

ABSTRACT
The function of histone deacetylase 2 (HDAC2) in transcriptional regulation and its role in oncogenesis have been well established. Here we discuss a transcription-independent HDAC2 pathway controlling cancer-related protein stability via the mouse double minute 2 homolog (MDM2) ubiquitin ligase. In synovial sarcoma, HDAC2 inactivation demonstrates significant therapeutic effect by degradation of the SS18-SSX driver oncoprotein.
KEYWORDS: HDAC2, MDM2, MULE, acetylation, ubiquitination, synovial sarcoma, targeted therapeutics
Synovial sarcoma is a high-grade soft tissue sarcoma for which traditional treatments provide limited benefit.1 Most patients are diagnosed between ages 15–35. The tumors commonly arise from the lower extremities,1,2 but they often metastasize to the lungs and lymph nodes.3,4 Pathohistological analysis of synovial sarcoma reveals that it consists of mesenchymal spindle-shaped cells with varying degrees of epithelial differentiation. This feature has also been seen in other sarcoma types, such as fibrosarcoma and leiomyosarcoma, but what makes synovial sarcoma unique is a translocation between chromosomes X and 18, t(X;18), that leads to the fusion of the synovial sarcoma translocation, Chromosome 18 (SS18) gene and either synovial sarcoma, X-Chromosome-related 1 (SSX1) or 2 (SSX2) (very rarely SSX4).5 The resulting transcript, referred to as SS18-SSX, is evidently a driver of synovial sarcoma, given the fact that overexpression of SS18-SSX can induce transformation of noncancerous rat fibroblast cells6 and replicates the pathogenesis of human synovial sarcoma in transgenic mice.7,8 More importantly, SS18-SSX provides an Achilles’ heel for synovial sarcoma therapy, as human synovial sarcoma cells have been observed undergoing apoptosis in response to RNA interference (RNAi)–mediated depletion of the fusion oncogene.9–11
Over the decades, promising progress has been made in understanding the molecular mechanisms behind SS18-SSX fusion protein-promoted tumorigenesis. Originally, many of the studies were focused on the transcriptional regulatory activity of SS18-SSX based on its ability to interact with mammalian SWItch/Sucrose Non–Fermentable (SWI/SNF) and Polycomb (Pc) complexes,6,12–16 yet it has become clear that SS18-SSX also possesses transcription-independent functions. For example, SS18-SSX can induce β-catenin stabilization and accumulation within the nucleus, which then aberrantly activates the canonical Wingless–related integration (WNT) pathway to support synovial sarcoma formation.17–19 Due to the complexity of SS18-SSX action, therapies that systematically target the synovial sarcoma biology remain in short supply. In addition, there has been no success in developing drugs that can directly bind SS18–SSX to inhibit its oncogenic functions.
Through unbiased screening of 900 compounds comprising 100 different drug classes, a recent study has identified histone deacetylase (HDAC) inhibitors (ex. SB939 and Quisinostat) as the most potent agents to kill human synovial sarcoma cells in tissue culture.20 Preclinical experiments have further shown the prominent efficacy of HDAC inhibition against tumor growth in a transgenic mouse model closely mimicking human synovial sarcomagenesis.20–22 Surprisingly, immunofluorescence staining and western blot analyses revealed that SS18-SSX protein levels were reduced in the tumor tissues from HDAC inhibitor-treated mice.22 A similar effect was witnessed in human synovial sarcoma cell lines in response to various HDAC inhibitors with distinct structures.20–22 Notably, the levels of SS18-SSX mRNA remained unaffected under the same conditions. Consistent with this, HDAC inhibitor-induced reduction of SS18-SSX protein levels was indeed reversed by MG132 proteasome inhibitor. Additionally, the higher levels of SS18-SSX protein were conjugated with polyubiquitin chains, suggesting that the fusion oncoprotein undergoes proteasome-dependent proteolysis after HDAC inhibitor treatment.
Using mass spectrometry, a 500 kDa protein (named Mcl-1 ubiquitin ligase E3, or MULE;23 also known as HUWE1,24 ARF-BP1,25 UREB1,26 LASU1,27 and HectH928) was identified that had specifically copurified with SS18-SSX in HDAC inhibitor-treated synovial sarcoma cells.22 MULE belongs to a major class of E3 enzymes that contain a characteristic homologous to the E6-AP carboxyl terminus (HECT) domain with catalytic activity for ubiquitin transfer to substrate proteins.29 The ability of MULE to ubiquitinate SS18-SSX was readily confirmed in both in vitro ubiquitination and human embryonic kidney (HEK293) cell-based expression systems. Significantly, by depleting MULE, the levels of endogenous SS18-SSX ubiquitination in human synovial sarcoma cells were remarkably reduced upon HDAC inhibitor treatment, which correlated with a dramatic increase in SS18-SSX protein levels. These results indicate that MULE is a major E3 ligase for SS18-SSX, and this ubiquitination activity contributes at least in part to HDAC inhibitor-induced SS18-SSX protein degradation. Coimmunoprecipitation analysis of serial MULE fragments further revealed that two protein domains, tryptophan-glutamate repeats and ubiquitin-binding motif (WWE and UBM, respectively), specifically interact with the SS18-SSX fusion oncoprotein. WWE is involved in mediating protein–protein interactions,30 while UBM is a monoubiquitin-binding motif with unknown biological significance.31
Interestingly, MULE binds to SS18-SSX through specific recognition of its C-terminal region, which contains a conserved domain (SSX repression domain, or SSXRD) known for chromatin association and transcriptional repression.32,33 However, MULE-mediated ubiquitination occurs at the N-terminus of SS18-SSX, involving a single lysine residue (lysine 23, or K23), mutation of which could almost completely abrogate HDAC inhibitor-induced conjugation of polyubiquitin chains to the fusion oncoprotein. It is now clear that MULE degradation of SS18-SSX protein relies on at least two levels of specificity– protein interaction with the SSX region, and ubiquitination position in SS18 (Figure 1). This mechanism therefore allows for more precise control of MULE-mediated destruction toward the fusion oncoprotein compared to its wild-type partners. Beyond these molecular observations, a critical problem raised is how MULE recognizes SS18-SSX in synovial sarcoma cells given that MULE is mostly located in the cytoplasm27,34 and SS18-SSX is an exclusively nuclear protein. Undeniably, the interaction between MULE and SS18-SSX was clearly detected in the cytoplasm of synovial sarcoma cells by in situ proximity ligation assay (PLA) and immunofluorescence microscopy.22 Therefore, future study is necessary to uncover additional regulatory mechanisms underlying nuclear-cytoplasmic shuttling of the SS18-SSX fusion protein and to elucidate the potential role of HDAC activity and HDAC inhibitor treatment during this process.
Figure 1.
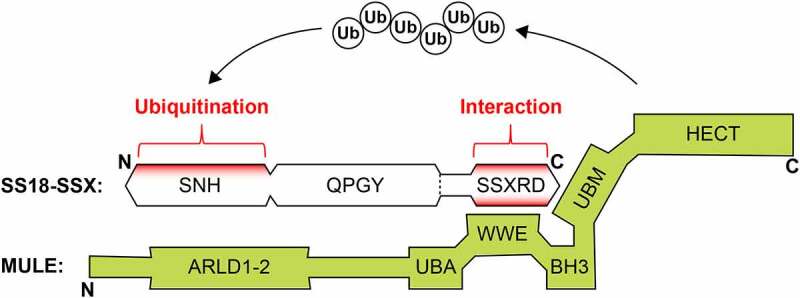
Overview of SS18-SSX ubiquitination by Mcl-1 ubiquitin ligase E3 (MULE)
The E3 ligase MULE binds to the C-terminal SSXRD domain of SS18-SSX through two distinct protein domains, WWE and UBM. Polyubiquitin chains are attached to a specific lysine residue (lysine 23, or K23) within the N-terminal SNH domain of SS18-SSX for protein degradation. SS18-SSX protein domains include SNH, SS18 N-terminal homology domain (residues 20–73); QPGY, glutamine-proline-glycine-tyrosine-rich domain (residues 187–379); SSXRD, SSX repression domain (residues 424–457). MULE protein domains include: ARLD, Armadillo repeat‐like domain (residues 104–815); UBA, ubiquitin–associated domain (residues 1316–1355); WWE, tryptophans–glutamate repeats (residues 1603–1680); BH3, Bcl–2 homology region 3 (residues 1976-1990); UBM, ubiquitin–binding motif (residues 2963-3082); HECT, homologous to the E6–AP carboxyl terminus (residues 3993–4374).
MULE is almost undetectable in synovial sarcoma, although its protein levels are increased after addition of MG132 proteasome inhibitor. This is consistent with recent studies that show MULE is unstable in human epidermal growth factor receptor 2 (HER2)-positive breast cancer cells, due to ubiquitin-mediated degradation by the oncogenic E3 ligase, mouse double minute 2 homolog (MDM2).35 Notably, an inverse relationship between MULE and MDM2 expression in breast cancer and liposarcoma patient specimens has also been demonstrated by immunohistochemistry-based tissue microarrays.36 Synovial sarcoma cells likely adopt a similar mechanism to degrade MULE and maintain its protein at low levels. Indeed, a lysine-rich region of MDM2 (residues 460–476) directly binds the acidic domain of MULE (residues 2425-2469), although this interaction is then blocked by HDAC inhibitor treatment. Mass spectrometric analysis of the MDM2 protein further identified five lysine residues (K466, K467, K469, K470, and K473) within the aforementioned lysine-rich region, which were specifically acetylated upon HDAC inhibition.
The relevance of these findings was highlighted with the generation of acetylation-defective MDM2 mutants that interacted with MULE, in a similar manner to what’s seen with wild-type MDM2, but that no longer respond to HDAC inhibitor-induced dissociation.22 Based on protein structure analysis, it becomes clear that the lysine-rich region of MDM2 forms a positively charged docking site for MULE through the attraction to its negatively charged acidic domain (Figure 2a). This binding interface is abolished upon HDAC inhibitor treatment, largely because of the charge neutralization of MDM2’s lysine-rich region by acetylation (Figure 2b). Curiously, lysine acetylation was also found to function as the binding signal for so-called “reader” proteins that are generally characterized by the presence of evolutionarily conserved bromodomains.37 Therefore, the effects of HDAC inhibitor treatment on MDM2 might be more complicated than simple dissociation of MULE. It will be important to determine whether site-specific acetylation signaling provides a widespread mechanism for differentially regulating the fate of MDM2 substrates under physiological and pathological conditions.
Figure 2.
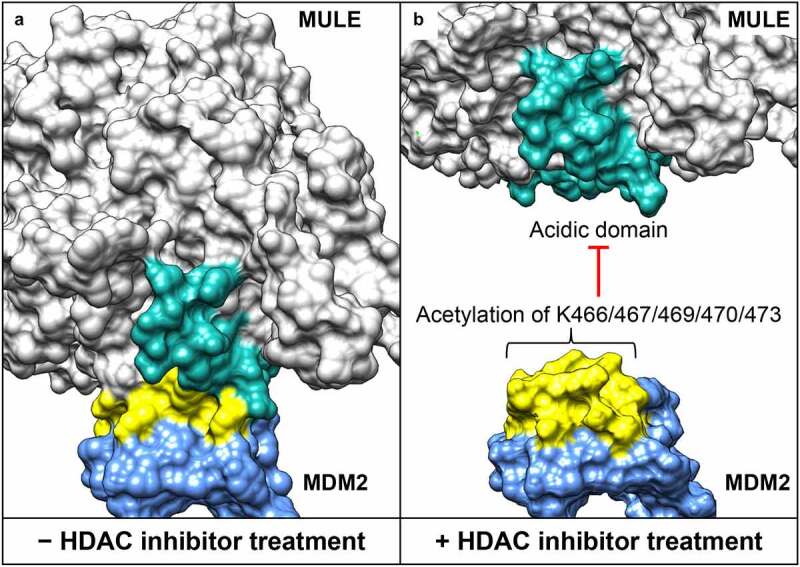
Structural interface between the acidic domain of Mcl-1 ubiquitin ligase E3 (MULE) and the lysine-rich domain of mouse double minute 2 homolog (MDM2). (a) Surface presentation of the MULE–MDM2 interaction. Models for MULE (residues 2261–2970) and MDM2 (residues 418-491) are generated by high-resolution comparative modeling with the Robetta server,39 and docking analysis performed using the ClusPro server.40 (b) Histone deacetylase (HDAC) inhibitor–induced lysine acetylation of MDM2 (at position 466, 467, 469, 470 and 473), leading to MULE dissociation. The acidic domain of MULE (residues 2432-2465) is highlighted in cyan, and the lysine–rich domain of MDM2 (residues 460-476) highlighted in yellow.
Lysine acetylation is a fully reversible post-translational modification; HDACs and sirtuins (SIRTs) are an important class of enzymes that can catalyze the removal of acetyl groups from proteins.39 Among eleven HDAC family members, only histone deacetylase 1 (HDAC1) and 2 (HDAC2) are substantially expressed in synovial sarcoma cell lines and patient specimens.22,38 Notably, HDAC1 and HDAC2 share considerable sequence homology (approximately 85% identical), and their complex assemblies and activities are largely redundant in various biological contexts, only with a few exceptions such as early embryogenesis and brain development.41–43 Depletion of HDAC2 in synovial sarcoma cells strongly inhibited the MDM2-MULE interaction and greatly promoted SS18-SSX degradation, while depletion of HDAC1 showed no obvious effect. These studies highlight a MDM2-mediated ubiquitination pathway specifically connecting HDAC2 activity with synovial sarcoma biology, despite HDAC1 having a similar capability to bind MDM2. At this moment it is not known whether both HDACs exist in the same MDM2 complex, seeing as HDAC1 and HDAC2 can form heterodimers between each other,44 and they are seen together in a number of multiprotein complexes.45 Thus, future work is necessary to untangle the mechanistic detail of MDM2 regulation, which may involve distinct sets of acetylated lysine residues targeted by HDAC1 and HDAC2, respectively.
In conclusion, HDAC2 is a novel component to the MDM2 ubiquitination pathway that serves a critical contribution in maintaining low levels of MULE protein inside human cancer cells. Intriguingly, MULE is also a ubiquitin E3 ligase and exhibits the tumor-suppressor function in synovial sarcoma by targeting the SS18-SSX driver oncoprotein for ubiquitination and degradation. Based on this information, drugs that either block the enzymatic activity of HDAC2 and MDM2 or inhibit their interaction might be especially valuable for synovial sarcoma treatment. For example, small-molecule HDAC inhibitors represent one possible therapeutic strategy that has displayed potent suppressive activity on synovial sarcoma cell proliferation in vitro and tumor growth in vivo that is being currently tested in clinical trials.46,47 In a broad perspective, elevated expression levels of both HDAC2 and MDM2 are found in a variety of tumor types, and their overexpression is often associated with aggressive tumor behavior (i.e. distant metastasis) and poor prognosis.48,49 Moreover, a significant correlation between HDAC2 and MDM2 has been highlighted with clinical evidence showing that HDAC2 is highly coexpressed with MDM2 in dedifferentiated liposarcoma samples.50 In keeping with this, two recent reports suggest a positive role of HDAC2 in regulation of the MDM2 gene expression in lung cancer and liposarcoma cells.50,51 However, neither HDAC2 inactivation nor HDAC inhibitor treatment can reduce MDM2 levels in synovial sarcoma,22 as well as in several other tumor types.52,53 In the future, it will be important to find the possibly druggable enzymes and/or cellular signaling molecules that participate in transcriptional induction of MDM2 by HDAC2, so that specific agents can be combined with HDAC inhibitor treatment more efficiently and effectively target various types of human cancers associated with the HDAC2-MDM2 pathway.
Funding Statement
This work was funded by the HudsonAlpha Foundation, the State of Alabama Cancer Fund, and St. Baldrick’s Childhood Cancer Research Grant [#429614] to LS.
Disclosure of potential conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Riedel RF, Jones R, Italiano A, Bohac C, Thompson J, Mueller K, Khan Z, Pollack S, Van Tine B.. Systemic anti-cancer therapy in synovial sarcoma: a systematic review. Cancers (Basel). 2018;10(11):1. doi: 10.3390/cancers10110417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison BA. Soft tissue sarcomas of the extremities. Proc (Bayl Univ Med Cent). 2003;16(3):285–5. doi: 10.1080/08998280.2003.11927915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krieg AH, Hefti F, Speth BM, Jundt G, Guillou L, Exner UG, Von Hochstetter AR, Cserhati MD, Fuchs B, Mouhsine E, et al. Synovial sarcomas usually metastasize after >5 years: a multicenter retrospective analysis with minimum follow-up of 10 years for survivors. Ann Oncol. 2011;22(2):458–467. doi: 10.1093/annonc/mdq394. [DOI] [PubMed] [Google Scholar]
- 4.De Necochea-campion R, Zuckerman LM, Mirshahidi HR, Khosrowpour S, Chen C-S, Mirshahidi S. Metastatic biomarkers in synovial sarcoma. Biomark Res. 2017;5:4. doi: 10.1186/s40364-017-0083-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ladanyi M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001;20(40):5755–5762. doi: 10.1038/sj.onc.1204601. [DOI] [PubMed] [Google Scholar]
- 6.Nagai M, Tanaka S, Tsuda M, Endo S, Kato H, Sonobe H, Minami A, Hiraga H, Nishihara H, Sawa H, Nagashima K. Analysis of transforming activity of human synovial sarcoma-associated chimeric protein SYT-SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha. Proc Natl Acad Sci U S A. 2001;98(7):3843–3848. doi: 10.1073/pnas.061036798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell. 2007;11(4):375–388. doi: 10.1016/j.ccr.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Haldar M, Hedberg ML, Hockin MF, Capecchi MR. A CreER-based random induction strategy for modeling translocation-associated sarcomas in mice. Cancer Res. 2009;69(8):3657–3664. doi: 10.1158/0008-5472.CAN-08-4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng C, Guo W, Yang Y, Zhao H. Downregulation of SS18-SSX1 expression by small interfering RNA inhibits growth and induces apoptosis in human synovial sarcoma cell line HS-SY-II in vitro. Eur J Cancer Prev. 2008;17(5):392–398 [DOI] [PubMed] [Google Scholar]
- 10.Cai W, Sun Y, Wang W, Han C, Ouchida M, Xia W, Zhao X, Sun B. The effect of SYT-SSX and extracellular signal-regulated kinase (ERK) on cell proliferation in synovial sarcoma. Pathol Oncol Res. 2011;17(2):357–367. doi: 10.1007/s12253-010-9334-y. [DOI] [PubMed] [Google Scholar]
- 11.Carmody Soni EE, Schlottman S, Erkizan HV, Uren A, Toretsky JA. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin Orthop Relat Res. 2014;472(3):874–882. doi: 10.1007/s11999-013-3065-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thaete C, Brett D, Monaghan P, Whitehouse S, Rennie G, Rayner E, Cooper CS, Goodwin G. Functional domains of the SYT and SYT-SSX synovial sarcoma translocation proteins and co-localization with the SNF protein BRM in the nucleus. Hum Mol Genet. 1999;8(4):585–591. doi: 10.1093/hmg/8.4.585. [DOI] [PubMed] [Google Scholar]
- 13.Soulez M, Saurin AJ, Freemont PS, Knight JC. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene. 1999;18(17):2739–2746. doi: 10.1038/sj.onc.1202613. [DOI] [PubMed] [Google Scholar]
- 14.Garcia CB, Shaffer CM, Eid JE. Genome-wide recruitment to Polycomb-modified chromatin and activity regulation of the synovial sarcoma oncogene SYT-SSX2. BMC Genomics. 2012;13:189. doi: 10.1186/1471-2164-13-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su L, Sampaio A, Jones K, Pacheco M, Goytain A, Lin S, Poulin N, Yi L, Rossi F, Kast J, et al. Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell. 2012;21(3):333–347. doi: 10.1016/j.ccr.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadoch C, Crabtree GR. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell. 2013;153(1):71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pretto D, Barco R, Rivera J, Neel N, Gustavson MD, Eid JE. The synovial sarcoma translocation protein SYT-SSX2 recruits beta-catenin to the nucleus and associates with it in an active complex. Oncogene. 2006;25(26):3661–3669. doi: 10.1038/sj.onc.1209413. [DOI] [PubMed] [Google Scholar]
- 18.Barham W, Frump AL, Sherrill TP, Garcia CB, Saito-Diaz K, VanSaun MN, Fingleton B, Gleaves L, Orton D, Capecchi MR, et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013;3(11):1286–1301. doi: 10.1158/2159-8290.CD-13-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrott JJ, Illum BE, Jin H, Zhu J-F, Mosbruger T, Monument MJ, Smith-Fry K, Cable MG, Wang Y, Grossmann AH, et al. Beta-catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget. 2015;6(26):22758–22766. doi: 10.18632/oncotarget.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laporte AN, Barrott JJ, Yao RJ, Poulin NM, Brodin BA, Jones KB, Underhill TM, Nielsen TO. HDAC and proteasome inhibitors synergize to activate pro-apoptotic factors in synovial sarcoma. PLoS One. 2017;12(1):e0169407. doi: 10.1371/journal.pone.0169407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laporte AN, Poulin NM, Barrott JJ, Wang XQ, Lorzadeh A, Vander Werff R, Jones KB, Underhill TM, Nielsen TO. Death by HDAC inhibition in synovial sarcoma cells. Mol Cancer Ther. 2017;16(12):2656–2667. doi: 10.1158/1535-7163.MCT-17-0397. [DOI] [PubMed] [Google Scholar]
- 22.Patel N, Wang J, Shiozawa K, Jones KB, Zhang Y, Prokop JW, Davenport GG, Nihira NT, Hao Z, Wong D, et al. HDAC2 regulates site-specific acetylation of MDM2 and its ubiquitination signaling in tumor suppression. iScience. 2019;13:43–54. doi: 10.1016/j.isci.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121(7):1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 24.Zhao X, Heng JIT, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol. 2008;10(6):643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen D, Kon N, Li M, Zhang W, Qin J, Gu W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell. 2005;121(7):1071–1083. doi: 10.1016/j.cell.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 26.Gu J, Dubner R, Fornace AJ, Iadarola MJ. UREB1, a tyrosine phosphorylated nuclear protein, inhibits p53 transactivation. Oncogene. 1995;11:2175–2178. [PubMed] [Google Scholar]
- 27.Liu Z, Miao D, Xia Q, Hermo L, Wing SS. Regulated expression of the ubiquitin protein ligase, E3(Histone)/LASU1/Mule/ARF-BP1/HUWE1, during spermatogenesis. Dev Dyn. 2007;236(10):2889–2898. doi: 10.1002/dvdy.21302. [DOI] [PubMed] [Google Scholar]
- 28.Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123(3):409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Kao SH, Wu HT, Wu KJ. Ubiquitination by HUWE1 in tumorigenesis and beyond. J Biomed Sci. 2018;25(1):67. doi: 10.1186/s12929-018-0470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aravind L. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem Sci. 2001;26(5):273–275. doi: 10.1016/S0968-0004(01)01787-X. [DOI] [PubMed] [Google Scholar]
- 31.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol. 2009;10(10):659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim FL, Soulez M, Koczan D, Thiesen H-J, Knight JC. A KRAB-related domain and a novel transcription repression domain in proteins encoded by SSX genes that are disrupted in human sarcomas. Oncogene. 1998;17(15):2013–2018. doi: 10.1038/sj.onc.1202122. [DOI] [PubMed] [Google Scholar]
- 33.Kato H, Tjernberg A, Zhang W, Krutchinsky AN, An W, Takeuchi T, Ohtsuki Y, Sugano S, De Bruijn DR, Chait BT, et al. SYT associates with human SNF/SWI complexes and the C-terminal region of its fusion partner SSX1 targets histones. J Biol Chem. 2002;277(7):5498–5505. doi: 10.1074/jbc.M108702200. [DOI] [PubMed] [Google Scholar]
- 34.Xu Y, Anderson DE, Ye Y. The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov. 2016;2:16040. doi: 10.1038/celldisc.2016.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci Signal. 2013;6(274):ra32. doi: 10.1126/scisignal.2003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canfield K, Wells W, Geradts J, Kinlaw WB, Cheng C, Kurokawa M. Inverse association between MDM2 and HUWE1 protein expression levels in human breast cancer and liposarcoma. Int J Clin Exp Pathol. 2016;9(6):6342–6349. [PMC free article] [PubMed] [Google Scholar]
- 37.Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat Rev Mol Cell Biol. 2015;16(4):258–264 [DOI] [PubMed] [Google Scholar]
- 38.Pacheco M, Nielsen TO. Histone deacetylase 1 and 2 in mesenchymal tumors. Mod Pathol. 2012;25(2):222–230. doi: 10.1038/modpathol.2011.157. [DOI] [PubMed] [Google Scholar]
- 39.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song Y, DiMaio F, Wang -R-R, Kim D, Miles C, Brunette TJ, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41(3):741–749. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 42.Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53(2–3):275–289. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- 43.Jaworska J, Ziemka-Nalecz M, Zalewska T. Histone deacetylases 1 and 2 are required for brain development. Int J Dev Biol. 2015;59(4–6):171–177. doi: 10.1387/ijdb.150071tz. [DOI] [PubMed] [Google Scholar]
- 44.Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, Huang S, Qiu Y. Trans-regulation of histone deacetylase activities through acetylation. J Biol Chem. 2009;284(50):34901–34910. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4(1):5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassier PA, Lefranc A, Y Amela E, Chevreau C, Bui BN, Lecesne A, Ray-Coquard I, Chabaud S, Penel N, Berge Y, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer. 2013;109(4):909–914. doi: 10.1038/bjc.2013.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chu QS, Nielsen TO, Alcindor T, Gupta A, Endo M, Goytain A, Xu H, Verma S, Tozer R, Knowling M, et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocation-associated recurrent/metastatic sarcomas-NCIC-CTG IND 200dagger. Ann Oncol. 2015;26(5):973–981. doi: 10.1093/annonc/mdv033. [DOI] [PubMed] [Google Scholar]
- 48.Onel K, Cordon-Cardo C. MDM2 and prognosis. Mol Cancer Res. 2004;2:1–8. [PubMed] [Google Scholar]
- 49.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280(2):168–176. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 50.Seligson ND, Stets CW, Demoret BW, Awasthi A, Grosenbacher N, Shakya R, Hays JL, Chen JL. Inhibition of histone deacetylase 2 reduces MDM2 expression and reduces tumor growth in dedifferentiated liposarcoma. Oncotarget. 2019;10(55):5671–5679. doi: 10.18632/oncotarget.27144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo SK, Hwang C-S, Choe T-B, Hong S-I, Yi JY, Hwang S-G, Lee H-G, Oh ST, Lee Y-H, Park I-C, et al. Selective inhibition of histone deacetylase 2 induces p53-dependent survivin downregulation through MDM2 proteasomal degradation. Oncotarget. 2015;6(28):26528–26540. doi: 10.18632/oncotarget.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67(7):3145–3152. doi: 10.1158/0008-5472.CAN-06-4397. [DOI] [PubMed] [Google Scholar]
- 53.Natarajan U, Venkatesan T, Radhakrishnan V, Samuel S, Rathinavelu A. Differential mechanisms of cell death induced by HDAC inhibitor SAHA and MDM2 inhibitor RG7388 in MCF-7 cells. Cells. 2018;8(1):8. doi: 10.3390/cells8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]