dependent Regulation of a Destabilized Amyloidogenic Protein, Cell Chem. Biol 26 (2019) 913–925.e4. 10.1016/j.chembiol.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, Su AI, Kelly JW, Wiseman RL, Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments, Cell Rep 3 (2013) 1279–1292. 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shurtleff MJ, Itzhak DN, Hussmann JA, Schirle Oakdale NT, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P, Vembar SS, Hernandez H, Cox J, Burlingame AL, Brodsky JL, Frost A, Borner GHH, Weissman JS, The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins, Elife 7 (2018). 10.7554/eLife.37018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bantscheff M, Boesche M, Eberhard D, Matthieson T, Sweetman G, Kuster B, Robust and Sensitive iTRAQ Quantification on an LTQ Orbitrap Mass Spectrometer, Mol. Cell. Proteomics 7 (2008) 1702–1713. 10.1074/mcp.M800029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ting L, Rad R, Gygi SP, Haas W, MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics, Nat. Methods 8 (2011) 937–940. 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pobre KFR, Poet GJ, Hendershot LM, The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends, J. Biol. Chem 294 (2019) 2098–2108. 10.1074/jbc.REV118.002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Otero JH, Lizák B, Feige MJ, Hendershot LM, Dissection of Structural and Functional Requirements That Underlie the Interaction of ERdj3 Protein with Substrates in the Endoplasmic Reticulum, J. Biol. Chem 289 (2014) 27504–27512. 10.1074/jbc.M114.587147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Meunier L, Usherwood Y-K, Chung KT, Hendershot LM, A Subset of Chaperones and Folding Enzymes Form Multiprotein Complexes in Endoplasmic Reticulum to Bind Nascent Proteins, Mol. Biol. Cell 13 (2002) 4456–4469. 10.1091/mbc.e02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams S-L, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sørensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, V Hogue CW, Figeys D, Tyers M, Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry., Nature 415 (2002) 180–3. 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- [39].Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrín-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MHY, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF, Global landscape of protein complexes in the yeast Saccharomyces cerevisiae, Nature (2006). 10.1038/nature04670. [DOI] [PubMed]
- [40].Gong Y, Kakihara Y, Krogan N, Greenblatt J, Emili A, Zhang Z, Houry WA, An atlas of chaperone–protein interactions in Saccharomyces cerevisiae : implications to protein folding pathways in the cell, Mol. Syst. Biol 5 (2009) 275. 10.1038/msb.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jansen G, Määttänen P, Denisov AY, Scarffe L, Schade B, Balghi H, Dejgaard K, Chen LY, Muller WJ, Gehring K, Thomas DY, An Interaction Map of Endoplasmic Reticulum Chaperones and Foldases, Mol. Cell. Proteomics 11 (2012) 710–723. 10.1074/mcp.M111.016550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schmidpeter PAM, Schmid FX, Prolyl Isomerization and Its Catalysis in Protein Folding and Protein Function, J. Mol. Biol 427 (2015) 1609–1631. 10.1016/j.jmb.2015.01.023. [DOI] [PubMed] [Google Scholar]
- [43].Ushioda R, Nagata K, Redox-Mediated Regulatory Mechanisms of Endoplasmic Reticulum Homeostasis, Cold Spring Harb. Perspect. Biol 11 (2019) a033910. 10.1101/cshperspect.a033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Marinko JT, Huang H, Penn WD, Capra JA, Schlebach JP, Sanders CR, Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis, Chem. Rev 119 (2019) 5537–5606. 10.1021/acs.chemrev.8b00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M, Comprehensive Characterization of Genes Required for Protein Folding in the Endoplasmic Reticulum, Science (80-.) 323 (2009) 1693–1697. 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shen X, Kan S, Hu J, Li M, Lu G, Zhang M, Zhang S, Hou Y, Chen Y, Bai Y, EMC6/TMEM93 suppresses glioblastoma proliferation by modulating autophagy, Cell Death Dis (2016). 10.1038/cddis.2015.408. [DOI] [PMC free article] [PubMed]
- [47].Lahiri S, Chao JT, Tavassoli S, Wong AKO, Choudhary V, Young BP, Loewen CJR, Prinz WA, A Conserved Endoplasmic Reticulum Membrane Protein Complex (EMC) Facilitates Phospholipid Transfer from the ER to Mitochondria, PLoS Biol 12 (2014) e1001969. 10.1371/journal.pbio.1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Guna A, Volkmar N, Christianson JC, Hegde RS, The ER membrane protein complex is a transmembrane domain insertase, Science (80-.) 359 (2018) 470–473. 10.1126/science.aao3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Coelho JPL, Stahl M, Bloemeke N, Meighen-Berger K, Alvira CP, Zhang Z-R, Sieber SA, Feige MJ, A network of chaperones prevents and detects failures in membrane protein lipid bilayer integration, Nat. Commun 10 (2019) 672. 10.1038/s41467-019-08632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Volkmar N, Thezenas M-L, Louie SM, Juszkiewicz S, Nomura DK, Hegde RS, Kessler BM, Christianson JC, The ER membrane protein complex promotes biogenesis of sterol-related enzymes maintaining cholesterol homeostasis, J. Cell Sci 132 (2019) jcs223453. 10.1242/jcs.223453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oikonomou C, Hendershot LM, Disposing of misfolded ER proteins: A troubled substrate’s way out of the ER, Mol. Cell. Endocrinol 500 (2020) 110630. 10.1016/j.mce.2019.110630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Soldà T, D’Antuono R, Raimondi A, Jung M, Melnyk A, Schorr S, Schreiber A, Simonelli L, Varani L, Wilson-Zbinden C, Zerbe O, Hofmann K, Peter M, Quadroni M, Zimmermann R, Molinari M, Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery, Nat. Cell Biol 18 (2016) 1173–1184. 10.1038/ncb3423. [DOI] [PubMed] [Google Scholar]
- [53].Carvalho P, Goder V, Rapoport TA, Distinct Ubiquitin-Ligase Complexes Define Convergent Pathways for the Degradation of ER Proteins, Cell 126 (2006) 361–373. 10.1016/j.cell.2006.05.043. [DOI] [PubMed] [Google Scholar]
- [54].Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY, Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation, Nat. Cell Biol 3 (2001) 24–29. 10.1038/35050524. [DOI] [PubMed] [Google Scholar]
- [55].Swanson R, A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha 2 repressor degradation, Genes Dev 15 (2001) 2660–2674. 10.1101/gad.933301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Wade Harper J, Kopito RR, Defining human ERAD networks through an integrative mapping strategy, Nat. Cell Biol 14 (2012) 93–105. 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fenech EJ, Lari F, Charles PD, Fischer R, Laétitia-Thézénas M, Bagola K, Paton AW, Paton JC, Gyrd-Hansen M, Kessler BM, Christianson JC, Interaction mapping of endoplasmic reticulum ubiquitin ligases identifies modulators of innate immune signalling, Elife 9 (2020). 10.7554/eLife.57306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Bolender RP, Weibel ER, A MORPHOMETRIC STUDY OF THE REMOVAL OF PHENOBARBITAL-INDUCED MEMBRANES FROM HEPATOCYTES AFTER CESSATION OF TREATMENT, J. Cell Biol 56 (1973) 746–761. 10.1083/jcb.56.3.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Bernales S, McDonald KL, Walter P, Autophagy Counterbalances Endoplasmic Reticulum Expansion during the Unfolded Protein Response, PLoS Biol 4 (2006) e423. 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Grumati P, Morozzi G, Hölper S, Mari M, Harwardt M-LI, Yan R, Müller S, Reggiori F, Heilemann M, Dikic I, Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy, Elife 6 (2017). 10.7554/eLife.25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, Wilkinson S, CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis, Dev. Cell 44 (2018) 217–232.e11. 10.1016/j.devcel.2017.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chino H, Hatta T, Natsume T, Mizushima N, Intrinsically Disordered Protein TEX264 Mediates ER-phagy, Mol. Cell 74 (2019) 909–921.e6. 10.1016/j.molcel.2019.03.033. [DOI] [PubMed] [Google Scholar]
- [63].Kelly JW, Pharmacologic Approaches for Adapting Proteostasis in the Secretory Pathway to Ameliorate Protein Conformational Diseases, Cold Spring Harb. Perspect. Biol (2019) a034108. 10.1101/cshperspect.a034108. [DOI] [PMC free article] [PubMed]
- [64].Hetz C, Axten JM, Patterson JB, Pharmacological targeting of the unfolded protein response for disease intervention, Nat. Chem. Biol 15 (2019) 764–775. 10.1038/s41589-019-0326-2. [DOI] [PubMed] [Google Scholar]
- [65].Zhu X, Zhang J, Sun H, Jiang C, Dong Y, Shan Q, Su S, Xie Y, Xu N, Lou X, Liu S, Ubiquitination of Inositol-requiring Enzyme 1 (IRE1) by the E3 Ligase CHIP Mediates the IRE1/TRAF2/JNK Pathway, J. Biol. Chem 289 (2014) 30567–30577. 10.1074/jbc.M114.562868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Urano F, Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1, Science (80-.) 287 (2000) 664–666. 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- [67].Kaushik S, Cuervo AM, Proteostasis and aging, Nat. Med 21 (2015) 1406–1415. 10.1038/nm.4001. [DOI] [PubMed] [Google Scholar]
- [68].Sepulveda D, Rojas-Rivera D, Rodríguez DA, Groenendyk J, Köhler A, Lebeaupin C, Ito S, Urra H, Carreras-Sureda A, Hazari Y, Vasseur-Cognet M, Ali MMU, Chevet E, Campos G, Godoy P, Vaisar T, Bailly-Maitre B, Nagata K, Michalak M, Sierralta J, Hetz C, Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1α, Mol. Cell 69 (2018) 238–252.e7. 10.1016/j.molcel.2017.12.028. [DOI] [PubMed] [Google Scholar]
- [69].Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D, Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response, Nat. Cell Biol 2 (2000) 326–332. 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- [70].Higgins R, Gendron JM, Rising L, Mak R, Webb K, Kaiser SE, Zuzow N, Riviere P, Yang B, Fenech E, Tang X, Lindsay SA, Christianson JC, Hampton RY, Wasserman SA, Bennett EJ, The Unfolded Protein Response Triggers Site-Specific Regulatory Ubiquitylation of 40S Ribosomal Proteins, Mol. Cell 59 (2015) 35–49. 10.1016/j.molcel.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dalemans W, Barbry P, Champigny G, Jallat S, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq J-P, Lazdunski M, Altered chloride ion channel kinetics associated with the ∆F508 cystic fibrosis mutation, Nature 354 (1991) 526–528. 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- [72].World Health Organisation, The molecular genetic epidemiology of cystic fibrosis, Hum. Genet. Program. Chronic Dis. Heal. Promot. World Heal. Organ (2004).
- [73].Pankow S, Bamberger C, Calzolari D, Bamberger A, Yates JR, Deep interactome profiling of membrane proteins by co-interacting protein identification technology, Nat. Protoc 11 (2016) 2515–2528. 10.1038/nprot.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ, Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive, Nature 358 (1992) 761–764. 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- [75].Hutt DM, Herman D, Rodrigues APC, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, Balch WE, Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis, Nat. Chem. Biol 6 (2010) 25–33. 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hutt DM, Loguercio S, Campos AR, Balch WE, A Proteomic Variant Approach (ProVarA) for Personalized Medicine of Inherited and Somatic Disease, J. Mol. Biol 430 (2018) 2951–2973. 10.1016/j.jmb.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wong MY, Shoulders MD, Targeting defective proteostasis in the collagenopathies, Curr. Opin. Chem. Biol. 50 (2019) 80–88. 10.1016/j.cbpa.2019.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].DiChiara AS, Taylor RJ, Wong MY, Doan N-D, Del Rosario AM, Shoulders MD, Mapping and Exploring the Collagen-I Proteostasis Network, ACS Chem. Biol 11 (2016) 1408–1421. 10.1021/acschembio.5b01083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Myllyharju J, Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis, Matrix Biol 22 (2003) 15–24. 10.1016/S0945-053X(03)00006-4. [DOI] [PubMed] [Google Scholar]
- [80].Doan ND, Hosseini AS, Bikovtseva AA, Huang MS, DiChiara AS, Papa LJ, Koller A, Shoulders MD, Elucidation of proteostasis defects caused by osteogenesis imperfecta mutations in the collagen-α2(I) C-propeptide domain, J. Biol. Chem (2020). 10.1074/jbc.RA120.014071. [DOI] [PMC free article] [PubMed]
- [81].Wright MT, Kouba L, Plate L, Thyroglobulin interactome profiling defines altered proteostasis topology associated with thyroid dyshormonogenesis., Mol. Cell. Proteomics (2020). 10.1074/mcp.RA120.002168. [DOI] [PMC free article] [PubMed]
- [82].Cohen AD, Comenzo RL, Systemic Light-Chain Amyloidosis: Advances in Diagnosis, Prognosis, and Therapy, Hematology 2010 (2010) 287–294. 10.1182/asheducation-2010.1.287. [DOI] [PubMed] [Google Scholar]
- [83].Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, Wiseman RL, Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain, Proc. Natl. Acad. Sci 111 (2014) 13046–13051. 10.1073/pnas.1406050111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP, Scampavia L, Brown SJ, Rosen H, Powers ET, Walter P, Hodder P, Wiseman RL, Kelly JW, Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation, Elife 5 (2016). 10.7554/eLife.15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Hafirassou ML, Meertens L, Umaña-Diaz C, Labeau A, Dejarnac O, Bonnet-Madin L, Kümmerer BM, Delaugerre C, Roingeard P, Vidalain PO, Amara A, A Global Interactome Map of the Dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors, Cell Rep (2017). 10.1016/j.celrep.2017.11.094. [DOI] [PubMed]
- [86].Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CKE, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R, Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites, Cell Host Microbe 5 (2009) 365–375. 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Puerta-Guardo H, Glasner DR, Espinosa DA, Biering SB, Patana M, Ratnasiri K, Wang C, Beatty PR, Harris E, Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism, Cell Rep 26 (2019) 1598–1613.e8. 10.1016/j.celrep.2019.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Puschnik AS, Marceau CD, Ooi YS, Majzoub K, Rinis N, Contessa JN, Carette JE, A Small-Molecule Oligosaccharyltransferase Inhibitor with Pan-flaviviral Activity, Cell Rep 21 (2017) 3032–3039. 10.1016/j.celrep.2017.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Coyaud E, Ranadheera C, Cheng D, Gonçalves J, Dyakov BJA, Laurent EMN, St-Germain J, Pelletier L, Gingras A-C, Brumell JH, Kim PK, Safronetz D, Raught B, Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus, Mol. Cell. Proteomics 17 (2018) 2242–2255. 10.1074/mcp.TIR118.000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L, Newton B, Hüttenhain R, Petit MJ, Baum T, Everitt A, Laufman O, Tassetto M, Shales M, Stevenson E, Iglesias GN, Shokat L, Tripathi S, Balasubramaniam V, Webb LG, Aguirre S, Willsey AJ, Garcia-Sastre A, Pollard KS, Cherry S, Gamarnik AV, Marazzi I, Taunton J, Fernandez-Sesma A, Bellen HJ, Andino R, Krogan NJ, Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis, Cell 175 (2018) 1931–1945.e18. 10.1016/j.cell.2018.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang X-P, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang H-Y, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, D’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, García-Sastre A, Shokat KM, Shoichet BK, Krogan NJ, A SARS-CoV-2 protein interaction map reveals targets for drug repurposing, Nature 583 (2020) 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Pankow S, Bamberger C, Yates JR, A posttranslational modification code for CFTR maturation is altered in cystic fibrosis, Sci. Signal 12 (2019) eaan7984. 10.1126/scisignal.aan7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, Lee YJ, Zakrzewska A, Kim SH, Kim ST, Kim SY, Lee HG, Soung NK, Ahn JS, Ciechanover A, Kim BY, Kwon YT, Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding, Nat. Cell Biol (2015). 10.1038/ncb3177. [DOI] [PMC free article] [PubMed]
- [94].Perera LA, Rato C, Yan Y, Neidhardt L, McLaughlin SH, Read RJ, Preissler S, Ron D, An oligomeric state-dependent switch in the ER enzyme FICD regulates AMPylation and deAMPylation of BiP, EMBO J 38 (2019). 10.15252/embj.2019102177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Nitika CM Porter, A.W. Truman, M.C. Truttmann, Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code, J. Biol. Chem 295 (2020) 10689–10708. 10.1074/jbc.REV120.011666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Yu J, Li T, Liu Y, Wang X, Zhang J, Wang X, Shi G, Lou J, Wang L, Wang C, Wang L, Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress, EMBO J (2020). 10.15252/embj.2019103841. [DOI] [PMC free article] [PubMed]
- [97].Ma Y, Yates JR, Proteomics and pulse azidohomoalanine labeling of newly synthesized proteins: what are the potential applications?, Expert Rev. Proteomics 15 (2018) 545–554. 10.1080/14789450.2018.1500902. [DOI] [PMC free article] [PubMed] [Google Scholar]