

Abstract
Background
Inflammatory bowel diseases are the most common chronic intestinal inflammatory conditions, and their incidence has shown a dramatic increase in recent decades. Limited efficacy and questionable safety profiles with existing therapies suggest the need for better targeting of therapeutic strategies. Adenosine monophosphate-activated protein kinase (AMPK) is a key regulator of cellular metabolism and has been implicated in intestinal inflammation. Macrophages execute an important role in the generation of intestinal inflammation. Impaired AMPK in macrophages has been shown to be associated with higher production of proinflammatory cytokines; however, the role of macrophage AMPK in intestinal inflammation and the mechanism by which it regulates inflammation remain to be determined. In this study, we investigated the role of AMPK with a specific focus on macrophages in the pathogenesis of intestinal inflammation.
Methods
A dextran sodium sulfate-induced colitis model was used to assess the disease activity index, histological scores, macroscopic scores, and myeloperoxidase level. Proinflammatory cytokines such as tumor necrosis factor-α, interleukin-6, and interleukin-1β were measured by enzyme-linked immunosorbent assay. Transient transfection of AMPKβ1 and LC3-II siRNA in RAW 264.7 cells was performed to elucidate the regulation of autophagy by AMPK. The expression of p-AMPK, AMPK, and autophagy markers (eg, LC3-II, p62, Beclin-1, and Atg-12) was analyzed by Western blot.
Results
Genetic deletion of AMPKβ1 in macrophages upregulated the production of proinflammatory cytokines, aggravated the severity of dextran sodium sulfate-induced colitis in mice, which was associated with an increased nuclear translocation of nuclear factor-κB, and impaired autophagy both in vitro and in vivo. Notably, the commonly used anti-inflammatory 5-aminosalicylic acid (ie, mesalazine) and sodium salicylate ameliorated dextran sodium sulfate-induced colitis through the activation of macrophage AMPK targeting the β1 subunit.
Conclusions
Together, these data suggest that the development of therapeutic agents targeting AMPKβ1 may be effective in the treatment of intestinal inflammatory conditions including inflammatory bowel disease.
Keywords: AMPK, macrophages, inflammation, autophagy, proinflammatory cytokines
Introduction
Inflammatory bowel disease (IBD) is a chronic, relapsing disease affecting the digestive tract that is broadly classified into ulcerative colitis and Crohn disease. They are the most common chronic intestinal inflammatory conditions, and their incidence and prevalence have shown a dramatic increase in the last decades.1-3 Although the exact etiology of IBD is not known, studies have provided evidence that dysregulated immune response, genetic factors, gut microbiota, and environmental factors contribute to the pathogenesis of IBD.4 The pathology of IBD involves a chronic inflammatory cycle of leukocyte infiltration, intestinal mucosal damage, ulceration, and regeneration. The current standard of care for IBD focuses on disease management and mitigation of symptoms to minimize complications (ie, surgical intervention) and decrease patient symptoms by virtue of their anti-inflammatory and immune modulatory activities. The novel strategy of “treat-to-target” that includes defining the target, monitoring disease, or controlling the inflammation by increasing or combining therapies has been adopted for patients with IBD.5 Even though there has been significant progress in developing improved therapies for IBD (eg, immunosuppressive agents, biologics), disadvantages such as limited efficacy and unwanted adverse effects still persist,6 which necessitate the development of better therapeutic strategies in IBD, potentially by identifying new targets.
Adenosine monophosphate-activated protein kinase (AMPK) is an αβγ-heterotrimer that regulates cellular metabolism to maintain energy homeostasis.7 Activation of AMPK requires the catalytic α-subunit and the regulatory β- and γ-subunits.8-10 Studies from AMPK β1-11 or AMPK β2-12 deficient mice have shown that the C-terminus of the β isoforms contains a subunit-binding sequence that is essential for tethering the γ and α subunits together and making an active heterotrimer. The deactivation of AMPK through genetic deletion of the α- and β-isoforms in different tissues has been shown to accelerate the development of numerous pathological processes, including insulin resistance, tumorigenesis, and aging.7 Inflammation has also been shown to reduce AMPK activity through a multitude of distinct mechanisms.7,13 Consistent with this concept is that AMPK is reduced in the colonic epithelial cells of mice with colitis14; however, direct genetic evidence establishing whether reductions in AMPK are a consequence or a cause of IBD does not currently exist.
It is possible that AMPK inhibits the development of IBD through multiple mechanisms. For example, the metformin-induced activation of AMPK has been shown to reduce experimental colitis by enhancing epithelial tight junction formation.14 In addition to these direct effects of AMPK on the gastrointestinal tract, AMPK may also exert beneficial effects on IBD through the inhibition of macrophage inflammation. Macrophages are a critical component of the innate immune system that perform a key role in the generation of gut inflammation.15 One of the hallmarks of IBD is a shift in the resident macrophages to a proinflammatory state, characterized by high levels of the proinflammatory marker inducible nitric oxide and inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-12, IL-23, and IL1β.16 It has been shown that reduced AMPK activity in bone marrow–derived macrophages (BMDM) from global AMPKβ1-deficient (AMPKβ1-/-) mice is associated with higher levels of the proinflammatory cytokines TNF-α, IL6, IL-12, IL-23, and IL1β.13 In addition, previous studies have found that pharmacological activation of AMPK using 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) suppresses acute and chronic colitis in dextran sodium sulfate (DSS)-treated mice by inhibiting macrophage nuclear factor (NF)-κB activation while reducing T helper type 1 (Th1) and Th17 immune responses.17 Despite the importance of previous studies showing the beneficial effects of metformin and AICAR on DSS-induced colitis, it is now established that these agonists exert effects on multiple pathways in addition to AMPK.7 Thus, additional evidence establishing the precise role of AMPK in suppressesing colitis and the possible mechanisms mediating these effects is required.
In this study, we explored the role of AMPK signaling in the pathogenesis of DSS-induced experimental colitis by utilizing global AMPKβ1-deficient (AMPKβ1-/-) and macrophage-specific AMPKβ1-deficient (AMPKβ1LysM) mice. Furthermore, we investigated the effects of salicylate, a direct AMPKβ1 activator in the DSS-induced colitis model.18 Our study shows that AMPK signaling plays a critical role in inhibiting the pathogenesis of experimental colitis by suppressing the inflammatory effects of macrophages, which may be mediated through the activation of autophagy.
MATERIALS AND METHODS
Mice
Age-matched (6–8 weeks old) male mice were used throughout the study. The generation and characterization of mice deficient for Prkab1 (which encodes the β1 subunit of AMPK) have been described previously.11,19 Hereafter, homozygous whole-body knockout mice are referred to as AMPKβ1-/-, and the wild-type littermates are shown as AMPKβ1+/+.
The Prkab1 floxed mice were generated from mice originally obtained from the Knock-Out Mouse Project. Briefly, gene-trap mice (C57BL/6N-Prkab1tm1a(KOMP)Wtsi) were initially crossed to FLPo recombinase-expressing mice (B6.Cg-Tg(Pgk1-flpo)10Sykr/J; JAX 011065). This process removed the LacZ reporter and neomycin cassette, yielding mice with loxP sites flanking exon 2. To obtain AMPKβ1 myeloid-deficient (AMPKβ1LysM) mice, Prkab1 floxed mice were paired with mice expressing Cre recombinase under the control of the LysM promoter (B6.129P2-Lyz2tm1(cre)Ifo/J; JAX 004781). Mice were maintained by breeding floxed mice that were Cre-positive with floxed mice that were Cre-negative, such that comparisons were made between littermate animals. Hereafter, the floxed mice are expressed as AMPKβ1fl/fl. The C57BL/6N mice were purchased from Taconic Biosciences (Rensselaer, NY).
All experimental animal procedures were in accordance with the guidelines and principles of the Canadian Council of Animal Care and were approved by the Animal Care Committee at the University of Ottawa and McMaster University.
Reagents
We obtained Dulbecco’s Modified Eagle’s Medium (DMEM) high glucose from Hyclone (GE Healthcare Life Sciences, Logan, UT). Dulbecco’s phosphate buffered saline without calcium and magnesium was purchased from Mediatech (catalog no. 21-031-CV). Fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Invitrogen Life Technologies (Carlsbad, CA) and trypsin/EDTA was obtained from Clonetics, Inc. (Walkersville, MD). Lipopolysaccharide (LPS), sodium salicylate (SS), and chloroquine (CQ) were purchased from Sigma-Aldrich (St. Louis, MO). An AMPKβ1 rabbit monoclonal antibody (1:1000; catalog no. 4178), phospho-AMPK-α (T172) (1:1000; catalog no. 2535), AMPKα (1:1000; catalog no. 5831), LC3 (1:1000; catalog no. 12741), Beclin-1 (1:1000; catalog no. 3495), Atg-12-5 (1:1000; catalog no. 4180), p62 (1:1000; catalog no. 5114), and β-actin (1:1000; catalog no. 4970) were purchased from Cell Signaling Technology, Inc. (Boston, MA). Rabbit polyclonal antibody TNF-α (1:1000; catalog no. ab9739), IL-1β (1:1000; catalog no. ab9722), NF-κB p65 (1:1000; catalog no. ab16502), and lamin B1 (1:1000; catalog no. ab65986) were purchased from Abcam (Cambridge, MA) and IL-6 (1:1000; catalog no. 250717) rabbit polyclonal antibody was obtained from Abbiotec (San Diego, CA). Finally, 5-aminosalicylic acid (5-ASA) was purchased from TCI America (Portland, OR).
Induction and Evaluation of DSS Colitis
We added DSS (mol wt. 36–54 kilodaltons; ICN Biomedicals Inc, Soho, OH) to drinking water at 5% weight/volume (w/v) for 5 days. The average DSS consumption per cage was recorded every day for the duration of the experiment. Mice were killed 5 days after the beginning of DSS administration to evaluate the severity of colitis using previously published scoring systems.20,21 The disease activity index (DAI) was calculated considering body weight loss, blood in feces, and stool consistency. Macroscopic scoring was performed immediately after the mice were killed based on rectal bleeding, rectal prolapse, diarrhea, and colonic bleeding. Colonic histological damage was scored based on the loss of crypt architectures, goblet cell depletion, crypt abscess, and inflammatory cell infiltration. The myeloperoxidase (MPO, an index of granulocyte infiltration and inflammation) level was determined using a published protocol.20
Peritoneal Macrophage Culture
Resident macrophages from the peritoneal cavity were collected and cultured from AMPKβ1+/+, AMPKβ1-/-, AMPKβ1fl/fl, and AMPKβ1LysM mice receiving DSS or drinking water as described previously with a slight modification.22 Briefly, mice were euthanized and 10 mL of cold calcium and magnesium-free Dulbecco’s phosphate-buffered saline was injected into the peritoneal cavity of each mouse. The fluid was then aspirated from the peritoneum slowly with syringe and needle. The fluid was dispensed into a 50 mL conical tube after removing the needle and centrifuged (400 × g for 10 min at 4°C). The supernatants were discarded and the collected cells were resespended in DMEM/F12 and supplemented with 10% FBS and 50 international units of penicillin, 50 μg streptomycin, and 2 mM glutamine per mL. Next, 2 × 105 cells were plated in 24-well plates. After 2 hours, the nonadhering cells were removed by gently washing 3 times with 1 × phosphate-buffered saline. Then 1 mL of the media was added and incubated overnight. Cultured cells were exposed to LPS (1 μg/mL) for 24 hours, the supernatants were collected, and IL-6, IL-1β, and TNF-α levels were measured by enzyme-linked immunosorbent assay (ELISA).
Cell Culture
The murine macrophage cell line RAW 264.7 (Passage 15–20), received from coauthor Bowdish’s laboratory, was cultured in DMEM/F12 supplemented with 10% FBS, 100 IU/mL of penicillin, and 100 μg/ml of streptomycin. Cells were maintained at 37°C in 5% CO2. After the cells reached 70% confluency, they were split at a 1:3 ratio.
Isolation of Macrophages From Colonic Lamina Propia and Bone Marrow
Macrophages from the colonic lamina propria were isolated by following the methods described in published protocol.23 Briefly, the dissected colons from the mice were cut into approximately 1.5 mm pieces and washed with calcium- and magnesium-free Hank’s balanced salt solution (HBSS), and the epithelial layer of the colon samples were removed by incubation in calcium- and magnesium-free HBSS containing FBS and 2 mM EDTA at 37°C for 20 minutes. The colon tissues were then digested in HBSS with 1.5 mg/mL collagenase type IV (Gibco) and 60 mg/mL DNase I (Sigma) for 40 minutes in a prewarmed orbital shaker at 37°C. Cells obtained after digestion were blocked with Fc block (anti-CD16/32; clone:93; isotype: rat IgG2a, λ; Biolegend no. 101302; 1:200) and stained with live/dead stain (7AAD; BD Biosciences no. 559925), CD45 FITC mAb (1:100; Biolegend no. 103108), MHC-II APC/Cy7 mAb (1:100; cell signaling no. 55413S), CD11b efluor mAb (1:200; eBioscience no. 48-0112-82), and CD11c APC mAb (1:100; eBioscience no. 17-0114-82). The stained cells were acquired on an LSR II cytometer, and CD103-F4/80+ cells were sorted as macrophages. The sorted cells were collected in a sterile fluorescence-activated cell-sorting tube precoated with FBS and immediately cultured in 24-well plates for further experiments.
We generated BMDM by slightly modifying a previously described method.24 First, AMPKβ1fl/fl and AMPKβ1LysM mice were euthanized to isolate the tibia and femur, and the ends of the bones were cut off and placed into a sterile 0.5 mL microfuge tube with a hole in the end punctured with an 18 gauge needle, which was then placed inside a 1.5 mL microfuge tube. Next, 100 μL of DMEM was added to a 0.5 mL tube and the bone marrow cells were collected by centrifuging the 1.5 mL tube at 2000 rpm for 4 minutes. The cells were resuspended in 100 mL DMEM supplemented with 10% FBS, 100 IU/mL of penicillin, and 100 μg/mL of streptomycin and plated in 6-well plates and placed in the incubator at 37°C in 5% CO2. Twenty-five ng/mL of mouse macrophage-colony stimulating factor purchased from R&D System Inc. was added to each well, and the cells were left to differentiate for 7 days. After the differentiation of the cells, the cells were treated as required, the supernatant was used for ELISA, and cells were lysed for Western blotting.
ELISA
Colon tissues were prepared as previously described,25 and the supernatants from the cells were collected after the completion of treatment. Cytokine (IL-1β, IL-6, and TNF-α) levels in the supernatants of cells and tissues were determined using commercially available ELISA kits from R&D System Inc. and expressed in units/mg of protein.
Western Blotting
The NE-PER nuclear and cytoplasmic extraction reagent kit (no. 78833, Thermo Scientific) was used to extract cytoplasmic and nuclear protein extracts as described in earlier work.26 Whole-cell lysates were extracted using a radioimmunoprecipitation assay buffer containing a 1× protease and phosphatase inhibitor cocktail and centrifuged at 12,000 rpm for 10 minutes at 4°C. The supernatants were collected, and the protein concentration was determined by the DC Protein Assay Kit (Bio-Rad Laboratories, Mississauga, Canada). Samples were separated by using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were then electrophoretically transferred onto nitrocellulose or polyvinylidene difluoride membranes. The membranes were incubated with 5% bovine serum albumin in 1× Tris-buffered saline Tween-20 at room temperature for 1 hour and then probed with primary antibodies overnight at 4°C. The membranes were then washed 3 times with Tris-buffered saline containing 0.1% Tween-20 followed by incubation with corresponding secondary antibodies for 1 hour at room temperature. Immunodetection was performed by visualization of the membrane using a chemiluminescent reagent (Thermo Scientific) and by exposure to a luminescent image analyzer, the ChemiDoc Touch Imaging System (Bio-Rad Laboratories).
siRNA Transfection
RAW 264.7 cells were seeded in 6-well plates at a density of 7 × 105 cells/well in antibiotic-free DMEM/F12 and were transfected with 50 nM of Map1lc3b and Prkab1 siRNA (Bioneer Inc, Oakland, CA) using the transfection reagent Dharmafect 4 (Dharmacon, Lafayette, CO). After 48 hours of transfection, the cells were pretreated with SS (3 mM) for 1 hour before the treatment with or without LPS (1 μg/mL) for 24 hours. The cells were then used for the Western blot and the supernatants were collected for ELISA.
Statistical Analysis
Data are expressed as mean ± standard error of mean. Where appropriate, comparison with 2 groups was made using the Student t test for unpaired data and the Student 1-way analysis of variance in GraphPad Prism version 5.0 (San Diego, CA) to determine the significance of intergroup differences. A value of P < 0.05 was considered statistically significant.
RESULTS
Germline Deletion of the β1 Subunit of AMPK and Myeloid-Specific AMPKβ1 Deficiency Enhance Colitis Severity in Mice
The AMPKβ1-/- mice and wild-type control (AMPKβ1+/+) mice were administered ad libitum with 5% DSS in drinking water for 5 days. The AMPKβ1-/- mice were utilized because genetic removal of this isoform reduces macrophage AMPK activity by >95%.13 The AMPKβ1-/- mice had significantly higher DAI (starting from day 3), macroscopic scores, MPO activity, and histological scores than the AMPKβ1+/+ mice on day 5 post-DSS (Figs. 1A–E). Increased severity of colitis in AMPKβ1-/- mice was associated with upregulation of several proinflammatory cytokines (IL-6, IL-1β, and TNF-α; Fig. 1F) and nuclear localization of NF-κB (Fig. 1G) in the colonic tissues.
Figure 1.

DSS-induced colitis was aggravated in AMPKβ1-/- and AMPKβ1LysM mice. Colitis was induced by 5% DSS in drinking water ad libitum in AMPKβ1+/+, AMPKβ1-/-, AMPKβ1fl/fl, and AMPKβ1LysM mice for 5 days, and mice were killed on day 5 post-DSS to assess inflammation in AMPKβ1+/+ and AMPKβ1-/- mice (A) DAI. (B) Macroscopic score. (C) MPO activity. (D) Representative micrographs of hematoxylin and eosin–stained colon cross-sections on day 5 post-DSS; bar = 100 μm. (E) Histological scores. (F) IL-6, IL-1β, and TNF-α levels. Data represent mean ± SEM (n = 5). *P < 0.05, compared with water-receiving mice, and #P < 0.05, compared with DSS-receiving AMPKβ1+/+ mice. Total proteins from colon tissues analyzed by Western blot. (G) Representative blot of nuclear translocation of NF-κB. Bar graph represents mean ± SEM for quantitation of NF-κB in the cytoplasm and the nucleus from 3 random mice in each group. *P < 0.05, compared with control mice, and #P < 0.05, compared with DSS-receiving AMPKβ1+/+ mice. (H) DAI to assess inflammation in AMPKβ1fl/fl and AMPKβ1LysM mice. (I) Macroscopic score to assess inflammation in AMPKβ1fl/fl and AMPKβ1LysM mice. (J) MPO activity to assess inflammation in AMPKβ1fl/fl and AMPKβ1LysM mice. (K) Representative micrographs of H&E stained colon cross-sections to assess inflammation in AMPKβ1fl/fl and AMPKβ1LysM mice; bar = 100 μm. (L) Histological damage score. (M) Proinflammatory cytokine levels in the colon, such as IL-6, IL-1β, and TNF-α. Data represent mean ± SEM (n = 4). *P < 0.05, compared with control mice, and #P < 0.05, compared with DSS-receiving AMPKβ1fl/fl mice. Total proteins from colon tissues were analyzed by Western blot. (N) Representative blot of nuclear translocation of NF-κB. Bar graph represents mean ± SEM for quantitation of NF-κB in the cytoplasm and the nucleus from 3 random mice in each group. H&E indicates hematoxylin and eosin; SEM, standard error of the mean.
Macrophages are critical in various immune responses, including those associated with colitis. To further investigate the role of AMPK in the regulation of macrophage function in the context of colitis, we utilized myeloid-specific AMPKβ1-deficient mice (AMPKβ1LysM). The AMPK β1fl/fl and AMPKβ1LysM mice were administered ad libitum with 5% DSS in drinking water for 5 days. Consistent with our previous observations in the germline AMPKβ1-/- mice, we observed an increased severity of DSS-induced colitis in the AMPKβ1LysM mice compared with the AMPKβ1fl/fl mice as evidenced by significantly higher DAI, MPO activity, proinflammatory cytokines (TNF-α, IL-6, and IL-1β), and nuclear translocation of NF-κB in the colonic tissues of the AMPKβ1LysM mice (Figs. 1H–N). These data suggest that the impairment of AMPK in macrophages plays an important role in the aggravation of intestinal inflammation in mice.
AMPKβ1 Deficiency Upregulates Proinflammatory Cytokine Production by Macrophages From the Peritoneal Cavity and Colonic Lamina Propria
To investigate whether macrophages directly play a role in increasing the susceptibility of DSS-induced colitis in AMPKβ1-/- and AMPKβ1LysM mice, peritoneal macrophages extracted from these mice including their wild-type counterparts treated with vehicle or DSS were stimulated with LPS for 24 hours. Compared to macrophages from vehicle-treated mice, those from DSS-treated mice showed enhanced production of proinflammatory cytokines in response to LPS; this inflammatory response was significantly greater in macrophages from AMPKβ1-/- and AMPKβ1LysM mice than those from AMPKβ1+/+ and AMPKβ1fl/fl mice (Figs. 2A–C). Furthermore, we sorted F4/80+ macrophages from the colonic lamina propria of AMPKβ1fl/fl and AMPKβ1LysM mice (Supplementary Fig. S1) and treated them with the AMPKβ1 activator SS before treatment with LPS (Figs. 2D–F). Activating AMPK reversed the effect of LPS on the secretion of proinflammatory cytokines in both DSS-treated and untreated macrophages from AMPKβ1fl/fl mice but did not reverse the effect in LPS-treated macrophages from AMPKβ1LysM mice. These results suggest that AMPKβ1 plays an important role in the regulation of inflammatory cytokine production by macrophage and that functional AMPK in macrophages contributes to the regulation of intestinal inflammation.
Figure 2.

Macrophages from the peritoneum and colonic lamina propria isolated from DSS-treated AMPKβ1-/- and AMPKβ1LysM mice showed enhanced response to LPS that was not reversed by SS. A-C, Macrophages were isolated from the peritoneal cavity from vehicle-treated and DSS-treated AMPKβ1+/+, AMPKβ1-/-, AMPKβ1fl/fl, and AMPKβ1LysM mice. The cultured macrophages were treated with LPS for 24 hours and the collected supernatants were assessed for the proinflammatory cytokines (A) IL-1β, (B) IL-6, and (C) TNF-α by ELISA. *P < 0.05, compared with control cells; #P < 0.05, compared with LPS-treated cells from AMPKβ1+/+ or AMPKβ1fl/fl mice in both vehicle- and DSS-treated group. Peritoneal macrophages were collected from at least 4 mice from each group with triplicate cultures presented as mean ± SEM. D-F, F4/80+ cells sorted from colonic lamina propria were pretreated with SS (3 mM) for 1 hour before treatment with LPS for 24 hours. The supernatants were then collected for analyzing the proinflammatory cytokines (D) IL-1β, (E) IL-6, and (F) TNF-α by ELISA. *P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells from vehicle-receiving AMPKβ1fl/fl or AMPKβ1LysM mice; $P < 0.05, compared with untreated cells and AMPKβ1fl/fl or AMPKβ1LysM mice; and & P < 0.05, compared with LPS-treated cells from DSS-receiving AMPKβ1fl/fl or AMPKβ1LysM mice. SEM indicates standard error of the mean.
Activation of AMPKβ1 Suppresses LPS-Induced Proinflammatory Response in Macrophage Cell Line (RAW 264.7 ) and BMDM
To investigate whether LPS-induced proinflammatory cytokine production could be inhibited by the activation of AMPK, we treated murine macrophages (RAW 264.7 cells) and BMDM from AMPKβ1fl/fl mice with or without SS for 1 hour before LPS treatment. Consistent with previous findings,13,27 we observed that LPS-treated macrophages showed a decreased phosphorylation of AMPK in both RAW 264.7 cells and BMDM (Fig. 3A) that was associated with an increase in the production of IL-6, IL-1β, and TNF-α (Figs. 3B–D). Notably, treatment of RAW 264.7 cells and BMDM with SS restored AMPK, activating phosphorylation, and prevented the LPS-induced production of IL-6, IL-1β, and TNF-α, suggesting an important role of AMPKβ1 in the regulation of inflammatory response in macrophages.
Figure 3.

AMPKβ1 regulates LPS-induced inflammatory response in macrophages. RAW 264.7 macrophage cells and BMDM cells were pretreated with SS (3 mM) for 1 hour before the treatment with LPS for 24 hours. (A) The extracted protein was analyzed for expression of phospho-AMPKα1, AMPKα1, and AMPKβ1. Bar graph represents the quantitative data.*P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells. The supernatants were collected and analyzed for (B) IL-6, (C) IL-1β, and (D) TNF-α. *P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells.
AMPK Activation Rescues Impaired Autophagy and Diminishes Production of Proinflammatory Cytokines in Macrophages
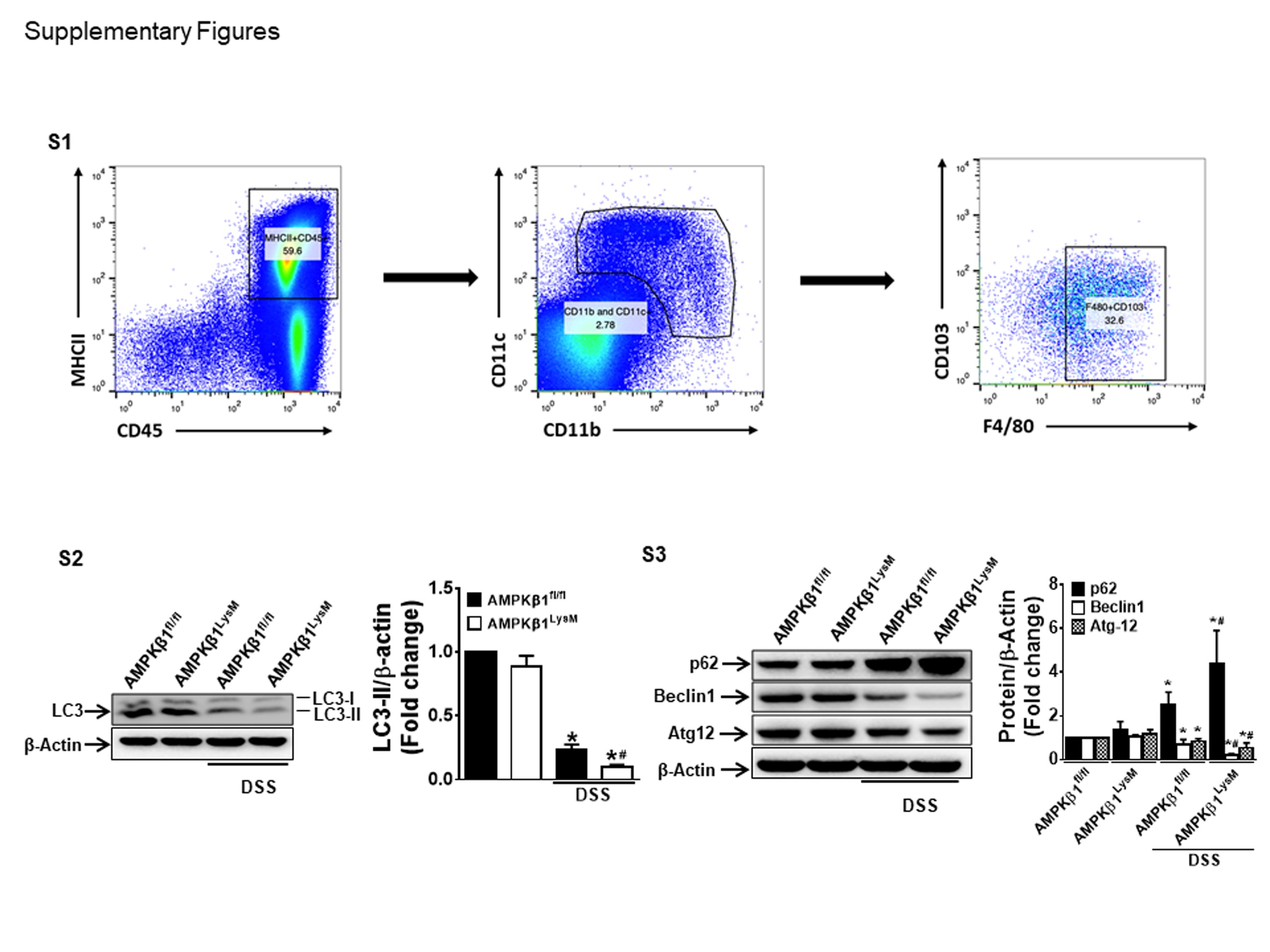
Impaired autophagy is an important mechanism contributing to increased inflammation in colitis.28,29 The activation of AMPK increases autophagy, which may be important for suppressing inflammation in macrophages.30,31 To investigate whether the activation of AMPK increases autophagy, RAW 264.7 cells were treated with LPS, SS, and CQ (an autophagy inhibitor). The LPS treatment reduced AMPK, activating phosphorylation, and downregulated the expression of autophagy markers (LC-3, Beclin-1, and Atg-12) while upregulating the expression of p62 in macrophages (Figs. 4A, B), suggesting an inhibitory effect of LPS on the autophagy processes. Notably, consistent with the restoration of AMPK phosphorylation, these effects were reversed by SS but were unaffected by CQ. We found that IL-1β (Fig. 4C) and TNF-α (Fig. 4D) production were inhibited by both CQ and SS but that the effect was immense with SS without any synergistic effect in LPS-treated RAW 264.7 cells. Consistent with these in vitro findings, Western blot analysis of LC3-II and other autophagy markers such as Beclin-1 and Atg-12 showed that these markers were reduced and that p62 was accumulated in the colonic tissues of DSS-treated AMPKβ1LysM mice compared with AMPKβ1fl/fl mice (Supplementary Figs, S2, S3). These results suggest that AMPK plays an important role in limiting inflammation in colitis via the regulation of autophagy.
Figure 4.

AMPK regulates autophagic flux in murine macrophage during inflammation. RAW 264.7 macrophage cells were treated with vehicle, LPS, SS (3 mM), or CQ (50 μM) or cotreated as indicated in the figure for 24 hours. The extracted protein was analyzed for expression of (A) p-AMPKα1, AMPKα1, and LC3 and (B) p62, Beclin-1, and Atg-12. Data show representative blot of 3 independent experiments and bar graph represents mean ± SEM. *P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells; $P < 0.05, compared with LPS- and SS-treated cells; γP < 0.05, compared with LPS- and CQ-treated cells; and &P < 0.05, compared with SS- and CQ-treated cells. The supernatants were collected to measure the expression of (C) IL-1β and (D) TNF-α. *P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells; and $P < 0.05, compared with LPS- and CQ-treated cells. SEM indicates standard error of the mean.
AMPK-Autophagy Axis Plays an Important Role in Regulation of Proinflammatory Cytokine Production in Macrophages
To further investigate whether defects in autophagy limit the anti-inflammatory effects of AMPK activation and that the effect is dependent on AMPK, RAW 264.7 cells were transiently transfected with nontargeted siRNA, LC3-II, or AMPKβ1 siRNA. Cells were then were treated with SS for 1 hour before the treatment with LPS for 24 hours. Transfection with LC3-II siRNA did not reverse the effect of LPS in cells pretreated with SS, indicating that the anti-inflammatory effect obtained by activation of AMPK is limited by impaired autophagy (Figs. 5A–D). The AMPKβ1 siRNA transfected cells showed a dramatically reduced expression of both AMPK β1 and AMPKα, thereby eliminating the effect of SS, suggesting an important role of AMPKβ1 in the stabilization and activation of AMPK. However, the basal level of LC3-II was not affected in the AMPKβ1 siRNA transfected cells, and the effect of the LPS-induced decreased LC3-II expression and increased proinflammatory cytokine secretion was not reversed by AMPK activation. (Figs. 5A–D). Moreover, BMDM isolated from AMPKβ1fl/fl and AMPKβ1LysM mice showed similar results in that the effect of LPS on LC3-II expression and proinflammatory cytokine secretion was reversed by SS in BMDM from AMPKβ1fl/fl but not in AMPKβ1LysM (Figs. 5E–H). These data suggest that the anti-inflammatory action of activated AMPK during inflammation is mediated by autophagy.
Figure 5.

AMPK-autophagy axis regulates proinflammatory cytokine secretions in macrophages during inflammation. RAW 264.7 macrophage cells were transiently transfected with nontargeted siRNA, LC3-II siRNA, or AMPKβ1 siRNA for 48 hours. Transfected cells were pretreated with SS before treatment with LPS for 24 hours. (A) Proteins extracted from the cells were analyzed for the expression of AMPKβ1, phospho-AMPKα1, AMPKα1, and LC3. (B-D) The supernatant was analyzed for the secretion of IL-6, IL-1β, and TNF-α. The bar graphs represent the quantitated data. *P < 0.05, compared with untreated cells or nontargeted siRNA-treated cells; #P < 0.05, compared with LPS-treated cells. Data are represented as mean ± SEM of 3 independent experiments. BMDM isolated from AMPKβ1fl/fl and AMPKβ1LysM mice were pretreated with SS before treatment with LPS for 24 hours. (E) The proteins extracted from the cells were analyzed for the expression of LC3. (F-H) The supernatant was analyzed for the secretion of IL-6, IL-1β, and TNF-α. *P < 0.05, compared with untreated cells; #P < 0.05, compared with LPS-treated cells. Data are represented as mean ± SEM of 3 independent experiments. SEM indicates standard error of the mean.
SS-Ameliorated DSS-Induced Colitis in Mice
The inhibition of LPS-induced proinflammatory cytokine secretion by SS in macrophages led us to investigate the anti-inflammatory effect of SS in vivo. Intraperitoneal treatment with SS along with DSS administration in drinking water significantly reduced the severity of DSS-induced colitis in C57BL/6 mice. The DAI, MPO levels, macroscopic scores, and histological scores were significantly improved by treatment with SS (Figs. 6A–E). The production of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) in the colon tissues was significantly inhibited by SS, which was accompanied by suppression of the nuclear translocation of NF-κB (Figs. 6F–I). Moreover, the decrease in LC3-II, Beclin-1, and Atg-12 and the increase in p62 in the inflamed colon were abrogated by SS (Figs. 6J–K). These findings show that salicylate reduces the severity and markers of DSS-induced colitis.
Figure 6.

DSS-induced colitis was ameliorated by SS. 5% DSS in drinking water or drinking water alone was administered to C57BL/6 mice for 5 days. SS (300 mg/kg) was given intraperitoneally to mice from day 0 of DSS administration. Innflammation was assessed by (A) DAI, (B) macroscopic score, (C) MPO activity, and (D) histological score. (E) Representative micrographs of H&E stained colon cross-sections on day 5 post-DSS; bar = 100 μm. Proinflammatory cytokines (F) IL-6, (G) IL-1β, and (H) TNF-α. Data represent mean ± SEM (n = 5). *P < 0.05, compared with vehicle-treated mice; #P < 0.05, compared with DSS-receiving mice. Total proteins from the colon tissues were analyzed by Western blot. (I) Representative blot for cytoplasmic and nuclear NF-κB from 3 random mice from each group. (J) Representative blot of LC3 expression from 3 random mice in each group. (K) Representative blot of p62, Beclin-1, and Atg-12 from 3 random mice in each group. *P < 0.05, compared with vehicle-treated mice; #P < 0.05, compared with DSS-receiving mice. H&E indicates hematoxylin and eosin; SEM, standard error of the mean.
Generally Prescribed 5-ASA and SS Ameliorate DSS-Induced Colitis in AMPKβ1fl/fl but not in AMPKβ1LysM Mice
Clinicians widely prescribe 5-ASA (ie, mesalazine), a derivative of salicylate, for the treatment of colitis; however, the exact mechanisms by which it elicits beneficial effects are not fully understood. To investigate whether macrophage AMPKβ1 is essential for the anti-inflammatory effects of 5-ASA, AMPKβ1fl/fl and AMPKβ1LysM mice were orally gavaged with 5-ASA or SS for 5 days starting from day 0 of DSS. Treatment with both 5-ASA and SS ameliorated DSS-induced colitis in AMPKβ1fl/fl mice, and this was associated with a significant reduction in DAI, MPO levels, macroscopic and histological scores, and proinflammatory cytokines (TNF-α, IL-6, and IL-1β; Figs. 7A–H). However, both 5-ASA and SS did not lower any of the parameters measured in AMPKβ1LysM mice. These data indicate that both 5-ASA and SS ameliorate colitis by targeting macrophage AMPKβ1.
Figure 7.

Both 5-ASA and SS ameliorated DSS-colitis in AMPKβ1fl/fl mice but not in AMPKβ1LysM mice. 5% DSS in drinking water was administered ad libitum to AMPKβ1fl/fl and AMPKβ1LysM mice with or without oral dosage of 5-ASA and SS at 300 mg/kg w/v in 0.5% carboxymethylcellulose once daily for 5 days. Inflammation was assessed by measuring (A) DAI, (B) macroscopic score, (C) MPO activity, and (D) histological damage score. (E) Representative micrographs of H&E stained colon cross-sections on day 5 post-DSS; bar = 100 μm. Proinflammatory cytokines, such as (F) IL-6, (G) IL-1β, and (H) TNF-α. Data represent mean ± SEM (n = 5). *P < 0.05, compared with vehicle-treated AMPKβ1fl/fl mice. H&E indicates hematoxylin and eosin; SEM, standard error of the mean.
Discussion
Research has shown that AMPK is a critical molecule in energy homeostasis and contributes to many cellular processes, including autophagy and inflammation.32 However, the precise role of AMPK in the pathogenesis of IBD is largely unknown. Using AMPKβ1-/- mice, we observed significant upregulation in the severity of DSS-induced colitis. The detrimental effect of AMPKβ1 deficiency in DSS-induced colitis was observed for all the parameters studied, including both macroscopic and histological indices, MPO level, and proinflammatory cytokines. Given that AMPKβ1 is expressed in macrophage, subsequent experiments in mice lacking AMPKβ1 specifically in myeloid cells revealed parallel results to the whole-body deficient model and showed that myeloid AMPK is critical for reducing DSS-induced inflammation. Intestinal mucosa serves as the largest reservoir for macrophages and exists in close proximity to the large number of bacteria and the antigenic stimuli.15 In our subsequent studies involving mice and cultured macrophages, we demonstrated the cell-autonomouse effects of macrophage AMPK signaling inhibited DSS- and LPS-induced inflammation.
Dysregulation of the autophagic processes causes disruption of several aspects of the compromised intestinal barrier function and immune activation that can lead to inappropriate responses and subsequent inflammation.29 Genome-wide association studies have revealed that polymorphisms in autophagy genes contribute to the development of IBD.33 It has also been shown that autophagy gene (ATG16L1)-deficient mice are highly susceptible to DSS-induced colitis and produce more proinflammatory cytokines (IL-1β and IL-18).34 We found that a decrease in AMPK, activating phosphorylation upon LPS treatment, was associated with impaired autophagy; this result was reversed by SS but not by CQ. Notably, LPS-induced proinflammatory cytokine production was inhibited by SS to a larger extent than by CQ, and upon treatment with both drugs no synergism was observed, suggesting that they exert their anti-inflammatory action via different signaling pathways. The phosphorylation of AMPK by cotreatment using SS and CQ was similar to that of SS alone, but in CQ-treated cells the phosphorylation was similar to untreated cells. This finding suggests that increased DSS-induced inflammation and colitis in AMPKβ1-deficient mice may result from impaired autophagy in macrophages. Future studies directly testing this hypothesis in mouse models with deficient autophagy signaling will be important.
Previous studies using nonspecific AMPK agonists such as AICAR and metformin have shown ameliorative effects in mouse models of experimental colitis. However, the safety and effectiveness of these agents for treating IBD in humans has been questioned.35-37 In addition, these agents have been shown to act via multiple pathways in addition to activating AMPK.7 Aminosalicylates, such as 5-ASA (mesalazine), have been used for more than 50 years for the treatment of ulcerative colitis.38 Recently, 5-ASA was shown to inhibit trinitrobenzoic sulphonic acid-induced colitis by AMPK activation.39 However, the mechanism by which it ameliorates colitis has not been fully understood except that it inhibits the nuclear translocation of NF-κB. Research has shown that SS directly and allosterically activates AMPK by binding to the β1 subunit.40 In our study, we concurrently treated AMPKβ1fl/fl and AMPKβ1LysM mice with SS and 5-ASA. Although both drugs effectively inhibited DSS-induced colitis in AMPKβ1fl/fl, the anti-inflammatory effects were eliminated in AMPKβ1LysM mice suggesting macrophage AMPKβ1 as a potential target of 5-ASA as well.
Conclusions
We have shown a novel role of macrophage AMPKβ1 in the pathogenesis of colitis, which is direct evidence suggesting that impairment in AMPK activity increases the severity of intestinal inflammation by inhibiting the autophagy process in macrophages. Activation of AMPKβ1 by SS treatment upregulates the autophagy process. This enhancement of autophagy by AMPKβ1 activation along with the reduction in the production of proinflammatory cytokines from macrophages ameliorates DSS-induced intestinal inflammation (Fig. 8). These findings provide important information on the mechanism of intestinal inflammation and suggest the development of targeted therapies aimed at activating macrophage AMPKβ1 for the treatment of various intestinal inflammatory conditions such as IBD.
Figure 8.

Schematic representation of the potential mechanism by which AMPKβ1 activation (by salicylates) reduces the severity of intestinal inflammation. Loss in regulatory function of AMPK aggravates DSS-induced colitis. Deactivation of AMPK or impaired AMPK activity increases the severity of intestinal inflammation by promoting production of proinflammatory cytokines and by impairing autophagy. Targeted activation of AMPKβ1 ameliorates intestinal inflammation by reducing the production of cytokines and by activating the autophagy process.
Supplementary Data
Supplementary data are available at Inflammatory Bowel Diseases online.
Supplementary Figure S1. Gating strategy for F4/80+ macrophages from the colonic lamina propria. Large intestinal lamina propria live CD45+MHCII+ samples were analyzed for CD11b and CD11c populations and were further separated for the CD103-F4/80+ population for macrophages.
Supplementary Figure S2. Colonic protein samples from AMPKβ1fl/fl and AMPKβ1LysM mice were analyzed for LC3. Data show representative blot, and bar graphs represent mean ± standard error of the mean for quantitation of each protein from 3 random mice in each group. *P < 0.05, compared to water-receiving mice; #P < 0.05, compared to DSS-receiving AMPKβ1fl/fl mice.
Supplementary Figure S3. Colonic protein samples from AMPKβ1fl/fl and AMPKβ1LysM mice were analyzed for p62, Beclin-1, and Atg-12. Data show representative blot, and bar graphs represent mean ± standard error of the mean for quantitation of each protein from 3 random mice in each group. *P < 0.05, compared to water-receiving mice; #P < 0.05, compared to DSS-receiving AMPKβ1fl/fl mice.
{kind=link}
Acknowledgments
The authors thank Dr. Walter Reinisch (McMaster University, Canada and Universitätsklinik für Innere Medizin III, Vienna, Austria) for his valuable suggestions.
Author contributions: SB, GRS, and WIK conceived and designed research. SB, HW, YHK, JG, PG, SH, and FMNH performed experiments. SB, DMB, JAK, MDF, and WIK analyzed and interpreted the results of experiments. SB and WIK drafted the manuscript. SB, GRS, MDF, DC, and WIK edited and revised the manuscript.
Supported by: This work was supported by grants from the Canadian Institutes of Health Research (CIHR) to GRS (201709FDN-CEBA-116200), MDF (PJT148634), and WIK (PJT156262). SB is supported by a Post-Doctoral Fellowship Award from Farncombe Family Digestive Health Research Institute, McMaster University. GRS is supported by a Canada Research Chair and the J. Bruce Duncan Endowed Chair in Metabolic Diseases. MDF is supported by a CIHR New Investigator award (MSH141981) and is the recipient of an Early Research Leadership Initiative grant from the Heart and Stroke Foundation of Canada and its partners and an Ontario Ministry of Research, Innovation, and Science Early Researcher Award.
Conflicts of interest: The authors declare no conflict of interest to disclose.
References
- 1. Podolsky DK. Inflammatory bowel disease. N Engl J Med. 1991;325:1008–1016. [DOI] [PubMed] [Google Scholar]
- 2. Molodecky NA, Soon IS, Rabi DM, et al. . Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54.e42. [DOI] [PubMed] [Google Scholar]
- 3. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12:720–727. [DOI] [PubMed] [Google Scholar]
- 4. Arnett HA, Viney JL. Gatekeepers of intestinal inflammation. Inflamm Res. 2010;59:1–14. [DOI] [PubMed] [Google Scholar]
- 5. Park SH, Aniwan S, Loftus EV Jr. Advances in the use of biologics and other novel drugs for managing inflammatory bowel disease. Curr Opin Pharmacol. 2017;37:65–71. [DOI] [PubMed] [Google Scholar]
- 6. Argollo M, Fiorino G, Hindryckx P, et al. . Novel therapeutic targets for inflammatory bowel disease. J Autoimmun. 2017;85:103–116. [DOI] [PubMed] [Google Scholar]
- 7. Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019:18:527–551. [DOI] [PubMed] [Google Scholar]
- 8. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009;89:1025–1078. [DOI] [PubMed] [Google Scholar]
- 9. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dzamko N, van Denderen BJ, Hevener AL, et al. . AMPK β1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem. 2010;285:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steinberg GR, O’Neill HM, Dzamko NL, et al. . Whole body deletion of AMP-activated protein kinase β2 reduces muscle AMPK activity and exercise capacity. J Biol Chem. 2010;285:37198–37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Galic S, Fullerton MD, Schertzer JD, et al. . Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest. 2011;121:4903–4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen L, Wang J, You Q, et al. . Activating AMPK to restore tight junction assembly in intestinal epithelium and to attenuate experimental colitis by metformin. Front Pharmacol. 2018;9:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith PD, Ochsenbauer-Jambor C, Smythies LE. Intestinal macrophages: unique effector cells of the innate immune system. Immunol Rev. 2005;206: 149–159. [DOI] [PubMed] [Google Scholar]
- 16. Soufli I, Toumi R, Rafa H, et al. . Overview of cytokines and nitric oxide involvement in immuno-pathogenesis of inflammatory bowel diseases. World J Gastrointest Pharmacol Ther. 2016;7:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bai A, Yong M, Ma AG, et al. . Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther. 2010;333:717–725. [DOI] [PubMed] [Google Scholar]
- 18. Hawley SA, Fullerton MD, Ross FA, et al. . The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336:918–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quinn JM, Tam S, Sims NA, et al. . Germline deletion of AMP-activated protein kinase beta subunits reduces bone mass without altering osteoclast differentiation or function. Faseb J. 2010;24:275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim JJ, Shajib MS, Manocha MM, et al. . Investigating intestinal inflammation in DSS-induced model of IBD. J Vis Exp. 2012;(60): 3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwon YH, Wang H, Denou E, et al. . Modulation of gut microbiota composition by serotonin signaling influences intestinal immune response and susceptibility to colitis. Cell Mol Gastroenterol Hepatol. 2019;7:709–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;83:14.1.1–14.1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harusato A, Geem D, Denning TL. Macrophage isolation from the mouse small and large intestine. Methods Mol Biol. 2016;1422:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fullerton MD, Ford RJ, McGregor CP, et al. . Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J Lipid Res. 2015;56: 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghia JE, Blennerhassett P, Collins SM. Impaired parasympathetic function increases susceptibility to inflammatory bowel disease in a mouse model of depression. J Clin Invest. 2008;118:2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banskota S, Regmi SC, Kim JA. NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP-7. Mol Cancer. 2015;14:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sag D, Carling D, Stout RD, et al. . Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu K, Zhao E, Ilyas G, et al. . Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11:271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haq S, Grondin J, Banskota S, et al. . Autophagy: roles in intestinal mucosal homeostasis and inflammation. J Biomed Sci. 2019;26:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim J, Kundu M, Viollet B, et al. . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fan K, Lin L, Ai Q, et al. . Lipopolysaccharide-induced dephosphorylation of AMPK-activated protein kinase potentiates inflammatory injury via repression of ULK1-dependent autophagy. Front Immunol. 2018;9:1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He C, Li H, Viollet B, et al. . AMPK suppresses vascular inflammation in vivo by inhibiting signal transducer and activator of transcription-1. Diabetes. 2015;64:4285–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hampe J, Franke A, Rosenstiel P, et al. . A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–211. [DOI] [PubMed] [Google Scholar]
- 34. Saitoh T, Fujita N, Jang MH, et al. . Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456: 264–268. [DOI] [PubMed] [Google Scholar]
- 35. Stang M, Wysowski DK, Butler-Jones D. Incidence of lactic acidosis in metformin users. Diabetes Care. 1999;22:925–927. [DOI] [PubMed] [Google Scholar]
- 36.Cuthbertson DJ, Babraj JA, Mustard KJW, et al. 5-Aminoimidazole-4-carboxamide-1-β -D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes. 2007;562078–2084. [DOI] [PubMed] [Google Scholar]
- 37. Misbin RI, Green L, Stadel BV, et al. . Lactic acidosis in patients with diabetes treated with metformin. N Engl J Med. 1998;338:265–266. [DOI] [PubMed] [Google Scholar]
- 38. Nielsen OH, Munck LK. Drug insight: aminosalicylates for the treatment of IBD. Nat Clin Pract Gastroenterol Hepatol. 2007;4:160–170. [DOI] [PubMed] [Google Scholar]
- 39. Park H, Kim W, Kim D, et al. . Mesalazine activates adenosine monophosphate-activated protein kinase: implication in the anti-inflammatory activity of this anti-colitic drug. Curr Mol Pharmacol. 2019;12:272–280. [DOI] [PubMed] [Google Scholar]
- 40. Steinberg GR, Dandapani M, Hardie DG. AMPK: mediating the metabolic effects of salicylate-based drugs? Trends Endocrinol Metab. 2013;24:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.