

Abstract
[URE3] is a prion of the nitrogen catabolism controller, Ure2p, and [PSI+] is a prion of the translation termination factor Sup35p in S. cerevisiae. Btn2p cures [URE3] by sequestration of Ure2p amyloid filaments. Cur1p, paralogous to Btn2p, also cures [URE3], but by a different (unknown) mechanism. We find that an array of mutations impairing proteasome assembly or MG132 inhibition of proteasome activity result in loss of [URE3]. In proportion to their prion—curing effects, each mutation affecting proteasomes elevates the cellular concentration of the anti-prion proteins Btn2 and Cur1. Of >4,600 proteins detected by SILAC, Btn2p was easily the most overexpressed in a pre9Δ (α3 core subunit) strain. Indeed, deletion of BTN2 and CUR1 prevents the prion—curing effects of proteasome impairment. Surprisingly, the 15 most unstable yeast proteins are not increased in pre9Δ cells suggesting altered proteasome specificity rather than simple inactivation. Hsp42, a chaperone that cooperates with Btn2 and Cur1 in curing [URE3], is also necessary for the curing produced by proteasome defects, although Hsp42p levels are not substantially altered by a proteasome defect. We find that pre9Δ and proteasome chaperone mutants that most efficiently lose [URE3], do not destabilize [PSI+] or alter cellular levels of Sup35p. A tof2 mutation or deletion likewise destabilizes [URE3], and elevates Btn2p, suggesting that Tof2p deficiency inactivates proteasomes. We suggest that when proteasomes are saturated with denatured/misfolded proteins, their reduced degradation of Btn2p and Cur1p automatically upregulates these aggregate-handling systems to assist in the clean-up.
Keywords: Tof2, Btn2, Cur1, [URE3], prion, proteasome, Pre9, SILAC, Irc25, Poc4
Introduction
[URE3] is an infectious protein (prion) of the Ure2 protein, based on self-propagating linear β- sheet-rich polymers (amyloid) of Ure2p. [PSI+] is likewise an amyloid-based prion of Sup35p. In each case, the prion (amyloid) form of the protein is inactive for the function of the normal (soluble) form. Ure2p is a repressor of genes for utilization of poor nitrogen sources (like allantoate) when a good source (like ammonia) is present. [URE3] is detected by the activity of the DAL5 (allantoate transporter) promoter in spite of the presence of ammonia. Sup35 is a subunit of the translation termination factor, and [PSI+] produces increased read-through of premature termination codons (such as ade2-1) (reviewed in Wickner 2006; Liebman and Chernoff 2012; Wickner et al. 2015).
These yeast prions are models for mammalian prion diseases, including not only the classical spongiform encephalopathies based on the PrP protein, but also Alzheimer’s disease (Aβ peptide), Parkinson’s disease (α-synuclein), type II diabetes (amylin), and other human amyloid diseases that are increasingly showing infectious protein features (Jaunmuktane et al. 2015; Mukherjee 2017; Kim et al. 2019; Sampson et al. 2020).
Different prion isolates (called “prion variants”) have distinct properties, varying in stability of propagation, intensity of the prion phenotype, sensitivity to overabundance or deficiency of various cellular components (often chaperones), ability to propagate with sequence variants of the prion protein (interspecies barrier or intraspecies barrier), “seed/propagon” number, lethality/toxicity to the host, and sensitivity or resistance to the various anti-prion systems (Derkatch et al. 1996; Tanaka et al. 2004; Edskes et al. 2009; Chen et al. 2010; McGlinchey et al. 2011; Wickner et al. 2014). A given prion isolate rather stably propagates as cells grow, although quite a few cases of ‘prion mutation’ have been reported, particularly when there is selective pressure to do so (reviewed in Wickner et al. 2019).
Like the human amyloid diseases, the yeast prions [URE3] and [PSI+] are detrimental to their hosts. Most variants of [URE3] and [PSI+] dramatically slow cell growth or are lethal (McGlinchey et al. 2011), but, of course, the mildest variants are usually used in lab work. Even mild variants are rare in wild cells, indicating that they too are detrimental to the host (Nakayashiki et al. 2005; Kelly et al. 2012). The parts of Ure2p and Sup35p that actually form the amyloid are called the prion domains (Ter-Avanesyan et al. 1994; Masison and Wickner 1995; King et al. 1997; Taylor et al. 1999), but have nonprion functions (Hosoda et al. 2003; Shewmaker et al. 2007). Prion domain sequence varies more rapidly in evolution than does the rest of the molecule (Chernoff et al. 2000; Santoso et al. 2000; Nakayashiki et al. 2001; Edskes and Wickner 2002; Resende et al. 2003; Edskes et al. 2009; Bateman and Wickner 2012). Because these changes produce barriers to prion transmission (Chernoff et al. 2000; Santoso et al. 2000; Nakayashiki et al. 2001; Edskes and Wickner 2002; Edskes et al. 2009; Bateman and Wickner 2012), the selection for rapid variation observed is evidence that infection with a prion is detrimental (Edskes et al. 2009; Bateman and Wickner 2012; Kelly et al. 2012).
As would be expected for detrimental infectious elements, there are several cellular systems that cure most of the prions as they arise. The paralogs Btn2p and Cur1p, working with Hsp42p, cure most variants of [URE3] as they arise (Kryndushkin et al. 2008; Wickner et al. 2014). Btn2p acts by collecting and sequestering Ure2p amyloid filaments at one site in the cell (Kryndushkin et al. 2008, 2011), an activity that also works on nonamyloid aggregates (Kryndushkin et al. 2012; Malinovska et al. 2012; Miller et al. 2015). Hsp104 has a disaggregase activity that is essential for prion propagation, cleaving amyloid filaments to make new propagons (Chernoff et al. 1995; Jung et al. 2002; Ness et al. 2002; Haslberger et al. 2010), and a distinct activity producing asymmetric segregation of prion propagons at cell division and thus curing [PSI+] on Hsp104 overproduction (Chernoff et al. 1995; Hung and Masison 2006; Ness et al. 2017). Without Hsp104 overproduction, this second activity cures many [PSI+] variants that arise in its absence (Gorkovskiy et al. 2017). The Upf1, Upf2, and Upf3 proteins are responsible for nonsense-mediated mRNA decay, and are normally ribosome-bound in complex with Sup35p (Czaplinski et al. 1998; Ivanov et al. 2008; He and Jacobson 2015). Most of the [PSI+] variants arising in the absence of Upf1, 2, or 3 are cured by simply restoring the normal amounts of the Upf proteins (Son and Wickner 2018). Upf1p can inhibit amyloid formation by Sup35p in vitro, and binds to Sup35p amyloid filaments in vivo. It is believed that the Upf proteins either compete for Sup35p monomers with the amyloid filaments, or block filament elongation by binding to the ends of the filaments (Son and Wickner 2018). Siw14p, a pyrophosphatase that cleaves the 5-pyrophosphate of several inositol polyphosphates (Steidle et al. 2016), also has a prion-curing action whose mechanism is as yet unclear (Wickner et al. 2017). The ribosome-associated chaperones, Ssb1/2p, Ssz1p, and Zuo1p insure proper folding of nascent proteins (Nelson et al. 1992; Pfund et al. 1998). These chaperones working together keep down the frequency of [PSI+] prion generation (Chernoff et al. 1999; Amor et al. 2015; Kiktev et al. 2015), and also cure most of the [PSI+] variants arising in their absence (Son and Wickner 2020). The frequency of spontaneous prion generation is elevated 2- to 15-fold in mutants defective any of these anti-prion genes (Wickner et al. 2014, 2017; Gorkovskiy et al. 2017; Son and Wickner 2018).
In addition to components that cure most prion variants as they arise or block prion generation, there are cellular components that prevent some variants from being toxic or lethal to the cell. Sis1p has activities nonessential for cell growth in the absence of prions that become essential if the [PSI+] prion is acquired (Kirkland et al. 2011). We used Hermes transposon mutagenesis to screen for genes preventing [URE3] lethality, finding genes, particularly LUG1/YLR352W, that can be disrupted in [ure-o], but are essential in [URE3] strains (Edskes et al. 2018). In the course of that study, a strain with the Hermes transposon inserted in tof2 was noted to destabilize the [URE3] prion.
Materials and methods
Genetic manipulations
Yeast media are as described by Sherman (1991), except ½ YPD is 5 g Yeast Extract, 20 g Peptone, 20 g dextrose, and 20 g agar per liter; YES+W is 5 g yeast extract, 30 g dextrose, and 30 mg tryptophan and 20 g agar per liter. Strains of S. cerevisiae are listed in Supplementary Table S1. Yeast transformation was performed by the Li acetate method (Geitz 2014). PCR was performed using Q5 polymerase (New England Biolabs) or Titanium Taq (Clonetech) using primers as shown in Supplementary Table S2. Knockout constructs were obtained by PCR amplification from the Saccharomyces Genome Deletion Collection (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). Hermes is marked by nourseothricin resistance (Gangadharan et al. 2010).
Detecting site of Hermes insertion
Genomic DNA was prepared from strain YES01 using the YeaStar Genomic DNA Kit from Zymo Research. Genomic DNA (1.5 μg) was digested with MseI in a total volume of 100 μl. The Qiagen MinElute Reaction Cleanup Kit was used and DNA was eluted with 15 μl water. Linkers HE968 and HE969 (20 pmol/μl each) were annealed in a thermocycler as follows: 95 °C 15 min, 75 °C 13 min, 55 °C 13 min, 35 °C 3 min 30 s, 20 °C 5 min, hold at 4 °C. Annealed linkers (100 pmol each) were ligated to 750 ng digested genomic DNA in a total volume of 20 μl using 1 μl of NEB ligase (4 units/μl) at 16 °C (there is only a 2 nuclotide overhang) for 3 h. Ligase and linkers were then removed using a Qiagen MinElute Kit and elution was done with 15 μl of water. The Hermes/genomic DNA junction was amplified using Titanium Taq (Clonetech) polymerase with 1 μl MseI-digested, linker-ligated genomic DNA, and 1 pmol/μl each of oligo HE967 and oligo HE970. The cycle was (1) 94 °C 3 min, (2) 94 °C 20 s, (3) 55 °C 15 s, (4) 68 °C 25 s, (5) go to 2 for 30 cycles, (6) 68 °C 5 min. The PCR product was then cloned into pCR-2.1-TOPO (ThermoFisher) and sequenced, revealing one junction of Hermes and chromosomal DNA.
Hermes insertion in TOF2
Genomic DNA of strain YES01 (the originally identified Hermes insertion isolate) was prepared using YeaStar Genomic DNA Kit from Zymo Research and the full Hermes insertion in TOF2 was amplified using primers HE1025 and ES02 using Titanium Taq (Clonetech). The PCR product was cloned into pCR2.1-TOPO creating pES01 and sequenced. The Hermes insertion replaced nt 1045–1054 of TOF2.
Tof2::Hermes was amplified from plasmid pES01 using oligos HE1025 and ES02.
Tof2::G418 was amplified from genomic DNA of the knockout strain using primers ES03 OR ES04 AND primers ES05 OR ES06. PCR products were transformed into strains BY241, Σ1278b, and 779-6A, and transformants were selected on 1/2YPD (to monitor [URE3]) containing the appropriate antibiotic. Hermes is marked by nourseothricin resistance (Gangadharan et al. 2010). Mutations were confirmed using oligos ES08, ES09, ES10, ES11, HE267, and HE268.
Gene deletion
Using the deletion collection as the source of knockout constructs, PCR amplification was performed using Q5 polymerase (New England Biolabs). For example, irc25::kanMX was amplified with combinations of ES21, ES22, ES23, and ES24, and rpn4::kanMX was amplified using primers ES37 and ES39 or ES38 and ES40. PCR reactions for each of the genes were mixed and used to transform YHE1609. Transformants were selected on 1/2YPD plates containing G418, and confirmed by PCR of DNA purified from candidate clones.
Overexpression of TOF2
TOF2 with 500 bp upstream UTR was amplified from genomic DNA using oligos ES12 and ES16. The PCR product was cloned into pRS424 as a BamHI-HindIII fragment resulting in plasmid pES04. Transformation of plasmid pES04 into YHE1609 or YHE1608 did not give any transformants while many transformants were obtained using pRS424. This suggests that overexpression of Tof2 is toxic to these cells.
Template for TRP1-based cassette used to disrupt BTN2 and CUR1
TRP1 containing 278 nt 5′ UTR and 46 nt 3′ UTR and with loxP sites (ATAACTTCgTATAgCATACATTATACgAAgTTAT) added was amplified with primers ES69 and ES70 from pCW037 to knock out BTN2 or ES71 and ES72 to knock out CUR1. pCW037 is identical to pCW036 (Edskes et al. 2014, Genetics) except for the BsmBI site being replaced with a SapI site. To create strains in which both BTN2 and CUR1 were deleted TRP1 was removed by expression of CRE from plasmid YEp351-cre-cyh (DELNERI et al. 2000) after the first knockout.
Assay of prions
When a good nitrogen source, such as ammonia, is present in the medium, Ure2p represses genes needed for utilizing poor nitrogen sources, such as allantoate (Cooper 2002). DAL5 encodes the allantoate transporter, which is ∼100-fold repressed on ammonia through the action of Ure2p. The prion form of Ure2p is essentially inactive in nitrogen catabolite repression, and this effect is detected in [URE3] cells as an Ade+ phenotype and white colony color (on adenine-limiting plates) in cells whose ADE2 gene has a DAL5 promoter; [ure-o] cells are Ade- and red (Schlumpberger et al. 2001; Brachmann et al. 2005).
Western blotting
Lysis Buffer was 8 M urea, 1 mM DDT, 1 mM EDTA, 1 mM PMSF, 1x protease inhibitor (Roche), 5% glycerol, in PBS. Rabbit polyclonal anti-Cur1p antibody (Bezsonov et al. 2021) was affinity purified using E. coli-produced Cur1-His6p. Rabbit polyclonal anti-Btn2p antibody is described by Bezsonov et al. Mouse monoclonal anti-Sup35p antibody BE4 (Bagriantsev and Liebman 2006) was a gift from S. Liebman. Mouse monoclonal anti Pgk1p antibody (ab113687) was purchased from Abcam. Anti-rabbit IgG(Fc)-alkaline phosphatase secondary antibody (S3738) was purchased from Promega. Anti-mouse IgG-alkaline phosphatase secondary antibody (S3562) was purchased from Millipore Sigma. In order to quantitate Btn2p levels in pre9Δ or proteasome chaperone mutants the sample was diluted to avoid signal saturation.
SILAC methods
SILAC strains:
Because our attempts to delete ARG4 in YHE1608 were unsuccessful, we created an arg1::TRP1 lys2::LEU2 strain to use in SILAC experiments. YMD74 was white on adenine limiting medium likely due to CAN1 being controlled by the DAL5 promoter. CAN1 was restored by transformation with a CAN1 containing DNA fragment and screening transformants for production of red pigment on adenine limiting medium. Disruption of PRE9 in YMD134 by kanMX resulted in YMD144.
Cells were grown in synthetic complete minimal glucose medium. Medium for isotope labeling contained 20 mg/L L-lysine-2HCl [13C6 99%; 15N2 99%] and 30 mg/L L-arginine-2HCl [13C6 99%; 15N4 99%] instead of the 50 mg/L used for the nonlabeling medium. In addition to the labeled amino acids, 200 mg/L of L-Proline was also present to reduce the metabolic conversion of arginine to proline. A starter culture was grown in media without labeled amino acids from which 12 ul was used to inoculate 60 ml media with or without labeled amino acids. When cells reached an OD600 between 0.6 and 0.7 they were collected by centrifugation at 4 °C and washed 3 times with ice-cold water before resuspending in a volume of water proportional to final OD600 of the cultures. Cells were stored frozen at −80°C.
Equal amount of cells from one labeled and one unlabeled culture were mixed and pelleted. Cells were resuspended in lysis buffer [1% sodium dodecanoate, 8 M urea, 1 mM DTT, 1 mM EDTA, 1 mM PMSF, 1× proteinase inhibitor (Roche), 5% glycerol, 10 mM Tris-HCl pH7.4] and broken with glass beads using a Biospec Mini-Beadbeater. The extract was clarified by centrifugation at 4 °C. Further processing is described in the supplementary information: after reduction and alkylation of thiols samples were digested with trypsin, subjected to off-line high pH concatenated separation, and subjected to LC/MS/MS with data analysis using MaxQuant (Rappsilber et al. 2007; Cox and Mann 2008; Wang et al. 2011).
Statistical analysis:
The distribution of Hermes inserts in Table 1 follows the binomial distribution, because each data point expresses two alternative results, finding the Hermes transposon in the indicated gene open reading frame or finding it elsewhere in the genome. The number of total insertions in the culture identified (N), the probability of finding an insertion in the indicated gene (p) and of finding it elsewhere (q) are used in calculations. The results should be approximately normally distributed as long as Npq ≫ 1, which was true for all our data. We want to calculate the probability that two sets of data ([URE3] culture vs. [ure-o] culture) could be samples from the same distribution of insertions, differing only because of random fluctuations on sampling. If p3, q3, N3 vs. po, qo, No are the parameters for [URE3] and [ure-o], respectively, then, combining populations, the overall chance of insertion in the particular gene is p = (p3•N3+po•No)/(N3+No), and the expected insertion number in the [URE3] clones is p•N3 with standard deviation S = [(N3+No)p(1-p)]1/2. The difference p•N3—p3•N3 between expected and observed number of mutant clones that lost the prion, is divided by the standard deviation S, and the probability of this difference (z) or greater from the expected result is obtained from a normal distribution table. The Mann-Whitney U-test was used for Table 2 to confirm that each mutant seed number was less than that of the wild-type.
Table 1.
Hermes insertions recovered in proteasome-related genes
| Gene | Gene size (nt) | [URE3] –Ade |
[ure-o] +Ade |
||
|---|---|---|---|---|---|
| Insertions | Unique | Insertions | Unique | ||
| TOF2 | 2316 | *4120 | **21 | 3723 | 67 |
| RPN4 | 1596 | **458 | **8 | 3734 | 67 |
| IRC25 | 540 | **50 | *1 | 269 | 16 |
| PBA1 | 917 | **1006 | **8 | 6913 | 46 |
| POC4 | 447 | **0 | *0 | 134 | 17 |
| HMS3 | 1443 | **179 | **5 | 4019 | 52 |
| RPN10 | 807 | **0 | *0 | 2665 | 24 |
| ADD66 | 804 | **349 | *5 | 1574 | 27 |
| RPN14 | 1254 | 10226 | *55 | 7513 | 106 |
| NAS6 | 687 | **2472 | 14 | 3499 | 29 |
| NAS2 | 664 | 2538 | 10 | 1940 | 24 |
| PRE9 | 777 | ** 60 | 1 | 558 | 11 |
Note: The Hermes transposon was mobilized in isogenic [ure-o] and [URE3] cells. After growth, DNA was isolated and insertion sites amplified by PCR, cloned, and sequenced (>57 × 106 reads for [URE3] and >45 × 106 for [ure-o] cultures; Edskes et al. 2018). “Insertions” means total reads within the indicated ORF, while “Unique” indicates distinct sites represented among the insertions in that ORF. Decreased recovery of insertions in a gene in the [URE3] culture could be due to failure of the insertion mutants to stably propagate [URE3] (as is the case for PRE9, for example) or their failure to prevent toxicity of the prion [as was shown for LUG1 (Edskes et al. 2018)]. Data from Edskes et al. (2018). Insertion recovery frequency was less in [URE3] cultures with *P < 0.005, **P < 0.0001.
Table 2.
[URE3] prion propagon numbers
| w.t. (YHE1609) | tof2Δ | irc25Δ | poc4Δ |
|---|---|---|---|
| 99 | 9 | 12 | 31 |
| 3980 | 14 | 7 | 98 |
| 126 | 437 | 27 | 66 |
| 2568 | 338 | 107 | 68 |
| 363 | 22 | 50 | 38 |
| 158 | 8 | 55 | 18 |
| 185 | 19 | 71 | 63 |
| 81 | 3 | 22 | 53 |
| 945 ± 1488 | 106 ± 175* | 44 ± 34** | 54 ± 25** |
Note: Eight whole colonies from YPAD plates with guanidine were replated on -Ade. Number of Ade+ colonies formed reflects number of seeds or propagons in the founder cell of the original colony (see Materials and Methods). The mean ± standard deviation is shown in the last row. Using the Mann-Whitney U-test, we found that the mutant seed numbers were lower than the w.t. with *P = 0.01 or **P < 0.001.
Results
Insertion or deletion mutations of TOF2 destabilize [URE3]
The [URE3] prion assay uses the red color developed by ade2 auxotrophs and a DAL5: ADE2 fusion gene. Ure2p negatively regulates DAL5 (and so ADE2 in our strains), but is inactive in [URE3] cells because it is converted to amyloid filaments. Thus, on adenine-limited media (e.g., YES+W plates) [ure-o] strains are red Ade- (DAL5 shut off, cells are ade2) while [URE3] strains are white Ade+ (DAL5 derepressed and ADE2 actively transcribed). In our Hermes transposon mutagenesis screen (Edskes et al. 2018), we compared insertion locations of a [URE3] culture grown without adenine to a [ure-o] culture grown with adenine. Failure to find insertions in a given gene preferentially in the [URE3] cells could be due to lethality of the prion in the absence of that gene (our goal in that work) or to that gene being essential/important for the propagation of the prion. To monitor loss of [URE3], cultures were plated on YES+W plates. In most cases, re-plating red/white sectored colonies produced only red and white colonies, but one exceptional colony produced many sectored subclones, even after several sub-clonings (Figure 1A). Red colonies produced only red subclones, but white or red/white sectored colonies produced more sectored colonies. This continuous production of [ure-o] sectors during mitotic growth on solid media, suggested that the gene disrupted by the transposon was important for prion propagation.
Figure 1.

tof2::Hermes or tof2::kanMX destabilize [URE3]. (A) The original BY241 [URE3-1] strain with a Hermes insertion (shown here to be tof2::Hermes) destabilizing [URE3-1]. A white [URE3-1] single colony of the strain in which [URE3-1] was unstable was streaked for single colonies on YES+W plates. White or pink colonies or sectors are [URE3] and red colonies are [ure-o]. (B) The original Hermes insertion or a deletion/insertion with kanMX (G418-resistance) in each of three strain backgrounds show the same effect as the original Hermes mutant. BY241 is the background that was utilized for the Hermes screen. Σ1278b is often used in studies of nitrogen regulation. Strain 779-6A has been used in studies of [PSI+] and [URE3].
The site of the Hermes insertion was identified by a PCR-based method (EDSKES et al. 2018), and proved to have replaced nt 1045–1054 (relative to the AUG) of the 2316 nt TOF2 ORF. This same Hermes insertion or the tof2::kanMX knockout was introduced into each of three backgrounds (BY241, Sigma 1278b or 779-6A). In each case, [URE3] was destabilized (Figure 1B).
First identified as a topoisomerase I interacter (Park and Sternglanz 1999), Tof2p is required for rDNA silencing as a part of a complex including Csm1 and Lrs4 (cohibin) and Cdc14, Sir2 and Net1 (RENT) (reviewed in Mekhail and Moazed 2010; Gartenberg and Smith 2016; Srivastava et al. 2016). This complex bridges between Fob1p bound to special sites in rDNA (RFB sites) and CLIP complexes (Heh1p and Nur1p) on the inner nuclear membrane. But what is the connection of Tof2p to instability of [URE3]?
Cmr1p (changed mutation rate) is a nuclear protein that responds to DNA damage or replication stress by changing from an even distribution in the nucleus to localization in a discrete site at the periphery (but inside) the nucleus (Gallina et al. 2015). Co-localizing with Cmr1p in the discrete particles are certain DNA replication, cell cycle, and chromatin remodeling proteins, and, notably, the Hsp104 disaggregase, the Hsp40 family protein Apj1p and Btn2p (Gallina et al. 2015). The curing of [URE3] by Btn2p requires Hsp42 (Wickner et al. 2014), and similarly, the localization of Cmr1p requires Btn2p and Hsp42 (Gallina et al. 2015). Remarkably, Cmr1p localization occurred at high frequency in tof2 mutants, as well as in the proteasome-related mutants, irc25, rpn4, and san1 (Gallina et al. 2015). This finding led us to examine whether these or other proteasome factors (Budenholzer et al. 2017) affect propagation of [URE3-1].
Proteasome-related genes necessary for the stability of [URE3]
In strain BY241 carrying [URE3-1] we introduced irc25Δ or rpn4Δ, and found that, like tof2Δ, each of these mutations destabilized [URE3-1] (Figure 2A). Consistent with this result, in our Hermes transposon screen (Edskes et al. 2018), insertions in either IRC25 or RPN4 or TOF2 (slightly), but not other proteins involved in the rDNA silencing complex, were unusual in [URE3] cells grown without adenine (selecting for the presence of the prion) compared to [ure-o] cells (Table 1). We previously showed that san1Δ does not destabilize [URE3-1] (Wickner et al. 2014). We examined other proteasome-related genes for their effects on [URE3]. In the Hermes screen, insertions in PBA1, POC4, HSM3, ADD66, and RPN10 were less common in [URE3] cells, but for RPN14, NAS6, or NAS2 there was little or no such effect (Table 1; Edskes et al. 2018). Rpn4p is a transcription factor stimulating the expression of proteasome-related genes (Xie and Varshavsky 2001), while Irc25p(Pba3p) and Poc4p(Pba4p) form a heterodimeric chaperone involved in assembly of alpha subunits into the 20S core proteasome (Le Tallec et al. 2007). Add66p(Pba2p) and Pba1p form another heterodimer involved in 20S particle assembly (Le Tallec et al. 2007) (Supplementary Figure S1 reviewed in Budenholzer et al. 2017). Hsm3p, Nas2p, Nas6p, and Rpn14p are assembly factors for the base of the 19S regulatory particle, while Rpn10 links the base of the 19S particle to its lid.
Figure 2.

Proteasome effects on [URE3-1] (A) [URE3-1] is destabilized by deleting genes for some proteasome chaperones or PRE9 encoding a proteasome subunit. Strain BY241 [URE3-1] with the indicated knockout, except for the [ure-o] strain, was grown as a patch on –Ade plates, then streaked for single colonies on YES+W plates. Red sectors or colonies are [ure-o]. (B) Strain BY241 [URE3-1] was grown in rich medium for 2 days with just solvent (DMSO) or 50 μM MG132 added and plated for single colonies on ½ YPD.
We directly tested the proteasome assembly and subunit genes for effects on stability of [URE3-1] by making knockouts in BY241 [URE3-1] (Figure 2A). Deletions of RPN4, IRC25, POC4, HSM3, and RPN10 clearly destabilized [URE3-1] while those of PBA1 and PBA2 only weakly or very weakly did so, respectively. Of the proteasome 20S core particle subunits, only α3, encoded by PRE9, is nonessential, and pre9Δ cells rapidly lost the prion.
If any of these genes were producing red colonies by merely affecting the prion phenotype without losing the prion, we would expect an even colony color. Sectoring at this high frequency implies loss of the prion. To confirm this conclusion, we mated the rpn4Δ, irc25Δ, poc4Δ, and rpn10Δ red colonies with the [ure-o] strain YHE1635. The diploids formed were mostly red and Ade-, implying that [URE3] had been lost from most of the cells. In many cases, some white prion-containing diploid papillae were also detected on –Ade plates implying that there were still a few [URE3] cells in the red colonies, and that the prion was being gradually lost during growth of the mutants.
In confirmation of the above, we find that MG132, a specific inhibitor of proteasome activity, cures [URE3-1] (Figure 2B). These results imply that a stable proteasome is required for maintenance of [URE3].
Propagon numbers are reduced in tof2Δ, irc25Δ, and poc4Δ
Propagons (or seeds) are the minimal unit of heritable prion amyloid (Cox et al. 2003). New propagons are made by Hsp104, Hsp70, and Hsp40 removing a monomer from the middle of a filament (reviewed by Mogk et al. 2015). To measure propagon number, cells are plated on solid medium containing 5 mM guanidine HCl, which effectively inhibits Hsp104. Cells then grow, with propagons (seeds) segregating into separate cells of the clone, until there is only one (or none) per cell. The number of propagons in the founding cell of each clone is then estimated by plating the entire colony on -Ade medium and counting the number of clones (Cox et al. 2003). We found that tof2Δ, irc25Δ, and poc4Δ mutations each reduced the propagon number of [URE3-1] (Table 2).
Proteasome mutants do not have evident effects on [PSI+]
We tested many of our proteasome mutants for effects on [PSI+] propagation in the 779-6 A background (Supplementary Figure S2). We found no evidence of sectoring for any of these strains. Note that in this background, tof2Δ (Figure 1) or pre9Δ (Supplementary Figure S3) destabilize [URE3] as much or more than they do in BY241. It has been reported that pre9Δ strains have decreased frequency of [PSI+] generation and lowered levels of Sup35p in the 74D-694 background (Tyedmers et al. 2008; Manogaran et al. 2011). We did not find any change in Sup35p levels in either the BY241 (Supplementary Figure S4) or 779-6A (Supplementary Figure S5) background for pre9Δ, poc4Δ, irc25Δ, add66Δ, or pab1Δ mutants compared to wild-type. Our experiments included log phase harvesting or stationary phase harvesting, and examination of both [PSI+] and [psi-] cells in rich or minimal media. In addition, in our SILAC experiments (see below) Sup35p levels were similar in the WT and pre9Δ cells. We note that the presence of [PSI+] adds to defects in proteasome genes to mildly increase the induction of Btn2p (Supplementary Figure S6).
Loss of [URE3] in proteasome mutants is prevented by btn2Δ and cur1Δ mutations or hsp42Δ
The pre1-1ts mutation (proteasome β4 subunit) results in elevated levels of Btn2p (Malinovska et al. 2012) and as the elevation of Btn2p cures [URE3] (Kryndushkin et al. 2008), this suggests that the destabilization of [URE3-1] in our proteasome mutants occurs via Btn2p. We found that in either btn2Δ pre9Δ or cur1Δ pre9Δ, [URE3-1] was partially stabilized compared to a pre9Δ strain, but only in the cur1Δ btn2Δ pre9Δ combination was [URE3-1] fully stabilized (Figure 3). These results indicate that the pre9Δ proteasome mutation acts through both Btn2p and Cur1p. In contrast, the btn2Δ deletion was sufficient to suppress the lower degree of [URE3-1] instability produced by irc25Δ or poc4Δ (Figure 3).
Figure 3.
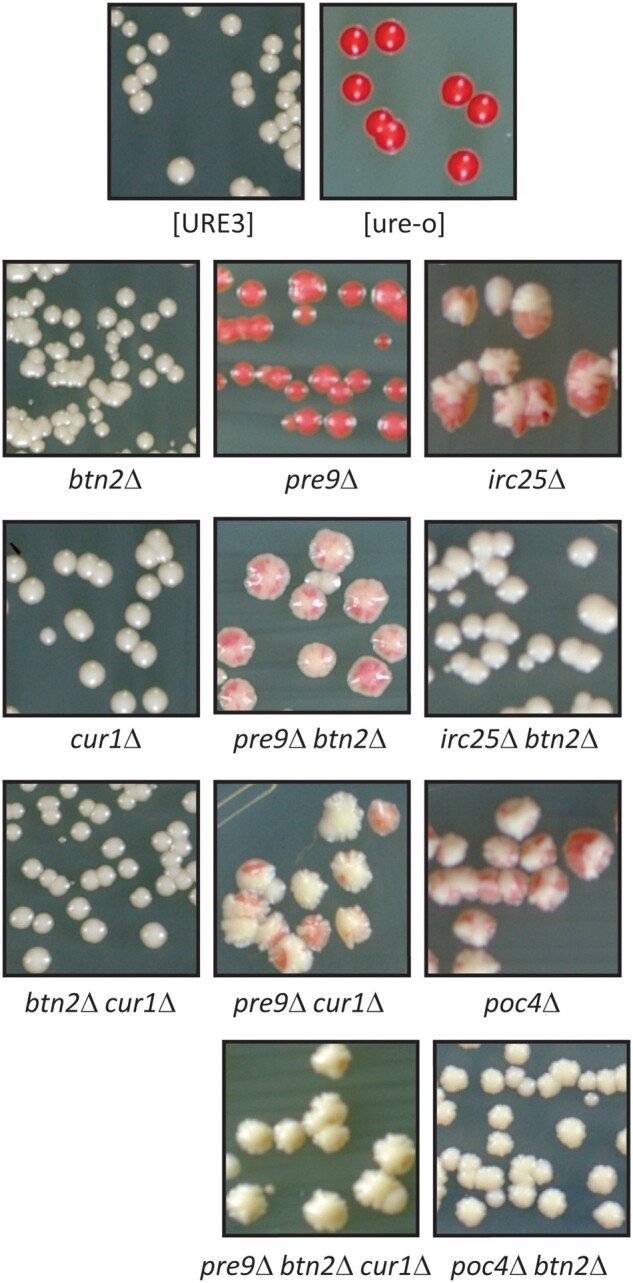
[URE3-1] loss in proteasome chaperone mutants and pre9Δ strains is prevented by btn2Δ and cur1Δ mutations. Red sectors or colonies are [ure-o]. All mutants were made in the BY241 background.
Hsp42 is necessary for curing of [URE3] by overproduction of Btn2p or Cur1p, and overproduction of Hsp42 is itself sufficient to cure [URE3] (Wickner et al. 2014). If proteasome mutant curing of [URE3] is due to overproduction of Btn2p, then we expect that that curing should be blocked in hsp42Δ strains. Indeed, pre9Δ hsp42Δ cells propagate [URE3] stably (Figure 4).
Figure 4.
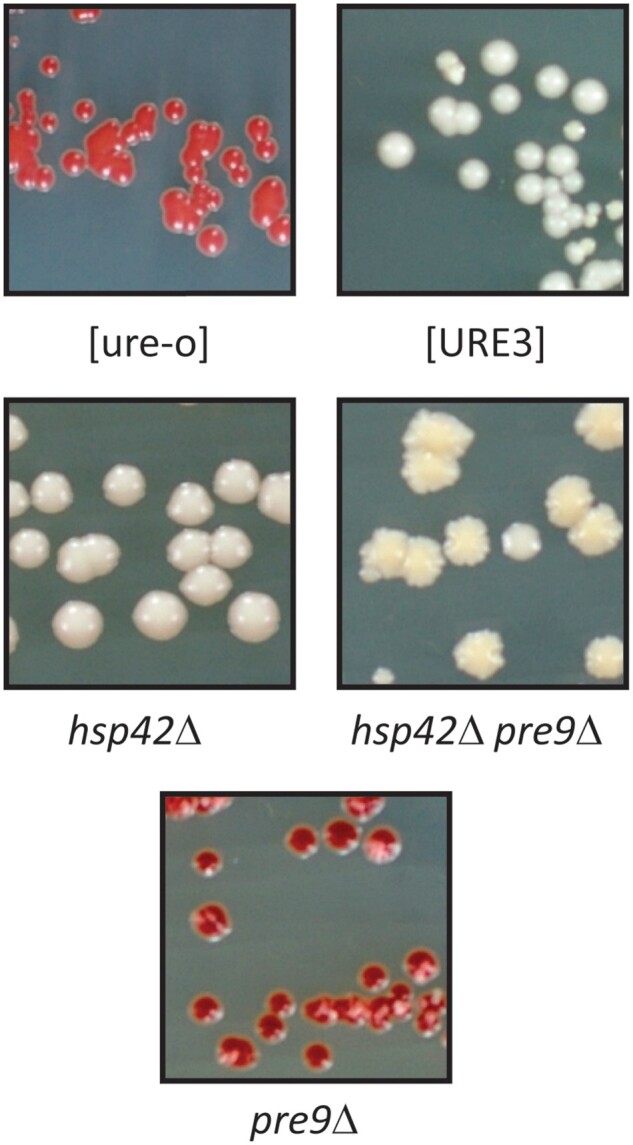
Loss of [URE3-1] in pre9Δ cells requires Hsp42p. Red sectors or colonies are [ure-o]. All mutants were made in the BY241 background. hsp42Δ causes flocculation.
Anti-Btn2p and anti-Cur1p used to quantitate cellular levels
The protein abundance database (https://pax-db.org/), summarizing a large number of studies, gives the abundance of Cur1p as 1.4 ppm (∼140 molecules/cell) and Btn2p as 6.1 ppm (∼300 molecules/cell). We obtained polyclonal antibodies to both Btn2p and Cur1p, and calibrated them with purified recombinant proteins made in E. coli (Supplementary Figure S7). Even using affinity—purified antibody, Cur1p levels in the wild-type were difficult to measure because of the presence of cross-reacting bands very close to the Cur1p band (Supplementary Figures S7 and S8) and the very low level of Cur1p in the wild-type (Figure 5). It is also clear that there are multiple bands reactive with anti-Cur1p, absent in a cur1Δ strain, and dramatically elevated in proteasome mutants (Figure 5, Supplementary Figure S8). However, we estimate that Cur1p comprises 1 ppm of cell protein in strain 779-6A (110 molecules per cell) and less in strain BY241. We find that Btn2p comprises 2.7 ppm in strain 779-6A (170 molecules per cell) and less in strain BY241. Given the substantial errors in the measurements and the variability between strains, both of our measurements are consistent with earlier estimates cited above.
Figure 5.

Elevation of Btn2p and Cur1p in proteasome mutants. Extracts of [ure-o] strains of BY241 with the indicated genotypes were analyzed by western blot. Fold-increase of Btn2p was measured relative to the wild-type with correction for loading using the Pgk1p control. Accurate measurements of Cur1p in the wild-type were impaired by the presence of nearby cross-reacting bands, but the elevation of Cur1p in the proteasomal mutants appears to be of the same order of magnitude as for Btn2p.
Btn2p and Cur1p expression is elevated in proteasome mutants
The suppression by btn2Δ and cur1Δ of the effects of proteasome mutants on [URE3] stability suggests that the abrogation of proteasome activity allows higher levels of Btn2p and Cur1p which, in turn, cures [URE3]. We therefore examined the effects of proteasome mutants on the levels of Btn2p and Cur1p. We find that the un-tagged Btn2p and Cur1p each are expressed at dramatically higher levels in the proteasome mutants than in the wild-type in both strain BY241 and in strain 779-6A (Figure 5, Supplementary Figure S5). In addition, Btn2p levels are also elevated in TOF2 knockout strains (Supplementary Figure S9). Moreover, the degree of overexpression is roughly proportional to the destabilization of [URE3]. Mutants pba1Δ and add66Δ have substantially increased Btn2p and Cur1p (19- to 24-fold in Figure 5 for Btn2p) but do not destabilize [URE3-1], while irc25Δ (89x), poc4Δ (92x) and pre9Δ (125x) result in even higher levels and do produce prion loss. The fold-increase varies somewhat (Supplementary Figures S5, S6, and S9) but the relative increase is quite consistent. We detect no sign of ubiquitin-conjugated Btn2p, even though the proteasome (and not the ubiquitin-conjugating system) is impaired. See, for example, pre9Δ cells in Supplementary Figure S10. Our data suggest that in pre9Δ cells the ubiquitin tag is removed but the defective proteasomes then do not degrade the Btn2p.
SILAC screen for other mediators of proteasome effects on [URE3]
The experiments described above show that proteasome mutants lead to elevated Btn2p and Cur1p, and that these two proteins, as well as Hsp42, are both needed for the instability of the [URE3] prion in those strains. We sought other proteins involved in these effects by comparing the levels of proteins in a pre9Δ with an isogenic wild-type using the SILAC method (Ong et al. 2002; see Materials and Methods).
The data showed the expected ∼20-fold elevation of Btn2p in the pre9Δ strain, but Cur1p was below the detection limit even in the mutant due to its very low levels. A range of other proteins were significantly elevated or depressed in the pre9Δ strain (Table 3, Supplementary Tables S3 and S4), but except for Btn2p, none of these changes had evident relation to the curing process. A total of 162 proteins had levels elevated 1.5-fold or more in the pre9Δ strain (44 proteins >2.0; Supplementary Table S3). The largest group of proteins (44) in this class are related to the proteasome and ubiquitination (Table 4.). A group of 17 proteins acts in protein folding. One hundred and seven proteins were reduced 1.5-fold or more in abundance in the pre9Δ strain (32 proteins >2.0; Supplementary Table S4). Somewhat surprisingly, 11 permeases and transporters were among those decreased in the mutant (Table 4).
Table 3.
Protein level changes in pre9Δ cells (SILAC)
| pre9Δ/WT | StdDev | Gene | Function |
|---|---|---|---|
| 0.14 | 0.06 | PTR2 | Transporter of di- and tri-peptides |
| 0.33 | 0.11 | SST2 | GTPase-activating protein for Gpa1p; regulates desensitization to alpha-factor pheromone |
| 0.36 | 0.15 | STE6 | Plasma membrane ATP-binding cassette (ABC) transporter; required for the export of a-factor |
| 0.36 | 0.15 | STE2 | Receptor for alpha-factor pheromone |
| 0.36 | 0.17 | INO1 | Inositol-3-phosphate synthase |
| 0.37 | 0.07 | CWP1 | Cell wall mannoprotein that localizes to birth scars of daughter cells |
| 2.53 | 0.45 | OTU1 | Deubiquitylation enzyme that binds to the chaperone-ATPase Cdc48p |
| 2.56 | 0.39 | ECM29 | Scaffold protein that assists in association of the proteasome core particle with the regulatory particle |
| 2.57 | 0.69 | APJ1 | HSP40 chaperone with a role in SUMO-mediated protein degradation |
| 2.62 | 1.01 | RTS3 | Putative component of the protein phosphatase type 2A complex |
| 2.71 | 0.55 | HBN1 | Protein of unknown function; similar to bacterial nitroreductases |
| 2.74 | 1.00 | YOR052C | AN1-type zinc finger protein |
| 2.82 | 0.42 | EDC1 | RNA-binding protein that activates mRNA decapping directly |
| 2.94 | 1.67 | SSA3 | HSP70 chaperone |
| 2.94 | 0.50 | CUZ1 | Interacts with ubiquitinated proteins, Cdc48p, and the proteasomal regulatory particle |
| 3.33 | 1.21 | YKR011C | Protein of unknown function |
| 3.37 | 0.73 | HLR1 | Protein involved in regulation of cell wall composition and integrity |
| 3.42 | 1.13 | BIO2 | Biotin synthase; catalyzes the conversion of dethiobiotin to biotin |
| 3.66 | 0.66 | YPL113C | Glyoxylate reductase; acts on glyoxylate and hydroxypyruvate substrates |
| 4.11 | 3.35 | ARO10 | Phenylpyruvate decarboxylase; catalyzes decarboxylation of phenylpyruvate to phenylacetaldehyde |
| 4.39 | 4.60 | BNA2 | Tryptophan 2,3-dioxygenase or indoleamine 2,3-dioxygenase; required for de novo biosynthesis of NAD from tryptophan |
| 4.52 | 3.24 | PDC5 | Minor isoform of pyruvate decarboxylase |
| 4.94 | 1.85 | TMA10 | Protein of unknown function |
| 4.95 | 0.67 | MAG1 | 3-methyl-adenine DNA glycosylase; involved in protecting DNA against alkylating agents |
| 4.97 | 3.56 | SNZ1 | Pyridoxal-5-phosphate synthase subunit |
| 5.02 | 1.07 | LPL1 | Phospholipase that maintains lipid droplet (LD) morphology |
| 6.44 | 1.49 | UMP1 | Chaperone required for correct maturation of the 20S proteasome |
| 21.43 | 6.42 | BTN2 | Aggregase and v-SNARE binding protein |
Note: Proteins changed 2.5-fold or more in the pre9Δ strain compared to the WT strain.
Table 4.
Proteins changed in pre9Δ cells (SILAC) grouped by description
| Increased >1.5-fold in pre9Δ | # | Decreased >1.5-fold in pre9Δ | # |
|---|---|---|---|
| Proteasome/ubiquitin | 44 | Transporter | 19 |
| Unknown | 23 | Mitochondria | 17 |
| Chaperone | 17 | Lipids | 15 |
| Carbohydrate | 17 | Carbohydrate | 12 |
| Mitochondria | 16 | Unknown | 11 |
| Golgi/ER | 15 | Phosphate | 9 |
| Cell cycle | 14 | Cell wall | 8 |
| Amino acids | 13 | Mating | 6 |
| Cell wall | 6 | — | — |
| Vacuole | 5 | — | — |
| NAD | 5 | — | — |
Note: Nine proteins within the proteasome/ubiquitin group are linked to the Cdc48 complex; 8 of these proteins are also present in the cell cycle group and 4 are also present in the golgi/ER group.
Several proteins whose overexpression or deficiency are known to affect [URE3] propagation were detectable in our SILAC experiments (Table 5). Significantly, Hsp42 was only slightly elevated in the pre9Δ strain (1.32 ± 0.25 fold in six measurements). Overexpression of Hsp42 can cure [URE3-1], and we find that hsp42Δ prevents destabilization of [URE3] by proteasome mutations, but our result suggests that, in this case, elevated Hsp42 is not responsible for curing the prion, but is a required co-factor. It is striking that Btn2p is easily the most dramatically elevated protein in our SILAC data, and the Western blot data indicates that Cur1p is similarly increased. Notably, none of the other detected proteins known to affect [URE3] propagation were substantially altered (Table 5).
Table 5.
Levels of proteins known to affect [URE3] propagation
| Protein | pre9Δ/WT | StdDev |
|---|---|---|
| Btn2p | 21 | 6 |
| Ssa1p | 1.54 | 0.23 |
| Ssa2p | 1.54 | 0.29 |
| Hsp104p | 1.53 | 0.28 |
| Fes1p | 1.42 | 0.26 |
| Sse1p | 1.40 | 0.20 |
| Sis1p | 1.33 | 0.17 |
| Hsp42p | 1.33 | 0.25 |
| Ydj1p | 1.20 | 0.22 |
| Hsp26p | 1.09 | 0.21 |
| Cpr7p | 1.04 | 0.15 |
| Zuo1p | 0.98 | 0.19 |
| Ssz1p | 0.96 | 0.17 |
| Upf3p | 0.96 | 0.16 |
| Swa2p | 0.85 | 0.14 |
| Ssb2p | 0.84 | 0.20 |
| Ssb1p | 0.81 | 0.17 |
Note: Summary of SILAC data for proteins whose overproduction or deficiency is known to affect the propagation of [URE3]. Except for Btn2p, the values shown are the average and standard deviation of 6 biological duplicates of the ratio of the given protein in pre9Δ cells to that in isogenic wild-type cells. For Btn2p, the amount in the wild-type was so low that it was only accurately measurable in three of the six experiments.
The overall client spectrum of the proteasome has not been mapped. Many papers focusing on the proteasome use model substrates. How and if the activity measured using these model substrates matches with the normal clientele is unknown. Our SILAC data offers a first glimpse into the normal functioning of the proteasome. For example, impairment of proteasome function improves growth of fzo1Δ strains defective in mitochondrial fusion (Shirozu et al. 2016), suggesting that there is an interaction between the proteasome system and mitochondrial fission/fusion. Our SILAC data show that 16 mitochondrial proteins have increased abundance in a pre9Δ strain whereas the abundance of 13 proteins is decreased (on ammonium medium; Table 5). Among those that are decreased are 5 of the 12 members of the cytochrome c complex. As discussed below, these changes may be explained by a posssibly changed proteasome specificity of the α4-α4 proteasomes produced in pre9Δ, irc25Δ, and poc4Δ strains (Velichutina et al. 2004; Kusmierczyk et al. 2008).
Discussion
Beginning with our observation that a tof2::Hermes mutation destabilized [URE3-1], and the fact that tof2Δ, like proteasome-deficient mutants irc25Δ and rpn4Δ, showed frequent intranuclear focus formation by Cmr1p (Gallina et al. 2015), we tested these and other mutants affecting proteasomes for effects on [URE3-1] stability. We found that many mutants in proteasome assembly destabilized [URE3] as much or more than the tof2::Hermes mutant, particularly pre9Δ, deficient in the α3 proteasome core component. Btn2p and Cur1p are known anti-prion proteins acting on [URE3] (Kryndushkin et al. 2008; Wickner et al. 2014), and the report of elevated Btn2-GFP and Cur1-GFP fusion proteins in pre1-1ts strains (Malinovska et al. 2012) led us to test whether btn2Δ or cur1Δ would suppress the destabilization of [URE3] by proteasome mutants. We found the curing by proteasome deficiency required Btn2p and Cur1p in an additive way, suggesting that these are two distinct anti-prion systems, and that the proteasome effect works through Btn2p and Cur1p. Previous work has shown that although Btn2p and Cur1p are paralogs, Btn2p collects Ure2p amyloid filaments in the process of curing, while Cur1p does not (Kryndushkin et al. 2008), consistent with their having different curing mechanisms.
We found that all of the proteasome deficient mutants had elevated levels of Btn2p and Cur1p, but only those with the highest levels destabilized [URE3]. In the opposite direction, ubr2Δ, known to increase proteasome activity by decreasing degradation of Rpn4p (a transcription factor promoting proteasome gene transcription) prevents curing of [URE3] by Btn2p or Cur1p overproduction (Bezsonov et al. 2021). The tof2::Hermes and tof2Δ strains also had similarly elevated levels of Btn2p and Cur1p, suggesting that these mutants also have reduced proteasomal activity. The previously known activities of Tof2p involve interaction with topoisomerase I (Top1p; Park and Sternglanz 1999), silencing of rDNA by tethering sites on rDNA to the nuclear membrane (along with other proteins; reviewed in Mekhail and Moazed 2010; Gartenberg and Smith 2016; Srivastava et al. 2016) and regulation of Cdc14 (Geil et al. 2008). It remains unclear what role Tof2p has in proteasome activity.
While the elevation of Btn2p and Cur1p in proteasome mutants is clear, we expected to see some accumulation of ubiquitinylated forms, at least for Btn2p. Conceivably, ubiquitin modification obscures the site recognized by our antibody, or it is not ubiquitinylation that tags Btn2p for proteasomal degradation. Most likely, the deubiquitinase (proteasome associated or not) is unaltered in pre9Δ cells and can remove the polyubiquitin chains, but the proteasomes do not degrade the protein. The N-terminal tails of the proteasome alpha ring proteins fan out over the hole in the center of the proteasome core particle thus limiting access. The N-terminal tail of Pre9p makes contact with the N-terminal tails of all other proteasome alpha ring proteins. Deletion of the N-terminal domain of Pre9p opens the center of the proteasome core particle. This has fueled speculation that these “opened” core particles differ in the client spectrum that the proteasome handles (Groll et al. 2000).
[PSI+] prions are generally not sensitive to overproduced Btn2p or Cur1p (Kryndushkin et al. 2008; Bezsonov et al. 2021). The sequestration mechanism for Btn2p curing should be (and is Wickner et al. 2014) more effective on variants of lower seed number. Most variants of [PSI+] have a higher seed number than do any known [URE3] variants, and this may explain why most variants of [PSI+] are not cured by even overproduced Btn2p. However, while the absence of an effect of proteasome mutations on [PSI+] is consistent with their acting on [URE3] via Btn2p and Cur1p, the wide variety of proteasome actions would preclude the converse inference.
pre9Δ, irc25Δ, and poc4Δ cells contain proteasomes with two α-4 subunits instead of one α-3 and one α-4 subunit in their α rings.
The only nonessential subunit of the proteasome core is α-3, encoded by PRE9 (Emori et al. 1991). The pre9Δ strains have replaced α-3 subunits with an extra copy of α-4, located in the α-3 position, and have several fold reduced activity as measured by model substrates and Matα2 stability (Velichutina et al. 2004). Mutants lacking the proteasome assembly chaperones Irc25 or Poc4 make about half of their proteasomes with the same α-4 for α-3 substitution (Kusmierczyk et al. 2008). Interestingly, human cells normally make a fraction of this same double α-4 proteasome lacking α-3 in a process regulated by the α-4 to α-3 ratio with α-4 levels regulated by the oncogenic tyrosine kinases ABL and ARG and the tumor suppressor BRCA1 (Padmanabhan et al. 2016). In a variation on this theme, mouse spermatogenesis (but not oogenesis) requires the substitution of the normal α-4 with another α-4, called α-4 s, without which substitution the process is arrested in meiosis I (Zhang et al. 2019). Whether yeast physiologically makes the (α-4)2 proteasome is not yet known. Thus, while we have interpreted our results as due to decreased proteasome activity, the real (interesting) possibility exists that it is due to altered proteasome specificity.
Confirming the extremely low levels of Btn2p and Cur1p in normal cells found in general surveys (https://pax-db.org/), we find that Btn2p and Cur1p are present at 170 and 110 molecules per cell compared to the reported high abundance of Sis1p of about 5 × 104 molecules per cell. The fact that Btn2p and Cur1p can each cure most variants of [URE3] at normal levels of either protein (Wickner et al. 2014) indicates that they are not, in this case, acting by sequestering Sis1p, as has been suggested for their prion-curing action on overproduction (Malinovska et al. 2012; Barbitoff et al. 2017).
We find that proteasome mutants destabilizing [URE3] lower the propagon number of the prion. Note that in measuring propagon number, colonies formed in the presence of guanidine that, when replated on –Ade plates produce no Ade+ colonies are not included in the averaging process. The diminished propagon number is as expected for curing mediated by Btn2p, which acts by collecting the many dispersed Ure2p filaments (Edskes et al. 1999) into one place in the cell, coincident with Btn2p itself (Kryndushkin et al. 2008). Even in the cells not yet cured by elevated Btn2p, the number of propagons or seeds should be reduced by their sequestration into one or a few sites in the cell, so they no longer separate as the cell forms a colony. Conversely, the seed number of [URE3-1] was raised by introducing btn2Δ and cur1Δ deletions (Kryndushkin et al. 2008).
Btn2p is overexpressed >200-fold in a pre9Δ strain based on quantitation of Western blots (Figure 5). Quantitation of Cur1p in a wild-type strain is difficult because of nearby cross-reacting bands and multiple Cur1p species, but it is clearly massively overproduced in pre9Δ and other proteasome-related mutants. The massive overproduction of Btn2p in proteasome mutants as judged by western blots is confirmed by SILAC, but Cur1 levels are too low in the wild-type to register above background by the latter method. Remarkably, these are easily the two most increased proteins compared to the other 4,600 proteins detected by SILAC. This suggests that pre9Δ not only lowers proteasome activity, but also changes its specificity because of the α4 for α3 substitution. For example, among the 15 most short-lived proteins in S. cerevisiae (Christiano et al. 2014), none were significantly different according to our SILAC data (see Supplementary Table S3). The most strongly elevated proteins comprise an eclectic array with widely varying functions. Lpl1, a type B phospholipase removing fatty acyl groups from phospholipids (Weisshaar et al. 2017), is among those most strongly increased in the pre9Δ strain.
We infer that interfering with the proteasomal degradation of Btn2p and Cur1p results in their elevated levels. One caveat to this conclusion is that the pre9Δ mutation may be sensed as a cellular stress, and part of the elevated Btn2p and Cur1p may be increased stress-induced synthesis. Of course, stress should also induce an array of chaperones and other proteins, such as Hsp26 and Hsp42, neither of which showed substantial elevations in our SILAC data for pre9Δ cells (Table 5). Ho et al. (2019) have reported that in hsp104Δ fes1Δ strains, Btn2, Hsp26, and Hsp42 are each present at substantially elevated levels, suggesting that the elevation was due to an Hsf1p-mediated heat shock response. Heat shock induces TSL1, HSP78, and UBI4 by 135-, 41-, and 26-fold, respectively (Gasch et al. 2000), but our SILAC data for pre9 cells show them induced by 1.007-, 0.57-, and 0.79-fold, respectively. It is also a general feature of heat-shock and other stress responses that they are transient, and with continued growth under the shock condition, levels soon return to near normal. Our mutants are growing in log phase and have been under the stress of the mutation for many generations by the time they are harvested. Our results are, by these criteria, not due to induction of a heat shock response.
Are the phenomena described here merely pathology of rare mutants or are they part of the physiological response to stress? Btn2p and Cur1p are stress-inducible (Malinovska et al. 2012; Ho et al. 2019), but it is possible that their stress-regulation is mediated by the proteasome. When heat or other stress produces large amounts of denatured proteins occupying the proteasome’s attention, less Btn2p and Cur1p are degraded allowing the Btn2p sequestration activity (and the unknown Cur1p activity) to aid in recovery. One assumes that degradation of irretrievably denatured proteins and recycling of their amino acids is preferable to merely sequestering them. Perhaps only when the proteasome is swamped with denatured proteins does the cell resort to sequestration as a Plan B.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of this article are represented fully within the article and its tables, figures, and supplementary files, the latter deposited in figshare: https://doi.org/10.25386/genetics.14161517. The Supplementary Information includes extended SILAC methods, strains of S. cerevisiae (Supplementary Table S1), primers used (Supplementary Table S2), proteins increased 1.5-fold or more in abundance in pre9Δ (Supplementary Table S3), Proteins decreased 1.5-fold or more in abundance in pre9Δ (Supplementary Table S4), a diagram of the proteasome α ring (Supplementary Figure S1), evidence that proteasome—related knockouts do not affect [PSI+] propagation (Supplementary Figure S2), evidence that [URE3-1] is less stable in 779-6A pre9Δ than in BY241 pre9Δ (Supplementary Figure S3), data showing that levels of Sup35p are not affected by proteasome—related mutations in strain BY241 (Supplementary Figure S4), data showing Btn2p levels in proteasome mutants are elevated by the presence of [PSI+] (Supplementary Figures S5 and S6), measuring the absolute levels of Btn2p and Cur1p in strain 779-6A by comparison with purified recombinant Btn2-His6 and Cur1-His6 (Supplementary Figure S7), the multiple bands of Cur1p (Supplementary Figure S8), the elevation of Btn2p in tof2 mutants (Supplementary Figure S9) and the absence of a ubiquitin ladder of Btn2p in a pre9Δ strain (Supplementary Figure S10). Supplementary Table S5 compares protein turnover rates with their levels by SILAC in pre9Δ strains. Supplementary material available at figshare: https://doi.org/10.25386/genetics.14161517.
Acknowledgments
The authors thank Eric Anderson (NIDDK Advanced Mass Spectrometry Core) for carrying out the SILAC experiments.
Funding
This work was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, project DK024950.
Conflicts of interest
None declared.
Literature cited
- Amor AJ, Castanzo DT, Delany SP, Selechnik DM, Van Ooy A, et al. 2015. The ribosome-associated complex antagonizes prion formation in yeast. Prion 9:144–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev S, Liebman SW.. 2006. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbitoff YA, Matveenko AG, Moskalenko SE, Zemlyanko OM, Newnam GP, et al. 2017. To CURe or not to CURe? Differential effects of the chaperone sorting factor Cur1 on yeast prions are mediated by the chaperone Sis1. Mol Microbiol. 105:242–257. [DOI] [PubMed] [Google Scholar]
- Bateman DA, Wickner RB.. 2012. [PSI+] prion transmission barriers protect Saccharomyces cerevisiae from infection: intraspecies ‘species barriers’. Genetics 190:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezsonov EE, Edskes HK, Wickner RB.. 2021. Innate immunity to yeast prions: Btn2p and Cur1p curing of the [URE3] prion is prevented by 60S ribosomal protein deficiency or ubiquitin/proteasome system overactivity. Genetics (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann A, Baxa U, Wickner RB.. 2005. Prion generation in vitro: amyloid of Ure2p is infectious. Embo J. 24:3082–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budenholzer L, Cheng CL, Li Y, Hochstrasser M.. 2017. Proteasome structure and assembly. J Mol Biol. 429:3500–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Bruce KL, Newnam GP, Gyoneva S, Romanyuk AV, et al. 2010. Genetic and epigenetic control of the efficiency and fidelity of cross-species prion transmission. Mol Microbiol. 76:1483–1499., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, et al. 2000. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol Microbiol. 35:865–876. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Lindquist SL, Ono B-I, Inge-Vechtomov SG, Liebman SW.. 1995. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268:880–884. [DOI] [PubMed] [Google Scholar]
- Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD.. 1999. Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI+] prion. Mol Cell Biol. 19:8103–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiano R, Nagaraj N, Frohlich F, Walther TC.. 2014. Global proteome turnover analyses of the yeasts S. cerevisiae and S. pombe. Cell Rep. 9:1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TG. 2002. Transmitting the signal of excess nitrogen in Saccharomyces cerevisiae from the Tor proteins to the GATA factors: connecting the dots. FEMS Microbiol Rev. 26:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BS, Ness F, Tuite MF.. 2003. Analysis of the generation and segregation of propagons: entities that propagate the [PSI+] prion in yeast. Genetics 165:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M.. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b. - range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- Czaplinski K, Ruiz-Echevarria MJ, Paushkin SV, Han X, Weng Y, et al. 1998. The surveillance complex interacts with the translation release factors to enhance termination and degrade aberrant mRNAs. Genes Dev. 12:1665–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delneri D, Tomlin GC, Wixon JL, Hutter A, Sefton M, et al. 2000. Exploring redundancy in the yeast genome: an improved strategy for use of the cre-loxP system. Gene. 252:127–135., [DOI] [PubMed] [Google Scholar]
- Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW.. 1996. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144:1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HE, Mukhamedova M, Edskes BK, Wickner RB.. 2018. Hermes transposon mutagenesis shows [URE3] prion pathology prevented by a ubiquitin-targeting protein: evidence for carbon/nitrogen assimilation cross-talk and a second function for Ure2p. Genetics 209:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Gray VT, Wickner RB.. 1999. The [URE3] prion is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc Natl Acad Sci USA. 96:1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Mccann LM, Hebert AM, Wickner RB.. 2009. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics 181:1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edskes HK, Wickner RB.. 2002. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full - length protein. Proc Natl Acad Sci USA. 99:16384–16391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emori Y, Tsukahara T, Kawasaki H, Ishiura S, Sugita H, et al. 1991. Molecular cloning and functional analysis of three subunits of yeast proteasome. Mol Cell Biol. 11:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallina I, Colding C, Henriksen P, Beli P, Nakamura K, et al. 2015. Cmr1/WDR76 defines a nuclear genotoxic stress body linking genome integrity and protein quality control. Nat Commun. 6:6533.[10.1038/ncomms7533] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL.. 2010. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Natl Acad Sci USA. 107:21966–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartenberg MR, Smith JS.. 2016. The nuts and bolts of transcriptionally silent chromatin in Saccharomyces cerevisiae. Genetics 203:1563–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, et al. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11:4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geil C, Schwab M, Seufert W.. 2008. A nucleolus-localized activator of Cdc14 phosphatase supports rDNA segregation in yeast mitosis. Curr Biol. 18:1001–1005. [DOI] [PubMed] [Google Scholar]
- Geitz RD. 2014. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol Biol. 1205:1–12. [DOI] [PubMed] [Google Scholar]
- Gorkovskiy A, Reidy M, Masison DC, Wickner RB.. 2017. Hsp104 at normal levels cures many [PSI+] variants in a process promoted by Sti1p, Hsp90 and Sis1p. Proc Natl Acad Sci USA. 114:E4193–E4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, et al. 2000. A gated channel into the proteasome core particle. Nat Struct Biol. 7: 1062–1067. [DOI] [PubMed] [Google Scholar]
- Haslberger T, Bukau B, Mogk A.. 2010. Towards a unifying mechanism for the ClpB/Hsp104-mediated protein disaggregation and prion propagation. Biochem Cell Biol. 88:63–75. [DOI] [PubMed] [Google Scholar]
- He F, Jacobson A.. 2015. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet. 49:339–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C-T, Grousl T, Shatz O, Jawed A, Ruger-Herreros C, et al. 2019. Cellular sequestrases maintain basal Hsp70 capacity ensuring balanced proteostasis. Nat Commun. 10:4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda N, Kobayashi T, Uchida N, Funakoshi Y, Kikuchi Y, et al. 2003. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J Biol Chem. 278:38287–38291. [DOI] [PubMed] [Google Scholar]
- Hung GC, Masison DC.. 2006. N-terminal domain of yeast Hsp104 chaperone is dispensable for thermotolerance and prion propagation but necessary for curing prions by Hsp104 overexpression. Genetics 173:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE.. 2008. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. Embo J. 27:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunmuktane Z, Mead S, Ellis M, Wadsworth JD, Nicoll AJ, et al. 2015. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525:247–250. [DOI] [PubMed] [Google Scholar]
- Jung G, Jones G, Masison DC.. 2002. Amino acid residue 184 of yeast Hsp104 chaperone is critical for prion-curing by guanidine, prion propagation, and thermotolerance. Proc Natl Acad Sci USA. 99:9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Masison DC.. 2001. Guanidine hydrochloride inhibits Hsp104 activity in vivo: a possible explanation for its effect in curing yeast prions. Curr Microbiol. 43:7–10. [DOI] [PubMed] [Google Scholar]
- Kanneganti V, Kama R, Gerst JE.. 2011. Btn3 is a negative regulator of Btn2-mediated endosomal protein trafficking and prion curing in yeast. Mol Biol Cell 22:1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly AC, Shewmaker FP, Kryndushkin D, Wickner RB.. 2012. Sex, prions and plasmids in yeast. Proc Natl Acad Sci USA. 109:E2683–E2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiktev DA, Melomed MM, Lu CD, Newnam GP, Chernoff YO.. 2015. Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol Microbiol. 96:621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kwon S-H, Kam T-I, Panicker N, Karuppagounder SS, et al. 2019. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron 103:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C-Y, Tittmann P, Gross H, Gebert R, Aebi M, et al. 1997. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 94:6618–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland PA, Reidy M, Masison DC.. 2011. Functions of yeast Hsp40 chaperone Sis1p dispensable for prion propagation but important for prion curing and protection from prion toxicity. Genetics 188:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock M, Nunes MM, Hermann M, Kube S, Dohmen RJ, et al. 2015. Proteasome assembly from 15S precursors involves major conformational changes and recycling of the Pba1–Pba2 chaperone. Nat Commun. 6:6123. [DOI] [PubMed] [Google Scholar]
- Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F.. 2012. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol Microbiol. 86:1531–1547. [DOI] [PubMed] [Google Scholar]
- Kryndushkin D, Shewmaker F, Wickner RB.. 2008. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. Embo J. 27:2725–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusmierczyk AR, Kunjappu MJ, Funakoshi M, Hochstrasser M.. 2008. A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat Struct Mol Biol. 15:237–244. [DOI] [PubMed] [Google Scholar]
- Le Tallec B, Barrault M-B, Courbeyrette R, Guerois R, Marsolier-Kergoat M-C, et al. 2007. 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol Cell 27:660–674. [DOI] [PubMed] [Google Scholar]
- Liebman SW, Chernoff YO.. 2012. Prions in yeast. Genetics 191:1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovska L, Kroschwald S, Munder MC, Richter D, Alberti S.. 2012. Molecular chaperones and stress-inducible protein-sorting factors coordinate the spaciotemporal distribution of protein aggregates. Mol Biol Cell 23:3041–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manogaran AL, Hong JY, Hufana J, Tyedmers J, Lindquist S, et al. 2011. Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS Genet. 7:e1001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masison DC, Wickner RB.. 1995. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science 270:93–95. [DOI] [PubMed] [Google Scholar]
- Mcglinchey R, Kryndushkin D, Wickner RB.. 2011. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A. 108:5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekhail K, Moazed D.. 2010. The nuclear envelope in genome organization, expression and stability. Nat Rev Mol Cell Biol. 11:317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SB, Ho CT, Winkler J, Khokhrina M, Neuner A, et al. 2015. Compartment-specific aggregases direct distinct nuclear and cytoplasmic aggregate deposition. Embo J. 34:778–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogk A, Kummer E, Bukau B.. 2015. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci. 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Salvadores N, Moreno-Gonzales I, Gonzales C, et al. 2017. Inductionn of IAPP amyloid deposition and associated diabetic abnormalities by a prion-like mechanism. J Exp Med. 214:2591–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayashiki T, Ebihara K, Bannai H, Nakamura Y.. 2001. Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell 7:1121–1130. [DOI] [PubMed] [Google Scholar]
- Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB.. 2005. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci USA. 102:10575–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA.. 1992. The translation machinery and 70 kDal heat shock protein cooperate in protein synthesis. Cell 71:97–105. [DOI] [PubMed] [Google Scholar]
- Ness F, Cox BS, Wongwigkarn J, Naeimi WR, Tuite MF.. 2017. Over-expression of the molecular chaperone Hsp104 in Saccharomyces cerevisiae results in the malpartition of [PSI+] propagons. Mol Microbiol. 104:125–143. [DOI] [PubMed] [Google Scholar]
- Ness F, Ferreira P, Cox BS, Tuite MF.. 2002. Guanidine hydrochloride inhibits the generation of prion “seeds” but not prion protein aggregation in yeast. Mol Cell Biol. 22:5593–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, et al. 2002. Stable isotope labeling by amion acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1:376–386. [DOI] [PubMed] [Google Scholar]
- Padmanabhan A, Vuong SA, Hochstrasser M.. 2016. Assembly of an evolutionarily conserved alternative proteasome isoform in human cells. Cell Rep. 14:2962–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Sternglanz R.. 1999. Identification of TOF2 using the two-hybrid system as a DNA topoisomerase I interacting protein. Yeast 15:35–41. [DOI] [PubMed] [Google Scholar]
- Pfund C, Lopez-Hoyo N, Ziegelhoffer T, Schilke BA, Lopez-Buesa P, et al. 1998. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. Embo J. 17:3981–3989., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y.. 2007. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2:1896–1906. [DOI] [PubMed] [Google Scholar]
- Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF.. 2003. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol Microbiol. 49:1005–1017. [DOI] [PubMed] [Google Scholar]
- Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, et al. 2020. A gut bacterial amyloid promotes α−synuclein aggregation and motor impairment in mice. Elife 9:e53111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoso A, Chien P, Osherovich LZ, Weissman JS.. 2000. Molecular basis of a yeast prion species barrier. Cell 100:277–288. [DOI] [PubMed] [Google Scholar]
- Schlumpberger M, Prusiner SB, Herskowitz I.. 2001. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 21:7035–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Masison DC.. 2008. Functionally redundant isoforms of a yeast Hsp70 chaperone subfamily have different antiprion effects. Genetics 179:1301–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F. 1991. Getting started with yeast. In: Guthrie C, Fink GR editors. Guide to Yeast Genetics and Molecular Biology. San Diego: Academic Press, p. 3–21. [Google Scholar]
- Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB.. 2007. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics. 176:1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirozu R, Yahiroda H, Murata S.. 2016. Proteasome impairment induces recovery of mitochondrial membrane potential and an alternative pathway of mitochondrial fusion. Mol Cell Biol. 36:347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Wickner RB.. 2018. Nonsense-mediated mRNA decay factors cure most [PSI+] prion variants. Proc Natl Acad Sci USA. 115:E1184–E1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son M, Wickner RB.. 2020. Normal levels of ribosome-associated chaperones cure two groups of [PSI+] variants. Proc Natl Acad Sci USA. 117:26298–26306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Srivastava R, Ahn SH.. 2016. The epigenetic pathways to ribosomal DNA silencing. Microbiol Mol Biol Rev. 80:545–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtmueller BM, Kish-Trier E, Ferrell K, Petersen CN, Robinson H, et al. 2012. Structure of a proteasome Pba1-Pba2 complex: implications for proteasome assembly, activation and biological function. J Biol Chem. 287:37371–37382., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidle EA, Chong LS, Wu M, Crooke E, Fiedler D, et al. 2016. A novel inositol pyrophosphate phosphatase in Saccharomyces cerevisiae: Siw14 protein selectively cleaves the β-phosphate from 5-diphosphoinositol pentakisphosphate (5PP-IP5). J Biol Chem. 291:6772–6783., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Chien P, Naber N, Cooke R, Weissman JS.. 2004. Conformational variations in an infectious protein determine prion strain differences. Nature 428:323–328. [DOI] [PubMed] [Google Scholar]
- Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB.. 1999. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283:1339–1343. [DOI] [PubMed] [Google Scholar]
- Ter-Avanesyan MD, Dagkesamanskaya AR, Kushnirov VV, Smirnov VN.. 1994. The SUP35 omnipotent suppressor gene is involved in the maintenance of the non-Mendelian determinant [psi+] in the yeast Saccharomyces cerevisiae. Genetics 137:671–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Madariaga ML, Lindquist S.. 2008. Prion switching in response to environmental stress. PLoS Biol. 6:e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velichutina I, Connerly PL, Arendt CS, Li X, Hochstrasser M.. 2004. Plasiticity in eucaryotic 20S proteasome ring assembly revealed by a subunit deletion in yeast. EMBO J. 23:500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, et al. 2011. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 11:2019–2026., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisshaar N, Welsch H, Guerra-Moreno A, Hanna J.. 2017. Phopholipase Lpl1 links droplet function with quality control protein degradation. Mol Biol Cell 28:716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB. 2006. Viruses and Prions of Yeast, Fungi and Parasitic Microorganisms in Fields Virology. 5th ed. Lippincott, Williams & Wilkins. [Google Scholar]
- Wickner RB, Bezsonov E, Bateman DA.. 2014. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci USA. 111:E2711–E2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Kelly AC, Bezsonov EE, Edskes HE.. 2017. Prion propagation is controlled by inositol polyphosphates. Proc Natl Acad Sci USA. 114:E8402–E8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, et al. 2015. Yeast prions: structure, biology and prion-handling systems. Microbiol Mol Biol Rev. 79:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Son M, Edskes BK.. 2019. Prion variants of yeast are numerous, mutable, and segregate on growth, affecting prion pathogenesis, transmission barriers and sensitivity to anti-prioin systems. Viruses 11:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Varshavsky A.. 2001. RPN4 is a ligand, substrate and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc Natl Acad Sci USA. 98:3056–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiroda H, Mizushima T, Okamoto K, Kameyama T, Hayashi H, et al. 2008. Crystal structure of a chaperone complex that contributes to the assembly of yeast 20S proteasomes. Nat Struct Mol Biol. 15:225–236. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ji SY, Busayavalasa K, Shao J, Yu C.. 2019. Meiosis I progression in spermatogenesis requires a type of testis-specific 20S core proteasome. Nat Commun. 10:3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of this article are represented fully within the article and its tables, figures, and supplementary files, the latter deposited in figshare: https://doi.org/10.25386/genetics.14161517. The Supplementary Information includes extended SILAC methods, strains of S. cerevisiae (Supplementary Table S1), primers used (Supplementary Table S2), proteins increased 1.5-fold or more in abundance in pre9Δ (Supplementary Table S3), Proteins decreased 1.5-fold or more in abundance in pre9Δ (Supplementary Table S4), a diagram of the proteasome α ring (Supplementary Figure S1), evidence that proteasome—related knockouts do not affect [PSI+] propagation (Supplementary Figure S2), evidence that [URE3-1] is less stable in 779-6A pre9Δ than in BY241 pre9Δ (Supplementary Figure S3), data showing that levels of Sup35p are not affected by proteasome—related mutations in strain BY241 (Supplementary Figure S4), data showing Btn2p levels in proteasome mutants are elevated by the presence of [PSI+] (Supplementary Figures S5 and S6), measuring the absolute levels of Btn2p and Cur1p in strain 779-6A by comparison with purified recombinant Btn2-His6 and Cur1-His6 (Supplementary Figure S7), the multiple bands of Cur1p (Supplementary Figure S8), the elevation of Btn2p in tof2 mutants (Supplementary Figure S9) and the absence of a ubiquitin ladder of Btn2p in a pre9Δ strain (Supplementary Figure S10). Supplementary Table S5 compares protein turnover rates with their levels by SILAC in pre9Δ strains. Supplementary material available at figshare: https://doi.org/10.25386/genetics.14161517.