

Abstract
Motivation
The 3D structure of chromatin in the nucleus is important for gene expression and regulation. Chromosome conformation capture techniques, such as Hi-C, generate large amounts of data showing interaction points on the genome but these are hard to interpret using standard tools.
Results
We have developed CSynth, an interactive 3D genome browser and real-time chromatin restraint-based modeller to visualize models of any chromosome conformation capture (3C) data. Unlike other modelling systems, CSynth allows dynamic interaction with the modelling parameters to allow experimentation and effects on the model. It also allows comparison of models generated from data in different tissues/cell states and the results of third-party 3D modelling outputs. In addition, we include an option to view and manipulate these complicated structures using Virtual Reality (VR) so scientists can immerse themselves in the models for further understanding. This VR component has also proven to be a valuable teaching and a public engagement tool.
Availabilityand implementation
CSynth is web based and available to use at csynth.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
It is now well established that the three-dimensional structure of the genome is important for cellular function (Lieberman-Aiden et al., 2009) and with the increasing amount of high resolution and throughput chromosome conformation capture (3C) data becoming available, such as Hi-C (Lieberman-Aiden et al., 2009), Promoter Capture Hi-C (Schoenfelder et al., 2018), Capture-C (Hughes et al., 2014) and Tri-C (Davies et al., 2017), there is a need to understand chromatin structure beyond visualizing data on a 2D genome browser and using heatmaps. The advent of sophisticated microscope imaging of chromatin to observe these structures using super resolution microscopy (Prakash, 2017) and electron microscopy (Ou et al., 2017) offers the ultimate means of visualising and understanding 3D genome architecture but these methods are laborious and expensive. Computational modelling offers a way to gain a better understanding of the complexity of chromatin in the nucleus and how the differences in structure cause enhancer/promoter/gene interactions in different cell types and disease. There are a number of methods for modelling chromatin 3D structures from 3C data but still a lack of easy to use tools (Oluwadare et al., 2019) so C data may be better understood by bench scientists.
Here, we present CSynth, an easy to use web-based portal that allows uploading of multiple 3C datasets, PDB models, annotations and quantitative data to generate 3D models of chromatin structure. The models and their parameters are interactive and may be manipulated in real time and compared in a high-quality fully rendered 3D genome browser that can be shared online and used in publications.
2 Materials and methods
2.1 Using CSynth
CSynth was developed to lower the barriers to the interrogation of complex multi-genomics data in the 3D rather than 2D genome. While the generation of genome-scale 3C data, such as Hi-C, is becoming commonplace, the computational barriers to generate 3D models from such data remain high. Even more limiting are the options to interact with such models in a dynamic nature and in concert with other classes of genomics data, such as ChIP-seq, ATACseq and RNA-seq. CSynth provides a flexible platform for the generation of restraint-based models as well as a fully featured environment to interact with these or externally generated models in a publication quality, fully rendered, interactive graphical 3D genome browser (Fig. 1).
Fig. 1.
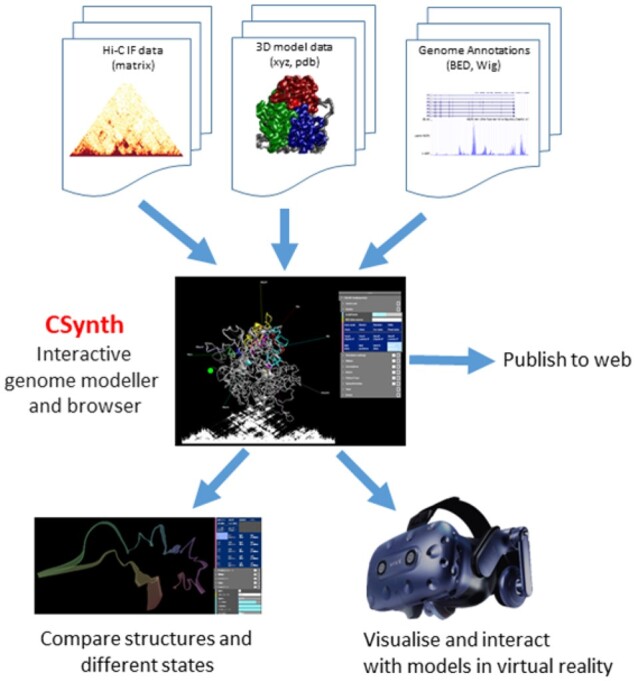
Overview of CSynth features. The CSynth website can upload interaction frequency data from the results of chromatin capture experiments, pre-existing 3D models generated by other methods and genome annotations files. Once loaded a 3D model is built based on the input IF data. Multiple models from different cell types, tissues or modelling techniques may be loaded and compared visually and statistically. CSynth also has a fully featured VR mode
Using CSynth’s web portal, users may register and upload their interaction frequency (IF) C data (e.g. Hi-C, Capture C and ChiaPet) and genome annotations via file upload or simply drag and drop these into the CSynth window. Once uploaded, the model is generated on the fly. Its orientation may be controlled by a handheld device [such as a mouse or Virtual Reality (VR) controller] or via a touch screen (depending on what is available). CSynth simultaneously shows the 3D model and 2D heatmap view underneath, allowing visualization and understanding of Hi-C interaction frequency data (IF) and their relationship between 2D and 3D space, so interesting features on the heatmap may be more easily understood in the 3D model. The generated model is easy to share via the internet (with a simple URL) with collaborators. Examples of such publicly available models can be seen on the CSynth website in the ‘Examples’ section (csynth.org/examples).
A key feature of CSynth is the ability to upload files, allowing the user to generate multiple different models, for example, looking at chromatin loop topology in different cell types or comparing multiple models made using different parameters. CSynth’s physics engine smoothly interpolates between the models so the viewer can more easily identify differences between structures. The various models and parameter settings are stored in the portal, allowing experiments to be tracked.
Also, we implement a VR mode which offers an alternative way of viewing and interacting with these complex datasets, allowing new perspectives on the data that would not be afforded via a 2D screen.
This feature has also been extensively used for teaching and public engagement. The VR mode is implemented using WebXR and available in many browsers. For example, complex chromatin loops can be observed at different points of view while actually ‘in’ the structure. The experience is tailored for use with the HTC Vive headset but other hardware could be tailored for on request.
3 Results
3.1 Comparison to other 3D visualization tools
There are currently several 3D genome browser implementations suitable for looking at 3D chromatin structure. See Supplementary Table S1 for an overview of the 3D genome viewing tools available. A key problem CSynth is addressing is to make a high-quality 3D modelling accessible that is easy and fast to run so that any person generating 3C data will use it routinely to gain further insight from their experiments. Another key factor is the results should be high quality so they can be used in publications.
Genome3d (Asbury et al., 2010) is a downloadable C++ application, which requires a computer running the Windows OS and the installation of software which makes it more limited for general use. GMOL (Nowotny et al., 2016) does not handle Hi-C data, but more recently the author has released GenomeFlow (Trieu et al., 2019) which offers a full Hi-C analysis pipeline. However, using Java requires the user to install the relevant Java version as opposed to using the desktop browser, again causing a barrier to entry to anyone who wants to rapidly and easily visualize their analysis (data). Tadkit (Serra et al., 2017) is web based and shows a 3D chromatin view in the context of a 2D browser based on IGV (Nicol et al., 2009) but there is no possibility provided to show different states (e.g. in different tissues).
3.2 Examples of CSynth modelling
In Figure 2, we show data generated from Capture-C data (Oudelaar et al., 2018) at the alpha globin region in mouse captured in erythroid cells at 4 kb resolution. Clearly visible is the chromatin looping of the α-globin (mm9, chr11:32 000 000–32 300 000) self-interacting domain. The coloured sections of the model represent genes loaded as Browser Extensible Data (BED) format and the ChIP-Seq data uploaded as WIG format. A video showing uploading and general features of CSynth can be seen in via the ‘Media section’ on the CSynth website at csynth.org or directly on YouTube (see https://youtu.be/SMgw_cfeH6Q and https://youtu.be/yO6W10Y1o04). More details on the modelling may be found in Supplementary Section S1.
Fig. 2.
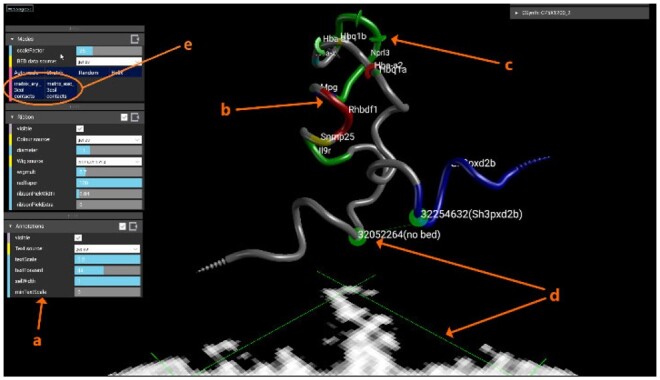
Modelling the mouse alpha globin locus. On uploading of IF data, CSynth generates both the heatmap and model to be inspected. ‘a’ A sub selection of menus that can be used to adjust the visualization. ‘b’ Colouring shows genes uploaded as BED format file for the region. ‘c’ Radius variation shows H3k4me1 data that has been uploaded in WIG (wiggle) format, the size of which may be adjusted using the wigmult parameter. ‘d’ The pair of green lines represents a point selected on the heatmap and the corresponding points on the 3D model which is useful to investigate patterns leading to structures seen in the heatmap and provides a direct way to interact between the 2D heatmap and proposed 3D structure
In Figure 3, we show an example of loading a large Hi-C dataset at 2 kb resolution from Schizosaccharomyces pombe Chromosome I, comparing the difference between mitosis and interphase states (Kakui et al., 2017) using CSynth’s dynamic GPU modelling. To find the parameters for modelling, we used the distance between certain chromosomal locations (Petrova et al., 2013). In interphase (Fig. 3a), chromatin fibre forms a characteristic structure and its telomeres are located in the vicinity as expected from Rabl orientation within the interphase nucleus in S.pombe (Funabiki, 1993). This is where the centromeric region (the centre of which is visible between the green and red arms in Fig. 3) and telomeres attach to the nuclear lamina which causes the overall structure to bend at this point. Here, CSynth shows it has several interesting folding patterns and shows looping that is not obvious in the heatmap view. In mitosis (Fig. 3b), one can see the structure is more compact, folding into the characteristic structure and each arm becomes individualized.
Fig. 3.
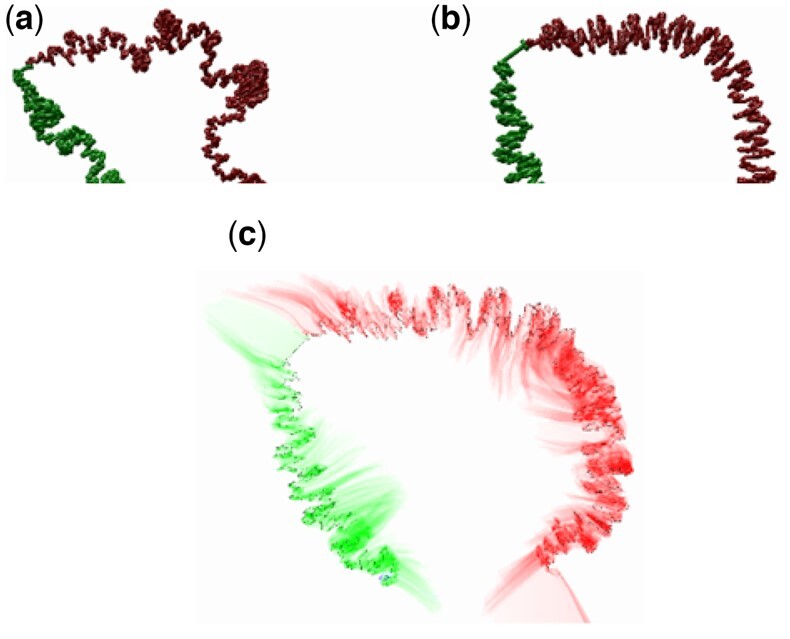
Schizosaccharomyces pombe chromosome I, comparing differences between (a) interphase and (b) mitosis states and the (c) transition between a and b. Red and green colouring show the two arms of the chromosome. Note the more compact and wide structure observed in mitosis. The same model parameters were used for both states
3.3 Modelling methods
There are a large number of 3D genome modelling methods available that can be represented by polymer, spheres or point-based models (Oluwadare et al., 2019). Benchmarking all available modellers is beyond the scope of this article but we compare CSynth’s modelling to ones that apply a similar point-based approach used by Chromosome3D(Adhikari et al., 2016) and LorDG(Trieu and Cheng, 2017). A key point is CSynth’s modelling, which is done quickly, in real time and is interactive which encourages the user to explore and gain an intuitive feel for the model by varying parameters. CSynth’s model is constantly being recalculated so transitions between states are animated and there is direct feedback when the user adjusts parameters on the model. An overview of the modelling process is shown in Figure 4.
Fig. 4.
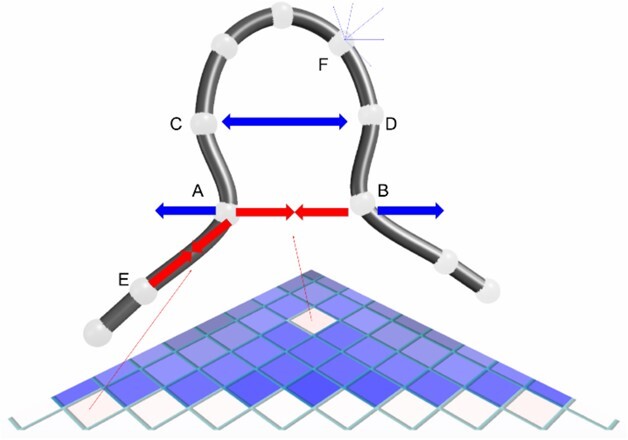
Schematic of the 3D model and IF heatmap showing the balance of IF attraction forces (red) and global repulsion forces (blue). The IF forces for particles A and B balance with the repulsion force. Pair C/D has no recorded IF value, so the particles are pushed further apart. The backbone is held together by IF forces along the upper diagonal of the heatmap, as indicated for pair A/E. Each particle is repulsed by all the other particles, this is indicated for particle F. In a real case, each particle will have IF forces attracting it to many other particles (not shown)
CSynth uses simple forces to seek conformations that best satisfy the known IFs. This builds on the work we used in FoldSynth (Todd et al., 2015) which is software we developed for interacting with protein structures. For example, unsatisfied IFs can have an effect on very long distances. This allows simpler (to compute) dynamics. Our dynamics are inspired by Poing (Jefferys, Kelley and Sternberg, 2010) largely based on spring-like forces. Some of the forces used in our dynamics may be related to real physical forces but the relationship is usually indirect; our dynamics are better thought of as an emulation rather than a simulation or modelling. The various forces we have built in CSynth are detailed in the Supplementary Section 1. Our dynamics work directly from IF or distance map inputs, which are held in sampler buffers on the GPU. The modelling system is based on particles (also referred to as beads), which are represented using the size of the fragment from the capture experiment. The particles generally match the Hi-C bands one to one, but we permit the use of multiple particles per cell for more refined modelling. The particles are assumed to be joined in a backbone chain (or chains). The modelling operates in conventional Newtonian dynamics steps, where in each step an overall force is computed on each particle; the force is then applied to the velocity which is used to compute a new position.The yeast model shown in figure 3 (2798 particles) are produced in less than 30 seconds on a 3.4GHz i7 machine with 16GB RAM and a GTX 1080 graphics card whereas most modelling packages take many minutes to several hours. The number of particles is limited by GPU texture constraint which is typically, as of writing, 16,000 particles. In tests, we have resolved models of 6284 particles (3 chromosomes of yeast at 2k resolution, see Supplementary Section 2) in a few seconds on an NVidia GTX 1080.
3.4 Model and data comparison
The main purpose of CSynth is for interactive 3D modelling of IF data, and comparison of states from multiple IF sources. It can also be used to visualize and compare data from other sources and can import and display static data in xyz or pdb format. CSynth does this by creating distance-based spring models from the distance data implicit in xyz data. These models permit inbetweening of different datasets to visualize their differences and similarities. Such model-based inbetweening is smoother and more informative than simple linear inbetweening of xyz coordinates; and also, naturally aligns the visual output. Furthermore, CSynth can move smoothly between its own models of IF data and imported distance-based models. For example, with the mouse example (Fig. 2), we have both IF data for erythroid and non-erythroid states (embryonic stem cell), and also a xyz data from an independently derived polymer model (Chiariello et al., 2020). Loading all four datasets (2 IF, 2 xyz imported) into CSynth allows the visualization of the differences between the states for each of the models, and between the CSynth and external models for each of the states. The differences can be visualized by transitions between the states or by history trace view similar to that shown in Figure 3.
3.5 Comparison with other modellers using simulated data
The principal feature of CSynth is the ability to visualize and interact with the modelling, better to understand both the data and the modelling. We do not make strong claims for the CSynth modelling, but illustrate here that it is comparable with other recent restraint-based modellers. We carried out tests to compare with LorDG and Chromosome3D, using the LorDG simulated datasets to permit some statistical verification. We added code to the modelling framework based on the LorDG Lorentzian function to allow these comparisons to be conveniently done in CSynth itself. We used chr20 from chainDres25 from the MissouriBox dataset, and loaded the IF data plus the resulting 10 pdb results files, 5 from LorDG modelling and 5 from Chromosome3D modelling which we show graphically using the history trace view shown in Figure 5. As in the section above, we could perform visual comparisons between the different models, and different runs from the imported models. Visual comparison immediately showed that 3 of the LorDG results were almost identical (apart from orientation), and brought out the differences with the rest. We were able to vary the parameters of both the LorDG model (such as c and alpha) and CSynth, and see their impact, and that visual differences between our model and the LorDG model with corresponding parameters were very small.
Fig. 5.

Exploration of LorDG data. GUI layout, ‘history trace’ view to compare results of modelling in 5 runs. Selecting the dark blue ‘dists’ buttons on the right-hand side selects the different imported LorDG solutions. Rainbow colouring, with hue changing along the chromosome. The ‘narrow’ regions show the techniques have very little conformation change between the solutions, the ’wide' regions have much more variation
We also applied statistics using multiple runs and comparing results with their ‘definitive’ simulated data. The statistics of this single experiment indicates that CSynth modelling gives marginally better results than either LorDG or Chromosome3D (Table 1). The differences are very small, and the statistical methods, scale of the experiment and the use of simulated data limit what conclusions we can safely draw.
Table 1.
Performance comparison using simulated data over multiple runs (RMSE, Root Mean Square Error; WRMSE, Weighted RMSE)
| RMSE | WRMSE | Pearson | Spearman | |
|---|---|---|---|---|
| CSynth | 6.260 | 0.230 | 0.730 | 0.939 |
| LorDG | 6.397 | 0.238 | 0.721 | 0.930 |
| Chromosome3D | 6.380 | 0.238 | 0.722 | 0.930 |
3.6 Availability
CSynth can run directly in Chrome and Firefox and has been tested on all major operating systems (including tablets). It is available from csynth.org where there are several example models and instructions for use. For larger models (more than 500 contact points), it is advised to use a discrete graphics card. The absolute limit typically is 16 000, but depends on the maximum texture size depending on the browser's WebGL implementation. Data can either be uploaded to the CSynth portal (https://csynth.molbiol.ox.ac.uk) for later use and for sharing, or can be directly drag-dropped from the local file system for quick viewing. Code is open source and available at https://github.com/csynth/csynth.
4 Discussion
4.1 Potential enhancements
The range of features CSynth supports adds complexity to the user interface. We aim to provide simplified interfaces for common applications based on user feedback. We are extending CSynth documentation of several existing features: scripting and API (JavaScript or Python via websockets). We plan to extend CSynth VR HTC Vive support to other eXtended Reality platforms.
4.2 Summary
CSynth provides a high quality, interactive, user friendly and powerful way of visualizing chromatin interaction data, by combining model, heatmap and genome annotations in one display in a standard web browser. These features are critical when trying to understand how structure and biological activity are interconnected in genome function. A key improvement in CSynth, in comparison to other currently available tools, is that modelling is done on the GPU dynamically. This allows the user to load chromosome capture matrices quickly and vary model parameter values for a better understanding of their effect on the modelling process. Another unique feature of CSynth is the facility to view and compare models between any number of different samples (e.g. tissues or cell types) or even other modelling systems. Finally, we use VR to view and interact with these complex 3D structures which helps get a better intuition for the 3D modelling and is also useful for teaching and public engagement. We foresee that CSynth has the potential to be an invaluable tool to understand the structure and dynamics of more complex systems, such as data generated from different samples from existing and new 3C-based techniques such as single-cell Hi-C (Stevens et al., 2017).
Supplementary Material
Acknowledgements
The authors acknowledge the use of the MRC WIMM Centre of Computational Biology computational infrastructure which hosts this work.
Funding
This research was funded in part by Medical Research Council [MC_UU_12025] and Wellcome Trust [106130/Z/14/B].
Conflict of Interest: none declared.
Contributor Information
Stephen Todd, Department of Computing, Goldsmiths, University of London, London, UK; London Geometry, Ltd., London, UK.
Peter Todd, London Geometry, Ltd., London, UK.
Simon J McGowan, Analysis, Visualization and Informatics, MRC Weatherall Institute of Molecular Medicine, Oxford, UK.
James R Hughes, Genome Biology Group, MRC WIMM Centre for Computational Biology, MRC Weatherall Institute of Molecular Medicine, Oxford, UK.
Yasutaka Kakui, The Francis Crick Institute, Chromosome Segregation Laboratory, London, UK.
Frederic Fol Leymarie, Department of Computing, Goldsmiths, University of London, London, UK; London Geometry, Ltd., London, UK.
William Latham, Department of Computing, Goldsmiths, University of London, London, UK; London Geometry, Ltd., London, UK.
Stephen Taylor, Analysis, Visualization and Informatics, MRC Weatherall Institute of Molecular Medicine, Oxford, UK.
References
- Adhikari B. et al. (2016) Chromosome3D: reconstructing three-dimensional chromosomal structures from Hi-C interaction frequency data using distance geometry simulated annealing. BMC Genomics, 17, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asbury T.M. et al. (2010) Genome3D: a viewer-model framework for integrating and visualizing multi-scale epigenomic information within a three-dimensional genome. BMC Bioinformatics, 11, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiariello,A. M. et al. (2020) A Dynamic Folded Hairpin Conformation Is Associated with α-Globin Activation in Erythroid Cells. Cell Reports, 30, 2125–2135.e5. doi: 10.1016/j.celrep.2020.01.044 [DOI] [PubMed] [Google Scholar]
- Davies J.O.J. et al. (2017) How best to identify chromosomal interactions: a comparison of approaches. Nat. Methods, 14, 125–134. [DOI] [PubMed] [Google Scholar]
- Funabiki H. et al. (1993) Cell cycle-dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J. Cell Biol., 121, 961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.R. et al. (2014) Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet., 46, 205–212. [DOI] [PubMed] [Google Scholar]
- Jefferys B.R. et al. (2010) Protein folding requires crowd control in a simulated cell. J. Mol. Biol., 397, 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakui Y. et al. (2017) Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat. Genet., 49, 1553–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E. et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol J.W. et al. (2009) The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics, 25, 2730–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotny J. et al. (2016) GMOL: an interactive tool for 3D genome structure visualization. Sci. Rep., 6, 20802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oluwadare O. et al. (2019) An overview of methods for reconstructing 3-D chromosome and genome structures from Hi-C data. Biol. Proced. Online, 21, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudelaar A.M. et al. (2018) Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat. Genet., 50, 1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou H.D. et al. (2017) ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science, 357, eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova B. et al. (2013) Quantitative analysis of chromosome condensation in fission yeast. Mol. Cell. Biol., 33, 984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash K. (2017) Chromatin Architecture: Advances from High-Resolution Single Molecule DNA Imaging. Springer. https://www.springer.com/gp/book/9783319521824. [Google Scholar]
- Schlick T. (2002) Molecular Modeling and Simulation: An Interdisciplinary Guide. Springer. https://www.springer.com/gp/book/9781441963505. [Google Scholar]
- Schoenfelder S. et al. (2018) Promoter capture Hi-C: high-resolution, genome-wide profiling of promoter interactions. J. Vis. Exp., doi:10.3791/57320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra F. et al. (2017) Automatic analysis and 3D-modelling of Hi-C data using TADbit reveals structural features of the fly chromatin colors. PLoS Comput. Biol., 13, e1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T.J. et al. (2017) 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature, 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd S. et al. (2015) FoldSynth: interactive 2D/3D visualisation platform for molecular strands. In: VCBM, pp. 41–50. https://dl.acm.org/doi/10.5555/2853955.2853962. [Google Scholar]
- Trieu T. et al. (2019) GenomeFlow: a comprehensive graphical tool for modeling and analyzing 3D genome structure. Bioinformatics, 35, 1416–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu T., Cheng J. (2017) 3D genome structure modeling by Lorentzian objective function. Nucleic Acids Res., 45, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.