

Abstract
Platelets play a pivotal role in hemostasis. Activated platelets are classified into two groups, according to their agonist response: aggregating and procoagulant platelets. Aggregating platelets consist of activated integrin αIIbβ3 and stretch out pseudopods to further attract platelets to the site of injury by connecting with fibrinogen. They mainly gather in the core of the thrombus and perform a secretory function, such as releasing adenosine diphosphate (ADP). Procoagulant platelets promote the formation of thrombin and fibrin by interacting with coagulation factors and can thus be considered as the connector between primary and secondary hemostasis. In addition to their functions in blood coagulation, procoagulant platelets play a proinflammatory role by releasing platelet microparticles and inorganic polyphosphate. Considering these important functions of procoagulant platelets, this subpopulation warrants detailed study to analyze their potential in preventing human diseases. This review summarizes the generation and important characteristics of procoagulant platelets, as well as their potential for preventing the adverse effects associated with current antiplatelet therapies.
Keywords: aggregating platelets, coagulation, hemostasis, procoagulant platelets, proinflammatory
Platelets are critical mediators of thrombosis and hemostasis. Upon activation, platelets polarize in different directions. Different populations of platelets play different roles in the process of thrombus formation. Procoagulant platelet which precisely localizes the thrombin burst to the wound sites, can be seen as a connector between primary and secondary hemostasis. Procoagulant platelet serves as a scaffold which supports the assembly of prothrombinase and tenase, resulting in the thrombin burst, which causes fibrin formation and aggregation to generate a fragile fibrin network. Accordingly, dysfunction of procoagulant platelet can cause hemostasis failure. And using procoagulant platelet as a novel marker to monitor and using it as a novel target to cure disease are promising. We summarized the mechanisms concerning about the procoagulant platelet formation by reading classical and novel papers. By summarizing the multi‐characteristics of procoagulant platelet, we considered these features can be used as markers to measure the population of platelets and provided some methods to distinguish it which was already used and proven by other scholars or is under discussed now.
1. INTRODUCTION
Hemostasis is a critical physiological process that stops bleeding at the site of a vascular injury and maintains the integrity of vessels. Cooperation between platelets, coagulation factors, and endothelial cells is critical to this complex process. Hemostasis is initiated by wounded vessels, which have exposed extracellular matrix, and further activates platelet adhesion, aggregation, secretion, and a cascade of coagulation factors. Platelet activation and the coagulation system function in concert to seal the damaged site; thus, their imbalance results in bleeding or thrombosis.
The way in which platelets interact with coagulation factors and accurately localize to the damaged site is of considerable interest. Activated platelets can be classified into at least two forms, procoagulant and aggregating platelets. 1 , 2 , 3 , 4 , 5 These two subpopulations have different manifestations. For example, the most abundant integrin, αIIbβ3, exhibits an “open” form on aggregating platelets and a “closed” form on procoagulant platelets, which display exposed phosphatidylserine (PS). 6 Aggregating platelets contain numerous pseudopods, whereas procoagulant platelets are balloon‐shaped without pseudopods. 3 , 6 In addition, aggregating platelets strongly secrete bioactive molecules such as ADP, adenosine triphosphate (ATP), and serotonin, whereas the secretory function of procoagulant platelets is weak. 6 While the exact role of aggregating platelets in inflammation is not yet completely understood, procoagulant platelets are known to exert proinflammatory effects and can release inflammation mediators such as inorganic polyphosphate and microparticles. 7 , 8 , 9 , 10 We show the morphological differences between resting platelet, aggregating platelet and procoagulant platelet in Figure 1.
FIGURE 1.

Different states of platelets viewed using scanning electron microscopy. A, Resting platelets are smooth and flat. B, Aggregating platelets contain many pseudopods. C, Procoagulant platelets are characterized by balloon‐shaped morphology and do not contain pseudopods. Bar = 2 μm. Images were taken from Agbani et al 11
In this review, we focus on the function of procoagulant platelets in recruiting coagulation factors to the damaged site and serving as a bridge between primary and secondary hemostasis. 7 , 11 First, we summarize the mechanisms of procoagulant platelet formation. Second, we discuss the attributes pivotal to procoagulant platelets, and finally, we highlight recent studies on their therapeutic use as clinical biomarkers.
2. MECHANISMS OF PROCOAGULANT PLATELET FORMATION
Procoagulant platelets are generally considered to be apoptotic. 12 , 13 , 14 However, recent studies have revealed that procoagulant platelets undergo necrosis. The suppression of caspase, a modulator of apoptosis, does not affect the procoagulant function of platelets, 15 , 16 whereas the inhibition of cyclophilin D (CypD), which modulates necrosis by inducing the opening of mitochondrial permeability transition pores (mPTP), greatly reduces their procoagulant function. 17 Jackson et al 18 compared the characteristics of apoptosis, necrosis, and procoagulant platelets (summarized in Table 1). However, upon inhibiting CypD, procoagulant platelets are still formed. In a study in CypD knockout mice, platelets externalized PS, and there were no major differences in the tail transection bleeding times between CypD+/+ and CypD−/− mice. 17 These findings suggest that other pathways may be involved in the generation of procoagulant platelets.
TABLE 1.
Characteristics of procoagulant platelets during apoptosis and necrosis (summarized from Jackson et al 18 )
| Characteristics | Apoptosis | Necrosis | Procoagulant platelets |
|---|---|---|---|
| Stimulus | Ratio of Bclxl to Bax/Bak | Cyclophilin D | Activation of GP VI receptor and/or protease‐activated receptors (e.g., thrombin, collagen, collagen‐related peptide) |
| Ca+ Dependence | Independent of Ca+ | Dependent on Ca+ | Dependent on Ca+ |
| Morphology | Cell shrinkage and apoptotic body formation | Cell swelling | Cell swelling and ballooning |
| Membrane integrity | Loss in the late stage | Loss in the early stage | Loss of membrane integrity |
| Mitochondrial transmembrane potential (ΔΨm) | Loss in the late stage | Loss in the early stage | Loss of ΔΨm |
| PS exposure | Exposed in the late stage | Exposed in the early stage | Exposed |
| Formation of MPTP | No | Yes | Yes |
| Method of inhibition | Bax/Bak knockout | Cyclophilin D knockout | Cyclophilin D knockout |
Abbreviations: GP VI, glycoprotein VI; mPTP, mitochondrial permeability transition pore; PS, phosphatidylserine.
Platelets vary in their responses to agonist stimulation, and various multifactorial processes determine the fate of platelets. Rheological and topographical factors within the thrombi in vivo have been proposed to affect the heterogeneity in platelet activation states. 19 Fluid dynamic studies have shown that platelets within the thrombus core undergo longer exposure to solute agonists than those on the outer thrombus shell. The thrombus core is less porous and has stronger platelet–cell adhesion than the outer thrombus shell, where convective flow currents and the relative increase in permeability result in decreased exposure to agonists. 20 Environmental factors such as different adhesive surfaces, and intrinsic platelet characteristics, such as age, surface protein expression, and subcellular components, may play a role in determining the heterogeneity in platelet response after activation. 20 , 21
In the context of those undergoing necrosis, we describe the classical mechanisms underlying the formation of procoagulant platelets. Detailed mechanisms can be found in Figure 2.
FIGURE 2.
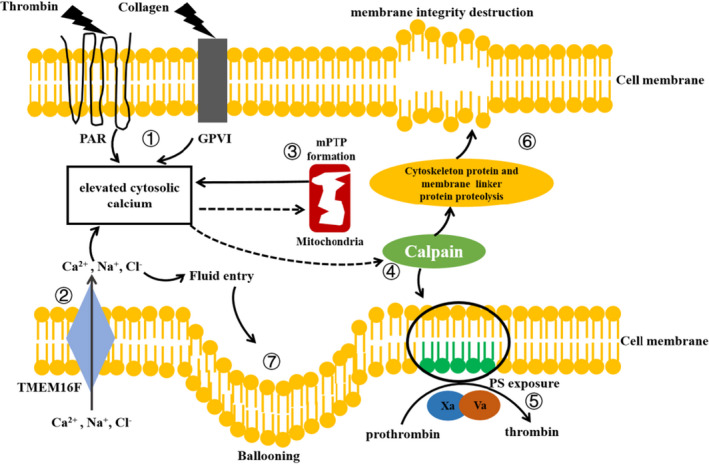
Mechanisms of procoagulant platelet formation. (1) Protease‐activated receptor and glycoprotein VI are located on the surface of platelets. After stimulating the platelets with agonists, such as thrombin and collagen, cytosolic Ca2+ concentration increases. (2) TMEM16F located on the platelet's membrane allows the entry of Na+, and Cl–, and especially Ca2+. This mechanism, to some extent, promotes higher Ca2+ concentration. (3) Elevated cytosolic Ca2+ concentration triggers the formation of mPTPs, which in turn leads to the release of Ca2+ stored in the mitochondria. (4) Elevated cytosolic Ca2+ activates calpain, which causes the redistribution of phospholipids and PS exposure. (5) PS exposure is a prerequisite for procoagulant activity. Coagulation factors Xa and Va aggregate on the PS surface and convert prothrombin into thrombin. (6) Activated calpain also hydrolyzes cytoskeleton proteins and membrane linker proteins, which damages the membrane integrity. (7) The entry of salt ions into platelets leads to high osmotic pressure and further fluid entry, thereby resulting in ballooning
2.1. Agonist demand
Although many agonists can activate platelets, only strong agonists can induce the formation of procoagulant platelets. Agonists include physiological and non‐physiological agonists. Among the physiological agonists, the co‐stimulation of thrombin or thrombin receptor‐activating peptide and glycoprotein VI agonists such as collagen, convulxin, and collagen‐related peptides, along with calcium (Ca2+) in the solution, produces the maximum activation effect. 11 , 22 , 23 , 24 ADP had little effect on the generation of procoagulant platelets according to Pasalic et al 23 Hua et al 11 evaluated the proportion of procoagulant platelets upon treatment with various agonists, including thrombin, collagen, or thrombin plus collagen, and showed that co‐stimulation with collagen and thrombin helped achieve the highest procoagulant platelet ratio (59.21%), followed by collagen activation alone (45.76%), with thrombin activation the least effective (20.78%). However, Pasalic et al 23 found that co‐stimulation with collagen and thrombin had the highest efficiency in producing procoagulant platelets, regardless of the sample type. Compared with the results of Hua et al 11 , those of Pasalic et al 23 indicated that thrombin was more efficient than collagen in generating procoagulant platelets. Pasalic et al 23 also compared washed platelets, platelet‐rich plasma, and whole blood, in terms of procoagulant platelet generation, and found that whole blood is the preferred sample type, whereas washed platelets are not ideal, as there are fewer steps, such as washing and centrifugation, required when whole blood samples are used compared to those when washed platelets are used.
In our laboratory, we have attempted to detect pro‐platelets using whole blood samples and observed that a high number of pre‐treatment steps can lead to excessive activation and loss of platelets. We detected the platelets using forward scattering light, side scattering light (SSC), and CD41a, a specific marker for platelets, and defined procoagulant platelets as those that were positive for the CD62P and GSAO [4‐(N‐(S‐glutathionylacetyl)amino)phenylarsonous acid] markers for necrotic platelets (R. Qiao, 2021, unpublished data).
Among non‐physiological agonists, Ca2+ ionophores, such as A23187 or ionomycin, have been used to induce a high and sustained rise in cytosolic Ca2+. 22 A previous study showed that almost all platelets can become procoagulant upon Ca2+ ionophore treatment. The methods of detecting procoagulant platelets in vitro employed in several studies, including the types of samples, agonist types and concentration, activation time, and the ratio of procoagulant platelets, are summarized in Table 2.
TABLE 2.
Methods used to generate procoagulant platelets described in the literature
| Sample types | Agonists | Conditions | Procoagulant platelet ratio |
|---|---|---|---|
| Washed platelets (5 × 104/μL)7 | Thrombin (0.1 U/ml) | 37°C, 10 min | 59.2% |
| Collagen (5 µg/ml) | 37°C, 10 min | 45.76% | |
| Thrombin (0.1 U/ml) + collagen (5 μg/ml) | 37°C, 10 min | 20.78% | |
| Whole blood 25 | Thrombin + collagen (final concentrations are variable) | Room temperature [25−27°C], 10 min | 29.7% |
| Whole blood 23 | Thrombin (2 U/ml) | Room temperature [25−27°C], 10 min | 13.15% |
| Collagen (10 μg/ml) | Room temperature [25−27°C], 10 min | 1.2% | |
| Thrombin (2 U/ml) + collagen (10 μg/ml) | Room temperature [25−27°C], 10 min | 17.3% | |
| Whole blood (5 × 104/µl) 29 | Thrombin (2 U/ml) + collagen (10 μg/ml) | 37°C, 10 min | 60% |
Tan et al 25 measured the percentage of procoagulant platelets in whole blood using CD45, a specific marker of leukocytes, CD41a, CD62P, and GSAO. Platelets that take up GSAO and express surface P‐selectin represent the procoagulant subpopulation (Figure 3).
FIGURE 3.
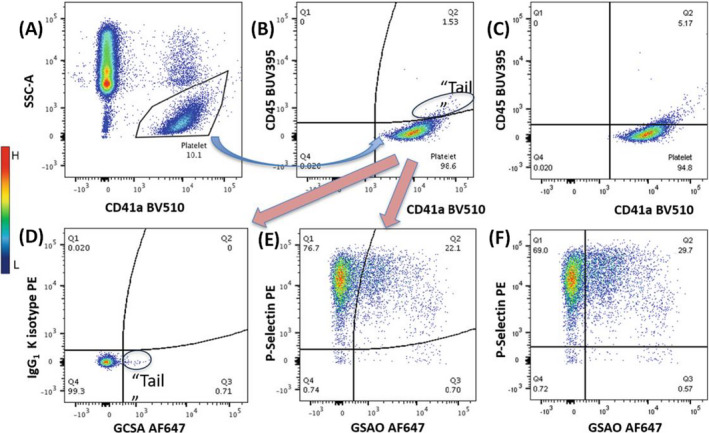
Detection of procoagulant platelets using flow cytometry. A, Platelets are first identified using polygonal gating on the SSC‐A versus CD41a plot and as presenting low SSC and CD41a positivity, and subsequent analyses are based on this gate. B, Gating is further refined in the lower right quadrant by selecting CD41a+/CD45– cells using curved quadrants, and subsequent analyses are based on this gate. The “tail” population (CD41a+/CD45+) is excluded, as it represents aggregates of platelets and leukocytes. C, Applying straight quadrants to the same platelet population and thresholds for both axes shown in (B) yields slightly different results. D, Thresholds for CD62P and GSAO are set by PE and GSCA isotype controls. E, Procoagulant platelets are identified in the upper‐right quadrants as CD62P+/GSAO+ cells in a stimulated sample. F, Applying straight quadrants to the same platelet population and thresholds for both axes shown in (E) yields slightly different procoagulant platelet percentages. Images were taken from Tan et al 25
2.2. Ca2+ dependency
Cytosolic Ca2+ concentration is increased in “procoagulant platelets” and “non‐procoagulant platelets,” which are both activated platelets. The key determinant of procoagulant platelets is maintained, extremely high cytosolic Ca2+ levels. The process by which this can be attained can be divided into three stages.
First, through intracellular release and extracellular entry, cytosolic Ca2+ concentration is increased, with the endoplasmic reticulum (ER) having the highest Ca2+ concentration among the cellular organelles. Three important molecules, stromal interaction molecule 1 (STIM1), Orai1, and TRPC6, are involved in extracellular Ca2+ entry.
2.2.1. STIM1
STIM1, a type 1A transmembrane Ca2+ sensor in the ER of platelets, can detect the release of Ca2+ from intracellular stores and regulate Orai1. 4 , 26 STIM1 senses the Ca2+ content of intracellular stores using a Ca2+‐binding EF‐hand domain that protrudes into the lumen of the store. Upon store depletion, Ca2+ is released from the EF‐hand domain, and the plasma membrane store‐operated calcium entry (SOCE) channel is activated. Varga‐Szabo et al 27 reported that STIM1 deficiency markedly reduced Ca2+ entry, and mice deficient in STIM1 showed severe defects in thrombus formation under flow, but only had a limited risk of bleeding complications. In addition, SOCE channels in mice carrying the STIM1 mutant were permanently opened in vivo, which led to sustained Ca2+ influx from the extracellular space and a bleeding phenotype in mice in vitro. 28 These findings indicate the key regulatory role of STIM1 in maintaining Ca2+ levels via SOCE channels in platelets.
2.2.2. Orai1 and TRPC6
Extracellular Ca2+ ions mainly enter through the plasma membrane channels Orai1 and TRPC6, which are the most abundant channels on the platelet surface. 26 , 29 Orai1 is a type of SOCE channel. 26 Using Synta‐66, an Orai1 blocker, Abbasian et al 29 found that the percentage of procoagulant platelets and the median fluorescence of Fluo‐4, which has high affinity for Ca2+, was reduced in whole platelets. The median fluorescence of Fluo‐5 N, which has a low affinity for Ca2+, in procoagulant platelets was unaffected. The authors speculated that Orai1 contributes to the elevation of cytosolic Ca2+ but is not directly involved in the generation of supramaximal Ca2+. 24
TRPC6 is an unselective cation channel. Upon platelet activation, Na+ enters the platelets via TRPC6, and Ca2+ entry occurs subsequently via Na+/Ca2+ exchange. SOCE channels, especially the major SOCE channel Orai1, have been proposed to be potential antithrombotic targets. 28 , 30 However, such targeting has been associated with serious adverse effects, such as severe combined immune deficiency in patients with a missense mutation in Orai1. 31 To address this problem, individualized genetic testing should be performed in patients carrying a missense mutation in Orai1.
The second stage is the entry of cytosolic Ca2+ into the mitochondria, and the opening of mitochondrial permeability transition pores (mPTPs). Because the cytosolic Ca2+ concentration in the first stage is different in procoagulant and non‐procoagulant platelets, their mitochondrial Ca2+ concentration also varies. Ca2+ enters the mitochondria through the mitochondrial calcium uniporter (MCU). 32 The pharmacologic inhibition of MCU might effectively abrogate the formation of procoagulant platelets without affecting other aspects of platelet activation, thus representing a novel pharmacologic target specific to the prevention of procoagulant platelets without affecting normal hemostasis. 33 Similar to the formation of action potentials, which are activated only when stimuli reach or exceed the threshold, mPTP opening also has a threshold. mPTPs open only when Ca2+ uptake into mitochondria reaches this threshold; thus, below the threshold of mPTP opening, platelets do not possess procoagulant properties.
In the third stage, mPTP opening leads to the cytoplasmic transport of mitochondrial Ca2+, generating extremely high cytosolic Ca2+ concentrations and subsequently, the formation of procoagulant platelets. 12 , 29 , 33
In summary, we speculate that mPTP opening, which leads to a high cytosolic Ca2+ concentration, is crucial for procoagulant platelet formation. Therefore, we can describe procoagulant platelet formation as an “all‐or‐nothing” process. Increased cytosolic Ca2+ concentration is required for the formation of both procoagulant and non‐procoagulant platelets. In particular, higher and more sustained cytosolic Ca2+ concentration generates procoagulant platelets, and lower and less sustained cytosolic Ca2+ concentration generates non‐procoagulant platelets.
2.3. TMEM16 and chloride ions (Cl–) in water entry
The transmembrane protein TMEM16 is also called anoctamin (ANO). TMEM16A (ANO1), TMEM16B (ANO2), and TMEM16F (ANO6) are the most studied members of the TMEM16 family.
2.3.1. TMEM16A and TMEM16B
Cl− blockers are used to inhibit Cl− entry in physiological agonist‐activated platelets. Cl− entry via Cl− channels is speculated to be necessary for membrane hyperpolarization by providing the driving force for Ca2+ entry and triggering full PS exposure. 34 TMEM16A and TMEM16B are Ca2+‐activated Cl− channels (CaCCs). 35 Increased cytosolic free Ca2+ triggers the opening of CaCCs and allows the entry of Cl− through these channels. In turn, increased anion concentration inside cells promotes Ca2+ entry into cells.
2.3.2. TMEM16F
To date, there are three known functions of TMEM16F. In 2012, it was described as a Ca2+‐activated channel for the entry of cations such as Ca2+, Na+, Li+, K+, Rb+, Ba2+, and Cs+, with increased permeability to Ca2+ and Ba2+. 36 Second, TMEM16F was described as a Ca2+‐activated channel for the entry of anions, such as Cl−. Lastly, it was also denoted as a Ca2+‐activated phospholipid scramblase that translocates PS to the platelet surface, thus facilitating blood coagulation. 35 , 36 , 37 , 38
There are two major types of phospholipid scramblases: caspase‐dependent lipid scramblase, such as Xkr8, that induce PS externalization, and Ca2+‐activated phospholipid scramblase, that mediates PS externalization in response to an intracellular increase in Ca2+. 39 Yang et al 35 generated TMEM16F knockout mice and reported that TMEM16F is important for thrombin generation and the activity of Ca2+‐activated scramblase in blood cells. TMEM16F knockout mice not only had impaired hemostasis as evidenced by a prolonged bleeding time, but also had impaired carotid artery thrombus formation, revealing the crucial role of TMEM16F in thrombosis. 4 , 35
In 1979, the first patient with Scott syndrome, a rare bleeding disorder, was reported. The patient had a normal platelet level, structure, adhesion, secretion, and aggregation, while a notable defect in procoagulant activity was observed. This phenomenon was due to decreased Ca2+‐dependent PS exposure, which is regulated by TMEM16F. 38 To date, only six cases of Scott syndrome have been reported. 40 TMEM16F may serve as a new pharmaceutical target to treat human hemostatic and thrombotic disorders, such as stroke and heart attack. 37
2.3.3. Water entry
The entry of salts, such as Ca2+ and Cl−, provides an osmotic drive for water entry and subsequently causes ballooning, a characteristic of procoagulant platelets. Agbani et al 7 investigated whether water entry was required for ballooning. Using sucrose to increase extracellular osmolality, they reported the blocking of water entry and ballooning along with significant inhibition of thrombus formation.
2.4. Role of calpain
Sustained elevation of cytosolic Ca2+ concentration can activate the thiol protease calpain. Calpain hydrolyzes membrane linker proteins, such as actin, vinculin, and myosin, which cause cytoskeleton remodeling and disruption and PS exposure. 41 , 42 Calpain also participates in the release of microparticles. 1 , 42 Microparticles, the “dust” of platelets, possess higher procoagulant activity than platelets, carry inflammatory substances, and can be delivered remotely.
3. FEATURES OF PROCOAGULANT PLATELETS
3.1. PS exposure
A key characteristic of procoagulant platelets is PS exposure, which is necessary for coagulation assembly and thrombin generation. In normal resting platelets, the phospholipid bilayer of the cell membrane is asymmetric, comprising phosphatidylcholine and sphingomyelin mainly on the outer surface, whereas PS and phosphoethanolamine are located on the inner part. Two enzymes maintain this asymmetry: flippase transports lipids from the outer surface to the inner surface, whereas floppase has the opposite activity. 43 PS exposure is dependent on the Ca2+‐dependent scramblase and calpain. 1 , 37 , 39 , 41
3.2. Balloon‐like membrane morphology
Ballooning is one of the properties of procoagulant platelets. Ballooning is an irreversible process that increases the area for coagulation assembly [for example, prothrombinase complex (FXa‐FVa) and tenase complex (FIXa‐FVIIIa)]. Ballooning also affects PS exposure, and the release of microvesicles decreases when ballooning is inhibited.
3.3. Glycoprotein IIb/IIIa (GPIIb/IIIa) inactivation
Platelets in the thrombus can be aggregating or procoagulant platelets. The former subpopulation participates in the retraction of fibrin clots, which require fibrin formation and integrin “outside‐in” signaling. During signaling, aggregating platelets possess an activated GPIIb/IIIa. Owing to ballooning and cytoskeleton disruption, procoagulant platelets lose their adhesive function, showing “closed” GPIIb/IIIa. 4
The presence of an activated GPIIb/IIIa conformation is confirmed by PAC‐1 antibodies for human platelets. 4 Anna et al utilized PAC‐1, which can bind the active conformation of GPIIb/IIIa, to detect aggregatory platelets. PAC‐1 binds to or near to the fibrinogen site, and its affinity to the active GPIIb/IIIa conformation is higher than that to fibrinogen. Munnix et al 6 found that platelets that bind annexin A5, a marker for PS detection, failed to bind PAC‐1. They concluded that the manifestation is reasonable because the round morphology and lack of pseudopods is compatible with a low integrin activation state, and thus diminished adhesion. This generates a balance, in which procoagulant activity suppressed integrin activation, whereas aggregate formation reduced the procoagulant activity of platelets. After activation, normal‐sized platelets bound PAC‐1 with a negative combination of annexin V, a marker for procoagulant platelets that binds to PS, and were defined as “aggregatory.” The authors speculated that the reduction in PAC‐1 binding in PS‐positive platelets is due to the down‐regulation of fibrinogen receptors via the reduction in aggregatory response. 7 , 11
3.4. Procoagulant spreading
The link between ballooning and procoagulant spreading was first identified in 2015. 7 In this study, most ballooning platelets had spread membrane structures, and all procoagulant spreading platelets had ballooned. This observation suggests that ballooning is required for procoagulant spreading but not vice versa. 7 However, procoagulant spreading only occurs when platelets adhere to agonist‐coated surfaces, and those in suspension did not exhibit procoagulant spreading. 7 , 15
3.5. Platelet‐derived microparticles
Microparticles ranging between 0.1 and 1 μm are derived from various cell types in the plasma. Platelets are the major source of microparticles in circulation. 44 The formation of microparticles is a consequence of membrane remodeling. 2 Through phospholipid scramblases and calpain, procoagulant platelets release platelet‐derived microparticles (PMPs). 7 Proteins that mediate platelet function related to hemostasis, inflammation, and immunity are also found in platelet PMPs. PMPs serve as a surface for the assembly of tenase and prothrombinase complexes to boost thrombin formation and coagulation. 45 The coagulation capacity of PMPs is suggested to be 50‐ to 100‐fold higher than that of activated platelets. 46 Platelets also express inflammasomes, that can hydrolyze and release inflammatory mediators, such as IL‐1α and IL‐1β, which are mostly wrapped in PMPs. 47 PMPs can then interact with inflammatory leukocytes, such as monocytes and neutrophils, and induce endothelial cells to express adhesion molecules, such as intercellular cell adhesion molecule‐1 (ICAM‐1). All these effects trigger an inflammatory response. 48 , 49
3.6. Release of polyP
Platelet dense granules contain polyP, which has a mean chain length of 80 units. Verhoef et al 50 proposed that polyP promotes contact system activation and coagulation. They showed that polyP released from dense granules aggregates into nanoparticles that accumulate on the platelet surface and are of sufficient size to promote FXII activation. 50 Meanwhile, Fredenburgh et al 51 speculated that in addition to FXI and FXII inhibitors, neutralizing agents of polyP attenuate thrombosis without disrupting hemostasis. Long‐chain polyP located on the platelet surface initiates FXII contact activation and leads to coagulation, whereas short‐chain polyP, which is soluble and does not bind to platelets, has minor effects on FXII activation and may serve other functions, such as interaction with tissue factor pathway inhibitor (TFPI). 52 PolyP has been recognized as a procoagulant platelet‐derived inflammatory mediator. PolyP‐driven FXII activation causes the promotion of the kallikrein‐bradykinin pathway. Following its activation, kallikrein hydrolyzes kininogen to induce bradykinin formation. The binding of bradykinin to its receptor then results in vessel dilation, neutrophil chemotaxis, and increased vascular permeability. 9
4. THERAPEUTIC APPLICATIONS OF PROCOAGULANT PLATELETS
4.1. Clinical biomarkers
There is always a need for suitable markers and/or identity criteria for the clinical assessment of thrombotic or bleeding events. As procoagulant platelets participate in thrombin generation and hemostasis promotion, altering their proportions may result in varying levels of risk of thrombosis and bleeding. Pasalic et al 23 found that patients with coronary heart disease had a higher platelet procoagulation potential than healthy controls in response to thrombin plus collagen (18.2 ± 2.2% vs. 25.2 ± 3.9%, p < 0.05). Lacunar stroke represents approximately 25% of all ischemic strokes and is associated with smaller infarct size and lower recurrence risk than those of non‐lacunar stroke. A previous study showed that patients with lacunar stroke (21.8 ± 11.4%) had the lowest levels of coated platelets (a population of procoagulant platelets) among those with cortical stroke (39.4 ± 12.7%, p < 0.001) or controls (31.6 ± 13.2%, p = 0.008). 53 In another study, the level of procoagulant platelets (>40%) was associated with recurrent cerebral infarction. 54 A recent study demonstrated that the ability of coated platelets to predict recurrent ischemic events following lacunar stroke (AUC: 0.835 ± 0.08, p < 0.001) had a sensitivity of 0.75 (0.35–0.97; 95% CI), specificity of 0.92 (0.85–0.97), positive predictive value of 0.43 (0.26–0.62), and negative‐predictive value of 0.98 (0.93–0.99). 55
Apart from their hemostatic function, procoagulant platelets also exhibit a proinflammatory function. PMPs released primarily by procoagulant platelets are proinflammatory mediators associated with various clinical manifestations. For example, PMPs are elevated in patients with rheumatoid arthritis and infectious diseases, such as HIV, dengue, and malaria. PMPs are also related to disease propagation. 56 Once released, PMPs are widely distributed. Thus, we speculate that procoagulant platelets can be used as biomarkers for inflammatory diseases. Although there are only a few studies on this topic, this is a promising research direction to explore. We can use experimental approaches based on the features introduced above to detect procoagulant platelets in vitro.
4.2. PS exposure is an assistive measure, but not sufficient, for procoagulant platelet detection
Although PS exposure is a prerequisite for the formation of a procoagulant platelet subpopulation, it is not entirely correlated with procoagulant platelets. Along with the imperfect correlation between PS exposure and the presence of coagulation factors on the platelet surface, even after strong activation, only some platelets expose PS, and a small subset of them is procoagulant. 11 Thus, PS exposure alone is not sufficient to conclude that procoagulant platelets will be formed. 25
4.3. Detecting procoagulant platelets using a combination of GSAO and P‐selectin
The combination of GSAO and P‐selectin can detect procoagulant platelets. 23 , 25 GSAO is a cell death marker that can pass through the procoagulant platelet membrane with elevated permeability, covalently bind to proteins with closely spaced dithiols, and resist washout. When tagged with a reporter compound, flow cytometry can analyze procoagulant platelets using a GSAO‐fluorescein marker. P‐selectin is normally stored in the α‐granule in resting platelets, and is a marker of activated platelets, which express surface P‐selectin upon activation.
GSAO in combination with P‐selectin can discriminate a population of platelets that present necrotic features and, functionally, a procoagulant phenotype. Positive test results for both GSAO and P‐selectin define procoagulant platelets. 25 Patients without coronary artery disease (CAD) had a synergistic increase in the ratio of procoagulant platelets when collagen was added to thrombin (thrombin vs. thrombin plus collagen: 12.2 ± 3.4% vs. 18.2 ± 2.2%, p < 0.05); however, platelets from patients with CAD reached their maximal procoagulant potential with thrombin stimulation alone, and no further increase was observed upon the addition of collagen, suggesting that patients with CAD have hypersensitivity to thrombin. 23
4.4. A novel target for antithrombosis
Antiplatelet drugs are used to treat thrombotic diseases. For example, aspirin, which blocks thromboxane A2 (TXA2) formation, is usually prescribed to prevent cardiovascular diseases, and P2Y12 inhibitors such clopidogrel, prasugrel, and ticagrelor, are used to block the secretion of ADP and ATP from platelets. Dual antiplatelet therapy, which refers to the combination of aspirin with a P2Y12 inhibitor, is the current treatment for the secondary prevention of atherothrombotic events in patients with acute coronary syndrome or those undergoing elective percutaneous coronary intervention. Aspirin and P2Y12 inhibitors resemble GPIIb/IIIa inhibitors such as abciximab and eptifibatide, and target aggregating platelets. 3 However, their clinical applications remain limited, because the inhibition of aggregating platelets is usually accompanied by bleeding, which can range from serious events, such as intracranial hemorrhage, to minor skin bruising. 1 More than 25% of patients using antiplatelet drugs experience ischemic events. 3
Targeting aggregating platelets corresponds to blocking platelet thrombus formation; thus, the bleeding site cannot be effectively blocked. When procoagulant platelets are targeted, thrombin formation can be inhibited without affecting the function of aggregating platelets, indicating its potential use as an antithrombotic method without bleeding risk. However, there is a lack of drugs that specifically target procoagulant platelets. A study on aquaporin‐1 (AQP1) knockout mice found that: (1) AQP1 is a critical mediator that regulates procoagulant platelet formation; (2) AQP1 inhibition decreases procoagulant spreading, microvesiculation, PS exposure, and thrombus formation time; and (3) AQP1 inhibition has minimal effects on the secretion and aggregation of platelet granules. These results suggest that AQP1 antagonists may represent novel antithrombotics targeting procoagulant platelets. 57 Unfortunately, there are still no AQP1‐specific inhibitors. Carbonic anhydrase (CA) inhibitors, such as acetazolamide and methazolamide, are clinically used as mild diuretics, and may be potent anti‐procoagulant agents. 58 Acetazolamide and methazolamide attenuate intracellular Cl– entry and suppress the procoagulant response of activated platelets in vitro and thrombosis in vivo. Zhang et al 59 propose that CA inhibitors can block AQPs. In conclusion, CA and AQP1 inhibitors may serve as potential therapeutic options for antithrombosis without bleeding risk.
4.5. A novel target for anti‐inflammation
Procoagulant platelets also mediate inflammation by releasing PMPs and polyP, suggesting their potential as targets for anti‐inflammation. PMPs and polyP also exert procoagulant effects; thus, they may also be potential targets for curing inflammatory and thrombotic disorders. A few studies have attempted to inhibit inflammation by inhibiting platelet function. However, annexin A5, a physiological anticoagulant protein, binds to and endocytoses PS, thereby inhibiting the release of PMPs. 60 This finding suggests that the human body has its own protective mechanism to resist thrombosis and inflammation, to a certain extent. Thus, the development of supplementary medicine based on this property is a promising research field but still has a long way to go.
CONFLICT OF INTEREST
The authors declare that they have no competing interest.
AUTHOR CONTRIBUTIONS
Yaxin Chu collected and summarized the literature. Han Guo and Yuncong Zhang structured and edited this review. Rui Qiao conceptualized this review and reviewed the final manuscript.
Funding information
This work was supported by grants from the Beijing Natural Science Foundation [grant number 7192222] and the Peking University Third Hospital Clinical Key Project [grant number BYSY2017008]
DATA AVAILABILITY STATEMENT
All data used in this review are presented in the paper.
REFERENCES
- 1. Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130:2171‐2179. [DOI] [PubMed] [Google Scholar]
- 2. Agbani EO, Williams CM, Hers I, Poole AW. Membrane ballooning in aggregated platelets is synchronised and mediates a surge in microvesiculation. Sci Rep. 2017;7:2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol. 2019;16:166‐179. [DOI] [PubMed] [Google Scholar]
- 4. Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet‐based coagulation: different populations, different functions. J Thromb Haemost. 2013;11:2‐16. [DOI] [PubMed] [Google Scholar]
- 5. Baaten CCFMJ, Ten Cate H, van der Meijden PEJ, Heemskerk JWM. Platelet populations and priming in hematological diseases. Blood Rev. 2017;31:389‐399. [DOI] [PubMed] [Google Scholar]
- 6. Munnix IC, Kuijpers MJ, Auger J, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27:2484‐2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agbani EO, van den Bosch MT, Brown E, et al. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation. 2015;132:1414‐1424. [DOI] [PubMed] [Google Scholar]
- 8. El‐Gamal H, Parray AS, Mir FA, Shuaib A, Agouni A. Circulating microparticles as biomarkers of stroke: a focus on the value of endothelial‐ and platelet‐derived microparticles. J Cell Physiol. 2019;234:16739‐16754. [DOI] [PubMed] [Google Scholar]
- 9. Müller F, Mutch NJ, Schenk WA, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. 2009;139:1143‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hua VM, Abeynaike L, Glaros E, et al. Necrotic platelets provide a procoagulant surface during thrombosis. Blood. 2015;126:2852‐2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kile BT. The role of the intrinsic apoptosis pathway in platelet life and death. J Thromb Haemost. 2009;7(Suppl 1):214‐217. [DOI] [PubMed] [Google Scholar]
- 13. de Botton S, Sabri S, Daugas E, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100:1310‐1317. [DOI] [PubMed] [Google Scholar]
- 14. Perrotta PL, Perrotta CL, Snyder EL. Apoptotic activity in stored human platelets. Transfusion. 2003;43:526‐535. [DOI] [PubMed] [Google Scholar]
- 15. Kulkarni S, Jackson SP. Platelet factor XIII and calpain negatively regulate integrin alphaIIbbeta3 adhesive function and thrombus growth[J]. J Biol Chem. 2004;279:30697‐30706. [DOI] [PubMed] [Google Scholar]
- 16. Schoenwaelder SM, Yuan Y, Josefsson EC, et al. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663‐666. [DOI] [PubMed] [Google Scholar]
- 17. Jobe SM, Wilson KM, Leo L, et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood. 2008;111:1257‐1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood. 2010;116:2011‐2018. [DOI] [PubMed] [Google Scholar]
- 19. Nesbitt WS, Westein E, Tovar‐Lopez FJ, et al. A shear gradient‐dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665‐673. [DOI] [PubMed] [Google Scholar]
- 20. Munnix IC, Cosemans JM, Auger JM, Heemskerk JW. Platelet response heterogeneity in thrombus formation. Thromb Haemost. 2009;102:1149‐1156. [DOI] [PubMed] [Google Scholar]
- 21. Topalov NN, Yakimenko AO, Canault M, et al. Two types of procoagulant platelets are formed upon physiological activation and are controlled by integrinα(IIb)β(3). Arterioscler Thromb Vasc Biol. 2012;32:2475‐2483. [DOI] [PubMed] [Google Scholar]
- 22. Mattheij NJA, Gilio K, van Kruchten R, et al. Dual mechanism of integrin alpha(IIb)beta(3) closure in procoagulant platelets. J Biol Chem. 2013;288:13325‐13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pasalic L, Wing‐Lun E, Lau JK, et al. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J Thromb Haemost. 2018;16:1198‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abbasian N, Millington‐Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020;4:154‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tan CW, Bourcy M, Pasalic L, Chen VM. Flow cytometry assessment of procoagulant platelets using a dithiol‐reactive probe. Methods Mol Biol. 2019;1967:305‐321. [DOI] [PubMed] [Google Scholar]
- 26. Varga‐Szabo D, Braun A, Nieswandt B. Calcium signaling in platelets. J Thromb Haemost. 2009;7:1057‐1066. [DOI] [PubMed] [Google Scholar]
- 27. Varga‐Szabo D, Braun A, Kleinschnitz C, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205:1583‐1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Authi KS. Orai1: a channel to safer antithrombotic therapy. Blood. 2009;113:1872‐1873. [DOI] [PubMed] [Google Scholar]
- 29. Pasalic L, Pennings GJ, Connor D, Campbell H, Kritharides L, Chen VM. Flow cytometry protocols for assessment of patelet function in whole blood. Methods Mol Biol. 2017;1646:369‐389. [DOI] [PubMed] [Google Scholar]
- 30. van Kruchten R, Braun A, Feijge MA, et al. Antithrombotic potential of blockers of store‐operated calcium channels in platelets. Arterioscler Thromb Vasc Biol. 2012;32:1717‐1723. [DOI] [PubMed] [Google Scholar]
- 31. Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179‐185. [DOI] [PubMed] [Google Scholar]
- 32. Zhao H, Li T, Wang K, et al. AMPK‐mediated activation of MCU stimulates mitochondrial Ca(2+) entry to promote mitotic progression. Nat Cell Biol. 2019;21:476‐486. [DOI] [PubMed] [Google Scholar]
- 33. Kholmukhamedov A, Janecke R, Choo HJ, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost. 2018;16:2315‐2321. [DOI] [PubMed] [Google Scholar]
- 34. Harper MT, Poole AW. Chloride channels are necessary for full platelet phosphatidylserine exposure and procoagulant activity. Cell Death Dis. 2013;4:e969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang H, Kim A, David T, et al. TMEM16F forms a Ca2+‐activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151:111‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium‐dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834‐838. [DOI] [PubMed] [Google Scholar]
- 37. Jacobsen KS, Zeeberg K, Sauter DR, Poulsen KA, Hoffmann EK, Schwab A. The role of TMEM16A (ANO1) and TMEM16F (ANO6) in cell migration. Pflug Arch. 2013;465:1753‐1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shimizu T, Iehara T, Sato K, Fujii T, Sakai H, Okada Y. TMEM16F is a component of a Ca2+‐activated Cl‐ channel but not a volume‐sensitive outwardly rectifying Cl‐ channel. Am J Physiol‐Cell Physiol. 2013;304:C748‐C759. [DOI] [PubMed] [Google Scholar]
- 39. van Kruchten R, Mattheij NJA, Saunders C, et al. Both TMEM16F‐dependent and TMEM16F‐independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood. 2013;121:1850‐1857. [DOI] [PubMed] [Google Scholar]
- 40. Millington‐Burgess SL, Harper MT. Gene of issue: ANO6 and Scott Syndrome. Platelets. 2020;31:964‐967. [DOI] [PubMed] [Google Scholar]
- 41. Wei H, Malcor JDM, Harper MT. Lipid rafts are essential for release of phosphatidylserine‐exposing extracellular vesicles from platelets. Sci Rep. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strack K, Lauri N, Maté SM, et al. Induction of erythrocyte microvesicles by Escherichia Coli Alpha hemolysin. Biochem J. 2019;476:3455‐3473. [DOI] [PubMed] [Google Scholar]
- 43. Hua VM, Chen VMY. Procoagulant platelets and the pathways leading to cell death. Semin Thromb Hemost. 2015;41:405‐412. [DOI] [PubMed] [Google Scholar]
- 44. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. 2014;123:2759‐2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. P roc Natl Acad Sci USA. 2015;112:12800‐12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sinauridze EI, Kireev DA, Popenko NY, et al. Platelet microparticle membranes have 50‐to 100‐fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007;97:425‐434. [PubMed] [Google Scholar]
- 47. Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen‐dependent microparticle production. Science. 2010;327:580‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou Q, Lian Y, Zhang Y, et al. Platelet‐derived microparticles from recurrent miscarriage associated with antiphospholipid antibody syndrome influence behaviours of trophoblast and endothelial cells. Mol Hum Reprod. 2019;25:483‐494. [DOI] [PubMed] [Google Scholar]
- 49. Duchez AC, Boudreau LH, Naika GS, et al. Platelet microparticles are internalized in neutrophils via the concerted activity of 12‐lipoxygenase and secreted phospholipase A(2)‐IIA. Proc Natl Acad Sci USA. 2015;112:E3564‐E3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verhoef JJF, Barendrecht AD, Nickel KF, et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood. 2017;129:1707‐1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fredenburgh JC, Gross PL, Weitz JI. Emerging anticoagulant strategies. Blood. 2017;129:147‐154. [DOI] [PubMed] [Google Scholar]
- 52. Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. 2006;103:903‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Prodan CI, Joseph PM, Vincent AS, Dale GL. Coated‐platelets in ischemic stroke: differences between lacunar and cortical stroke. J Thromb Haemost. 2008;6:609‐614. [DOI] [PubMed] [Google Scholar]
- 54. Kirkpatrick AC, Stoner JA, Dale GL, Prodan CI. Elevated coated‐platelets in symptomatic large‐artery stenosis patients are associated with early stroke recurrence. Platelets. 2014;25:93‐96. [DOI] [PubMed] [Google Scholar]
- 55. Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J Thromb Haemost. 2020;18:660‐668. [DOI] [PubMed] [Google Scholar]
- 56. Rozmyslowicz T, Majka M, Kijowski J, et al. Platelet‐ and megakaryocyte‐derived microparticles transfer CXCR4 receptor to CXCR4‐null cells and make them susceptible to infection by X4‐HIV. AIDS. 2003;17:33‐42. [DOI] [PubMed] [Google Scholar]
- 57. Agbani EO, Williams CM, Li Y, et al. Aquaporin‐1 regulates platelet procoagulant membrane dynamics and in vivo thrombosis. JCI Insight. 2018;3:e99062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Agbani EO, Zhao XJ, Williams CM, et al. Carbonic anhydrase inhibitors suppress platelet procoagulant responses and in vivo thrombosis carbonic anhydrase inhibitors as Antithrombotics. Platelets. 2020;31:853‐859. [DOI] [PubMed] [Google Scholar]
- 59. Zhang JZ, An Y, Gao JW, et al. Aquaporin‐1 translocation and degradation mediates the water transportation mechanism of acetazolamide. PLoS One. 2012;7:e45976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. de Laat B, Wu XX, van Lummel M, Derksen RH, de Groot PG, Rand JH. Correlation between antiphospholipid antibodies that recognize domain I of beta 2‐glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood. 2007;109:1490‐1494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data used in this review are presented in the paper.