

Abstract
Background
Muscle‐invasive bladder cancer (MIBC) is a heterogeneous disease with varying clinical courses and responses to treatment. To improve the prognosis of patients, it is necessary to understand such heterogeneity.
Methods
We used single‐sample gene set enrichment analysis to classify 35 MIBC cases into immunity‐high and immunity‐low groups. Bioinformatics analyses were conducted to compare the differences between these groups. Eventually, single‐cell mass cytometry (CyTOF) was used to compare the characteristics of the immune microenvironment between the patients in the two groups.
Results
Compared with patients in the immunity‐low group, patients in the immunity‐high group had a higher number of tumor‐infiltrating immune cells and greater enrichment of gene sets associated with antitumor immune activity. Furthermore, positive immune response‐related pathways were more enriched in the immunity‐high group. We identified 26 immune cell subsets, including cytotoxic T cells (Tcs), helper T cells (Ths), regulatory T cells (Tregs), B cells, macrophages, natural killer (NK) cells, and dendritic cells (DCs) using CyTOF. Furthermore, there was a higher proportion of CD45+ lymphocytes and enrichment of one Tc subset in the immunity‐high group. Additionally, M2 macrophages were highly enriched in the immunity‐low group. Finally, there was higher expression of PD‐1 and Tim‐3 on Tregs as well as a higher proportion of PD‐1+ Tregs in the immunity‐low group than in the immunity‐high group.
Conclusion
In summary, the immune microenvironments of the immunity‐high and immunity‐low groups of patients with MIBC are heterogeneous. Specifically, immune suppression was observed in the immune microenvironment of the patients in the immunity‐low group.
Keywords: immune microenvironment, mass cytometry, muscle‐invasive bladder cancer, ssGSEA, tumor heterogeneity
In this study, we investigated the relationship between immune infiltration and cancer development in MIBC patients. We collected 35 MIBC cancerous samples and used their RNA profile data to calculate the scores of 29 immune signatures in each sample using single sample gene set enrichment analysis (ssGSEA). Bioinformatics analyses including differential gene expression analysis, GO molecular function enrichment analysis, GSEA were performed to investigate the differences between the immunity‐high and immunity‐low groups. We also analyzed the composition and function of infiltrating immune cells in MIBC tissues using single‐cell mass cytometry. Our findings reveal the presence of heterogeneity in the immune micro‐environment and provides guidance for the therapy of patients with MIBC.

1. INTRODUCTION
Bladder cancer (BC) is the 10th most common cancer globally, with an incidence rate four times higher in women than in men. 1 Smoking and occupational exposure to chemical and water pollutants are major risk factors for this type of cancer. 2 BC is mainly classified into non‐muscle‐invasive BC (NMIBC) and muscle‐invasive BC (MIBC), with MIBC accounting for 25% of the cases. 3 Although the majority of MIBCs are identified by the first diagnosis, 10%–20% of the cases result from progression of NMIBC. When compared to NMIBC, MIBC presents with a more aggressive phenotype characterized by a higher risk of metastasis and poor prognosis, with a 5‐year survival rate of 60% in patients without recurrence and <10% in patients who have already developed distant metastasis. 4
Surgery is the main treatment for MIBC. Besides, stage T2 BC can be treated with neoadjuvant chemotherapy preoperatively, with the decision to use systemic chemotherapy and/or radiotherapy postoperatively on the basis of pathologic findings. The use of platinum‐based chemotherapeutics as first‐line agents for advanced BC has been hampered by the presence of strong myelosuppressive toxic effects that reduce leukocyte and platelet levels in patients. Consequently, they are only used in half of all patients with BC. 5 Additionally, immune checkpoint inhibitors (ICIs) are emerging therapeutic agents that can effectively improve the survival time of patients with MIBC. 6 The inhibition of immune checkpoints promotes tumor regression by reactivating immune cytotoxicity in MIBC. 7 However, there are differences in the response to treatment among patients, mainly due to the heterogeneity that exists among individuals, even for the same drug. Indeed, some patients with MIBC show continued disease progression, even after receiving the corresponding treatments. 8 As a result, only 20%–30% of MIBC patients respond to treatment with ICIs. 9 Therefore, there is an urgent need to identify new and effective drug targets to enhance therapeutic outcomes for these patients.
The molecular characterization of samples is essential for the accurate prediction of responses to therapeutics such as ICIs. 10 Biomarkers that have been shown to correlate with responsiveness to immunotherapy in BC include tumor mutational burden, 11 tumor molecular subtype, 12 CD8+ tumor‐infiltrating lymphocytes (TILs), and PD‐L1 expression on other immune cells. 13 Transcriptome profiling facilitates the molecular classification of BC, leading to the adoption of more precise treatment regimens and accurate prediction of treatment outcomes.
Research on BC has shown significant molecular heterogeneity. 14 , 15 Tumor heterogeneity is one of the main factors affecting the efficacy of chemoradiotherapy and the prognosis of surgery in different patients. 16 A study on the heterogeneity between tumors will not only help in understanding disease pathogenesis, but will also act as a guide for personalized therapy, which may help improve patient survival and prognosis. Currently, single‐cell technologies enable the study of tumor heterogeneity by analyzing tumor evolutionary relationships. 17 Mass cytometry (CyTOF) applies the single‐cell theory by employing metal isotope‐labeled antibodies that allow the simultaneous detection of up to 40 parameters in a single cell. CyTOF overcomes the effects caused by the overlap of emission spectral signals between traditional flow channels, enabling precise analysis of cell subpopulations. 18 This technique has been developed for nearly 10 years, since the cellular fraction of human peripheral blood was first detected. 19 CyTOF plays an important role in the analysis of heterogeneity across multiple solid tumors, such as kidney cancer, 20 lung adenocarcinoma, 21 and breast cancer. 22
In this study, we investigated the relationship between immune infiltration and cancer development in patients with MIBC. We collected 35 MIBC samples and used their RNA profile data to calculate the scores of 29 immune signatures in each sample using single‐sample gene set enrichment analysis (ssGSEA). 23 Bioinformatics analyses, including differential gene expression analysis, GO molecular function enrichment analysis, and GSEA were performed to investigate the differences between the immunity‐high and immunity‐low groups. We also analyzed the composition and function of infiltrating immune cells in MIBC tissues using single‐cell CyTOF. We then searched for potential prognostic biomarkers by comparing the differences in subsets of immune cells between the two groups. Our findings reveal the presence of heterogeneity in the immune microenvironment and may provide guidance for the therapy of patients with MIBC.
2. MATERIALS AND METHODS
2.1. Patients
This study was approved by the Human Subject Committee of Guangxi Medical University (approval number: 2019 [KY‐E‐088]). The 35 MIBC tissue samples used in this study were collected at The First Affiliated Hospital of Guangxi Medical University in China from January 2018 to June 2019. Patients undergoing chemo‐ or radiotherapy before resection were excluded, and the pathology results were confirmed by two experienced pathologists. All participants provided informed consent.
2.2. Acquisition of RNA sequencing data
Total RNA was extracted from tissues using TRIzol® reagent (Invitrogen) according to the manufacturer's protocol, and RNA quality was evaluated using a Thermo Scientific Nanodrop 2000 spectrophotometer. Ribosomal RNA (rRNA) was then eliminated from total RNA using Ribo‐Zero rRNA removal kits (Illumina), according to the manufacturer's instructions. Next, cDNA libraries were constructed by reverse transcription of the purified mRNAs. The libraries were then amplified using PCR, followed by sequencing for 150 cycles on an Illumina HiSeq 4000 sequencer (Illumina). The quality of the raw sequencing data was assessed using FastQC software. Preprocessing of the raw data (including adapter trimming and quality filtering) was performed using fastp. 24 The clean data were then mapped to the human genome (hg19) using HISAT2. 25 Finally, StringTie was used to separately assemble the RNA sequences, 26 , 27 while Cufflinks was used to merge the data. 28
2.3. ssGSEA
Enrichment of 29 immune signatures for each MIBC sample was quantified using the ssGSEA score (enrichment level) as previously described. 23 The enrichment levels of the 29 immune signatures were then used to perform hierarchical clustering. Finally, the samples were divided into immunity‐high and immunity‐low groups, based on the results of clustering.
Identification of differentially expressed genes (DEGs), and pathway enrichment analysis.
The R package DESeq2 was used to analyze DEGs, using adjusted p value <0.05, and |Log2(foldchange)| >1.5, as the cut‐off criteria. Hierarchical clustering analysis was conducted using a heatmap. Gene ontology (GO) enrichment analysis was performed using clusterProfiler. 29 Statistical significance was set at p < 0.05.
2.4. Gene set enrichment analysis (GSEA)
GSEA was performed using expression profiles of 35 MIBC samples. The specific parameter settings were as follows: gene set database: c2.cp.kegg.v7.2.symbols.gmt; number of permutations: 1000; enrichment statistic: weighted; collapse dataset to gene symbols: False; metric for ranking genes: Signal2Noise. Normalized enrichment score (NES) >1 and nominal p value (NOM p‐val) <0.05, were considered to indicate significant differences.
2.5. CyTOF marker labeling and detection
The protocol used to dissect the tumor tissues was described previously. 30 The cell suspensions were preserved in liquid nitrogen prior to staining. Selected antibodies (listed in Figure 3B) were conjugated to isotopic tags using a MaxPar X8 Antibody labeling kit (Fluidigm) according to the manufacturer's instructions. Cell suspensions were removed from liquid nitrogen and 1.5 million living cells were taken from each sample. Cells were stained with cisplatin (Fluidigm) to a final concentration of 5 mmol/L for determining viability. The cell suspensions were then incubated with human Fc receptor blocking solution (Biolegend) for 10 min, followed by incubation with surface antibody cocktail for 1 h. Afterward, the cells were washed and incubated with nuclear antigen staining buffer working solution for 30 min at 25℃, followed by incubation with intracellular antibody cocktail for 1 h. After washing, the samples were incubated in a fresh solution of 1.6% paraformaldehyde at room temperature for 10 min and then stained with a DNA intercalator (Fluidigm) overnight at 4°C. Before the analysis on a CyTOF2 instrument (Fluidigm), cells were prepared with subsequent washes in cell staining buffer and deionized water to remove buffer salts. Finally, 10% EQ™ Four element calibration beads were used to resuspend the cells. Labeled samples were analyzed using the CyTOF2 instrument at a rate <500 events/s.
FIGURE 3.
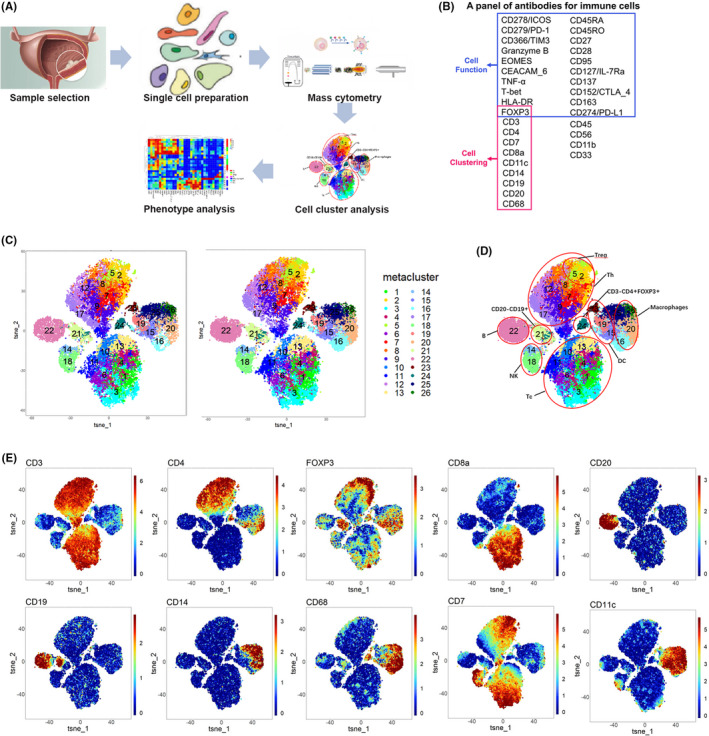
Description of immune microenvironment by CyTOF. A, Workflow of CyTOF. B, Antibodies panel of CyTOF. C, The t‐SNE maps of the immunity‐high and immunity‐low groups. D, The t‐SNE maps of antibodies for cell typing. E, The t‐SNE maps of tumor‐infiltrating immune cells in cancer tissues of 35 patients with MIBC
2.6. Analysis of CyTOF data
CyTOF software v6.7 was used to normalize and merge the resulting flow cytometry standard files (FCS). Analyzed data and FCS files were then uploaded to the online software Cytobank (https://www.cytobank.org/) or R packages (such as cytofkit, Rtsne, FlowSOM, cytofexplorer, and ggplot) to perform analysis and manual gating.
2.7. Statistical analysis
The mean values of independent samples were analyzed using either t‐ or Mann‐Whitney U tests. The log‐rank test was used for survival analysis. All analyses were conducted using SPSS (version 24.0; IBM Corp), and p values <0.05 were considered statistically significant.
3. RESULTS
3.1. Immunophenotyping based on ssGSEA
To investigate the presence of immune heterogeneity in MIBC, we collected 35 MIBC samples. ssGSEA was used to assess and grade the enrichment of 29 immune signatures for each sample. Through ssGSEA and cluster analyses, the 35 samples were classified into immunity‐high (n = 18) and immunity‐low (n = 17) groups (Figure 1A). We found that there was a higher enrichment of natural killer (NK) cells, neutrophils, helper T cells (Ths), B cells, mast cells, T cells (Tcs), TILs, macrophages, and dendritic cells (DCs) in the immunity‐high group than in the immunity‐low group. In addition, immune function associated gene sets such as type I and type II interferon response, immune checkpoint, Tc costimulation, cytolytic activity, and antigen‐presenting cells costimulation were also highly enriched in the immunity‐high group (Figure 1A). These results indicated that there was a significant difference in the infiltration of tumors by immune cells between the two groups. To determine if the above differences had an effect on the pathology of patients with MIBC, a comparative analysis of their clinical parameters was conducted. The results showed that there were no significant differences in the clinical parameters between the two groups (Figure 1B). Furthermore, the results of the log‐rank test and Kaplan‐Meier curves revealed that patients in the immunity‐high group had a longer overall survival (OS) time than patients in the immunity‐low group (Figure 1C). In summary, ssGSEA showed that there was a significant difference in the cell composition and activation status of the immune microenvironment in the immunity‐high and immunity‐low groups, which may contribute to the differences in prognosis between the two groups.
FIGURE 1.
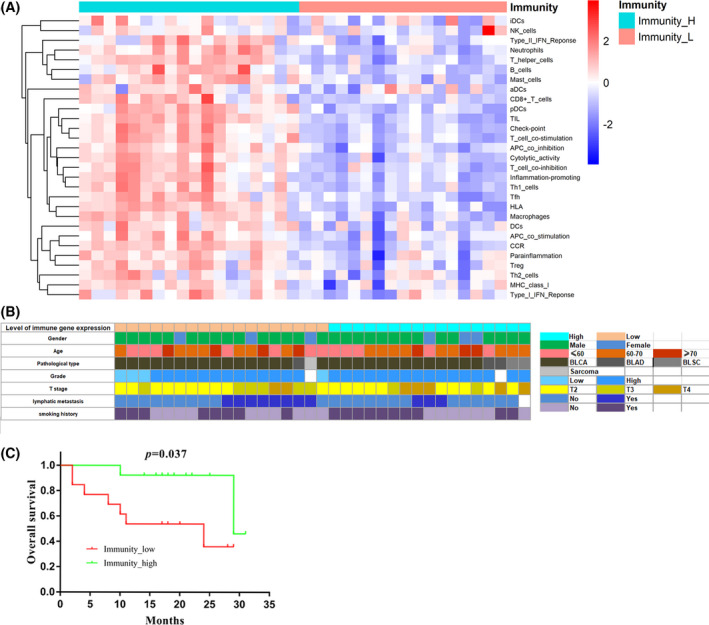
Immunophenotyping of MIBC based on ssGSEA. A, Clustering of 29 immune signatures. Immunity_H, immunity‐high; Immunity_L, immunity‐low. B, Clinical parameter of 35 patients with MIBC. C, Kaplan‐Meier curves for overall survival of MIBC patients (n = 27) by Immunophenotyping. Immunity‐high (n = 14); Immunity‐low (n = 13). p: p value of Log‐Rank test; p < 0.05 was considered to indicate statistical differences
To investigate the differences in molecular mechanisms between the immunity‐high and immunity‐low groups, we identified DEGs between the two groups. In total, 541 upregulated and 786 downregulated mRNAs in the immunity‐high group, compared with the immunity‐low group, were obtained from differential gene expression analysis (Figure 2A,B). Pathway enrichment analysis was conducted for the 541 mRNAs that were upregulated in the immunity‐high group. As shown in Figure 2C, the GO biological process terms revealed a high enrichment of genes functioning in the immune system, including Tc activation, regulation of Tc activation, leukocyte proliferation, migration, chemotaxis, cell‐cell adhesion, and mononuclear cell proliferation. GSEA results showed that the top ten gene sets (ranked by NES) that were enriched in the immunity‐high group, when compared to the immunity‐low group, included chemokine signaling pathway, cytokine to cytokine receptor interaction, cell adhesion molecules, hematopoietic cell lineage, primary immunodeficiency, systemic lupus erythematosus, leukocyte transendothelial migration, intestinal immune network for IgA production, calcium signaling pathway, and viral myocarditis gene sets. Furthermore, the B cell receptor signaling, Tc receptor signaling, and NK cell‐mediated cytotoxicity pathways were enriched in the immunity‐high group (Figure 2D). These results are consistent with our previous results showing that the immunity‐high group tended to have a more activated status of immunity. The above results suggested that the immunity‐high group and immunity‐low group immune microenvironments were heterogeneous.
FIGURE 2.
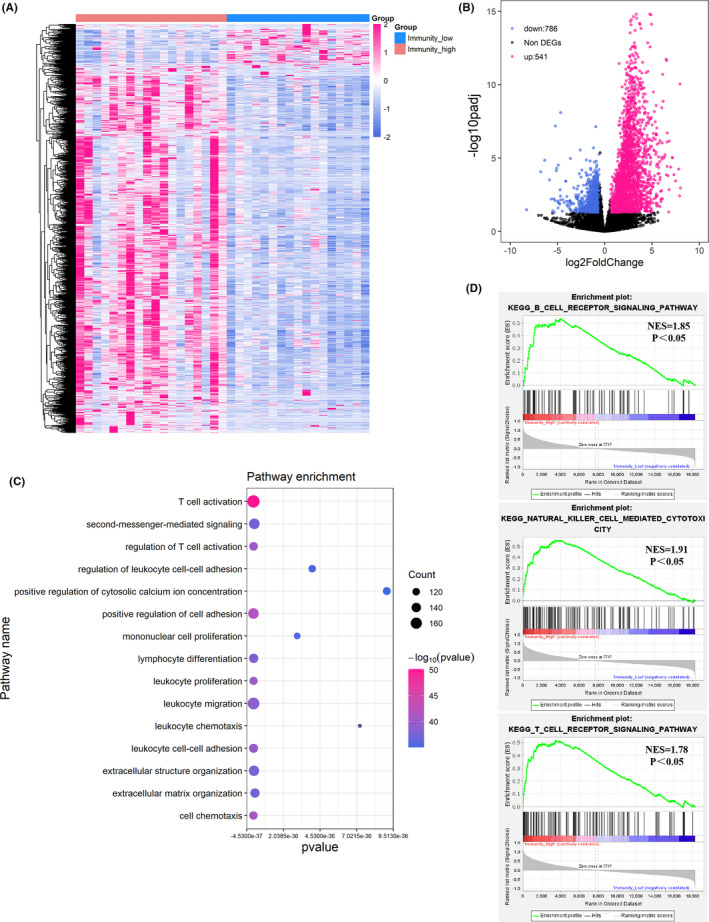
Differentially expressed genes and enrichment analysis. A, Heatmap of the differentially expressed genes (DEGs) between the immunity‐high and immunity‐low groups. Samples (column) and genes (row) were clustered by unsupervised hierarchical cluster analysis. B, Volcano plot showed the DEGs between the immunity‐high and immunity‐low groups. Red dots represented the significantly upregulated genes in the immunity‐high group compared with immunity‐low group (Log2(foldchange)) >1.5 and adjusted p < 0.05). Blue dots represented the significantly downregulated genes in the immunity‐high group compared with immunity‐low group (Log2(foldchange)) <−1.5 and adjusted p < 0.05). Black dots represented non‐DEGs. C, Go‐bubble plot showed top 15 pathways of GO enrichment analysis, ranked by p value. D, Significant pathways identified by GSEA
3.2. Characterization of the immune microenvironment of MIBC using CyTOF
CyTOF was used to further explore the heterogeneity of the immune microenvironment between the immunity‐high and immunity‐low groups. This technique can simultaneously detect over 40 cell markers at the single‐cell level; it was used to accurately characterize intratumoral immune cells of the 35 MIBC samples. The workflow of CyTOF is described in Figure 3A. First, single‐cell suspensions from tissues were acquired through manual dissociation. Then, they were labeled with 34 immune‐associated antibodies, including the cell typing and cellular function panels (Figure 3B). CyTOF was used to detect stained immune cells, followed by dimension reduction of the high‐dimensional data obtained. The cell proportions and the level of expression of cellular surface markers were determined and compared between the immunity‐high and immunity‐low groups. Based on our antibody panels, t‐distributed stochastic neighbor embedding (t‐SNE) was used to generate two‐dimensional images to visualize tumor infiltration by CD45+ lymphocytes. CD45+ lymphocytes were divided into 26 cell subsets (or clusters) based on the similarity in the expression of cellular surface markers (Figure 3C). Based on such expression, markers, we identified clusters 1, 3, 4, 6, 10, 11, and 13 as Tcs (CD3+ and CD8+); clusters 2, 5, 7, 8, 9, 12, and 17 as Ths (CD3+ and CD4+); clusters 2 and 5 as regulatory Tcs (Tregs; CD3+, CD4+, and FOXP3+); cluster 22 as B cells (CD19+ and CD20+); clusters 16, 20, 25, and 26 as macrophages (CD14+ and CD68+); clusters 15, 19, and 23 as DCs (CD3‐ and CD11c+); clusters 14 and 18 as NK cells (CD3‐ and CD56+); and clusters 21 and 24 as other cell types (Figure 3D,E). Cluster abundance volcano plots showed that cluster 16 (M2 macrophages; CD68+ and CD163+) and cluster 23 (DCs) were more enriched in the immunity‐low group, whereas cluster 10 (Tcs) was more enriched in the immunity‐high group (Figure 4A). After manual gating of CD45+ lymphocytes, Tcs, Ths, Tregs, B cells, macrophages, and DCs on Cytobank, the proportion of CD45+ lymphocytes was found to be higher in the immunity‐high group than in the immunity‐low group, which was consistent with our previous results (Figure 4B). We then used CyTOF to accurately characterize the intratumoral immune cells of the 35 MIBC samples and discovered that there were significant differences in the composition of tumor‐infiltrating immune cells between the two groups of patients. This lays a foundation for further analysis of the immune microenvironment of MIBC.
FIGURE 4.
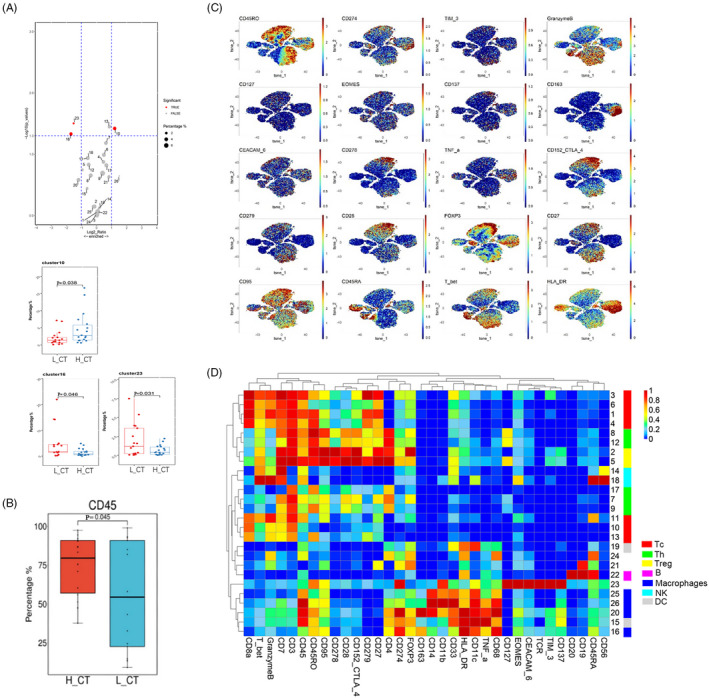
Differences of immune cell composition and function between the immunity‐high and immunity‐low groups A, Volcano plot and Box plots showed the differential expressed tumor‐infiltrating immune cell subsets between the immunity‐high and immunity‐low groups. H_CT: cancer tissues of immunity‐high group; L_CT cancer tissues of immunity‐low group. Red dots represented the significantly differential expressed cell subsets (|Log2(foldchange)| >1 and p < 0.05). Gray dots represented the non‐significantly differential expressed cell subsets (|Log2(foldchange)| <1 or p > 0.05). B, Box plots show differentially expressed of tumor‐infiltrating lymphocytes between the immunity‐high and immunity‐low groups. C, The t‐SNE maps of antibodies for cellular function. D, The heatmap showed expressions of 34 markers in 26 tumor‐infiltrating immune cell subsets. p: p value of t‐test or Mann‐Whitney U test; p < 0.05 was considered to indicate statistical differences
3.3. Immune suppression in patients of the immunity‐low group
Since there was a difference in the composition of TILs between the immunity‐high and immunity‐low groups, we speculated that there was also a difference in the cellular functions. To determine this, we examined the expression of functional markers associated with activation, differentiation, and exhaustion in the 26 cell subsets (Figure 4C). The majority of the Tc subsets belonged to memory Tcs (CD45RO+), and high PD‐1 expression was observed in Tc subsets (clusters 1, 3, and 4). Moreover, in the Ths subsets (clusters 2, 5, 8, and 12), we observed heterogeneous coexpression of PD‐1 with the inhibitory receptor CTLA4 and the activation markers CD28 and CD278 (Figure 4D). The coexpression of high levels of PD‐1 and CTLA4 may be associated with the exhaustion phenotype. Intriguingly, higher PD‐1 and Tim‐3 expression on Tregs and higher proportions of PD‐1+ Tregs were detected in the immunity‐low group (Figure 5A,B). This suggested that there was immunosuppression in the patients in the immunity‐low group compared to patients in the immunity‐high group, accounting for their poor clinical outcomes. Taken together, it appears that the tumor microenvironment of patients with MIBC in the immunity‐low group is under stronger immunosuppressive conditions than that of patients in the immunity‐high group. Therefore, we found that a decrease in the number and function of tumor‐infiltrating immune cells helps tumors to escape immune surveillance, promoting the development of cancer, and ultimately leading to the poor prognosis of patients with MIBC.
FIGURE 5.
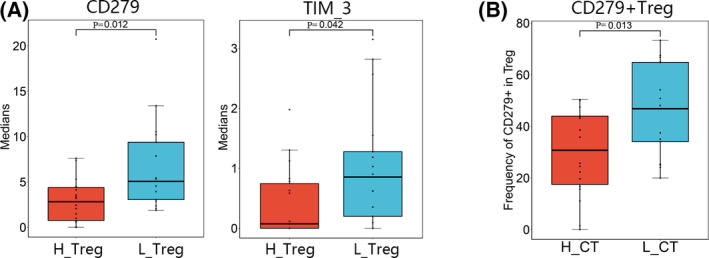
Differential maker expressions of Tregs and frequency of PD‐1+ Treg between the immunity‐high and immunity‐low groups. A, Box plots showed differentially expressed of CD279 (PD‐1) and TIM‐3 on Tregs between the immunity‐high and immunity‐low groups. H_Treg: Treg in the immunity‐high group; L_Treg: Treg in the immunity‐low group. B, Box plot showed differential frequency of CD279+ (PD‐1) Treg between the immunity‐high and immunity‐low groups. H_CT: cancer tissues of immunity‐high group; L_CT cancer tissues of immunity‐low group. p: p value of t‐test or Mann‐Whitney U test; p < 0.05 was considered to indicate statistical differences
4. DISCUSSION
MIBC is a subtype of BC that invades the detrusor muscle and has a high risk of metastasizing. 31 It is estimated that the majority (75%) of newly diagnosed BC patients have NMIBC, while only 25% have MIBC. 3 Targeted therapy and biomarker discovery for MIBC lag far behind other cancers, resulting in poor prognosis for patients. However, there has been a recent increase in the understanding of MIBC pathology based on genomic and transcriptomic profiling. 32 There have been many molecular characterizations of patients with MIBC, with different subtypes of patients showing different responses to the same treatment. This indicates that the molecular characterization of patients is critical for the development of therapeutic strategies. 33 , 34 Although molecular subtypes play a key role in guiding clinical therapy, there are still challenges in the application of molecular characterization in clinical practice. Studies have shown that intrinsically aggressive basal BCs are more sensitive to combination chemotherapy than low aggressive luminal subtypes. 35 Therefore, more studies are needed to verify the molecular characteristics of the different subtypes.
In addition to molecular characterization, the recent advent of immune checkpoint blockade has also improved the therapeutic outcomes of MIBC. 36 However, patients with similar clinical parameters and laboratory indices may have diverse responses to ICIs owing to the existence of molecular tumor heterogeneity. 37 Thus, an in‐depth study of heterogeneity is necessary to explain why only 20%–30% of MIBC cases have a favorable response to ICIs. 9 Dissection of the tumor immune microenvironment can partly explain this heterogeneity. A study reported that patients with high PD‐L1 expression on tumor‐infiltrating immune cells were associated with a better response to anti‐PD‐L1 therapy. 38 Another study 39 demonstrated that response to nivolumab, an inhibitor of PD‐1, was associated with an increase in Tc and NK cells, as well as a decrease in macrophages in patients with melanoma, indicating the importance of studying the immune microenvironment. Therefore, new knowledge on the regulatory mechanisms of the immune microenvironment of tumors based on accurate molecular typing may provide new insights into the treatment of MIBC.
In this study, we used ssGSEA to classify 35 MIBC cases into immunity‐high and immunity‐low groups. High infiltration of tumors by immune cells as well as more positive antitumor activities were observed in the immunity‐high group, which was consistent with previous findings. 23 It is known that Tcs can kill cancer cells. Thus, patients with higher infiltration of CD3+ or CD8+ Tcs in the tumor epithelium or invasive margin have a longer disease‐free survival or OS, indicating that this infiltration is a favorable prognostic factor for BCs. 40 Among immune cells, B cells are important members of humoral immunity and are considered to be independent favorable prognostic factors for MIBC. 41 Besides, NK cells exert antitumor effects under low major histocompatibility complex conditions, and it has been reported that CD56bright NK cells are positively correlated with prognosis. 42 Moreover, macrophages can differentiate into two types: pro‐inflammatory and tumoricidal M1 macrophages, and M2 macrophages, which inhibit inflammation. 43 M2 macrophages serve as “protumoral macrophages” which contribute to poor clinical prognosis and disease progression. 44 Additionally, DCs are antigen‐presenting cells that initiate an immune response by transmitting collected information to the adaptive immune system. 45 The infiltration of a tumor by some immune cells plays a positive antitumor role and improves the survival of patients. Consistently, the survival time of patients in the immunity‐high group was longer than that in the immunity‐low group. Moreover, GO and GSEA analyses showed that the immunity‐high group had a higher enrichment of several pathways related to the positive immune response than the immunity‐low group. The results of ssGSEA combined with the results of bioinformatics analysis suggest a more activated status of immunity in the patients of the immunity‐high group. Therefore, patients in the immunity‐high group had better survival and higher immune cell infiltration, consistent with previous results. 23
CyTOF overcomes the spectral overlap limitation associated with traditional flow channels while maintaining the high throughput of flow cytometry, and has been applied to uncover the heterogeneity of immune cells. 46 , 47 We used CyTOF, with an antibody panel that included cell typing and cellular function, to effectively divide the tumor‐infiltrating immune cells into 26 subsets that contain common immune cells. The analysis of cell abundance showed that clusters 16 (M2 macrophages) and 23 (DCs) were enriched in the immunity‐low group, and that cluster 10 was more enriched in the immunity‐high group. It is known that M2 macrophages have the ability to promote cancer progression as they play an antagonistic role in the immune process against tumors, whereas Tcs kill tumor cells. 43 In addition, the proportion of CD45+ lymphocytes was higher in the immunity‐high group. These results are consistent with our ssGSEA analysis findings. Results of manual gating showed that there was a higher number of Treg cells in the immunity‐low group. Tregs produce immunosuppressive cytokines, such as interleukin‐10 (IL‐10) and transforming growth factor‐β (TGF‐β), leading to the inhibition of Tcs in the tumor microenvironment. 48 Further analysis of the expression of functional markers revealed that TIM‐3 and PD‐1 were highly expressed on Tregs in the immunity‐low group. Noteworthy, it is known that TIM‐3 marked tumor‐associated Foxp3+ Tregs have a great inhibitory effect on CD8+ TILs, and targeting TIM‐3 is a potent immunotherapeutic approach. 49 Park and colleagues reported that a high expression of PD‐1 by Tregs increased the suppression of CD8+ Tcs compared to PD‐1− Tregs during chronic viral infection. 50 Altogether, our findings showed that the immunity‐low group had a relatively lower number of tumor‐infiltrating CD45+ lymphocytes. We suggest that there is immune suppression in the immune microenvironment of tumors of patients with MIBC in the immunity‐low group, which is conducive to the survival and development of the tumors. Our findings provide evidence to explain the tumor heterogeneity of MIBC and reveal more insights into its clinical treatment.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethics and Human Subject Committee of Guangxi Medical University. All experiments and methods were performed according to relevant guidelines and regulations.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Xi Wang, Lixin Pan, Qinchen Lu, Haoxuan Huang contributed equally to this work.
Funding information
This work was supported by Guangxi Clinical Research Center for Urology and Nephrology (No.2020AC03006). Guangxi Science and Technology Base and Talent Special Fund (2019AC17009, AD17195090). National Natural Science Foundation of China (81760454, 82060460). Guangxi Natural Science Fund for Innovation Research Team (2016GXNSFGA38006). Youth Science Foundation of Guangxi Medical University (GXMUYSF202108).
Contributor Information
Zhong Tang, Email: 18978868502@qq.com.
Yuanliang Xie, Email: 136641161@qq.com.
Tianyu Li, Email: 547370852@qq.com.
DATA AVAILABILITY STATEMENT
All original data throughout our manuscript are available upon reasonable request.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Islami F, Goding Sauer A, Miller KD, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. 2018;68(1):31‐54. [DOI] [PubMed] [Google Scholar]
- 3. Pang KH, Esperto F, Noon AP. Opportunities of next‐generation sequencing in non‐muscle invasive bladder cancer outcome prediction. Translational andrology and urology. 2017;6(6):1043‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kamoun A, de Reyniès A, Allory Y, et al. A consensus molecular classification of muscle‐invasive bladder cancer. Eur Urol. 2020;77(4):420‐433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nadal R, Bellmunt J. Management of metastatic bladder cancer. Cancer Treat Rev. 2019;76:10‐21. [DOI] [PubMed] [Google Scholar]
- 6. Jiang W, Zhu D, Wang C, Zhu Y. An immune relevant signature for predicting prognoses and immunotherapeutic responses in patients with muscle‐invasive bladder cancer (MIBC). Cancer Med. 2020;9(8):2774‐2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bellmunt J, Powles T, Vogelzang NJ. A review on the evolution of PD‐1/PD‐L1 immunotherapy for bladder cancer: the future is now. Cancer Treat Rev. 2017;54:58‐67. [DOI] [PubMed] [Google Scholar]
- 8. Massari F, Di Nunno V, Cubelli M, et al. Immune checkpoint inhibitors for metastatic bladder cancer. Cancer Treat Rev. 2018;64:11‐20. [DOI] [PubMed] [Google Scholar]
- 9. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet. 2017;389(10064):67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McConkey DJ, Choi W, Shen YU, et al. A prognostic gene expression signature in the molecular classification of chemotherapy‐naïve urothelial cancer is predictive of clinical outcomes from neoadjuvant chemotherapy: a phase 2 trial of dose‐dense methotrexate, vinblastine, doxorubicin, and cisplatin with bevacizumab in urothelial cancer. Eur Urol. 2016;69(5):855‐862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lv J, Zhu Y, Ji A, Zhang Q, Liao G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci Rep. 2020;40(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song BN, Kim SK, Mun JY, Choi YD, Leem SH, Chu IS. Identification of an immunotherapy‐responsive molecular subtype of bladder cancer. Ebiomedicine. 2019;50:238‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou TC, Sankin AI, Porcelli SA, Perlin DS, Schoenberg MP, Zang X. A review of the PD‐1/PD‐L1 checkpoint in bladder cancer: From mediator of immune escape to target for treatment. Urologic Oncol. 2017;35(1):14‐20. [DOI] [PubMed] [Google Scholar]
- 14. Czerniak B, Dinney C, McConkey D. Origins of bladder cancer. Annual Rev Pathol. 2016;11:149‐174. [DOI] [PubMed] [Google Scholar]
- 15. Bellmunt J, Orsola A, Leow JJ, Wiegel T, De Santis M, Horwich A. Bladder cancer: ESMO Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2014;25(Suppl 3):iii40‐iii48. [DOI] [PubMed] [Google Scholar]
- 16. Dagogo‐Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81‐94. [DOI] [PubMed] [Google Scholar]
- 17. Finotello F, Rieder D, Hackl H, Trajanoski Z. Next‐generation computational tools for interrogating cancer immunity. Nat Rev Genet. 2019;20(12):724‐746. [DOI] [PubMed] [Google Scholar]
- 18. Spitzer MH, Nolan GP. Mass cytometry: single cells. Many features. Cell. 2016;165(4):780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bendall SC, Simonds EF, Qiu P, et al. Single‐cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687‐696.21551058 [Google Scholar]
- 20. Chevrier S, Levine JH, Zanotelli VRT, et al. An immune atlas of clear cell renal cell carcinoma. Cell. 2017;169(4):736‐49.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single‐cell analyses. Cell. 2017;169(4):750‐65.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schulz D, Zanotelli VRT, Fischer JR, et al. Simultaneous multiplexed imaging of mRNA and proteins with subcellular resolution in breast cancer tissue samples by mass cytometry. Cell Syst. 2018;6(1):25‐36.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. He Y, Jiang Z, Chen C, Wang X. Classification of triple‐negative breast cancers based on Immunogenomic profiling. J Experimental Clin Cancer Res. 2018;37(1):327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884‐i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol. 2019;37(8):907‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nat Biotechnol. 2015;33(3):290‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript‐level expression analysis of RNA‐seq experiments with HISAT. StringTie and Ballgown. Nature protocols. 2016;11(9):1650‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghosh S, Chan CK. Analysis of RNA‐seq data using TopHat and cufflinks. Methods Mol Biol. 2016;1374:339‐361. [DOI] [PubMed] [Google Scholar]
- 29. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu Q, Gao J, Tang S, et al. Integrated RNA sequencing and single‐cell mass cytometry reveal a novel role of LncRNA HOXA‐AS2 in tumorigenesis and stemness of hepatocellular carcinoma. OncoTargets and therapy. 2020;13:10901‐10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sanli O, Dobruch J, Knowles MA, et al. Bladder cancer. Nature Rev Dis Primers. 2017;3:17022. [DOI] [PubMed] [Google Scholar]
- 32. Robertson AG, Kim J, Al‐Ahmadie H, et al. Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell. 2018;174(4):1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sjödahl G, Lauss M, Lövgren K, et al. A molecular taxonomy for urothelial carcinoma. Clin Cancer Res. 2012;18(12):3377‐3386. [DOI] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Choi W, Czerniak B, Ochoa A, et al. Intrinsic basal and luminal subtypes of muscle‐invasive bladder cancer. Nature Rev Urology. 2014;11(7):400‐410. [DOI] [PubMed] [Google Scholar]
- 36. Siefker‐Radtke AO, Apolo AB, Bivalacqua TJ, Spiess PE, Black PC. Immunotherapy with checkpoint blockade in the treatment of urothelial carcinoma. J Urol. 2018;199(5):1129‐1142. [DOI] [PubMed] [Google Scholar]
- 37. da Costa JB, Gibb EA, Nykopp TK, Mannas M, Wyatt AW, Black PC. Molecular tumor heterogeneity in muscle invasive bladder cancer: biomarkers, subtypes, and implications for therapy. Urologic Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 38. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387(10031):1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934‐49.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van Wilpe S, Gerretsen ECF, van der Heijden AG, de Vries IJM, Gerritsen WR, Mehra N. Prognostic and predictive value of tumor‐infiltrating immune cells in urothelial cancer of the bladder. Cancers. 2020;12(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li BO, Severson E, Pignon J‐C, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michel T, Poli A, Cuapio A, et al. Human CD56bright NK cells: an update. J Immunol. 2016;196(7):2923‐2931. [DOI] [PubMed] [Google Scholar]
- 43. Takeuchi H, Tanaka M, Tanaka A, Tsunemi A, Yamamoto H. Predominance of M2‐polarized macrophages in bladder cancer affects angiogenesis, tumor grade and invasiveness. Oncology Letters. 2016;11(5):3403‐3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12(4):265‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stern L, McGuire H, Avdic S, et al. Mass cytometry for the assessment of immune reconstitution after hematopoietic stem cell transplantation. Front Immunol. 2018;9:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winkler F, Bengsch B. Use of mass cytometry to profile human T cell exhaustion. Front Immunol. 2019;10:3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Speiser DE, Ho PC, Verdeil G. Regulatory circuits of T cell function in cancer. Nat Rev Immunol. 2016;16(10):599‐611. [DOI] [PubMed] [Google Scholar]
- 49. Das M, Zhu C, Kuchroo VK. Tim‐3 and its role in regulating anti‐tumor immunity. Immunol Rev. 2017;276(1):97‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Park HJ, Park JS, Jeong YH, et al. PD‐1 upregulated on regulatory T cells during chronic virus infection enhances the suppression of CD8+ T cell immune response via the interaction with PD‐L1 expressed on CD8+ T cells. J Immunol. 2015;194(12):5801‐5811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All original data throughout our manuscript are available upon reasonable request.