

ABSTRACT
The tumor protein p53 (TP53, best known as p53) transcription factor is a critical tumor suppressor, but those p53-inducible genes most important for tumor suppression have remained unclear. Using unbiased RNA interference and CRISPR (Clustered Regularly Interspersed Palindromic Repeats)/Cas9 (CRISPR-associated protein 9) screens, genetically engineered mouse models, human cancer genome analysis, and integrative eCLIP-sequencing and RNA-sequencing analyses, we reveal a new branch of p53-mediated tumor suppression involving the RNA splicing regulator Zinc finger Matrin-type 3, Zmat3.
KEYWORDS: p53, ZMAT3, tumor suppression, splicing, CRISPR screen
Inactivation of TP53, encoding the p53 transcription factor, is frequent in human cancer, underscoring the critical role of p53 in tumor suppression. Although p53 can transcriptionally induce a panoply of target genes encoding proteins with a range of cellular functions, the molecular pathways most central for p53-mediated tumor suppression have remained largely enigmatic. Delineating tumor-suppressive components downstream of p53 is important for defining potentially targetable pathways in p53-deficient tumors but is complicated by the breadth of p53-inducible genes. In our recent manuscript1 we therefore, sought to define transcriptional programs underlying p53-mediated tumor suppression using unbiased and sensitive RNA interference and CRISPR (Clustered Regularly Interspersed Palindromic Repeats)/Cas9 (CRISPR-associated protein 9) pooled screens in vivo.
Our screens were based on previous findings in which we generated mice expressing the p53 transactivation domain mutant, p53L25Q,W26S, which activates only ~10% of p53-inducible genes, yet is nonetheless a robust tumor suppressor in mouse models2, allowing us to pinpoint those p53-regulated genes most tightly associated with tumor suppression. We identified genes efficiently activated by both wild-type p53 and the p5325,26 mutant, transduced short hairpin RNA (shRNA) or single guide RNA (sgRNA) libraries targeting these genes into oncogene-expressing mouse embryonic fibroblasts, and performed allograft tumor assays in immunocompromised mice. We then queried which shRNAs and sgRNAs became enriched in emerging tumors – an indicator of targeting a tumor suppressor gene. Strikingly, the most enriched shRNAs and sgRNAs in both screens targeted the same gene, Zmat3 (zinc finger matrin-type 3), encoding a zinc finger RNA-binding protein, suggesting that Zmat3 is an important tumor suppressor. We showed further that CRISPR/Cas9-mediated Zmat3 knockout in autochthonous mouse models of lung adenocarcinoma and hepatocellular carcinoma enhanced tumorigenesis, underscoring a broad role for Zmat3 in tumor suppression in different tissues. Reinforcing these findings was our observation that while expression of wild-type ZMAT3 inhibited proliferation in p53-deficient human cancer cells, the human tumor-derived ZMAT3-R99Q mutant was unable to do so, suggesting that ZMAT3 can be functionally inactivated in human tumors. Moreover, analysis of Project Achilles CRISPR/Cas9 screen data in human cancer cells revealed that ZMAT3 inactivation enhances proliferation of cells with wild-type p53, further supporting a role for ZMAT3 in suppressing human cancer. Taken together, these data highlight a tumor-suppressive p53-ZMAT3 axis in both mouse and human cells.
To gain insight into the mechanisms of Zmat3 tumor suppressor function, we analyzed the RNA-binding profile of Zmat3 by performing eCLIP (enhanced crosslinking and immunoprecipitation). While ZMAT3 was previously reported to regulate RNA stability by binding the 3ʹ untranslated region of target mRNAs3, we found that Zmat3 binds mRNAs at a stereotypical position upstream of the 3ʹ splice site, suggesting a role for Zmat3 in regulating splicing (Figure 1). Indeed, RNA-sequencing analysis revealed Zmat3-dependent alternative splicing of hundreds of transcripts, including some transcripts directly bound by Zmat3. The transcripts encoding the p53 negative regulators Mdm2 (transformed mouse 3T3 cell double minute 2) and Mdm4 (transformed mouse 3T3 cell double minute 4) were both Zmat3-bound and alternatively spliced, with the most dramatic effect on Mdm4 isoform expression. Zmat3-expressing cells predominantly expressed Mdm4-S (Mdm4 short) in which exon 6 is skipped and a premature stop codon is introduced, destabilizing the transcript through nonsense-mediated RNA decay. Importantly, Mdm4 exon 6 skipping is known to trigger p53 stabilization4, and an Mdm2 isoform lacking sequences involved in p53 binding is also enriched in Zmat3-expressing cells. Therefore, an important aspect of Zmat3 action in tumor suppression may be to reinforce p53 activity through the regulation of Mdm4 and Mdm2 splicing. However, Zmat3 overexpression inhibits the proliferation of p53 null cells, indicating that Zmat3 has tumor-suppressive activities beyond feeding back to p53. Accordingly, we found that Zmat3 directly binds and regulates the splicing of transcripts encoding proteins involved in varied cellular processes, such as adhesion (e.g. Dst, dystonin) and polarity signaling (e.g. Dlg1, discs large MAGUK scaffold protein 1). Similarly, ZMAT3 was recently shown to contribute to p53-mediated transformation suppression in human colorectal cancer cells by driving alternative splicing of CD44 (CD44 molecule), which encodes a cell adhesion protein5. Zmat3 also directly binds and directs alternative splicing of transcripts encoding splicing modulators such as Dhx9 (DEAH (Asp-Glu-Ala-His) box polypeptide 9). These observations suggest that Zmat3 can also indirectly modulate splicing programs to broadly affect cellular biology, consistent with our observations that many alternatively spliced transcripts affected by Zmat3 status are not directly bound by Zmat3.
Figure 1.
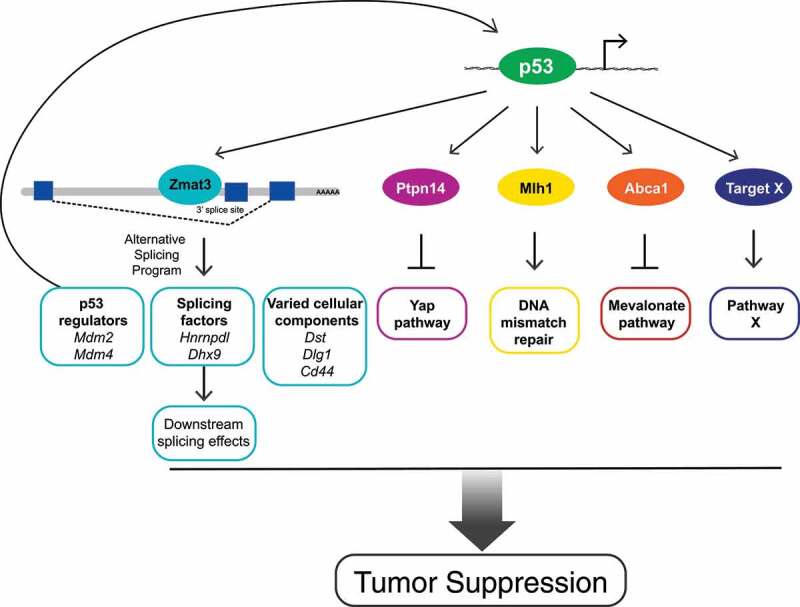
Potential mechanism of p53 tumor suppression via Zmat3 and other target genes. p53 transcriptionally activates numerous target genes with roles in tumor suppression, including Zmat3 (zinc finger matrin-type 3), encoding an RNA-binding protein. Direct Zmat3 binding to transcripts encoding negative regulators of p53 positions Zmat3 in a positive feedback loop with p53, amplifying the p53 network. In addition, Zmat3 binds and regulates alternative splicing of transcripts encoding proteins involved in various cellular processes that modulate transformation. Other tumor-suppressive p53 target genes include Ptpn14 (protein tyrosine phosphatase non-receptor type 14), which inhibits the Yap pathway; Mlh1 (mutL homolog 1), which promotes DNA repair; and Abca1 (ATP binding cassette subfamily A member 1), which inhibits the mevalonate pathway. “X” represents additional unknown target genes which may vary with context. Together, these targets comprise p53 tumor suppression function
The plethora of p53-inducible genes involved in diverse p53 responses suggests that p53-mediated tumor suppression requires the coordinated action of multiple downstream effectors. Indeed, in an shRNA screen in cultured human cancer cells using a library targeting p53 and its downstream target genes, the only shRNAs enriched with passage in vitro were against p53, suggesting that no single p53 target gene displayed the potent anti-proliferative effect of p536. Similarly, in our study, attenuation of Zmat3 expression only accounted for part of the p53-deficient phenotype, suggesting that the combined action of multiple target genes is required for tumor suppression. Moreover, the specific p53 transcriptional program important for tumor suppression may be dependent on context, such as the cell type or oncogenic signal. Remarkably, ZMAT3 expression is highly p53-dependent in various cell types, suggesting that it may be part of a “core” p53 tumor suppressor program. Indeed, Zmat3 emerged as a tumor suppressor not only in our screens and in lung and liver cancer mouse models but also in a lymphoma mouse model7. In addition to Zmat3, several p53 target genes involved in diverse cellular processes, including Abca1 (ATP binding cassette subfamily A member 1), Mlh1 (mutL homolog 1), and Ptpn14 (protein tyrosine phosphatase non-receptor type 14)8, have been recently implicated in tumor suppression (Figure 1). However, whether these genes act in a context-specific manner or are core elements of the p53 pathway remains unclear.
Deconstructing the pathways downstream of p53 is ultimately important for developing therapies for p53-deficient tumors. For example, the discovery of Ptpn14 and Abca1 as components of p53-mediated tumor suppression suggests that inhibiting Yap1 (Yes1 associated transcriptional regulator) with drugs like verteporfin and the mevalonate pathway with drugs like statins may be effective therapeutic strategies in p53-deficient pancreas and liver cancers, respectively (Figure 1). Splicing dysregulation has recently emerged as an important driver of cancer, and our work on Zmat3 provides a link between p53 and regulation of RNA splicing, as well as revealing an alternative splicing program that could be exploited therapeutically. Our findings demonstrate that Zmat3 opposes expression of the full-length Mdm4-fl isoform by promoting exon 6 skipping, suggesting the potential of modulating this pathway. Splicing regulators, such as PRMT5 (protein arginine methyltransferase 5), can promote Mdm4-fl accumulation9, and thus PRMT5 inhibitors could drive Mdm4-S isoform expression, mimicking Zmat3, representing a promising opportunity for therapeutic intervention through p53 activation in cancers with intact p5310. Identifying drugs that could mimic other ZMAT3-dependent splicing changes could reveal therapies that are effective in p53-deficient tumors. Further investigation into p53 regulation of splicing and the tumor suppression mechanisms of other p53 effectors may reveal new starting points for the development of improved cancer treatments.
Acknowledgments
We thank Anthony M. Boutelle for the critical reading of the commentary.
Funding Statement
This work was supported by the American Cancer Society [PF-12-195-01-TGB]; National Institutes of Health [R35 CA197591]; National Institutes of Health [F32 CA162681-02]; Tobacco-Related Disease Research Program [28IP-0037].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, Zhu C, Houlihan SL, Mello SS, Yee BA, et al. Zmat3 is a key splicing regulator in the p53 tumor suppression program. Mol Cell. 2020;80(3):1–3. doi: 10.1016/j.molcel.2020.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brady CA, Jiang D, Mello S, Johnson T, Jarvis L, Kozak M, Broz D, Basak S, Park E, McLaughlin M, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145(4):571–3. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bersani C, Huss M, Giacomello S, Xu L-D, Bianchi J, Eriksson S, Jerhammar F, Alexeyenko A, Vilborg A, Lundeberg J, et al. Genome-wide identification of Wig-1 mRNA targets by RIP-Seq analysis. Oncotarget. 2016;7(2):1895–1911. doi: 10.18632/oncotarget.6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardot B, Bouarich-Bourimi R, Leemput J, Lejour V, Hamon A, Plancke L, Jochemsen AG, Simeonova I, Fang M, Toledo F, et al. Mice engineered for an obligatory Mdm4 exon skipping express higher levels of the Mdm4-S isoform but exhibit increased p53 activity. Oncogene. 2015;34(22):2943–2948. doi: 10.1038/onc.2014.230. [DOI] [PubMed] [Google Scholar]
- 5.Muys BR, Anastasakis DG, Claypool D, Pongor L, Li XL, Grammatikakis I, Liu M, Wang X, Prasanth KV, Aladjem MI, et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev. 2021;35(1–2):102–116. doi: 10.1101/gad.342634.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrysik Z, Galbraith MD, Guarnieri AL, Zaccara S, Sullivan KD, Pandey A, MacBeth M, Inga A, Espinosa JM.. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017;27(10):1645–1657. doi: 10.1101/gr.220533.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janic A, Valente LJ, Wakefield MJ, Di Stefano L, Milla L, Wilcox S, Yang H, Tai L, Vandenberg CJ, Kueh AJ, et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med. 2018;24(7):947–953. doi: 10.1038/s41591-018-0043-5. [DOI] [PubMed] [Google Scholar]
- 8.Boutelle AM, Attardi LD.. p53 and tumor suppression: it takes a network. Trends Cell Biol. 2021. doi: 10.1016/j.tcb.2020.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, Guccione E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013;27(17):1903–1916. doi: 10.1101/gad.219899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerhart SV, Kellner WA, Thompson C, Pappalardi MB, Zhang X-P, Montes De Oca R, Penebre E, Duncan K, Boriack-Sjodin A, Le B, et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci Rep. 2018;8(1):9711. doi: 10.1038/s41598-018-28002-y. [DOI] [PMC free article] [PubMed] [Google Scholar]