

Abstract
Crohn’s disease (CD), a form of inflammatory bowel disease (IBD), is characterized by heterogeneity along multiple clinical axes, which in turn impacts disease progression and treatment modalities. Using advanced data integration approaches and systems biology tools, we studied the contribution of CD susceptibility variants and gene expression in distinct peripheral immune cell subsets (CD14+ monocytes and CD4+ T cells) to relevant clinical traits. Our analyses revealed that most clinical traits capturing CD heterogeneity could be associated with CD14+ and CD4+ gene expression rather than disease susceptibility variants. By disentangling the sources of variation, we identified molecular features that could potentially be driving the heterogeneity of various clinical traits of CD patients. Further downstream analyses identified contextual hub proteins such as genes encoding barrier functions, antimicrobial peptides, chemokines, and their receptors, which are either targeted by drugs used in CD or other inflammatory diseases or are relevant to the biological functions implicated in disease pathology. These hubs could be used as cell type–specific targets to treat specific subtypes of CD patients in a more individualized approach based on the underlying biology driving their disease subtypes. Our study highlights the importance of data integration and systems approaches to investigate complex and heterogeneous diseases such as IBD.
Keywords: Crohn’s disease, heterogeneity, clinical phenotypes, blood, monocytes, CD4+ T cells, gene expression, genetics, data integration, systems biology, networks, hubs, drug targets, drug repurposing
INTRODUCTION
Crohn’s disease (CD) is an inflammatory bowel disease (IBD) characterized by variable degrees of disease heterogeneity, including disease location and behavior, perianal involvement, disease progression, extraintestinal manifestations, and the need for and response to different therapies.1–4 Clinical, endoscopic, and histological findings are used in the proper diagnosis and clinical management of patient subgroups.5 However, the underlying molecular features that dictate and contribute to the CD disease spectrum are greatly unknown. Associations between the observed heterogeneity of specific clinical traits and variations in molecular (genomic and transcriptomic) and cellular (cell-type specific) features have been documented.6–8 In particular, various subtypes of immune cells such as CD8+, CD4+ T cells, and CD14+ monocytes have been associated with variation in activity, prognosis, and severity of disease.7,9–13 Nevertheless, a comprehensive analysis of molecular features that contributes to the vast panels of clinical heterogeneity from a systemic and network biology perspective has not been carried out. In this study, we use a combinatorial approach (Fig. 1A) driven by unsupervised data integration, co-expression-based modularization of genes, pathway analysis, and integration with interaction networks to discover and interpret various features in a cell type–specific manner. These findings represent some of the possible mechanisms driving the observed phenotypic heterogeneity in CD and help identify novel drug targets or provide targets toward repurposing drugs to treat particular CD subtypes.
FIGURE 1.

A, Illustrative representation of the workflow used in the study to analyze and integrate the data sets. B, Variance inferred per latent factor and distributed across the data sets as inferred by Multi-Omic Factor Analysis.16
MATERIALS AND METHODS
Patient Selection
The current study was conducted at the University Hospitals of Leuven (Leuven, Belgium). We cross-sectionally collected peripheral blood mononuclear cell from 33 genotyped CD patients with endoscopy-proven active disease (presence of ulcerations). From those patients, disease features including disease location and behavior, perianal and upper gastrointestinal (GI) involvement, previous therapies, age, smoking status, and disease duration were collected (supplementary material online).
Bioinformatic Analyses
In this study, we used various computational and bioinformatic tools to analyze, integrate, and interpret heterogeneous data sets. Furthermore, a list of bioinformatics terms are provided in the appendix in supplementary material online.
Multi-omic factor analysis (MOFA)16 is a data integration tool to help interpret the sources of variation in each of the 3 data sets (monocyte gene expression, CD4+ gene expression, and genetics) or a combination thereof. Multi-omic factor analysis, which is a methodology for the latent variable model-driven integration of multi-omic data sets, is based on the mathematical principles of factor analysis and Bayesian modeling for parameter inference. MOFA can be considered as a statistically advanced version of a principal component analysis (PCA), which helps interpret representative variables that drive the variance in a data set. However in contrast to PCA, MOFA enables the dissection of latent factors (which are low-dimensional representative variables of heterogeneous high-dimensional data sets and analogous to principal components in a PCA) to help identify hidden factors (features such as genes, proteins, metabolites, etc in -omics data sets) or a combination of them that drive the cumulative variance. In the literature, there are various tools such as iCluster17 and Group Factor Analysis (GFA)18 for the integration of heterogeneous -omic data sets in the latent variable model category. Though MOFA was found to be more efficient in terms of computational power and time, both GFA and iCluster were less efficient in inferring nonredundant/unique latent factors and factors with contributions from multiple -omic data sets.16 In general, in contrast to other data integration methods,19–24 MOFA has several added functionalities and advantages such as the ability to handle missing values by imputation, interpretation of user-friendly solutions, and flexibility to adopt appropriate likelihood models based on the distribution patterns of the different data sets.16
After the identification of the latent factors and ranking of features (explained in detail in succeeding sections), CEMiTool25 was used to identify co-expression modules and integrate gene expression signatures with benchmarked molecular interaction networks. Functionally coherent genes tend to be co-expressed (ie, having similar expression patterns) due to common underlying drivers of expression.102 The power of co-expression network analysis can be harnessed to identify sets of genes that act in concert with each other. Moreover, co-expression networks also tend to form modules (ie, communities/groups of co-expressed genes) with overrepresented functions, share topological (ie, the structure of the network) similarity with real-world networks, capture underlying molecular mechanisms, and highlight hubs (ie, genes with a large number of neighbors).26–28 Various tools have been developed to deal with co-expression network analysis. Some of the prominent ones include WGCNA,29 PETAL,30 CoP,31 GNET,32 DiffCoEx,33 CoExpress,34 DICER,35 and DINGO.36 In comparison with these, CEMiTool offers several added advantages such as automatic gene filtering, overrepresentation analysis of the modules with respect to the phenotypes, gene set enrichment analysis, integration with the interactome, inclusion of multiple phenotypic classes, module merging based on similarity measures, availability as an R package, automation, and computational efficiency.25
For identifying the upstream transcriptional regulators of a given set of genes (in our case, genes within the CEMiTool identified co-expression modules associated with clinical phenotypes), CheA337 was used. Several online and standalone databases/tools that help users identify the transcriptional regulators for a given set exist in the literature. These include VIPER,38 DoRothEA,39 BART,27 TFEA.ChIP,40 oPOSSUM,41 and MAGICACT.42 Based on various benchmarking analysis with different data sets, CheA3 was found to perform better.37
The underlying sets of physical interactions between molecules are the basic rule-based structures which drive the phenotypic responses to stimuli. As a source of molecular interactions, we used Parsimonious Composite Network (PCNet),43 which is a composite molecular interaction network resource compiled by merging 21 different interaction databases. PCNet was benchmarked on 446 disease gene sets for its performance based on disease gene recovery.43 Although various molecular interaction network resources exist, we chose PCNet because of its performance attributes reported in the benchmarking analysis.43
Although PCNet is a good source of molecular interactions, it does not provide the context in terms of signal flow/signal transduction (ie, signaling pathways). To address this, Reactome,44 which is a compendium of curated signaling pathways comprising protein-protein interactions, was used to identify the signaling pathways overrepresented in the gene sets investigated.
Data Integration
Multi-omic factor analysis16 was used to integrate the 3 data sets—namely the gene expression from CD4+ T cells and CD14+ monocytes (protocols used for cell separation, RNA isolation, sequencing, and genotyping are included in supplementary material online), and the single nucleotide polymorphism profiles. The top 2500 genes with the highest variance in expression were selected. Default model training options were used to construct the MOFA model object with the exception of the DropFactorThreshold (representative of variance cutoff) and maxiter (number of iterations), which were set at 0.02 and 5000, respectively. The weights and variance contributions were retrieved from the converged MOFA model. Latent factors (LFs) with no weight contributions from any of the patients were discarded. Multiple regression was used to identify the explanatory LFs that contribute to the clinical traits. Latent factor trait relationships with a nominal P value ≤0.1 were considered to be significant.
The data set corresponding to the explanatory LFs were identified based on their variance contributions to the LFs. If the ratio between the variance contribution of the first and second data set was ≥6, only the first data set was considered as having contributed to the explanatory LF, or else both the data sets were considered as having contributed. Latent factors that associated with gender were discarded to remove any sex-specific signals. Phenotypes with class imbalances were discarded.
Co-expression and Network Analysis
The top 1000 genes (ranked by the weights assigned to the gene by the corresponding eLF) corresponding to the strongest -omic layer(s) of the explanatory LFs were used for the co-expression analysis which was performed using the CEMiTool.25 Correlation-based associations between genes were assigned if the Pearson correlation coefficient (PCC) was ≥0.8 or ≤−0.8. Modules with less than 20 genes were discarded. The soft threshold beta was selected by testing different beta values and evaluating the resulting fitness of the networks/modules to the scale-free topology.45, 46 The maximum beta value, at which the R2 corresponding to the fitness of the networks/modules to the scale-free topology levels off, was used as the optimal beta.25
The activity of the gene modules with respect to the clinical phenotypes was carried out using the mod_gsea function of the CEMiTool followed by the mapping of the normalized enrichment scores (NES) to the modules. If the modules corresponding to the explanatory LFs are not related to phenotypes, such LFs were not considered for further analysis. To check for the overrepresentation of the modules with specific functions such as Reactome Signaling Pathways, the mod_ora function was used. Gene sets corresponding to Reactome Signaling Pathways were retrieved from the Molecular Signatures Database v7.0.47 Reactome Signaling Pathway gene sets with less than 10 genes were not considered for the overrepresentation analysis. Gene sets with false discovery rate (FDR) (adjusted P value) ≤0.05 were deemed to be significant.
For the network analysis, we used PCNet as the interaction network resource. The plot_interactions function was used to superimpose the module-wise co-expression networks onto the physical interaction network represented by PCNet. The top 50 hubs based on their connectivities in the co-expression network were retrieved to be compared with functional gene lists representative of a priori knowledge.
Drug Target Analysis
Lists of molecules targeted by drugs in the case of CD, ulcerative colitis (UC), ankylosing spondylitis (AS), rheumatoid arthritis (RA), psoriasis (PS) and primary sclerosing cholangitis (PSC) and genes associated (based on genetic associations, somatic mutations, RNA expression, text mining, and animal models) with CD were retrieved from the Open Targets database as of November 19, 2019.48 In the case of genes for which the CD associations were inferred from their transcriptomic expression levels, the expression measurements were confined to samples derived from the colon, intestine and rectum. The expression quantitative trait loci (eQTL) data were retrieved from Di Narzo et al,49 and information on genes corresponding to barrier function, antimicrobial peptides, cell adhesion molecules/chemokines/chemokine receptors, and IBD susceptibility loci was obtained from Vancamelbeke et al,50 Arijs et al,51 Arijs et al,52 and Mirkov et al,53 respectively. Regulator prioritization was performed by using the ChEA3 tool.37
RESULTS
Identifying Strongest Axes of Variation Linked to Clinical Traits
Using MOFA to integrate the various -omic layers, we identified 13 latent factors that captured independent sources of phenotypic variation spread across the different -omic layers (Fig. 1B, Supplementary Fig. 1, and supplementary material online). The 13 identified LFs together explained about 58% of the variation in the CD14+ (monocytes) gene expression, 67% of the CD4+ gene expression data, and 3% of the variance in the mutational data.
Notably, the strongest LF (LF1) was active in both transcriptomic data sets (CD14 + 18.46%; CD4 + 9.81% variance contribution, respectively), whereas LF6 was associated with both CD4+ gene expression and mutational data (Fig. 1B, supplementary material online). We also identified explanatory LFs (eLFs), which are defined as LFs that explain the heterogeneity of the patients in terms of their clinical phenotypes (Fig. 2A, supplementary material online). Though 7 of the 13 LFs could significantly (P < 0.1) explain at least 1 of the 13 clinical traits considered in this study, 2 LFs correlated with at least 2 traits each, barring LF6 which was gender-associated. Latent factor 5, with a strong contribution from monocyte gene expression for example, was associated with the number of antitumor necrosis factor (TNF) agents used and previous exposure to vedolizumab (VDZ). Conversely, LF11 was active in both CD4+ and monocyte gene expression and was associated with 1 trait only—disease behavior. Other prominent examples of single trait LFs include LF12, which is active along monocyte gene expression and is associated with disease location (Fig. 2A). Both LF11 and LF12 were associated uniquely to disease behavior and disease location, respectively, making them specific in capturing the signals that could be driving these phenotypes. Furthermore, no other eLFs were found to be associated with the 2 axes of clinical phenotypes. Analyzing the weights and factors (supplemental material online) corresponding to the LFs revealed the functional characteristics captured by each of the LFs across different data sets. We observed significant (FDR 5%) enrichment of signaling pathways (with a minimum of at least 30 pathways) in 10 of the 13 LFs associated with CD4+ gene expression (Supplementary Fig. 2A).
FIGURE 2.


A, Table depicting the explanatory LFs, which are defined as LFs associated with at least one clinical trait. The figure also shows the respective data sets that contribute to the variance explained by the LFs. By tracing the variance contributions to the LFs associated with the clinical traits, we could identify the -omic data sets contributing to the clinical trait. Latent factors associated with gender were discarded to exclude sex-specific signals. B, Summary of modules (derived from co-expression analysis, see methods section “Bioinformatic analyses” for more details), their relevance to the corresponding phenotypes (indicated by the column “Module activity in phenotype”), and the overlap between the hubs (top 50 genes) in each of the modules and targets of drugs used in intestinal inflammatory disorders such as Crohn’s disease and ulcerative colitis, other inflammatory disorders such as ankylosing spondylitis, rheumatoid arthritis, psoriasis, and sclerosing cholangitis, associations with CD, role as CD eQTLs or genes relevant to CD in terms of their activity (antimicrobial peptide functions, barrier functions, chemokine functions, and cell adhesion). ^ - if the total number of genes in a module was less than 50, all the genes in the module were considered as hubs. In the last column, the active modules with the highest percentage of unique overlapping hubs are indicated.
Latent factor 7 (along CD4+ transcriptomics), with the highest number of overrepresented gene sets for example, was enriched with signaling pathways associated with Fc epsilon receptor, MyD88, Toll-like receptors, and induction of interferon-mediated responses among others (Supplementary Fig. 2B). Interleukin (IL) signaling pathways were found to be top overrepresented gene sets as weighted by LF3, the second strongest LF along the CD14+ gene expression layer (Fig. 1B, Supplementary Fig. 2C, D).
Unsupervised Clustering Along Explanatory Latent Factors Identifies Patient Groups With Distinct Clinical Traits
To confirm the relevance of the eLFs in segregating the patients along the corresponding clinical traits, we performed unsupervised clustering of patients along the eLFs, that is, using the weights assigned to the samples by the eLFs. With this step, we could identify clusters that were aligned with corresponding clinical traits associated with the eLF (Table 1, supplementary material online). In 7 of the 8 LF-trait associations, we observed significant correlation between the cluster membership and clinical traits of samples, suggesting that the eLFs help capture the underlying clinical classes. Three of the LF-trait associations were also characterized by the enrichment of the corresponding clinical traits within the identified clusters (Table 1, supplementary material online). For example, the 2 clusters obtained by sample aggregation along LF12, which aligned with monocyte gene expression, were explanatory of heterogeneity with respect to the occurrence of disease location. Similarly, clustering of samples based on the weights of LF11 which is dominant in both monocyte and CD4+ gene expression, could achieve the separation of patients along the axis of disease behavior (Table 1, supplementary material online). Dual trait-associated LFs include LF5 and LF9. Latent factor 9 was associated significantly—positively with the phenotype of perianal disease (R2 = 0.45) and inversely with smoking (R2 = −0.33), potentially capturing a link between perianal subtypes and incidence of smoking.
TABLE 1.
Results of the Unsupervised Clustering of Samples Based on the Explanatory Latent Factors Associated with the Traits.
| Trait | LFa | Dominantomic layer |
Correlation Between Cluster Membership and Clinical Rraits |
R2 | P | Optimal Clustering | Enrichment of Traits in Clustersb |
|---|---|---|---|---|---|---|---|
| Disease location | LF12 | CD14 | Yes | 0.42 | 0.0164 | k = 2 | No |
| Disease behavior | LF11 | CD14 | Yes | −0.29 | 0.0965 | k = 5 | No |
| Disease behavior | LF11 | CD4 | Yes | −0.29 | 0.0965 | k = 5 | No |
| Perianal disease | LF9 | CD14 | Yes | 0.45 | 0.00834 | k = 3 | Yes |
| Number of anti-TNF agents exposed to | LF5 | CD14 | Yes | 0.437 | 0.0109 | k = 2 | No |
| Exposure to Vedolizumab | LF3 | CD14 | Yes | 0.563 | 0.0006 | k = 2 | Yes |
| Exposure to Vedolizumab | LF5 | CD14 | Yes | 0.46 | 0.007 | k = 2 | Yes |
| Smoking | LF9 | CD14 | Yes | −0.3255 | 0.6453 | k = 2 | No |
aEnrichment was deemed as significant for those overrepresentation events with an adjusted P value of ≤ 0.1.
bLFs associated with gender were discarded to exclude sex specific signals.
Integration of Co-expression Modules and Interaction Networks Reveal Communities and Hubs Potentially Involved in Mediating Disease Heterogeneity
To identify communities (groups of genes that work in concert with each other) and hubs (highly connected genes) involved in mediating disease heterogeneity, we carried out modularization based on co-expression and integration thereof. Modularization was performed on the expression of ranked (based on the weights provided by the eLF) genes from the data set contributing strongly in terms of variance to the eLF. The modules obtained (Fig. 2B) were not only related to the corresponding phenotypes of the eLF but were also enriched with signaling pathways (supplementary material online). For instance, the activity of modules (Fig. 3A, supplementary material online) inferred (optimal soft threshold beta = 8, Fig. 3A) from the monocyte gene expression data sets corresponding to LF11 associated with disease behavior could distinguish the different subtypes of patients characterized by disease behavior—namely, inflammatory, fibrostenotic, and penetrating CD (Fig. 3B). Of the 10 determined modules that were generated in the previously mentioned case, 6 of them displayed activity to varying extents on at least 1 subtype of disease behavior. Though modules 4 and 5 had polarized effects on the fibrostenotic and inflammatory phenotypes, module 2 was active along the fibrostenotic and penetrating behaviors of CD.
FIGURE 3.

A, The “scale-free” aspect of the modules (inferred from the monocyte gene expression) obtained over a window of values for the soft threshold beta parameter. Modules corresponding to the beta value of 8 was chosen because there was a plateauing of the observed “scale-free”-ness or aspect. Many real biological networks display scale-free behavior captured by the scale-free topology model fit R2 value plotted on the y axis. B, The activity profile of the monocyte gene expression modules in relation to the disease behavioral phenotype. Only the modules active with respect to the phenotype are displayed. The NES score represents the z-score normalized expression of the samples within each phenotype and is used to assess the alterations of module activity with respect to the phenotype. C, Overrepresented Reactome gene sets at FDR ≤ 0.05 in module 5, (D) in module 8 (E), and in module 9. F, The top 10 genes (hubs) identified in module 9 after integrating the co-expression network derived from module 9 with the benchmarked interaction network PCNet.43 Hubs are defined as nodes (genes) with a high connectivity (ie, number of neighbors) in the network and represent nodes that could play an active role in the given context.
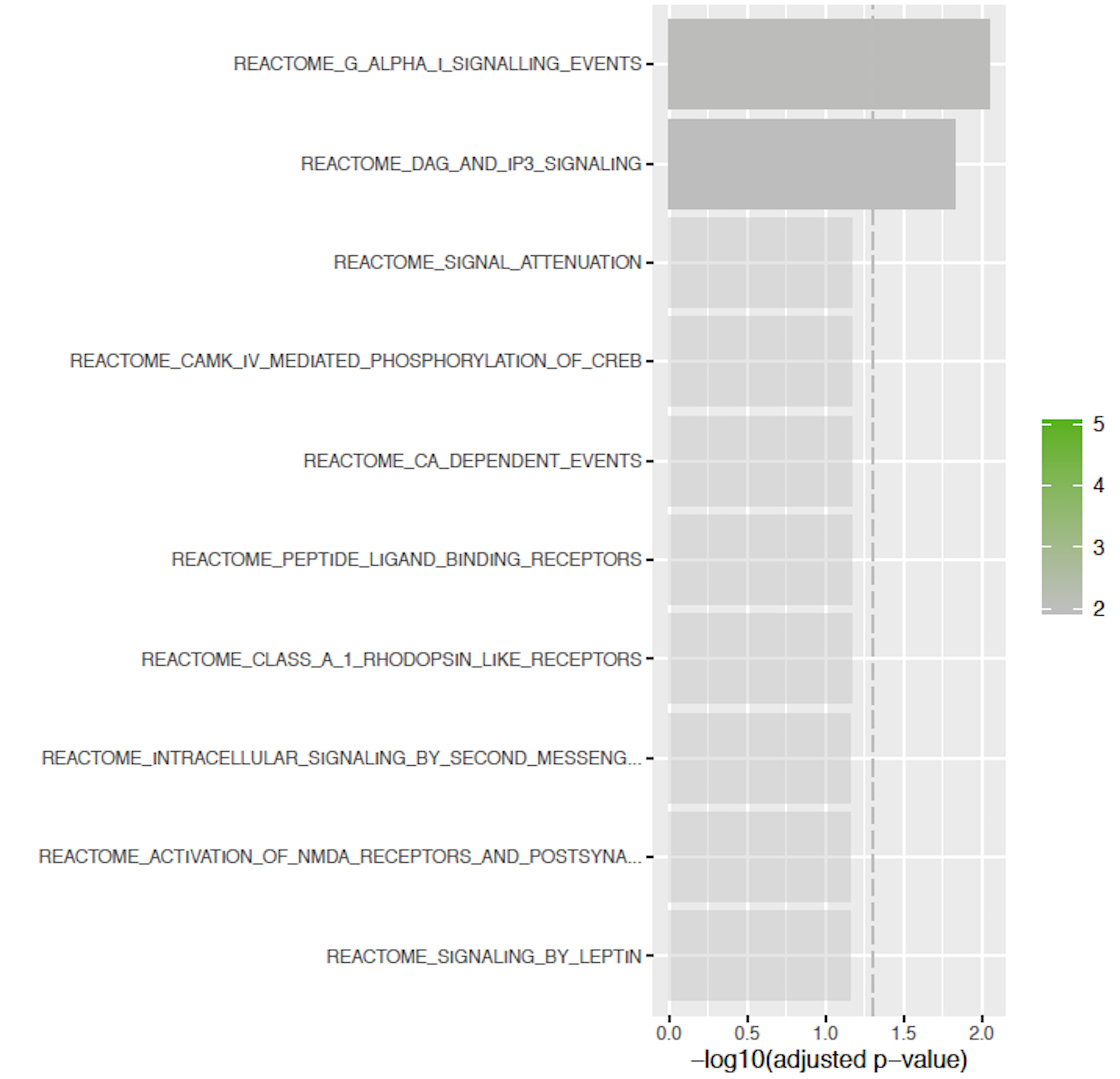
Upon deeper investigations inquiring in to the role of the genes that make up these modules, we gained a proper understanding of the functional landscape. Overrepresentation analysis of module 5 genes displayed an enrichment of pathways related to IL-1, IL-10, and IL-13 signaling, NLRP3 inflammasome formation, RIP (receptor-interacting serine/threonine-protein kinase)-mediated NFKB activation, TAK1 (nuclear receptor subfamily 2 group C member 2)-mediated activation of NFKB by phosphorylation, and activation of IKKs complex among others at the top (FDR ≤ 0.05; Fig. 3C). Module 4 meanwhile was enriched in G alpha (i) signaling (which is involved in the downstream segment of G-protein coupled receptor pathway) and the DAG/IP3 signaling pathway (Supplementary Figure 3). Interestingly, module 8, which did not display any activity on the sample classes based on disease behavior, was enriched with interferon alpha/beta signaling pathways and their induction (Fig. 3D). We also observed other co-expressed communities such as modules 1, 6, and 9, which were unique to particular disease behavior classes. For example, module 9 was active with respect to the penetrating phenotype (Fig. 3B) and was uniquely and prominently enriched with pathways associated with signaling and induction of interferon gamma, presentation of soluble exogenous antigens, and IL-12, IL-27 pathways (Fig. 3E). The network graph of module 9 (Fig. 3F) also highlights important network hubs (characterized by co-expression and interactions) such as STAT1, UBE2L6, and WARS, among the top 10.
As discussed previously, LF11 is associated with CD disease behavior and has a contribution from CD4+ expression, as well (supplementary material online). Interestingly, 5 of the 8 modules (modules 1, 2, 3, 7, and 8) derived from CD4+ expression were active across disease behavioral subphenotypes but were confined to the inflammatory and penetrating type of CD disease behavior (Fig. 4A). Genes within module 2, which is active only in the penetrating subtype, were enriched with pathways (Fig. 4B) corresponding to chemokines and their receptors, G-protein coupled receptor signaling, and IL-10 signaling, among others. The top 10 hubs within module 2 included GZMH, FGFBP2, and PRF1 (Fig. 4C). Modules 1, 3, and 7—despite being active—did not display any enrichment in terms of signaling pathways. However, module 3 harbored biologically relevant hubs (Fig. 4D), such as OGT, JMJD7-PLA2G4B, CDK5RAP3, LUC7L3, and DDX26B, which are relevant for orchestrating post-translational and post-transcriptional mechanisms.
FIGURE 4.

A, The activity profile of the CD4+ gene expression modules in relation to the disease behavioral phenotype. Only the modules active with respect to the phenotype are displayed. B, Overrepresented Reactome gene sets at FDR ≤ 0.05 in module 2. C, The top 10 genes (hubs) identified in module 2 and (D) module 3 after integrating the co-expression network derived from the corresponding modules with the benchmarked interaction network PCNet.43 Genes encoding proteins such as OGT (UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase) and which play important roles in mediating T-cell responses83 are identified as hubs.
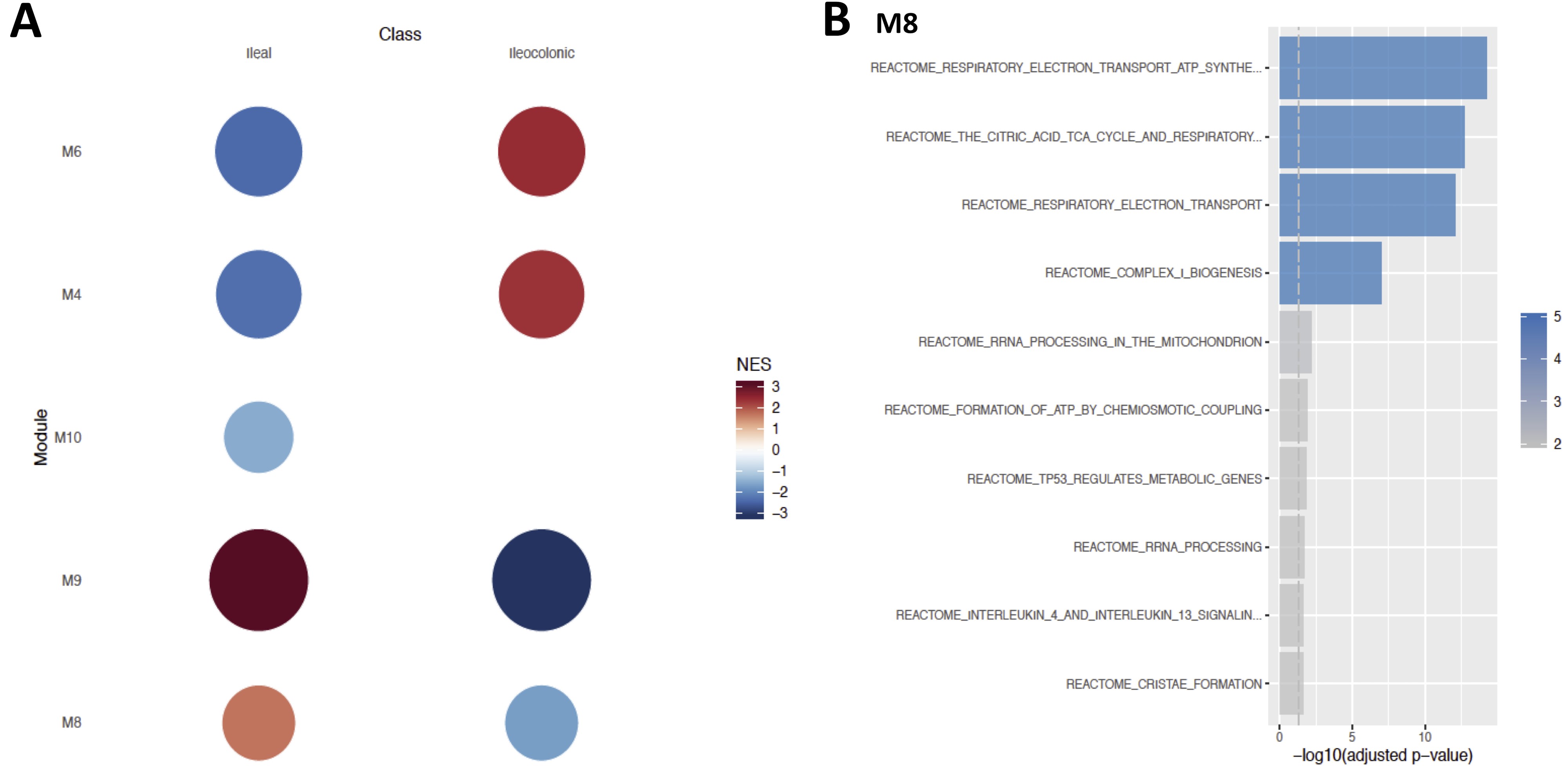
As for explaining variability in disease location, results from similar analysis revealed 11 different modules (Supplementary Fig. 4A), 4 of which were active across ileal and ileocolonic CD. Module 8 in particular was observed to have strong signals emerging from the expression of genes encoding proteins and enzymes especially in pathways such as the respiratory electron transport, tricarboxylic acid (TCA) cycle, and complex 1 biogenesis (NADH,-ubiquinone oxidoreductase or NADH dehydrogenase; Supplementary Fig. 4B).
Hubs From Phenotype-Related Modules Capture Novel/Known Therapeutic Targets and Disease-related Genes
The top hubs in modules also overlapped (Fig. 2B, supplementary material online) with previously known targets of drugs aimed to treat CD and ulcerative colitis (UC), in addition to other inflammatory diseases such as ankylosing spondylitis, rheumatoid arthritis, psoriasis, and primary sclerosing cholangitis. The hubs could also be annotated as disease relevant genes such as those known to be CD eQTL genes, IBD susceptibility loci and/or those encoding intestinal barrier proteins, antimicrobial peptides, cell adhesion molecules, chemokines, and their receptors. For a particular phenotype-module combination, modules active for the corresponding phenotype were bound to harbor the highest number of functional hubs (defined as high-ranking proteins known to be previous drug targets in UC, CD, AS, RA, PS, and PSC). In 4 of the 5 phenotype-module combinations portrayed in Figure 2B, the module with the highest percentage of functional hubs was active across the corresponding phenotype. For instance, 64% of the hubs in module 9, which is active across disease behavior in monocytes (Fig. 5, supplementary material online), were either annotated as already known drug targets and/or associated with CD and/or relevant to CD.
FIGURE 5.

The functional profile of the genes in the monocyte-gene expression derived module 9 active across the disease behavioral phenotype. Sixty-four percent of the module 9 hubs were annotated as being targets of drugs used to treat intestinal inflammatory disorders such as CD and UC, other inflammatory disorders such as ankylosing spondylitis, rheumatoid arthritis, psoriasis, and sclerosing cholangitis, associations with CD, role as CD eQTLs or genes relevant to CD in terms of their activity (antimicrobial peptide functions, barrier functions, chemokine functions and cell adhesion). Genes marked in orange denote transcription factors. **Indicates TFs that are identified as being relevant regulators (of the genes in the same modules) by ChEA3.37 The figure indicates that more than half of the genes were previously identified as involved in the pathogenesis of CD as recorded in the Open Targets database.48
One of the prominent module 9 hubs (expressed in monocytes) associated with disease behavior and a CD drug target is CXCL10 encoding the C-X-C motif chemokine. CXCL10 is not only known for its role in recruiting pathogenic T cells to inflamed sites54 but also in mediating the production of pro-inflammatory cytokines such as IL-12 and IL-23 in IFN-gamma primed monocytes.55 Interestingly, module 9 genes corresponding to disease behavior in monocytes were also overrepresented in IFN-gamma and IL-12 signaling pathways (Fig. 3E). Yet another intriguing CD drug target captured by our analysis includes TYK2, which was identified as a CD4+ hub gene in an active module (M7) associated with disease behavior. TYK2 is a nonreceptor tyrosine-protein kinase/Janus kinase involved in modulating pathways of various interleukins including IL-12 and IL-23.56, 57 TYK2 is targeted in the context of multiple disorders including rheumatoid arthritis58, 59 and CD60 by various anti-inflammatory drugs, but specific TYK2 inhibitors are now also under study in IBD.61
In addition to identifying hubs previously targeted in Crohn’s disease, we discovered several potential targets used in other inflammatory disorders. Of the 37 hubs that were known as targets used to treat ulcerative colitis, ankylosing spondylitis, rheumatoid arthritis, psoriasis, and sclerosing cholangitis, only 27% were previously targeted in CD (supplementary material online). The remaining 73% (27) of the hubs were found to be targeted only by any 1 of the UC, AS, RA, PS, or PSC drugs, thus throwing open the door for repurposing such hubs for CD. Two thirds (18 of 27) of these repurposable hubs were associated with monocyte gene expression modules active across the phenotypes of disease behavior or disease location. For instance, ADORA3, CXCR2, FLT3, PRKCE, IL1A, IL1R1, and CD40 belong to the previously mentioned category of repurposable hubs in monocyte gene expression modules which are active with respect to the phenotype of disease behavior (Table 2). Many of these hubs are either reported to play critical roles in inducing hyperinflammatory responses in monocytes or in mediating interactions of monocytes with other cell types (Table 2). In the case of disease location, repurposable hubs in monocytes included various subunits of the NADH-ubiquinone oxidoreductase enzymatic complex, FAAH, and SELL encoding the L-selectin protein (supplementary material online). As a calcium-dependent lectin that mediates cell-cell adhesion, L-selectin is involved in recruiting CD4+ T cells to chronically inflamed small intestinal tissues.62 It also plays a role in the pathogenesis of IBD by virtue of its role in the post-translational modifications of adhesions such as mucosal addressin cell adhesion molecule 1 (MAdCAM-1).63
TABLE 2.
List of Relevant Hubs in Monocyte Gene-expression Modules Associated With Disease Behavior to Potentially Repurpose for Use as CD Drug-targets.
| Hub Protein Name | Hub Protein Description | Functions | Module Harboring the Hub (No. pathways enriched) | Representativs Reactome Pathways Enriched in the Module |
Drugs Targeting the Hub (Disease* - Clinical trial ID) |
|---|---|---|---|---|---|
| ADORA3 | Adenosine receptor A3 | Proinflammatory activation of monocytes91 | M4 (2) | G Alpha I signaling events, DAG and IP3 signaling | Piclidenoson (PS - NCT03168256,NCT00428974,NCT01265667) Piclidenoson (RA - NCT02647762,NCT00280917,NCT00556894, NCT01034306) Caffeine (UC - NCT02760615^) Caffeine (RA - NCT01636557, NCT03131973) |
| CXCR2 | C-X-C chemokine receptor type 2 | Recruitment of atherogenic and inflammatory monocytes92 Involved in the adhesion of monocytes to tissues and other cell types93 |
M2 (2) | IL-4 and IL-13 signaling | Navarixin (PS - NCT00684593) |
| FLT3 | Receptor-type tyrosine-protein kinase FLT3 | Involved in normal granulocyte-monocyte progenitor development94 Proliferation of monocytes95 |
M4 (2) | G Alpha I signaling events, DAG and IP3 signaling | Lestaurtinib (PS - NCT00236119) Pexidartinib (RA - NCT01090570^) Elubrixin (UC - NCT00748410^) |
| PRKCE | Protein kinase C epsilon type | Upregulated in monocytes96 | M4 (2) | G Alpha I signaling events, DAG and IP3 signaling | Sotrastaurin (PS -NCT00885196) Sotrastaurin (UC -NCT00572585) |
| IL1A | Interleukin-1 alpha | Induces interleukin 1 receptor antagonist production in human monocytes97 Involved in the cross-talk between monocytes and stromal98 |
M5 (13) | IL-10 signaling, NLR signaling | Bermekimab (PS -NCT01384630) |
| IL1R1 | Interleukin-1 receptor type 1 | Expressed in monocytes in response to LPS exposure99 | M6 (-) | - | Anakinra (RA - NCT00111410,NCT00121043, NCT00117091,NCT00037700, NCT00537667^) AMG-108 (RA - NCT00293826, NCT00369473) |
| CD40 | Tumor necrosis factor receptor superfamily member 5 | Induction of IL-1β and tumor necrosis factor-α synthesis100 Adhesion of monocytes to other cell types101 |
M9 (18) | Interferon-gamma signaling, Antigen processing cross presentation, IL-12 family signaling | PG-102(RA - NCT00787137^) |
*PS, psoriasis; RA, rheumatoid arthritis; UC, ulcerative colitis.
^Suspended, terminated or withdrawn clinical trials.
Distinct Regulatory Programs Drive CD Subtypes by Modulating the Expression of Active Gene Modules
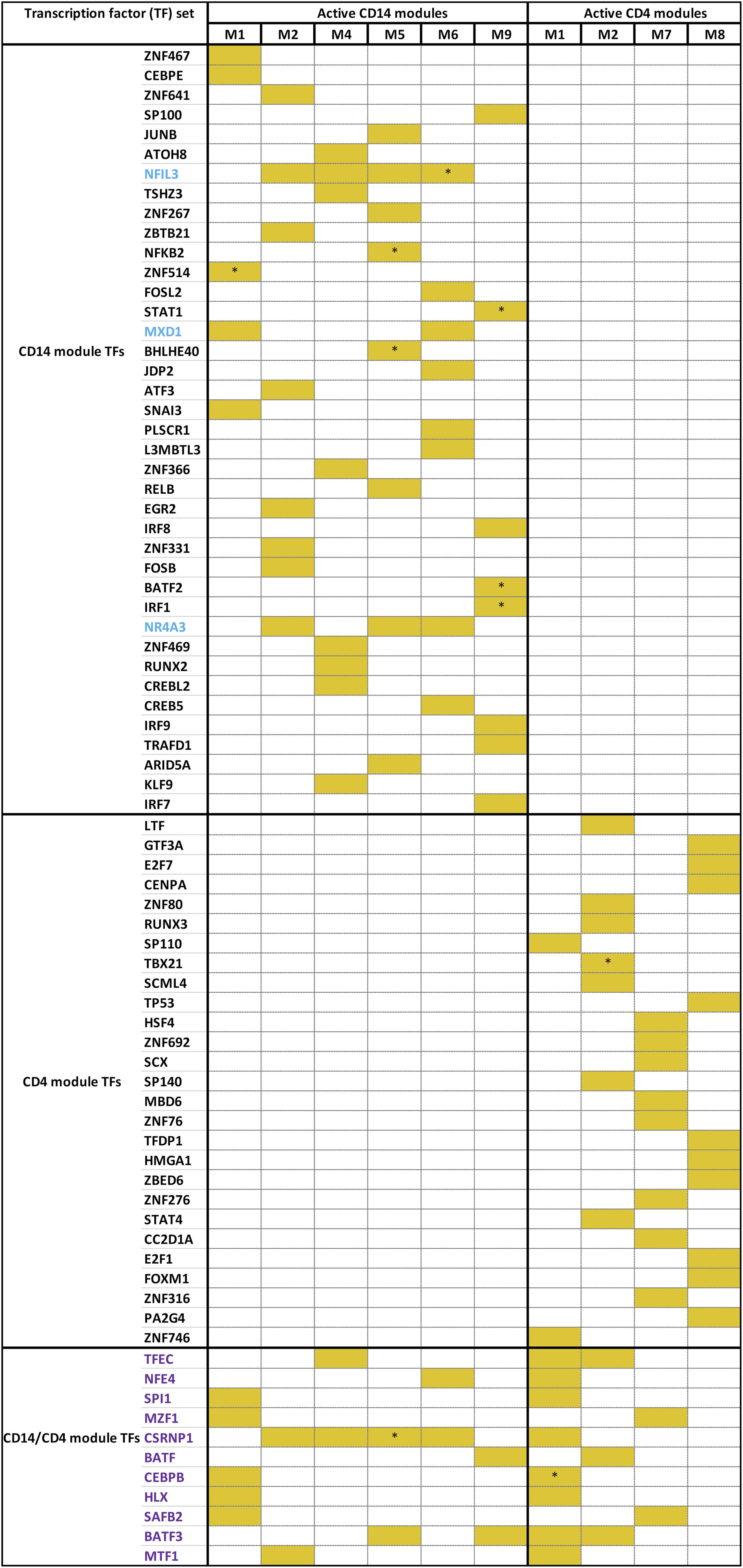
Distinct modules that drive the phenotypes are also expected to have specific regulatory programs driving their expression. By mining orthogonal libraries containing information on transcription factor-target gene information, we prioritized transcription factors governing the expression of the various modules (supplementary material online). Exclusive regulatory control was observed at 3 different levels: first at the level of traits, secondly between modules corresponding to the same clinical trait in the same cell type, and finally between modules corresponding to the same clinical trait from different cell types. At the level of individual traits overall, of the 126 transcription factors (TFs) that were identified as regulators of genes from all the modules active in the phenotypes as mentioned in Figure 2B, 70% of the TFs were specific to particular traits in a single cell type (Fig. 6A).
FIGURE 6.

A, Overlap of the transcription factors inferred for the active modules derived from gene expression in monocytes and CD4+ cells for the associated phenotypes. B, Transcriptional regulatory network showing the TFs modulating the expression of the monocyte modules active with respect to the disease behavior phenotype. Orange nodes represent the modules, purple nodes the TFs exclusively regulating the modules, and blue nodes the TFs regulating more than one module. The numbers within parentheses inside the orange nodes represent the number of Reactome signaling pathways enriched (FDR ≤ 0.05) within the genes found in the module. The thickness of the edges denote the percentage of genes of the module which are regulated by the TF. The figure indicates that the functionally distinct modules (as shown earlier by the set of Reactome pathways uniquely overrepresented in each of them) also have mostly distinct regulatory control, with very few transcription factors controlling the multiple modules. Visualization was performed using Cytoscape.90 C, Bar chart displaying the integrated mean ranks (from across orthogonal TF-target libraries) of the top 10 TFs for the genes in module 9 (enriched in interferon-gamma related pathways) derived from monocyte gene expression and active with respect to the disease behavioral phenotype. D, The distribution profile of genes in module 9 as targets of the most relevant TFs identified.
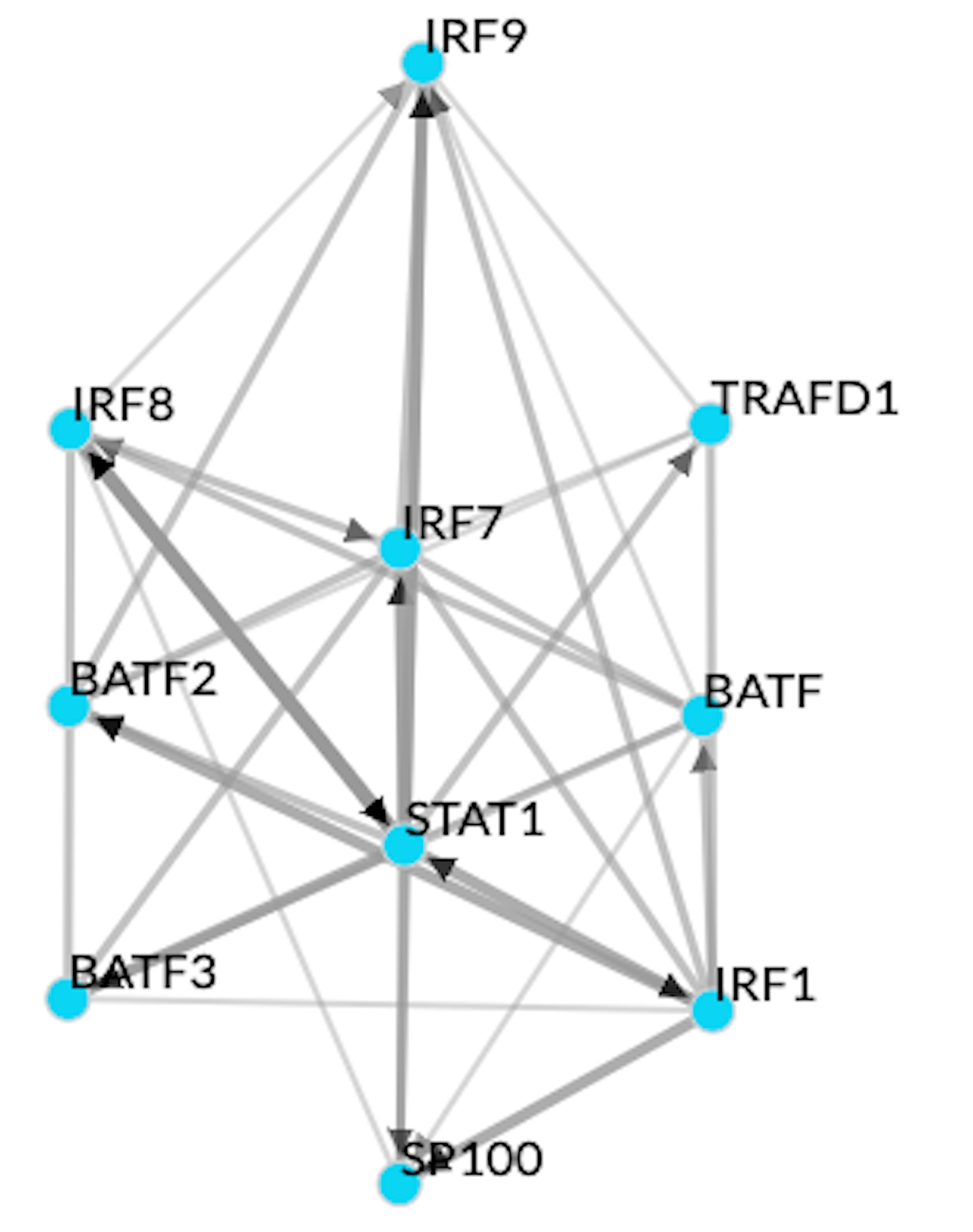
Within active modules in the same cell type, for example, in the case of monocytes in the context of disease behavior, we could point out a high degree of exclusivity in terms of the TFs that regulate the modules (Fig. 6B). Of the 50 TFs controlling the expression of the active modules, only 5 (MXD1, NR4A3, NFIL3, CSRNP1, BATF3) potentially modulated the expression of more than 1 active module. Modules with a greater number of enriched Reactome signaling pathways (M5 with 13 pathways, M9–11, M4, and M2 two each, and none in M1 or M6) were observed to have a higher proportion (M9, 55%; M5, 40%; M4 and M6, 16% each; M2, 17%; and M1, 12%) of their genes as targets of the identified TFs (Fig. 6B, supplementary material online). Among the functional hubs in module 9 (which has the highest coverage in terms of target coverage of transcription factors), 4 hubs (STAT1, BATF2, IRF1, ZNF595) were annotated as transcription factors, of which 3 (STAT1, BATF2, IRF1) were already identified (Fig. 6C, Supplementary data 13) as regulators of genes within the module. Cumulatively, STAT1, BATF2, IRF1, which were also known to be among the genes associated with CD, regulated 73% of the genes expressed in module 9 (Fig. 6D), possibly as a result of complex interplays among the regulators (supplementary material online). In effect, we identified STAT1, BATF2, and IRF1 acting as primary transcriptional modulators driving the expression of module 9 and thereby potentially the disease behavioral phenotype among CD patients.
As the third example of regulatory exclusivity, we investigated the active modules corresponding to different cell types and relevant for the same trait, as exemplified by disease behavior–associated modules in CD4+ cells and monocytes (supplementary material online). Of the 77 TFs inferred as regulators of the modules active with respect to disease behavior, the majority (86%) was specific to either monocyte or CD4+ associated modules, whereas the remaining TFs could regulate active gene modules in both CD4+ and monocytes. For instance, BATF3 was identified as a regulator of module 9 (enriched with interferon gamma signaling pathways) in monocytes and modules 1 and 2 (enriched with signaling pathways of chemokines including IL-10 and G-protein coupled receptor-mediated events; Supplementary Fig. 6, supplementary material online). On the other hand, and interestingly enough, master regulators such as NR4A3, STAT1, and EGR2 were confined to regulating the active modules solely in monocytes and not any of the active modules in CD4+ cells. Along with NR4A3, NFIL3 and MXD1 were found to regulate the expression of multiple active modules in monocytes (Supplementary Fig. 6).
DISCUSSION
The clinical heterogeneity of Crohn’s disease is well known. Due to the complexity of CD and the involvement of diverse immune and other cell types, untangling the mechanisms behind the observed heterogeneity has been challenging. However, the collection of electronic clinical records and the advent of cell type–specific data generation, next generation sequencing techniques, data integration tools, and biological networks has made it possible to investigate patient-derived data sets and provide biological insights into the pathogenesis of CD subtypes. In this study, we integrated genomics, CD4+ T cell and monocyte (CD14+) transcriptomics from CD patients to identify molecular features explaining CD heterogeneity such as disease behavior, disease location, and exposure to anti-inflammatory agents. Given that CD has underpinnings in both the innate and adaptive arms of the immune system,64 we focused on CD14+ monocytes as a cell type from the innate arm; and from the adaptive arm, we zoomed in on CD4+ T cells since CD is believed to be a disease concerning the Th1:Th17:Treg axis.
Using MOFA, we identified latent factors (defined as hidden variables inferred from the set of observed variables) and used them as proxies by which the variance within the -omic data sets is captured. Among the 5 identified eLFs, monocyte gene expression was the single largest contributor to variation. Two of the 5 eLFs were associated with multiple traits, suggesting that certain molecular profiles might have overlapping mechanisms. Synergistic traits (ie, traits which are related to each other in positive or negative manner) could have precipitated the emergence of multi-trait LFs. However, the large number of known and unknown clinical covariates or confounding factors could explain some of the not so significant LF-clinical trait associations in the study. Although sex-specific differences exist in terms of IBD incidence rates65 and may impact immune responses,66 we decided to focus on the most clinically relevant clinical phenotypes related to disease activity and disease state.
The fact that the genomic layer does not relate to any of the clinical axes of heterogeneity suggests that disease susceptibility single nucleotide polymorphisms do not contribute to disease heterogeneity, which is in agreement with previous observations made by Cleynen et al.8 In other words, nongenetic components such as gene expression seem to be more relevant to CD heterogeneity than genetic ones. The higher variance contribution from the dynamic gene expression layer could supersede the low contribution from the genomic layer due to network-wide propagation of disruptive effects of albeit a small number of mutations with inadequate effect sizes. However, it has to be mentioned that this bias could also be linked to the small number of features in the genomic layer (since we focused only on previously known disease susceptibility loci) compared with the 2 gene expression data sets. Furthermore, disease susceptibility loci are likely to be different from genes driving disease phenotypes and disease outcomes.67 In our cohort, for example, none of the patients harbored NOD2 mutations (known to be associated with CD subtypes68, 69) linked to CD susceptibility despite the presence of other nonsusceptibility-associated NOD2 mutations at a genome-wide level which were not considered in this analysis. To identify the molecular features that contribute to the heterogeneity, we looked into the individual data sets that contributed the strongest to the explanatory latent factors and identified features at the gene expression level that discriminated the different categories within the same axis of clinical heterogeneity. Some of the identified features at the gene expression level were previously known to be involved in Crohn’s disease pathogenesis, thus supporting our inferences.
Using modularization based on co-expression, functional analysis of the resulting modules followed by integration with interaction networks, we identified prominent pathways and hubs that drive the clinical heterogeneity. For example, the modulation of interferon-gamma expression and associated pathways in monocytes was associated with disease behavior as characterized by inflammatory, penetrating, and/or fibrostenotic subphenotypes. In addition, and unsurprisingly, some of the prominent hubs such as STAT1 when dysregulated result in inflammatory responses70 and in maintaining the balance between protective and pathogenic effects in the gut.71 Furthermore, other hubs such as GBP1 and GBP2 are involved in imparting protection against a broad range of pathogens including viruses.72, 73 Previous studies have also reported an increased interferon activation in mononuclear cells isolated from both the peripheral blood and lamina propria of CD patients.74, 75 Even though intestinal interferon production is under strong negative control in normal conditions and significantly upregulated in active CD,76 we report that there are differences in interferon expression in circulating monocytes as a function of disease behavior. Some of the differences might be due to the hyperactive state of the immune system in the inflammatory behavioral subtype of CD patients. Concomitant with the modulation of interferon-activity, pathways related to antiviral mechanisms were also overrepresented, suggesting that alterations in the virome77, 78 could trigger the hyperactive state of the immune system in such inflammatory phenotypes. Interferon-gamma, for example, is involved in mediating CD pathologies in a dextran sodium sulfate–induced mice colitis model as a result of the cumulative effects of particular host susceptibility genes and exposure to viral infections.79 Even though we did not evaluate the microbiome or the virome in this study, exploring the link between the virome and responses to anti-inflammatory drugs80 would help in understanding the basic mechanisms associated with drug responses in CD.
We also identified signatures of monocyte gene expression via the 4 different active modules (modules 4, 6, 8, and 9) that help explain a link with disease location. However, module 8 had an overrepresentation of pathways related to TCA cycle and electron transport chains. Previous studies have suggested that activity of the TCA cycle, among other functional categories of genes, could distinguish the ileal and colonic types of CD.6 Although this observation was made from intestinal tissue specimens, such underlying signatures in the intestine could perhaps explain the priming of monocytes with a similar metabolic pattern, given that certain fractions of monocytes are often accumulated81 and released into the bloodstream as a result of intestinal shedding82 after chronic inflammation which characterizes CD.
Yet another finding is the alignment of various post-translational and post-transcriptional regulators with the heterogeneity axis of disease behavior. Of notable mention is the identification as a top hub of OGT, which encodes the 110 kDa subunit of UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase in CD4+ cells. We know that OGT plays key roles in mediating the addition of glycosyl groups to serine/threonine residues of proteins in response to antigenic challenges, particularly in T cells.83 Not only does OGT contribute to the heterogeneity of CD4+ expression data along the axes of disease behavior but it also is expressed at a higher level in 75% of the patients with the inflammatory subtype of the disease. This could partly be explained by the fact that the inflammatory subtype of CD is exposed to a high load of antigens, which constantly challenges the immune cells, especially the CD4+ cells which then help the B cells to produce specific antibodies, elicit macrophages toward enhanced microbicidal activity, etc.84 In addition, OGT is also known to glycosylate proteins like NOD2 (whose activity has considerable effects in determining CD susceptibility) and the latter’s ability to initiate the inflammatory cascade via the NF-kB pathway.85 Furthermore, deletion of OGT predisposes the mammalian gut to inflammation.86 Thus by enhancing expression of post-translational regulators like OGT, CD4+ cells are able to elicit enhanced inflammatory responses to antigenic challenges in the gut.
To demonstrate the functional and therapeutic relevance of the identified hubs in monocytes and CD4+ cells, we checked if these hubs have previously been targeted in inflammatory diseases or if they are relevant to the disease in terms of the biological functions. Although some of the hubs (such as TYK2) we identified were previously targeted by therapeutics used in CD (Fig. 2B), their specificities with respect to the cell type and disease subtypes were not previously defined. In addition, we also point out the relevance of novel hubs (such as ADORA3, CXCR2, FLT3, PRKCE, IL1A, IL1R1, and CD40, among others) for drug repurposing based on drug target information from different nongut-related inflammatory disorders and also highlight their potential use as targets for different subgroups of CD patients. Such hubs can therefore be used to design drug repurposing strategies targeted toward specific CD subtypes.
Despite the promising CD subtype–specific signatures that we uncovered in this study, there exist many limitations. First, due to the complex nature of CD and the inflammatory cascades, there are several other immune and nonimmune cell types that propel the disease87 and the plethora of phenotypes associated with it. We have explored just 2 different cell types, namely the monocytes and CD4+ lymphocytes. Secondly, we have also not considered interactions between cell types88, 89 that dictate many of the dynamics in terms of the immune responses in CD. Thirdly, we have not profiled other -omic layers such as proteomics, metabolomics, etc. which capture molecular activities that may be closer to the phenotype. Also, our findings warrant validation in an independent cohort.
CONCLUSION
To summarize, we used a combination of different computational approaches including statistical and network-aided mechanistic data integration to interpret multiple -omic data sets (genotype and transcriptomic readouts from CD4+ T cells and CD14+ monocytes) from CD patients with different clinical profiles in terms of disease heterogeneity (Fig. 7). Although our study was constrained by the lack of longitudinal data from multiple cell types for a significantly larger number of patients to track potential progression of different subtypes of disease, we provided a first attempt into inferring the biological mechanisms that characterize CD heterogeneity in 2 specific cell types, namely CD4+ T cells and CD14+ monocytes. Although these 2 cell types are known to contribute to IBD pathogenesis,103–105 their roles in CD heterogeneity have not been explored. We identified sets of genes, pathways, and hubs that collectively distinguish CD subtypes and could be used to further our understanding of CD heterogeneity in addition to developing new therapeutic strategies.
FIGURE 7.

Graphical summary of the clinical, experimental, and bioinformatic workflow used in the study. Some of the prominent results are also depicted.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Supported by: This work was supported by the European Research Council Advanced Grant (ERC-2015-AdG, 694679, CrUCCial). Conflicts of interest: BV reports financial support for research from Pfizer; lecture fees from Abbvie, Ferring, Takeda Pharmaceuticals, Janssen and R Biopharm; consultancy fees from Janssen and Sandoz. JS reports lecture fees from Abbvie, Takeda, Janssen and Nestle Health Sciences. Sé.V reports financial support for research: MSD, AbbVie, Takeda, Pfizer, J&J; Lecture fee(s): MSD, AbbVie, Takeda, Ferring, Centocor, Hospira, Pfizer, J&J, Genentech/Roche; Consultancy: MSD, AbbVie, Takeda, Ferring, Centocor, Hospira, Pfizer, J&J, Genentech/Roche, Celgene, Mundipharma, Celltrion, SecondGenome, Prometheus, Shire, Prodigest, Gilead, Galapagos. Sé.V is senior clinical investigator of the Research Foundation Flanders (FWO). MF reports financial support for research: Amgen, Biogen, Janssen, Pfizer, Takeda, Consultancy: Abbvie, Boehringer-Ingelheim, MSD, Pfizer, Sandoz, Takeda and Thermo Fisher; Speakers fee: Abbvie, Amgen, Biogen, Boehringer-Ingelheim, Falk, Ferring, Janssen, Lamepro, MSD, Mylan, Pfizer, Sandoz, and Takeda.
REFERENCES
- 1. Gajendran M, Loganathan P, Catinella AP, et al. A comprehensive review and update on Crohn’s disease. Dis Mon. 2018;64:20–57. [DOI] [PubMed] [Google Scholar]
- 2. Ray K. IBD: Genotypes and phenotypes of IBD. Nat Rev Gastroenterol Hepatol. 2015;12:672. [DOI] [PubMed] [Google Scholar]
- 3. Bettenworth D, Lopez R, Hindryckx P, et al. Heterogeneity in endoscopic treatment of Crohn’s disease-associated strictures: an international inflammatory bowel disease specialist survey. J Gastroenterol. 2016;51:939–948. [DOI] [PubMed] [Google Scholar]
- 4. Silverberg MS, Satsangi J, Ahmad T, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19(Suppl A):5A–36A. [DOI] [PubMed] [Google Scholar]
- 5. Maaser C, Sturm A, Vavricka SR, et al. ; European Crohn’s and Colitis Organisation [ECCO] and the European Society of Gastrointestinal and Abdominal Radiology [ESGAR]. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: initial diagnosis, monitoring of known IBD, detection of complications. J Crohns Colitis. 2019;13:144–164. [DOI] [PubMed] [Google Scholar]
- 6. Weiser M, Simon JM, Kochar B, et al. Molecular classification of Crohn’s disease reveals two clinically relevant subtypes. Gut. 2018;67:36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chao K, Zhang S, Yao J, et al. Imbalances of CD4(+) T-cell subgroups in Crohn’s disease and their relationship with disease activity and prognosis. J Gastroenterol Hepatol. 2014;29:1808–1814. [DOI] [PubMed] [Google Scholar]
- 8. Cleynen I, Boucher G, Jostins L, et al. ; International Inflammatory Bowel Disease Genetics Consortium . Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chapuy L, Bsat M, Sarkizova S, et al. Two distinct colonic CD14+ subsets characterized by single-cell RNA profiling in Crohn’s disease. Mucosal Immunol. 2019;12:703–719. [DOI] [PubMed] [Google Scholar]
- 10. McKinney EF, Lee JC, Jayne DR, et al. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Funderburg NT, Stubblefield Park SR, Sung HC, et al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology. 2013;140:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smids C, Horjus Talabur Horje CS, Drylewicz J, et al. Intestinal T cell profiling in inflammatory bowel disease: linking T cell subsets to disease activity and disease course. J Crohns Colitis. 2018;12:465–475. [DOI] [PubMed] [Google Scholar]
- 13. Lee JC, Lyons PA, McKinney EF, et al. Gene expression profiling of CD8+ T cells predicts prognosis in patients with Crohn disease and ulcerative colitis. J Clin Invest. 2011;121:4170–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jostins L, Ripke S, Weersma RK, et al. ; International IBD Genetics Consortium (IIBDGC) . Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farh KK, Marson A, Zhu J, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Argelaguet R, Velten B, Arnol D, et al. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14:e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mo Q, Wang S, Seshan VE, et al. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc Natl Acad Sci U S A. 2013;110:4245–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leppäaho E, Ammad-ud-din M, Kaski S. GFA: exploratory analysis of multiple data sources with group factor analysis. J Mach Learn Res. 2017. [Google Scholar]
- 19. Bunte K, Leppäaho E, Saarinen I, et al. Sparse group factor analysis for biclustering of multiple data sources. Bioinformatics. 2016;32:2457–2463. [DOI] [PubMed] [Google Scholar]
- 20. Klami A, Virtanen S, Leppäaho E, et al. Group factor analysis. IEEE Trans Neural Netw Learn Syst. 2015;26:2136–2147. [DOI] [PubMed] [Google Scholar]
- 21. Khan SA, Virtanen S, Kallioniemi OP, et al. Identification of structural features in chemicals associated with cancer drug response: a systematic data-driven analysis. Bioinformatics. 2014;30:i497–i504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vasconcelos Y, De Vos J, Vallat L, et al. ; French Cooperative Group on CLL. Gene expression profiling of chronic lymphocytic leukemia can discriminate cases with stable disease and mutated Ig genes from those with progressive disease and unmutated Ig genes. Leukemia. 2005;19:2002–2005. [DOI] [PubMed] [Google Scholar]
- 23. de Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bayesian group factor analysis with structured sparsity.
- 25. Russo PST, Ferreira GR, Cardozo LE, et al. CEMiTool: a Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinformatics. 2018;19:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang Y, Han L, Yuan Y, et al. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat Commun. 2014;5:3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mähler N, Wang J, Terebieniec BK, et al. Gene co-expression network connectivity is an important determinant of selective constraint. PLoS Genet. 2017;13:e1006402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xulvi-Brunet R, Li H. Co-expression networks: graph properties and topological comparisons. Bioinformatics. 2010;26:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. [DOI] [PubMed] [Google Scholar]
- 30. Petereit J, Smith S, Harris FC Jr, et al. petal: Co-expression network modelling in R. BMC Syst Biol. 2016;10(Suppl 2):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ogata Y, Suzuki H, Sakurai N, et al. CoP: a database for characterizing co-expressed gene modules with biological information in plants. Bioinformatics. 2010;26:1267–1268. [DOI] [PubMed] [Google Scholar]
- 32. Desai AP, Razeghin M, Meruvia-Pastor O, et al. GeNET: a web application to explore and share Gene Co-expression Network Analysis data. Peerj. 2017;5:e3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics. 2010;11:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watson M. CoXpress: differential co-expression in gene expression data. BMC Bioinformatics. 2006;7:509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chiu DS, Talhouk A. diceR: an R package for class discovery using an ensemble driven approach. BMC Bioinformatics. 2018;19:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ha MJ, Baladandayuthapani V, Do KA. DINGO: differential network analysis in genomics. Bioinformatics. 2015;31:3413–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Keenan AB, Torre D, Lachmann A, et al. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019;47:W212–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alvarez MJ, Shen Y, Giorgi FM, et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garcia-Alonso L, Holland CH, Ibrahim MM, et al. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019;29:1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Puente-Santamaria L, Wasserman WW, Del Peso L. TFEA.ChIP: a tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq data sets. Bioinformatics. 2019;35:5339–5340. [DOI] [PubMed] [Google Scholar]
- 41. Kwon AT, Arenillas DJ, Worsley Hunt R, et al. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda). 2012;2:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roopra A. MAGIC: A tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput Biol. 2020;16:e1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang JK, Carlin DE, Yu MK, et al. Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst. 2018;6:484–495.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Croft D, O’Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barabási A-L. Scale-free networks: a decade and beyond. Science. 2009;325:412–413. [DOI] [PubMed] [Google Scholar]
- 46. Albert R. Scale-free networks in cell biology. J Cell Sci. 2005;118:4947–4957. [DOI] [PubMed] [Google Scholar]
- 47. Liberzon A, Birger C, Thorvaldsdóttir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carvalho-Silva D, Pierleoni A, Pignatelli M, et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 2019;47:D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Di Narzo AF, Peters LA, Argmann C, et al. Blood and intestine eQTLs from an anti-TNF-resistant Crohn’s disease cohort inform IBD genetic association loci. Clin Transl Gastroenterol. 2016;7:e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vancamelbeke M, Vanuytsel T, Farré R, et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:1718–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Arijs I, De Hertogh G, Lemaire K, et al. Mucosal gene expression of antimicrobial peptides in inflammatory bowel disease before and after first infliximab treatment. PLoS One. 2009;4:e7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Arijs I, De Hertogh G, Machiels K, et al. Mucosal gene expression of cell adhesion molecules, chemokines, and chemokine receptors in patients with inflammatory bowel disease before and after infliximab treatment. Am J Gastroenterol. 2011;106:748–761. [DOI] [PubMed] [Google Scholar]
- 53. Mirkov MU, Verstockt B, Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol Hepatol. 2017;2:224–234. [DOI] [PubMed] [Google Scholar]
- 54. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. [DOI] [PubMed] [Google Scholar]
- 55. Zhao Q, Kim T, Pang J, et al. A novel function of CXCL10 in mediating monocyte production of proinflammatory cytokines. J Leukoc Biol. 2017;102:1271–1280. [DOI] [PubMed] [Google Scholar]
- 56. Shimoda K, Kato K, Aoki K, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13:561–571. [DOI] [PubMed] [Google Scholar]
- 57. Ishizaki M, Akimoto T, Muromoto R, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J Immunol. 2011;187:181–189. [DOI] [PubMed] [Google Scholar]
- 58. Dowty ME, Lin TH, Jesson MI, et al. Janus kinase inhibitors for the treatment of rheumatoid arthritis demonstrate similar profiles of in vitro cytokine receptor inhibition. Pharmacol Res Perspect. 2019;7:e00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Boyle DL, Soma K, Hodge J, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signaling in rheumatoid arthritis. Ann Rheum Dis. 2015;74:1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Danese S, Grisham M, Hodge J, et al. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: a hub for multiple inflammatory cytokines. Am J Physiol Gastrointest Liver Physiol. 2016;310:G155–G162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Virtanen AT, Haikarainen T, Raivola J, et al. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rivera-Nieves J, Olson T, Bamias G, et al. L-selectin, alpha 4 beta 1, and alpha 4 beta 7 integrins participate in CD4+ T cell recruitment to chronically inflamed small intestine. J Immunol. 2005;174:2343–2352. [DOI] [PubMed] [Google Scholar]
- 63. Kobayashi M, Hoshino H, Masumoto J, et al. GlcNAc6ST-1-mediated decoration of MAdCAM-1 protein with L-selectin ligand carbohydrates directs disease activity of ulcerative colitis. Inflamm Bowel Dis. 2009;15: 697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dai C, Jiang M, Sun MJ. Innate immunity and adaptive immunity in Crohn’s disease. Ann Transl Med. 2015;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shah SC, Khalili H, Gower-Rousseau C, et al. Sex-based differences in incidence of inflammatory bowel diseases-pooled analysis of population-based studies from Western Countries. Gastroenterology. 2018;155:1079–1089.e3. [DOI] [PubMed] [Google Scholar]
- 66. Park HJ, Choi JM. Sex-specific regulation of immune responses by PPARs. Exp Mol Med. 2017;49:e364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee JC, Biasci D, Roberts R, et al. ; UK IBD Genetics Consortium . Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat Genet. 2017;49:262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schäffler H, Geiss D, Gittel N, et al. Mutations in the NOD2 gene are associated with a specific phenotype and lower anti-tumor necrosis factor trough levels in Crohn’s disease. J Dig Dis. 2018;19:678–684. [DOI] [PubMed] [Google Scholar]
- 69. Lesage S, Zouali H, Cézard JP, et al. ; EPWG-IBD Group; EPIMAD Group; GETAID Group . CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Thoeni C, Hamilton EA, Elkadri A, et al. The effects of STAT1 dysfunction on the gut. LymphoSign J. 2016;3:19–33. [Google Scholar]
- 71. Pott J, Stockinger S. Type I and III Interferon in the Gut: Tight Balance between Host Protection and Immunopathology. Front Immunol. 2017;8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nordmann A, Wixler L, Boergeling Y, et al. A new splice variant of the human guanylate-binding protein 3 mediates anti-influenza activity through inhibition of viral transcription and replication. Faseb J. 2012;26:1290–1300. [DOI] [PubMed] [Google Scholar]
- 73. Neun R, Richter MF, Staeheli P, et al. GTPase properties of the interferon-induced human guanylate-binding protein 2. FEBS Lett. 1996;390: 69–72. [DOI] [PubMed] [Google Scholar]
- 74. Fais S, Capobianchi MR, Silvestri M, et al. Interferon expression in Crohn’s disease patients: increased interferon-gamma and -alpha mRNA in the intestinal lamina propria mononuclear cells. J Interferon Res. 1994; 14:235–238. [DOI] [PubMed] [Google Scholar]
- 75. Sasaki T, Hiwatashi N, Yamazaki H, et al. The role of interferon gamma in the pathogenesis of Crohn’s disease. Gastroenterol Jpn. 1992;27:29–36. [PubMed] [Google Scholar]
- 76. Bocci V. Roles of interferon produced in physiological conditions. A speculative review. Immunology. 1988;64:1–9. [PMC free article] [PubMed] [Google Scholar]
- 77. Pérez-Brocal V, García-López R, Nos P, et al. Metagenomic analysis of Crohn’s disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm Bowel Dis. 2015;21:2515–2532. [DOI] [PubMed] [Google Scholar]
- 78. Norman JM, Handley SA, Baldridge MT, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cadwell K, Patel KK, Maloney NS, et al. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. De Vlaminck I, Khush KK, Strehl C, et al. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell. 2013;155:1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Franzè E, Caruso R, Stolfi C, et al. Lesional accumulation of CD163-expressing cells in the gut of patients with inflammatory bowel disease. PLoS One. 2013;8:e69839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Creamer B, Shorter RG, Bamforth J. The turnover and shedding of epithelial cells. I. The turnover in the gastro-intestinal tract. Gut. 1961;2:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lund PJ, Elias JE, Davis MM. Global analysis of O-GlcNAc glycoproteins in activated human T cells. J Immunol. 2016;197:3086–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hou CW, Mohanan V, Zachara NE, et al. Identification and biological consequences of the O-GlcNAc modification of the human innate immune receptor, Nod2. Glycobiology. 2016;26:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhao M, Xiong X, Ren K, et al. Deficiency in intestinal epithelial O-GlcNAcylation predisposes to gut inflammation. EMBO Mol Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cader MZ, Kaser A. Recent advances in inflammatory bowel disease: mucosal immune cells in intestinal inflammation. Gut. 2013;62:1653–1664. [DOI] [PubMed] [Google Scholar]
- 88. Courth LF, Ostaff MJ, Mailänder-Sánchez D, et al. Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci U S A. 2015;112:14000–14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Al-Ghadban S, Kaissi S, Homaidan FR, et al. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci Rep. 2016;6:29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Su G, Morris JH, Demchak B, et al. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:8.13.1–8.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yuryeva K, Saltykova I, Ogorodova L, et al. Expression of adenosine receptors in monocytes from patients with bronchial asthma. Biochem Biophys Res Commun. 2015;464:1314–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bernhagen J, Krohn R, Lue H, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. [DOI] [PubMed] [Google Scholar]
- 93. Papadopoulou C, Corrigall V, Taylor PR, et al. The role of the chemokines MCP-1, GRO-alpha, IL-8 and their receptors in the adhesion of monocytic cells to human atherosclerotic plaques. Cytokine. 2008;43:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Böiers C, Buza-Vidas N, Jensen CT, et al. Expression and role of FLT3 in regulation of the earliest stage of normal granulocyte-monocyte progenitor development. Blood. 2010;115:5061–5068. [DOI] [PubMed] [Google Scholar]
- 95. Kim SW, Choi SM, Choo YS, et al. Flt3 ligand induces monocyte proliferation and enhances the function of monocyte-derived dendritic cells in vitro. J Cell Physiol. 2015;230:1740–1749. [DOI] [PubMed] [Google Scholar]
- 96. Maffei R, Bulgarelli J, Fiorcari S, et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica. 2013;98: 1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Jenkins JK, Arend WP. Interleukin 1 receptor antagonist production in human monocytes is induced by IL-1 alpha, IL-3, IL-4 and GM-CSF. Cytokine. 1993;5:407–415. [DOI] [PubMed] [Google Scholar]
- 98. Di Paolo NC, Shafiani S, Day T, et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of Mycobacterium tuberculosis infection. Immunity. 2015;43:1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Vasilyev FF, Silkov AN, Sennikov SV. Relationship between interleukin-1 type 1 and 2 receptor gene polymorphisms and the expression level of membrane-bound receptors. Cell Mol Immunol. 2015;12:222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Suttles J, Milhorn DM, Miller RW, et al. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (il)-4 and il-10 anti-inflammatory action. J Biol Chem. 1999;274:5835–5842. [DOI] [PubMed] [Google Scholar]
- 101. Alderson MR, Armitage RJ, Tough TW, et al. CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med. 1993;178:669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. van Dam S, Võsa U, van der Graaf A, et al. Gene co-expression analysis for functional classification and disease-gene predictions. Brief Bioinform. 2018;19:575–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Imam T, Park S, Kaplan MH, et al. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol. 2018;9:1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Shale M, Schiering C, Powrie F. CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev. 2013;252:164–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schwarzmaier D, Foell D, Weinhage T, et al. Peripheral monocyte functions and activation in patients with quiescent Crohn’s disease. PLoS One. 2013;8:e62761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.