

Abstract
Motivation
Vast majority of human genetic disorders are associated with mutations that affect protein–protein interactions by altering wild-type binding affinity. Therefore, it is extremely important to assess the effect of mutations on protein–protein binding free energy to assist the development of therapeutic solutions. Currently, the most popular approaches use structural information to deliver the predictions, which precludes them to be applicable on genome-scale investigations. Indeed, with the progress of genomic sequencing, researchers are frequently dealing with assessing effect of mutations for which there is no structure available.
Results
Here, we report a Gradient Boosting Decision Tree machine learning algorithm, the SAAMBE-SEQ, which is completely sequence-based and does not require structural information at all. SAAMBE-SEQ utilizes 80 features representing evolutionary information, sequence-based features and change of physical properties upon mutation at the mutation site. The approach is shown to achieve Pearson correlation coefficient (PCC) of 0.83 in 5-fold cross validation in a benchmarking test against experimentally determined binding free energy change (ΔΔG). Further, a blind test (no-STRUC) is compiled collecting experimental ΔΔG upon mutation for protein complexes for which structure is not available and used to benchmark SAAMBE-SEQ resulting in PCC in the range of 0.37–0.46. The accuracy of SAAMBE-SEQ method is found to be either better or comparable to most advanced structure-based methods. SAAMBE-SEQ is very fast, available as webserver and stand-alone code, and indeed utilizes only sequence information, and thus it is applicable for genome-scale investigations to study the effect of mutations on protein–protein interactions.
Availability and implementation
SAAMBE-SEQ is available at http://compbio.clemson.edu/saambe_webserver/indexSEQ.php#started.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Mutations introduce diversity in genome that can be either advantageous or cause diseases. Their effect on molecular level is manifested as alterations of wild-type properties of the corresponding macromolecules such as proteins, DNAs and RNAs (Kucukkal et al., 2015; Petukh et al., 2015a,b). Of particular interest is the effect of mutations on protein–protein interactions, since protein–protein interactions are essential for a wide range of cellular processes such as signal transductions, cell metabolism, regulation of gene expression, transport and muscle contractions (Bustin, 2015; Jones and Thornton, 1996; Keskin et al., 2008). Therefore, understanding the effect of mutations on protein–protein interactions at molecular level is crucial for protein engineering (Orii and Ganapathiraju, 2012), developing novel therapeutics (Petta et al., 2016; Wells and McClendon, 2007) and revealing molecular mechanism of diseases (Kuzmanov and Emili, 2013; Nibbe et al., 2011). This prompted numerous investigations, both experimental (Fragoza et al., 2019) and computational (Das et al., 2012), to explore the impact of mutations on protein–protein interactions.
Computational methods for predicting the effect of mutations on protein–protein binding energy are alternative to experimental techniques, since they are less time consuming and do not require biochemical work to prepare the samples. Because of that, various computational methods (described below) were developed, however, most of them require structural information. This is a severe limitation for genome-scale approaches, since it is estimated that only about 6.5% of known human interactome has structural information (Mosca et al., 2013).
Among various computational methods, some are based on physical energy-based features or knowledge-based features, some use machine learning algorithms others linear combination of energy terms. For example, FoldX (Guerois et al., 2002; Schymkowitz et al., 2005), a machine learning method uses physical energies, such as van der Waals, electrostatic energy, hydrogen bond and solvation energy. In addition, this method considers conformational changes of side chains using rotamer approach. In addition to physical energy, knowledge-based energy terms were also used to determine ΔΔG. For example, SAAMBE (Petukh et al., 2015a,b, 2016) uses combination of MM/PBSA and knowledge-based energy terms. The specialty of SAAMBE is that it uses amino-acid-specific dielectric constants to mimic the conformational flexibility caused by mutation. BindProfX (Xiong et al., 2017) is another method, which combines conservation profile with the FoldX to improve the prediction of ΔΔG. In 2018, a statistical energy-based ΔΔG predictor based on a coarse-grained model, the BeAtMuSiC (Dehouck et al., 2013), was developed. All these methods based on either physical energy or knowledge-based potential or combination of both were reported to achieve Pearson correlation coefficient (PCC) ranging from 0.38 to 0.68 as benchmarked on SKEMPI v1.1 database (Moal and Fernández-Recio, 2012).
In recent years, several machine learning-based methods have been developed with structure-based features to predict ΔΔG upon mutations. The first developed machine learning-based predictor is mCSM (Pires et al., 2014), which uses atomic distance pattern surrounding the mutation site to represent the neighboring environment and achieved a high correlation of 0.80 on 2317 single mutations from SKEMPI v1.1 database. Recently published iSEE (Geng et al., 2019) method is based on 31 features involving position-specific scoring matrix, structure interface profile and energy-based features and utilizes a random forest model to predict ΔΔG caused by a given mutation. iSEE achieved a high correlation of 0.8 on single mutations in dimeric complexes from SKEMPI v1.1. MutaBind (Li et al., 2016) is another predictor, which obtained a correlation of 0.68 on the single point mutations in SKEMPI 1.1. Recently, MutaBind2 (Zhang et al., 2020) was developed and reported to achieve PCC of 0.82 against experimental ΔΔG from SKEMPI v2.0(Jankauskaite et al., 2019). It is important to mention that MutaBind2 can predict ΔΔG caused by multiple mutations as well. BindProfX (Xiong et al., 2017) combines its interface profile with the FoldX score to improve the prediction of ΔΔG using random forest model and achieved PCC of 0.74 on 1131 single mutations from SKEMPI v1.1. However, BindProfX can only predict ΔΔG for mutations located at the interface of the protein complexes. Recently, an improved version of mCSM method, called mCSM-PPI2 (Rodrigues et al., 2019), was reported. In mCSM-PPI2 method, the graph-based signature framework of mCSM is combined with additional inter-residue complex network, evolutionary information and energetic terms. Another recent innovative algorithm is TopNetTree (Wang et al., 2020), which integrates topological features and a deep learning algorithm, represented by a topology-based network tree. The method achieved a PCC of 0.82 on single mutations from SKEMPI v2.0 database. Our recently published method, SAAMBE-3D (Pahari et al., 2020) is a structure-based machine learning algorithm, which utilizes several knowledge-based features representing the physical environment surrounding mutation site. SAAMBE-3D is the fastest method available so far for predicting ΔΔG caused by single mutation and comes as stand-alone code as well. Moreover, in addition to predicting ΔΔG, the method predicts whether the mutation is disruptive or non-disruptive, which enables identification of disease-causing mutations.
The important thing to note here is that all the above-mentioned methods require a 3D structure of the protein complex as input to predict ΔΔG upon mutations. However, as mentioned above, only 6.5% of known human interactome has structural information (Mosca et al., 2013). Therefore, the applicability of these structure-based methods is limited. A partial solution that can extend their applicability is to predict the structures of protein complexes from sequence using homology modeling. However, generating high-quality 3D structures is not always possible which makes the predictions much less accurate. Therefore, it is crucial to develop a method, which can predict ΔΔG caused by mutations using only sequence information.
Currently, there is only one sequence-based method, the ProAffiMuSeq (Jemimah et al., 2019), which takes the sequence of two interacting chains as input. However, ProAffiMuSeq is intended to only predict ΔΔG caused by mutations located at the interfaces of the protein–protein complexes, and thus still requires structural information. Our attempt to use it for predicting ΔΔG for non-interfacial mutations resulted in negative PCC as benchmarked against experimental data (see Section 3). The ProAffiMuSeq is a machine learning-based method, achieves a PCC of 0.75 in benchmarking test (90% training and 10% testing sets) taken from 1173 interfacial mutations in protein–protein complexes from PROXiMATE database (Jemimah et al., 2017).
Here, we report a new development of SAAMBE, the SAAMBE-SEQ, which is a truly sequence-based machine learning algorithm to predict the binding affinity changes upon single mutation in protein–protein complexes. Unlike other existing methods, SAAMBE-SEQ does not either require a 3D complex structure as input or knowledge of interfacial residues. Therefore, this method can be applied to protein complexes without known structure. The prediction of ΔΔG using SAAMBE-SEQ is found to be either more accurate or comparable to leading structure-based methods. The method is available as a webserver as well as stand-alone code. SAAMBE-SEQ utilizes 80 features representing evolutionary information using position-specific scoring matrix, sequence-based features and change in some physical properties of mutation site. SAAMBE-SEQ is trained on 2398 single point mutations from 200 complexes taken from SKEMPI v2.0. The method uses the Gradient Boosting Decision Tree (GBDT) machine learning algorithm and achieves a PCC of 0.83. Furthermore, SAAMBE-SEQ is also trained to discriminate disruptive from non-disruptive mutations and achieves accuracy of 0.81, precision of 0.65, sensitivity and specificity of 0.81 as benchmarked against Cornell University dataset (Fragoza et al., 2019; Pahari et al., 2020).
2 Materials and methods
2.1 Dataset creation
The amino acid sequences and experimentally measured binding free energies in this work were taken from the recently updated version of SKEMPI, SKEMPI v2.0 database (Jankauskaite et al., 2019), which compiles experimentally measured binding affinity values for wild-type as well as mutant protein–protein complexes. SKEMPI 2.0 contains binding affinity data for 7085 mutations from 389 protein complexes. Only cases of single point mutations from dimeric complexes were considered, resulting in 2446 mutations from 207 different protein–protein complexes. Then, the binding free energy (ΔG) was calculated from the binding affinity:
| (1) |
where R is the ideal gas constant, is temperature in kelvin and is binding affinity of the given protein complex. The ΔG is calculated for both wild-type and mutant protein complexes. Then, the change in binding free energy upon mutation (ΔΔG) is calculated by subtracting ΔG for wild-type from that of mutant
| (2) |
For some mutations, multiple measurements were carried out, and all the measured binding affinity values were reported in SKEMPI v2.0 database. If the standard deviation of ΔΔG for a particular mutation is less than 1.0 kcal mol−1, we considered those cases and used average value for developing and benchmarking our model. We removed all mutations with standard deviation greater than 1 kcal mol−1. Further, we removed the complexes for which any chain contains less than 20 amino acid residues. Therefore, the final compiled dataset consists of 2398 single point mutations from 200 different dimeric complexes.
2.2 Model development
Our methodology of predicting binding free energy changes due to mutation in protein complexes incorporates only sequence-based features. Our machine learning model is based on GBDT algorithm. Overall, we used 80 features which include average Position-Specific Scoring Matrix (PSSM) for mutant and interaction chain, conservation score at mutation site, change in molar volume, hydrophobicity, flexibility, hydrogen bonds, polarity, mutation type, chemical nature and size of the mutated amino acid. Label encoding method is used for incorporating mutation type, change in polarity, chemical properties, hydrogen bond donor/acceptor and size features. We describe the features in detail in the following section. We also analyzed the importance of each feature using XGBoost machine learning software. To avoid overfitting and make a robust model, we carried out 100 times 5-fold cross validations. We created two models: one using 80% and another using 90% of the compiled dataset to train the model and remaining 20 or 10% is used for testing the performance of the model. For a more accurate estimation, we repeated the whole process 100 times and then averaged the PCC and Mean Square Error (MSE). Figure 1 represents a schematicillustration of the SAAMBE-SEQ method.
Fig. 1.
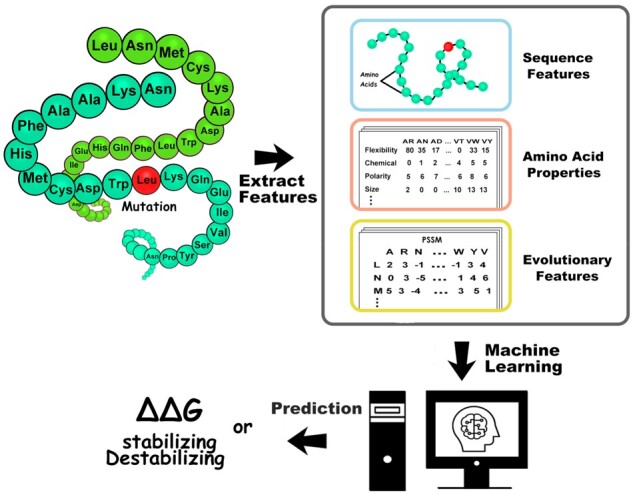
Schematic illustration of SAAMBE-SEQ method
2.3 Features
2.3.1. Features based on the position-specific scoring matrix (PSSM)
The corresponding protein sequence is utilized as input to search and align homologous sequences from Uniref50 (Suzek et al., 2014) database (https://www.uniprot.org/downloads) using the PSI-BLAST program (Camacho et al., 2009) with 3 iterations and a cutoff E-value of 0.001. Then the PSSM is constructed through a multiple sequence alignment of the highest scoring hits. As a result, we obtain an L × 20 PSSM for each protein sequence, where L is the length of each protein sequence. Each row of the PSSM matrix represents the log likelihood score for amino acid substitutions at the corresponding positions in the input sequence:
| (3) |
where Pi, j represents the score of the amino acid residue in the ith position of the protein sequence being changed to amino acid type j during the evolution process.
2.3.2. Evolutionary features of mutated and interaction chains
Protein sequences of different sizes have different lengths of PSSM. To make the PSSM descriptor become a size-uniform matrix, one approach is to represent a protein sample P by
| (4) |
where
| (5) |
and is the composition of the amino acid type j in the PSSM and represents the average score of the amino acid residues in the protein P being mutated to amino acid type j during the evolution process. All values in PSSM of each protein sequence are normalized to be between 0 and 1 by sigmoid function:
| (6) |
where x is the original value of PSSM.
Using this method, we obtained 20 uniform average conservation scores for mutated and interaction chains, respectively.
2.3.3. Conservation score at mutation site
We select the rows belonging to the given mutation site from PSSM to obtain 20 conservation scores features for the mutation site.
2.3.4. Sequence neighbors feature
We labeled ten residues including five on the left and five on the right side from the mutation site. There can be 20 possibility of each label representing 20 different amino acids. These are the ten residues which can have a significant influence on mutation site.
2.3.5. Features related to mutation site
We used nine features related to mutation site: net volume, net hydrophobicity, mutation type, net flexibility, chemical property, size, polarity, hydrogen bond and label_hydrophobicity. For detailed description of each feature, refer to our previous paper (Pahari et al., 2020) and Supplementary Material.
2.4 Feature importance analysis
We analyzed the importance of each selected feature for the prediction performance of SAAMBE-SEQ method. To evaluate the feature importance, we used XGBoost algorithm from python package. Figure 2 represents the importance level of each feature. Figure 2 reveals that average PSSM of mutant chain (MC avePSSM) and conservation score at mutation site (CS Mutation Site) are the two most important features in our model. The third highest contributing feature is average PSSM for interaction chain (IC avePSSM). These three features capture the evolutionary conservation of a given amino acid at the mutation site as well as of surrounding of mutation site and its change upon mutation. PSSM has already been established for providing crucial information in hotspot (Moreira et al., 2017) and binding site prediction (Walia et al., 2014). The next important feature is sequence neighbor, where we considered 10 amino acids near mutation site according to primary sequence. Sequence neighbor feature captures the influence of neighboring amino acid residues on the mutation site. The next three important features are mutation type, change in molar volume and hydrophobicity of amino acid residues upon the mutation. We applied feature selection protocol on the training set with 5-fold cross validation when tested on 20% of dataset. Supplementary Table S7 displays the performances of the models using additive feature groups in each iteration. The final model achieves a PCC of 0.83, higher than the 0.82 using all features. For comparison, we also trained our model by removing each feature from our final model and tested the robustness of SAAMBE-SEQ.
Fig. 2.
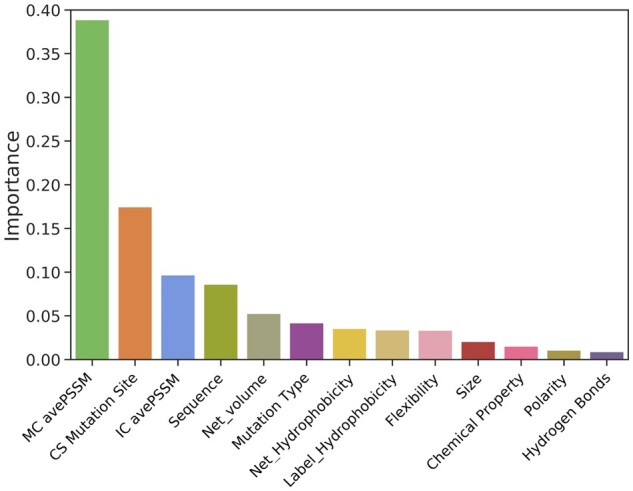
Importance level of each feature selected for SAAMBE-SEQ
2.5 no-STRUC dataset
From literature, we collected the experimental ΔΔG values upon mutation in protein–protein complexes, for which structure is not available. We utilized UniProt database to obtain the sequence from protein name and used these. We provided the Uniprot ID and the corresponding reference in Supplementary Tables S1 and S2 for homodimer and heterodimer complexes, respectively.
3 Results
We trained SAAMBE-SEQ on a large and diverse dataset containing experimental ΔΔG for 2398 single point mutations from 200 protein–protein dimeric complexes. For predicting ΔΔG upon a given mutation, we developed a regression model using 80 knowledge-based features, representing evolutionary information and physical environment surrounding the mutation site. In order to build a reliable and robust model, we performed 100 times 5-fold cross validation. Selection of the training and test sets were repeated 100 times randomly, and average PCC and MSE are considered. We trained our model against 80% as well as 90% of the 2398 mutations present in our compiled dataset and tested against the remaining 20 or 10% data. In 5-fold cross validation, our model shows a correlation of 0.83 and MSE of 1.44 kcal/mol when tested on 20% of the database (Fig. 3a). In contrast, we obtained a PCC of 0.91 and MSE of 0.90 kcal/mol when tested on 10% of the database (Fig. 3c). In Figure 3, we also plotted the distribution of both experimental as well as predicted ΔΔGs for the corresponding test sets. In both cases, it can be seen that the distribution of predicted ΔΔGs using SAAMBE-SEQ is remarkably similar to corresponding experimental ΔΔGs. To avoid any bias and overfitting, we chose the model, which is trained on 80% of the dataset for the rest of the article.
Fig. 3.
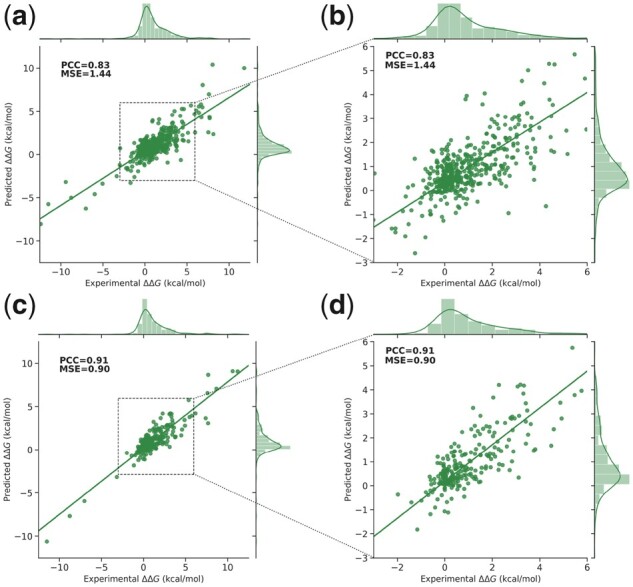
SAAMBE-SEQ predicted ΔΔG against experimental ΔΔG. (a, b) In case of 20% of mutations as test set and (c, d) in case of 10% of mutations as a test set. Panels on the left show the results over the entire data range, while panels of the right zoom at the range of 95% of the entries that have ΔΔG between −3.0 and 6.0 kcal/mol
Furthermore, a detailed comparison between predicted and experimental ΔΔGs upon mutations associated with different types of amino acids is plotted in Figure 4. All mutations from the 20% test set were evaluated. In Figure 4, x-axis and y-axis represent the amino acid residue type for wild-type and mutant, respectively. The value (in kcal/mol) of variance of ΔΔG upon each type of mutation is shown in color code—the darker the color the larger is the variance. We categorized the amino acid residue types in three categories depending on their physico-chemical characteristics: (i) size of the residue (small, medium and large); (ii) polarity (polar and non-polar) and (iii) hydrophobicity (hydrophobic, neutral and hydrophilic).
Fig. 4.

Comparison of the experimental and SAAMBE-SEQ predicted variance of ΔΔG due to mutations associated with different amino acid types on test set
One can see in Figure 4 that both experimental data as well as predicted data using SAAMBE-SEQ indicate that variance of binding energy changes associated with mutations from small residue type in wild-type to small residue type in mutant is usually low, while mutations from small to large residue type result in large change in binding energy. Another interesting fact is that if the amino acid residue type is alanine in the wild-type, then irrespective of any residue type in the mutant, the binding energy change is always small. One can also see that in both experimental as well as SAAMBE-SEQ predicted data, mutations involving hydrophobic to hydrophobic residue are usually associated with small binding energy change. Overall, comparing the patterns on the left and right panels in Figure 4, one can easily notice that they are very similar indicating that SAAMBE-SEQ predicted variances of ΔΔG is very similar to those of experimental data.
3.1 Performance comparison on blind datasets
For validation, we used three recently published datasets (for more details, see Geng et al., 2019): MDM2-p53, NM and s487 and one compiled by us, termed no-STRUC, which contains mutations from protein complexes whose 3D structures are not available.
3.1.1. Performance of SAAMBE-SEQ on MDM2-p53 dataset
MDM2-p53 dataset contains 33 mutations among which 7 were reported as mutation for which ΔΔG exceed experimental detection limit. Therefore, we removed these 7 entries from our validation test set, resulting in 26 mutations from a single protein complex (PDB ID is 1YCR, however, in our benchmarking, we used only sequence information). These mutations were not used in our training and test dataset. We compared the correlation between experimental and predicted ΔΔG on these 26 mutations. SAAMBE-SEQ achieved a PCC of 0.35 and MSE of 0.45 kcal/mol. We compared the performance of SAAMBE-SEQ with the only existing sequence-based method, ProAffiMuSeq, which obtained a PCC of 0.16 and MSE of 0.99 kcal/mol. We also compared the prediction of SAAMBE-SEQ with other existing high-performing structure-based methods such as iSee, mCSM, BindProfX, FoldX, mCSM-PPI2, MutaBind2 and SAAMBE-3D. Figure 5 shows the performance of SAAMBE-SEQ and other methods on MDM2-p53 validation dataset. We can see in Figure 5a that SAAMBE-SEQ outperforms iSee, FoldX, mCSM and MutaBind2 and achieved similar performances (PCC = 0.35) as of BindProfX (PCC = 0.36) and mCSM-PPI2 (PCC = 0.35). Figure 5a indicates that SAAMBE-3D outperforms all the existing methods and achieved a PCC of 0.41. However, we need to keep in mind that SAAMBE-SEQ is a sequence-based method and the comparable performances of the method with the already established high-performing structure-based methods make SAAMBE-SEQ an outstanding ΔΔG predictor. Moreover, Figure 5b reflects that SAAMBE-SEQ has the second lowest MSE of 0.45 kcal/mol after mCSM-PPI2. Furthermore, we carried out Fisher-T statistical significance test of correlation coefficient (Supplementary Table S3) for each method and also evaluated whether the difference in other methods compared to SAAMBE-SEQ is statistically significant or not (Supplementary Table S4) using Fisher-Z test.
Fig. 5.
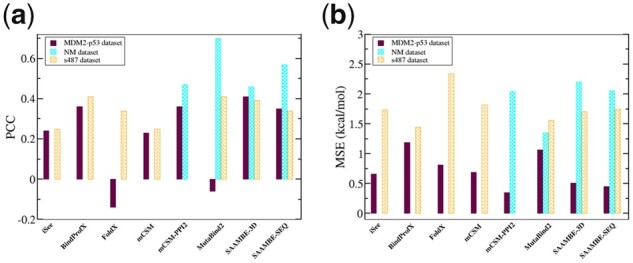
Performance comparison of SAAMBE-SEQ with other existing structure-based methods on three validation test set (MDM2-p53, NM and s487) in terms of (a) PCC and (b) MSE
3.1.2. NM dataset
The second validation dataset was taken from the NM dataset studied by Benedix et al. (2009). We only selected single mutations that were not present in our training dataset, and we removed cases in which more than two chains were present in the PDB structure. In this way, we filtered out 27 single mutations from a single protein complex (PDB ID: 1IAR, however, in the benchmark, we used only sequence information). Unfortunately, we could not compare the prediction performance of SAAMBE-SEQ with ProAffiMuSeq as these mutations from NM dataset are present in their training dataset. However, we compared the performance of SAAMBE-SEQ with existing structure-based methods, mCSM-PPI2 (Rodrigues et al., 2019), SAAMBE-3D and MutaBind2 (Zhang et al., 2020) on these selected 27 mutations. Figure 5a presents the correlation between experimental and predicted ΔΔG for the 27 mutations for all the above-mentioned methods. We could not calculate ΔΔG using iSee, mCSM and FoldX because the corresponding webservers were unavailable. Also, we were unable to compare the prediction with BindProfX method as this method only can predict ΔΔG for interfacial mutations. Figure 5a indicates that SAAMBE-SEQ outperforms SAAMBE-3D and mCSM-PPI2 in predicting ΔΔG upon mutations from NM dataset. SAAMBE-SEQ achieved PCC of 0.57 and MSE of 2.06 kcal/mol. It should be mentioned that MutaBind2 is the highest performer with PCC of 0.70 and MSE of 1.35 kcal/mol. The results of statistical significance are shown in Supplementary Tables S3 and S4.
3.1.3. S487 dataset
The third validation dataset is s487, compiled by Geng et al. (2019) The dataset contains 487 single mutations from 56 complexes and all mutations are located at protein–protein interfaces. Figure 5 represents a prediction comparison in the form of PCC and MSE obtained using different structure-based ΔΔG predictors along with SAAMBE-SEQ. The PCC and MSE values, achieved by iSee, BindProfX, FoldX, mCSM, MutaBind2 and SAAMBE-3D are taken from our previous paper (Pahari et al., 2020). We could not compare the prediction of mCSM-PPI2 as some or all of these 487 mutations are present on their training dataset (SKEMPI v2.0). As shown in Figure 5a, BindProfX and MutaBind2 both achieve the highest PCC of 0.41 followed by SAAMBE-3D (PCC = 0.39) and SAAMBE-SEQ (PCC = 0.34).
Unfortunately, we could not compare the ΔΔG prediction on the s487 validation dataset using SAAMBE-SEQ with the only existing sequence-based method, the ProAffiMeSeq, as some of these mutations are already included in their training dataset. Therefore, we considered their compiled validation dataset, s473, which is a combination of above-mentioned three datasets. They removed all the mutations which are not present at the protein–protein interfaces as ProAffiMeSeq is trained to predict ΔΔG only for interfacial mutations. Thus, on the s473 dataset, SAAMBE-SEQ achieved a PCC of 0.35 and MSE of 1.73 kcal/mol, whereas ProAffiMuSeq obtained a PCC of 0.20. Furthermore, the statistical significance was evaluated and the results are shown in Supplementary Tables S3 and S4.
3.1.4. no-STRUC dataset
The last validation test was done on no-STRUC dataset compiled by us (see Section 2 and Supplementary Tables S1 and S2). This dataset comprised experimentally measured changes of the binding free energy of protein–protein complexes for which there is no available experimentally determined 3D structure. Because of that, all the above-mentioned structure-based methods cannot be tested. The only methods that can handle such dataset are SAAMBE-SEQ and ProAffiMuSeq.
We divided the no-STRUC dataset into two categories: homodimer and heterodimer. ProAffiMuSeq was trained on some of the entries in homodimer dataset since some of the no-STRUC cases are taken from PROXiMATE database (Jemimah et al., 2017). Nevertheless, the benchmarking was carried out and the results are shown in Table 1. Among the 30 mutations in homodimer dataset, ProAffiMuSeq could not predict for 5 non-interfacial mutations. Therefore, we discarded those five mutations while comparing the performance of SAAMBE-SEQ with ProAffiMuSeq on homodimer dataset and reported in Table 1. SAAMBE-SEQ achieved a correlation of 0.35 and MSE of 1.42 kcal/mol when considered all the 30 mutations from homodimer dataset.
Table 1.
Comparison of prediction performance of SAAMBE-SEQ with ProAffiMuSeq on both homodimer and heterodimer protein complexes from no-STRUC dataset
| Dataset | SAAMBE-SEQ, PCC | SAAMBE-SEQ, MSE (kcal/mol) | ProAffiMuSeq, PCC | ProAffiMuSeq, MSE (kcal/mol) |
|---|---|---|---|---|
| Homodimers (Supplementary Table S1) | 0.37 | 1.34 | −0.10 | 3.74 |
| Heterodimers (Supplementary Table S2) | 0.47 | 0.73 | 0.19 | 2.91 |
One can see from Table 1 that SAAMBE-SEQ drastically outperforms ProAffiMuSeq, despite that some of the cases in the homodimer dataset were used for training of ProAffiMuSeq model. In Supplementary Table S5, we evaluated the statistical significance of the correlation obtained using SAAMBE-SEQ and ProAffiMuSeq for both homodimers and heterodimers. The P-value indicates that for homodimers, the correlation obtained using none of the two methods is statistically significant. However, the P-value for SAAMBE-SEQ is closer to be statistically significant. For heterodimers, correlation obtained using SAAMBE-SEQ is indeed statistically significant while this is not the case for ProAffiMuSeq. Further, we evaluated whether there is a significant statistical difference in the PCC of SAAMBE-SEQ and ProAffiMuSeq using Fisher-Z test. We obtained P-value of 3.63E-2 for homodimers and 1.20E-2 for heterodimers (Supplementary Table S6).
3.2 Performance of SAAME-SEQ on identifying disruptive and non-disruptive mutations both for homodimer as well as heterodimer
We explored the success of the prediction of SAAMBE-SEQ in classifying disruptive and non-disruptive mutations using only sequence information in case of both homodimer as well as heterodimer complexes. As mentioned in our previous paper (Pahari et al., 2020), Cornell University dataset contains 2500 single mutations from 300 homodimer protein complexes and 245 single mutations from 50 heterodimeric complexes. Yeast two-hybrid (Y2H) experiments were conducted at Cornell University (Fragoza et al., 2019) and the mutations were scored either disruptive or non-disruptive. The dataset was purged to remove identical sequences and cases where any of the two chains has less than 20 amino acid residues. We combined both homodimer and heterodimer complexes together and ended up with 342 mutations from 90 protein complexes. These 342 mutation entries were split into 80% training and 20% test sets. We used the same features for this classification as described in Section 2 for our SAAMBE-SEQ model. We carried out ROC analysis and found that our method is 84% successful in classifying disruptive and non-disruptive mutations for the 342 mutations for both homodimer and heterodimer complexes. We plotted ROC in Figure 6 and further prediction performance is measured by area under the curve, accuracy, precision and sensitivity. SAAMBE-SEQ achieved an accuracy of 0.81, precision of 0.65, sensitivity and specificity of 0.81 in classifying disruptive and non-disruptive mutation.
Fig. 6.

Prediction performance of SAAMBE-SEQ in identifying disruptive and non-disruptive mutations
3.3 Webserver
We implemented SAAMBE-SEQ as a user-friendly webserver, freely available at http://compbio.clemson.edu/saambe_webserver/indexSEQ.php#started. The server front end is built using JavaScript and backend using PHP. It is hosted on a Linux server running in Apache. SAAMBE-SEQ can be used in two different ways: (i) predict the effect of mutation specified by the user in the given boxes. User needs to provide FASTA sequence of the protein–protein complex, which can be provided by uploading the sequence in the FASTA format or by inputting the sequence in appropriate box. User must provide two sequences corresponding to two protein chains. User need to make sure that they are uploading or putting the sequence in the appropriate place corresponding to mutated chain and interaction chain. Then, user require to provide mutation details in three different boxes: in position box, corresponding residue number according to FASTA sequence file should be provided. It is important to remember that the first residue number starts with 1 not 0. In the ‘Original Amino Acid’ box, user must specify one-letter code for the wild-type residue as a string and similarly for ‘Mutated Amino Acid’, mutant residue in one-letter code must be mentioned. In this way, user can submit a single job. (ii) If user wants to submit multiple jobs at the same time, in addition to uploading or inputting sequences of mutated and interaction chain in FASTA format, user need to upload a file called ‘List_Mutation.txt’. The file must contain a list of mutations information in a text file for batch processing. A sample ‘List_Mutation.txt’ file is provided in the submission page to assist the user for submission of jobs. (iii) User can also directly download the SAAMBE-SEQ code by clicking the download option available via the top navigation bar. A readme file will also be downloaded which will guide the user how to use the code.
4 Conclusion
Machine learning methods are the alternative to the first principle-based approaches such as quantum mechanics (QM) modeling, molecular dynamics (MD) and Monte Carlo (MC) simulations (Klepeis et al., 2009; Paquet and Viktor, 2015), molecular mechanics PB/GB surface area (MM/PB/GBSA), multiscale and mesoscale methods. In terms of modeling, the effects of amino acid substitutions on protein stability, binding and dynamics, one should mention methods as free energy perturbation (FEP), thermodynamics integration (TI) and molecular mechanics Poisson-Boltzmann/Generalized Born surface area (MM/PB/GBSA) (Getov et al., 2016; Li et al., 2014; Petukh et al., 2015a,b). However, machine learning methods are more accurate in their predictions and require less computational time, making them primary choice for large-scale investigations. Indeed, the above-mentioned first principle-based methods frequently require days of computation for a single case and since they require 3D structure, any small structural imperfection could result in very wrong predictions.
It should be mentioned that one of the most indicative measure of methods performance is the MSE. The best SAAMBE-SEQ MSE is 0.90 kcal/mol when tested on 10% of the training set. Other methods mentioned in the article reported MSE ranging from 0.94 kcal/mol up to 2.89 kcal/mol. Thus, one should be careful in interpreting prediction results, since they come with an inherited error. In the simplest way, the predictions should be considered on the background of reported MSE. However, different MSEs were reported depending on the datasets used in the benchmarking. Therefore, the safest protocol should apply the largest reported MSE to investigations on new set of cases (for which there is no experimental data). Alternatively, one may want to utilize as many as possible predictors and seek a consensus.
Here, we reported a method, the SAAMBE-SEQ method, which predicts the change of the binding free energy caused by single mutations utilizing sequence information only. Combined its computational efficiency, accuracy and availability as a stand-alone code, the SAAMBE-SEQ is the only available method to be applied on genome-scale investigations. Indeed, genomic sequencing produces much more data than the efforts of structure determination, and this trend is not going to change. Therefore, there is a desperate need for machine learning methods that can make predictions using only genomic sequencing data, a need that SAAMBE-SEQ addresses for protein–protein interactions. Furthermore, it is demonstrated that SAAMBE-SEQ is capable of distinguishing disruptive from non-disruptive mutations. Since disruptive mutations are usually disease-causing, SAAMBE-SEQ can be used for early diagnosis by detecting the disruptive mutations.
Funding
This work was supported by a grant from National Institutes of Health [R01GM125639]. E.A. was supported by grants from National Institutes of Health [R01GM093937, P20GM121342].
Conflict of Interest: none declared.
Supplementary Material
Contributor Information
Gen Li, Department of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA.
Swagata Pahari, Department of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA.
Adithya Krishna Murthy, Department of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA.
Siqi Liang, Department of Computational Biology, Cornell University, Ithaca, NY 14850, USA.
Robert Fragoza, Department of Computational Biology, Cornell University, Ithaca, NY 14850, USA.
Haiyuan Yu, Department of Computational Biology, Cornell University, Ithaca, NY 14850, USA.
Emil Alexov, Department of Physics and Astronomy, Clemson University, Clemson, SC 29634, USA.
References
- Benedix A. et al. (2009) Predicting free energy changes using structural ensembles. Nat. Methods, 6, 3–4. [DOI] [PubMed] [Google Scholar]
- Bustin S. (2015) Molecular biology of the cell, sixth edition; ISBN: 9780815344643; and molecular biology of the cell, sixth edition, the problems book; ISBN 9780815344537. Int. J. Mol. Sci., 16, 28123–28125. [Google Scholar]
- Camacho C. et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J. et al. (2012) Genome-scale analysis of interaction dynamics reveals organization of biological networks. Bioinformatics, 28, 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck Y. et al. (2013) BeAtMuSiC: prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res., 41, W333–W339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoza R. et al. (2019) Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun., 10, 4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng C. et al. (2019) iSEE: interface structure, evolution, and energy-based machine learning predictor of binding affinity changes upon mutations. Proteins Struct. Funct. Bioinf., 87, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getov I. et al. (2016) SAAFEC: predicting the effect of single point mutations on protein folding free energy using a knowledge-modified MM/PBSA approach. Int. J. Mol. Sci., 17, 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerois R. et al. (2002) Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol., 320, 369–387. [DOI] [PubMed] [Google Scholar]
- Jankauskaite J. et al. (2019) SKEMPI 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics (Oxford, England), 35, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemimah S. et al. (2019) ProAffiMuSeq: sequence-based method to predict the binding free energy change of protein–protein complexes upon mutation using functional classification. Bioinformatics, 36, 1725–1730. [DOI] [PubMed] [Google Scholar]
- Jemimah S. et al. (2017) PROXiMATE: a database of mutant protein–protein complex thermodynamics and kinetics. Bioinformatics, 33, 2787–2788. [DOI] [PubMed] [Google Scholar]
- Jones S., Thornton J.M. (1996) Principles of protein–protein interactions. Proc. Natl. Acad. Sci. USA, 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keskin O. et al. (2008) Principles of protein−protein interactions: what are the preferred ways for proteins to interact? Chem. Rev., 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- Klepeis J.L. et al. (2009) Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol., 19, 120–127. [DOI] [PubMed] [Google Scholar]
- Kucukkal T.G. et al. (2015) Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol., 32, 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmanov U., Emili A. (2013) Protein–protein interaction networks: probing disease mechanisms using model systems. Genome Med., 5, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. (2014) Predicting the impact of missense mutations on protein–protein binding affinity. J. Chem. Theory Comput., 10, 1770–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. (2016) MutaBind estimates and interprets the effects of sequence variants on protein–protein interactions. Nucleic Acids Res., 44, W494–W501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moal I.H., Fernández-Recio J. (2012) SKEMPI: a Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics, 28, 2600–2607. [DOI] [PubMed] [Google Scholar]
- Moreira I.S. et al. (2017) SpotOn: high accuracy identification of protein–protein interface hot-spots. Sci. Rep., 7, 8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca R. et al. (2013) Interactome3D: adding structural details to protein networks. Nat. Methods, 10, 47–53. [DOI] [PubMed] [Google Scholar]
- Nibbe R.K. et al. (2011) Protein–protein interaction networks and subnetworks in the biology of disease. WIREs Syst. Biol. Med., 3, 357–367. [DOI] [PubMed] [Google Scholar]
- Orii N., Ganapathiraju M.K. (2012) Wiki-Pi: a web-server of annotated human protein–protein interactions to aid in discovery of protein function. PLoS One, 7, e49029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahari S. et al. (2020) SAAMBE-3D: predicting effect of mutations on protein–protein interactions. Int. J. Mol. Sci., 21, 2563–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet E., Viktor H.L. (2015) Molecular dynamics, Monte Carlo simulations, and Langevin dynamics: a computational review. Biomed. Res. Int., 2015, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I. et al. (2016) Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Therapy, 24, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukh M. et al. (2016) SAAMBE: webserver to predict the charge of binding free energy caused by amino acids mutations. Int J. Mol. Sci., 17, 547–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukh M. et al. (2015. a) On human disease-causing amino acid variants: statistical study of sequence and structural patterns. Hum. Mutat., 36, 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukh M. et al. (2015. b) Predicting binding free energy change caused by point mutations with knowledge-modified MM/PBSA method. PLoS Comput. Biol., 11, e1004276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D.E.V. et al. (2014) mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics, 30, 335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues C.H.M. et al. (2019) mCSM-PPI2: predicting the effects of mutations on protein–protein interactions. Nucleic Acids Res., 47, W338–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J. et al. (2005) The FoldX web server: an online force field. Nucleic Acids Res., 33, W382–W388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzek B.E. et al. ; the UniProt Consortium. (2015) UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics, 31, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia R.R. et al. (2014) RNABindRPlus: a predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLoS One, 9, e97725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. et al. (2020) A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat. Mach. Intell., 2, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells J.A., McClendon C.L. (2007) Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Xiong P. et al. (2017) BindProfX: assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts. J. Mol. Biol., 429, 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N. et al. (2020) MutaBind2: predicting the impacts of single and multiple mutations on protein–protein interactions. iScience, 23, 100939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.