

ABSTRACT
For recognition of specific regulatory sequences in the genome (i.e., response elements, REs), the tumor suppressor protein 53 kDa (p53) exhibits dose-dependent selectivity. In general, binding to REs linked to target genes involved in the positive regulation of cell death requires higher levels of p53 than those connected to cell survival. Our recent findings provide a mechanistic explanation for this phenomenon. Specifically, we demonstrate that subtle differences in DNA shape, encoded in RE DNA sequence, determine the utilization of two biochemically distinct DNA-binding modes, ultimately connected to different biological outcomes.
KEYWORDS: p53, DNA binding, genome recognition, cell fate
Cellular adaptation is essential to survival in a changing environment. Among the most potent mechanisms of adaptation is the reprogramming of gene expression patterns. This process is typically orchestrated by the signal-induced activation of various transcription factors, most of which are idiosyncratic and can only induce the induction of a single transcriptional program, and hence, a single phenotypic change. However, some transcription factors can interpret an array of upstream signals and selectively activate one of several gene expression programs, leading to distinct cell fates. For example, the sex determining region Y-box 2 transcription factor (SOX2) can drive either pluripotency or neuronal differentiation, depending on the context1. Similarly, the MYC proto-oncogene, and bHLH transcription factor can induce either cell cycle progression or cell death.2 Even more complex is the tumor suppressor protein 53 kDa (p53), which, in response to different stress signals can induce any of several biological pathways (e.g., cell cycle arrest, apoptosis, senescence, etc.).3 Each of these pathways is regulated by a distinct set of direct p53 transcriptional targets. For functionally promiscuous transcription factors like SOX2, MYC, and p53, a fundamental question is: How can a single, sequence-specific transcription factor selectively control the expression of distinct sets of target genes? For p53 specifically, the most prominent theory holds that it exhibits dose-dependent target gene activation, with response elements (REs) linked to pro-survival targets having a higher affinity for p53, and those connected to genes involved in activation of apoptosis having a lower affinity.4 Our recent studies provide an explanation for this phenomenon by uncovering an RE code which enforces different mechanisms of p53 binding at distinct classes of genomic targets.5
p53 target gene selectivity has been linked to the status of the lysine amino acid at position 120 (Lys120),6,7 which resides in the first highly flexible loop (L1). Specifically, activation of cell stress pathways results in acetylation of this residue,8 which in turn correlates with the induction of apoptosis. Furthermore, a conservative lysine-to-arginine (Arg) mutation (K120R) negatively impacts induction of this process, while preserving all the other p53 biological functions.6,7,9 However, the mechanism explaining this selectivity remained unknown. Based on the flexibility of Lys120 within the L1 loop, as well as degeneracy at RE positions contacting this residue, we hypothesized that the Lys120-dependent selectivity is linked to an RE code. To elucidate common sequence features of Lys120-dependent binding sites, we analyzed genome-wide binding of the wild-type (WT)- and K120R-p53.5 Chromatin immunoprecipitation followed by sequencing (ChIP-seq) revealed that over half of the WT-p53 binding sites in the genome were deficient in K120R-p53 binding. Not surprisingly, the group of K120R-unbound sites included the vast majority of REs linked to target genes involved in apoptosis. Computational analysis of the DNA sequence of REs revealed that the major sequence disparity between sites sensitive or insensitive to K120R mutation was in positions 3, 8, 13, and 18 in the 20-bp RE. More specifically, it demonstrated that K120R-unbound REs were enriched for G/C bps at those positions, while there was a preference for A/T bps in the K120R-bound sites. Most importantly, this distinction separated sites connected to different p53 effector pathways, by showing that most prominent “pro-survival” targets (e.g., p21/CDKN1A, PLK2, GDF15) are linked to A/T-rich, K120R-bound REs, while most “pro-apoptotic” targets (e.g., BAX, FAS, NOXA) have G/C-rich, K120R-unbound REs.
In vitro DNA-binding analysis confirmed this newly derived RE code, once again showing that the enrichment in G/C bps at positions 3, 8, 13, and 18 was connected to K120R-sensitivity and lower affinity*. In vitro DNA-binding experiments also lead us to a more significant advance – the discovery of two distinct p53 DNA-binding modes (Figure 1). Performing these experiments under two different salt concentrations (200 mM NaCl and 275 mM NaCl) allowed us to gain insight into the nature of the p53 binding mechanism utilized at different REs. Essentially, we found that p53 uses a non-electrostatic binding mode at A/T-rich, high-affinity REs (Figure 1, left) and an electrostatic binding mode at G/C-rich, low-affinity binding sites (Figure 1, right). Examining structural data, revealed the mechanistic explanation for this observation. Compering crystal structures of p53 in a complex with different DNA molecules showed that A/T vs. G/C content at positions 3, 8, 13, and 18 impacts the DNA shape. Enrichment in A/T bps leads to a narrower minor and a wider major groove, while G/C-rich REs are characterized by a wider minor and a narrower major groove. Structural analysis also showed that the Arg248 residue senses differences in the minor groove width, adapts different conformations, and makes biophysically different interactions with two groups of binding sites. Specifically, Arg248 makes hydrophobic interactions with A/T-rich REs and electrostatic interactions with G/C-rich sites. At the same time, the Lys120 residue senses the major groove width, explaining the inability of K120R-p53 mutant to bind the narrow major groove of G/C-rich, low-affinity REs. We confirmed our findings in vitro and in vivo by performing targeted mutations of a panel of natural and artificial REs. Remarkably, by changing the genomic BAX RE from G/C-rich to A/T-rich (using the CRISPR/Cas9 system in human cells), we transformed this RE to a higher affinity and K120R-bound site, while subsequently altering cell fate.
Figure 1.
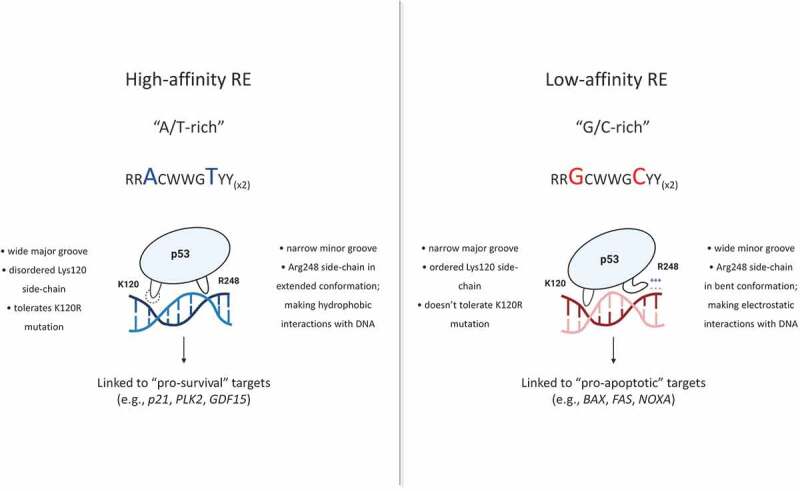
Differential recognition of high-affinity (“pro-survival”) and low-affinity (“pro-apoptotic”) responsive elements by p53. Differences in DNA shape guide distinct Arg248 conformations and interactions with distinct binding sites, ultimately regulating the DNA-binding mode utilized by p53
In summary, our findings offer a mechanistic explanation to a previously raised question – how can the p53 protein induce diverse gene expression programs in a dose-dependent manner? We show that the key player in this process is, unexpectedly, the DNA shape. To our knowledge, there have not been previous studies that identify the roles of local DNA shape recognition and differential arginine-minor groove interactions as essential regulators of sequence-specific transcription factor target gene selectivity. Our findings may represent a single example of a biophysical mechanism that may be used by other DNA-binding proteins to control cell fate decisions.
References
- 1.Zhang S, Bell E, Zhi H, Brown S, Imran SAM, Azuara V, Cui W.. OCT4 and PAX6 determine the dual function of SOX2 in human ESCs as a key pluripotent or neural factor. Stem Cell Res Ther. 2019;10(1):1. doi: 10.1186/s13287-019-1228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMahon SB. MYC and the control of apoptosis. Cold Spring Harb Perspect Med. 2014;4(7):a014407. doi: 10.1101/cshperspect.a014407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27(50):6507–3. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szak ST, Mays D, Pietenpol JA. Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol. 2001;21(10):3375–3386. doi: 10.1128/MCB.21.10.3375-3386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farkas M, Hashimoto H, Bi Y, Davuluri RV, Resnick-Silverman L, Manfredi JJ, Debler EW, McMahon SB. Distinct mechanisms control genome recognition by p53 at its target genes linked to different cell fates. Nat Commun. 2021;12(1):484. doi: 10.1038/s41467-020-20783-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24(6):841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24(6):827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 8.Mellert HS, McMahon SB. Biochemical pathways that regulate acetyltransferase and deacetylase activity in mammalian cells. Trends Biochem Sci. 2009;34(11):571–578. doi: 10.1016/j.tibs.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]