

ABSTRACT
The PIDDosome is a Caspase-2-activating platform assembling in response to centrosome amplification or genotoxic stress. We have recently shown that both stimuli depend on ANKRD26 (ankyrin repeat domain-containing protein 26)-mediated localization of PIDD1 (p53-inducible protein with death domain) at the centrosome, demonstrating how this organelle can directly influence cell fate.
KEYWORDS: PIDDosome, p53, centrosome, DNA damage
The Caspase-2-PIDDosome (hereafter PIDDosome) is a multiprotein complex composed of PIDD1 (p53-inducible protein with a death domain or LRDD), the adapter protein RAIDD (RIP-associated ICH1/CED3-homologous protein with death domain or CRADD) and Caspase-2 (or CASP2), whose assembly results into trans-autoproteolysis and activation of Caspase-2.1 Despite being highly evolutionarily conserved, the functional role of Caspase-2 remains poorly understood and its involvement in apoptosis ambiguous. In the early days, some evidence indicated that the PIDDosome could be an effector of the DNA damage response (DDR). PIDD1, encoding the core component of the complex, was first identified as a target gene of TP53 (tumor protein p53, best known as p53).2 Conversely, the PIDDosome was shown to contribute to p53 stabilization and transcription of its target genes,3 suggesting thereby that it can promote a positive feedback loop. Furthermore, PIDD1 and PIDDosome assembly are subjected to control by key DDR kinases, namely ATM (ataxia telangiectasia mutated) and CHK1 (checkpoint kinase 1),4,5 strongly suggesting an involvement of the complex in mediating cellular events in response to genotoxic stress. Surprisingly, however, mouse genetics casted strong doubts on the concept of PIDDosome as key mediator of the DDR, since PIDDosome-defective mice consistently lacked clear phenotypes in response to DNA-damaging agents.2
We previously showed that the PIDDosome can be activated by a centrosome-driven signal in response to cytokinesis failure.6 In this context, the PIDDosome appears as an essential and non-redundant p53 activator, whose assembly is engaged as a surveillance mechanism responsive to supernumerary centrosomes (Figure 1). Importantly, mouse genetics confirmed the functional relevance of PIDDosome activation in response to cell division failure, determining the ploidy distribution of the liver during organogenesis and regeneration.2,6 Strikingly, however, mechanistic aspects of this pathway as well as its potential crosstalk with the DDR remained unexplored.
Figure 1.
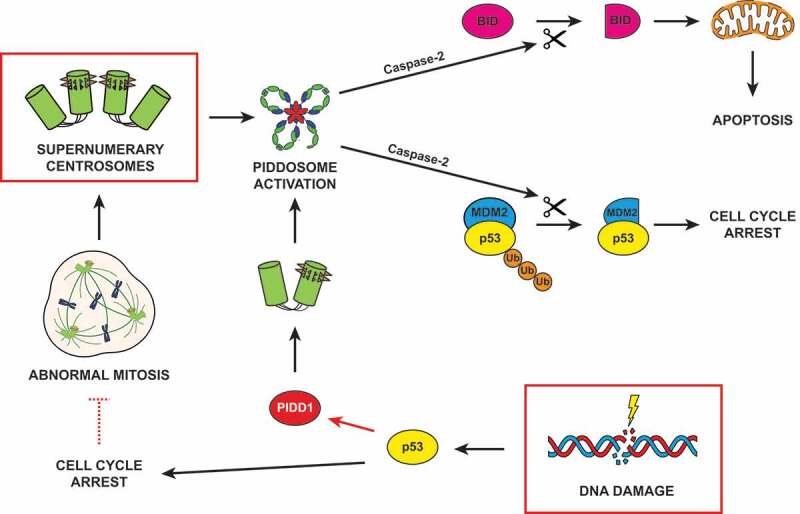
Sources and consequences of PIDDosome activation. Supernumerary centrosomes lead to Caspase-2 activation via the PIDDosome, resulting into either TP53 (tumor suppressor protein p53, best known as p53)-dependent cell cycle arrest or apoptosis. DNA damage can induce PIDDosome activation following two different paradigms. In the first one, p53 stabilization transactivates PIDD1 (the gene coding for PIDD1, p53-inducible protein with a death domain), resulting in a global increase in its cellular levels (red arrow). The centrosome, locally concentrating PIDD1 active moieties, promotes PIDDosome assembly. In the second scenario, cells which halted their cell cycle during DNA damage response can escape this proliferative block and reenter mitosis (red dashed inhibitory arrow). Proliferation in the presence of unrepaired DNA damage or an under-replicated genome frequently leads to abnormal cell division and accumulation of supernumerary centrosomes, which eventually drive PIDDosome assembly. Caspase-2 activation promotes the proteolytic cleavage of its two main substrates, MDM2 (mouse double minute 2) and BID (BH3 interacting-domain death agonist), culminating in cell cycle arrest or apoptosis, respectively
In our latest work, we shed light on how the PIDDosome activation signal is ignited at the centrosome. We identified the centriolar distal appendage protein ANKRD26 (ankyrin repeat domain-containing protein 26) as the centrosomal receptor for PIDD1, promoting its constitutive localization at the centrosome.7 By surgically removing the PIDD1 minimal interacting domain from ANKRD26 we demonstrated that lack of PIDD1 centrosomal localization (and not a more general impairment of centrosome function) determined an inability of cells to sustain PIDDosome assembly in response to supernumerary centrosomes. Importantly, Andrew Holland’s laboratory reached the same conclusion on ANKRD26 role in PIDDosome activation, performing an unbiased genome-wide screen for factors controlling cellular proliferation in the presence of supernumerary centrosomes.8 To our surprise, our ANKRD26-deficient cell lines revealed that PIDD1 centrosomal localization is necessary for PIDDosome activity also in the context of DDR, as ANKRD26 depletion completely suppressed activation of the complex upon DNA damage. In this specific condition, PIDDosome activation did not require the presence of supernumerary centrosomes, yet it relied on the p53-dependent elevation of PIDD1 levels (Figure 1). These data suggest that the centrosome acts as a hub concentrating a basal amount of PIDD1 active moieties, whose levels, however, do not reach a critical activation threshold in an unperturbed condition. Both an increase in centrosome number and a general p53-dependent boost in PIDD1 expression (such as upon DNA damage) can independently lead to exceeding this threshold, igniting PIDDosome activation.7
In summary, our work offers new insights into the regulation of PIDDosome activation, proposing a model which is able to reconcile several seemingly unrelated or contradictory observations in the field. Firstly, it clarifies that PIDDosome assembly can indeed occur following DNA damage, as a consequence of p53-dependent PIDD1 transactivation. Secondly, it explains why DNA damage triggers a robust PIDDosome activation in p53-defective cells only when they are forced into mitosis (e.g., through CHK1 inhibition).5 Conceivably, in this context PIDDosome assembly cannot rely on the p53-dependent elevation of PIDD1 levels but rather depends on the presence of supernumerary centrosomes, a common outcome of faulty mitoses arising in this experimental condition. Thirdly, it elucidates how the PIDDosome can be activated both upstream and downstream of p53. In the first scenario, PIDDosome assembly is triggered by the presence of extra centrosomes and results into p53 stabilization due to Caspase-2-dependent MDM2 (mouse double minute 2) inactivation. On the contrary, in the context of DNA damage, the PIDDosome is engaged at a later stage, subsequently to p53 activation. Importantly, when the DDR alone has failed to elicit a p53 output sufficient to halt cell division, PIDDosome activation becomes particularly prominent, imparting measurable changes to the dynamics of p53 accumulation.9 Considering that p53 accumulation dynamics are crucial to define the cellular fate during DDR, it appears that PIDDosome activation can act as an important fail-safe mechanism when the canonical DDR signaling proves incapable of effectively stopping the cell division cycle (Figure 1).
Taken together, we speculate that the most physiological trigger for PIDDosome activation is the lack of cytokinesis, invariably resulting into the acquisition of supernumerary mature centrosomes. Yet, it appears that DNA damage alone or in combination with a faulty mitosis can trigger different degrees of PIDDosome activation, invariably relying on the centrosome as activatory scaffold to kickstart PIDDosome assembly (Figure 1). Our model, however, does not clarify the contribution of p53-independent DDR events in defining PIDDosome activation. It has been previously shown that PIDD1 death domain becomes phosphorylated at Thr788 in an ATM-dependent manner,4 promoting RAIDD binding and Caspase-2 autocatalytic activation. It will be interesting to investigate whether this and other posttranslational modifications can concur to set the threshold for PIDDosome activation capacity also in the context of a centrosome-driven response, representing a general shared feature. Furthermore, an additional aspect that still remains elusive is what defines the output of PIDDosome activation. In fact, while in our experimental conditions the primary consequence of PIDDosome activation is a p53-dependent cell cycle arrest, Caspase-2 has also been shown to promote apoptosis in different settings. One might speculate that specific subcellular compartments possess peculiar PIDDosome regulators, thereby compartmentalizing Caspase-2 activity and concurring to determine distinct fates. Moreover, different cellular cues could be responsible for a distinctive posttranslational modification profile of PIDDosome components, ultimately resulting into differential proteolytic activities. Eventually, since cell cycle blockade depends on MDM2 cleavage whereas apoptosis relies additionally on the proteolytic processing of the proapoptotic BCL-2 family member BID (BH3 interacting-domain death agonist),10 we speculate that PIDDosome-dependent cell fate determination could be shaped by the relative abundance of Caspase-2 key substrates at its primary activation site.
Funding Statement
This work was supported by the Giovanni Armenise-Harvard Foundation [CDA 2017] and the University of Trento.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Reference
- 1.Tinel A, Tschopp J.. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304(5672):1–3. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- 2.Sladky V, Schuler F, Fava LL, Villunger A. The resurrection of the PIDDosome-emerging roles in the DNA-damage response and centrosome surveillance. J Cell Sci. 2017;130(22):3779–3787. doi: 10.1242/jcs.203448. [DOI] [PubMed] [Google Scholar]
- 3.Oliver TG, Meylan E, Chang GP, Xue W, Burke JR, Humpton TJ, Hubbard D, Bhutkar A, Jacks T. Caspase-2-mediated cleavage of Mdm2 creates a p53-induced positive feedback loop. Mol Cell. 2011;43(1):57–71. doi: 10.1016/j.molcel.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ando K, Kernan JL, Liu PH, Sanda T, Logette E, Tschopp J, Look AT, Wang J, Bouchier-Hayes L, Sidi S. PIDD death-domain phosphorylation by ATM controls prodeath versus prosurvival PIDDosome signaling. Mol Cell. 2012;47(5):681–693. doi: 10.1016/j.molcel.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133(5):864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017;31(1):34–45. doi: 10.1101/gad.289728.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burigotto M, Mattivi A, Migliorati D, Magnani G, Valentini C, Roccuzzo M, Offterdinger M, Pizzato M, Schmidt A, Villunger A, et al. Centriolar distal appendages activate the centrosome-PIDDosome-p53 signalling axis via ANKRD26. Embo J. 2020;40(4):e104844. doi: 10.15252/embj.2020104844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans LT, Anglen T, Scott P, Lukasik K, Loncarek J, Holland AJ. ANKRD26 recruits PIDD1 to centriolar distal appendages to activate the PIDDosome following centrosome amplification. Embo J. 2020;40(4):e105106. doi: 10.15252/embj.2020105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsabar M, Mock CS, Venkatachalam V, Reyes J, Karhohs KW, Oliver TG, Regev A, Jambhekar A, Lahav GA. Switch in p53 dynamics marks cells that escape from DSB-induced cell cycle arrest. Cell Rep. 2020;33(6):108392. doi: 10.1016/j.celrep.2020.108392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.López-García C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ, Horswell S, Grönroos E, Favero F, Rowan AJ, et al. BCL9L dysfunction impairs caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell. 2017;31(1):79–93. doi: 10.1016/j.ccell.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]