

STRUCTURED ABSTRACT
Objectives:
Identify the metabolites that are increased in the plasma of severely injured patients that developed ARDS versus severely injured patients that did not, and assay if these increased metabolites prime PMNs and induce pulmonary sequestration in an animal model of ARDS. We hypothesize that metabolic derangement due to advanced shock in critically injured patients leads to the pulmonary sequestration of neutrophils (PMNs), which serves as the first event in the acute respiratory distress syndrome (ARDS).
Summary Background Data:
Intracellular metabolites accumulate in the plasma of severely injured patients.
Methods:
Untargeted metabolomics profiling of 67 critically injured patients was completed to establish a metabolic signature associated with ARDS development. Metabolites that signficantly increased were assayed for PMN priming activity in vitro. The metabolites that primed PMNs were tested in a two-event animal model of ARDS to identify a molecular link between circulating metabolites and clinical risk for ARDS.
Results:
After controlling for confounders, four metabolites significantly increased: creatine, dehydroascorbate, fumarate, and succinate in trauma patients who developed ARDS (p<0.05). Succinate alone primed the PMN oxidase in vitro at physiologically relevant levels. Intravenous (IV) succinate-induced PMN sequestration in the lung, a first event, and followed by IV lipopolysaccharide, a second event, resulted in ARDS in vivo requiring PMNs. Succinate receptor (SUCNR1) inhibition abrogated PMN priming, PMN sequestration, and ARDS.
Conclusion:
Significant increases in plasma succinate post-injury may serve as the first event in ARDS. Targeted inhibition of the SUCNR1 may decrease ARDS development from other disease states to prevent ARDS globally.
Keywords: Trauma, injury, hemorrhagic shock, metabolomics, neutrophils
MINI-ABSTRACT
Succinate increases in the plasma of severely injured patients. Succinate occupancy of the SUCNR1 primes neutrophils and induces pulmonary sequestration as the first event in a two-event model of ARDS. The presented data provides an alternative pathogenesis of ARDS in the severely injured and a target for therapeutic intervention.
INTRODUCTION
The acute respiratory distress syndrome (ARDS) remains a morbid and lethal disease, characterized by hypoxemic respiratory failure due to an acute, diffuse inflammatory lung process resulting in increased alveolar capillary permeability.1,2 ARDS affects ~200,000 patients annually in the United States, over 3 million worldwide.1 Direct pulmonary insults may cause ARDS, including aspiration, pneumonia, and pulmonary contusion, and systemic diseases are also associated with its development: trauma, sepsis, and pancreatitis.1 Despite significant research into its pathogenesis, there are few effective therapies to prevent ARDS.1 While sepsis is the most common determinant of ARDS, severe injury, a leading cause of morbidity and mortality worldwide, is the second most common etiology.1-6 ARDS following severe injury defined as a new injury severity score (NISS) >15, occurs in 20-50% of patients 48 hours post-injury.5,7
Quantitative metabolite profiling via mass spectrometry following severe injury has been used to identify early markers of varying disease states.8 While most research has focused on global metabolic changes post-injury, there is evidence that certain metabolites play specific physiologic roles.9,10 In particular, the tri-carboxylic acid (TCA) cycle metabolite succinate activates both platelets and monocytes and has been implicated in ischemia/reperfusion injury.9-11 During severe shock, succinate accumulates due to reverse activity of respiratory complex II leading to a rewiring of cellular energy metabolism.9,11,12 Furthermore, when released from the mitochondrial plasma membrane via the succinate transporter SLC13A3, or by tissue injury, succinate may enter the circulation. Many tissue and cell types express SLC13A3, including lung, liver, kidney, endothelium, and circulating blood cells.11-19 Succinate may activate a distinct receptor, SUCNR1/GPR91, and directly modulates processes in blood cells, including leukocytes, leading to dysregulation of mitochondrial energy production, inflammation and tissue injury.9,11-17,20 Succinate accumulation following severe injury is associated with early mortality, <36 hours post-injury; however, much of this research investigated compounds covalently linked to succinate (α-tocopherol, cortocisteroids, etc.) to increase water solubility, which is disparate from receptor:ligand interactions of SUCNR1 and succinate.12,15,21-23
Mechanistically, the pathogenesis of ARDS consists of at least two events.24-26 The first event involves sequestration of neutrophils (PMNs) either by activation of the pulmonary endothelium (PMN adherence) or priming of PMNs which stiffens them resulting in dramatically reduced transit times.24,26-28 A second stimulus activates these primed, sequestered PMNs resulting in PMN-mediated endothelial damage, capillary leak, and ARDS.24-27 We hypothesize that metabolic derangement due to advanced shock and tissue injury in critically injured patients leads to plasma succinate accumulation, which also serves as the first event in a two-event animal model of ARDS through SUCNR1 activation.
METHODS
Materials:
Please see Methods, Supplemental Digital Content 1, for clarification.
Clinical sample collection:
67 critically injured patients (NISS>15) enrolled in The Control of Major Bleeding After Trauma (COMBAT) study, a clinical trial based at Denver Health Medical Center (ClinicalTrials.gov #NCT01838863) had whole blood collected in the field (76%) or in the emergency room within 4 hours post-arrival (24%) and then serially over a 7 day period (see Figure, Supplemental Digital Content 2).29,30 Patients were then stratified by the presence or absence of ARDS using the Berlin Criteria by two blinded physicians (Table 1).30
Table 1.
Demographic Data for ARDS patients.
| Variable | No ARDS (n=46) |
ARDS (n=21) |
|---|---|---|
| Age (years) | 33.5 (25-48.5) | 44 (24-50) |
| BMI | 25.1 (23.4-28.1) | 27.1 (24.7-29.5) |
| Male Sex (%) | 78% | 86% |
| New Injury Severity Score (NISS) | 12 (3.75-28.75) | 43 (34-57)*** |
| Max AIS Head | 0 (0-1) | 0 (0-4.5) |
| Max AIS Chest | 0 (0-3) | 3 (2-4.5)** |
| Penetrating Injury (%) | 50% | 29% |
| Admission Glasgow Coma Scale (GCS) | 15 (13-15) | 12 (3-15)* |
| Admission SBP | 98 (80-115) | 90 (70-106) |
| Admission HR | 108 (83-123) | 123 (91-136) |
| Admission WBC count (field, x106/μl) | 10.6 (6.4-16.9) | 7.1 (4.8-17.3) |
| Admission WBC count (ED, x106/μl) | 10.3 (4.6-22.6) | 9.1 (5.3-19.0) |
| Admission Base Deficit (mEq/L) | 7 (5-12.5) | 11 (7-12) |
| RBCs Transfused in 1st day (units) | 0 (0-2) | 8 (4-13)*** |
| Ventilator Free Days (VFDs) | 28 (27-28) | 21 (11-24)*** |
| Mortality (%) | 0% | 5% |
p<0.05
p<0.01
p<0.001 (Wilcoxon non-parametric test)
Metabolomics:
Mass spectrometry-based metabolomics analyses were performed on plasma isolated from collected whole blood as described (see Supplemental Digital Content, Methods for details).31,32 Absolute quantification using stable isotope-labeled standards was performed for metabolites that significantly changed in the initial patient plasma.33,34 Spermine and spermidine were measured in a previous data set of 500 trauma activation patients in the first sample collected in the Emergency Department (ED).35
PMN Priming:
Heparinized whole blood was collected from healthy donors and neutrophils (PMNs) isolated under approval from COMIRB.36 PMNs were incubated at 37°C with NS or various metabolites (Table 2) for 5 minutes or pre-incubated with DMSO, BAPTAAM [50 μM], or Gö6983 [50 nM] for 10 min followed by NS or disodium succinate [1000 μM] priming for 5 min, activated with 1 μM fMLF, and the maximal rate of O2− production measured.36,37
Table 2.
Metabolites of interest in ARDS patients controlling for confounders
| Metabolite | No Exclusions | Excluded Head AIS<3 |
Excluded Chest AIS<3 |
Excluded Baseline Lung Disease |
||||
|---|---|---|---|---|---|---|---|---|
| Fold Change |
p-value | Fold Change |
p-value | Fold Change |
p-value | Fold Change |
p-value | |
| Metabolites of Interest that met criteria (fold change>1.5 & p<0.05) | ||||||||
| Creatine | 2.14 | 0.0006 | 1.92 | 0.0255 | 2.00 | 0.0291 | 2.20 | 0.0010 |
| Succinate | 1.93 | 0.0016 | 2.28 | 0.0026 | 1.79 | 0.0203 | 1.97 | 0.0036 |
| Fumarate | 1.67 | 0.0109 | 1.78 | 0.0231 | 1.65 | 0.0163 | 1.59 | 0.0263 |
| Dehydroascorbate | 2.72 | 0.0013 | 2.74 | 0.0497 | 2.63 | 0.0291 | 2.72 | 0.0022 |
| Metabolites of Interest that did not meet criteria (fold change<1.5 & p>0.05) | ||||||||
| O-Propanoyl-Carnitine | 1.57 | 0.0168 | 1.69 | 0.0044 | 2.30 | 0.0646 | 1.60 | 0.0307 |
| sn-Glycerol-3-Phosphate | 4.04 | 0.0031 | 3.36 | 0.0683 | 4.42 | 0.0116 | 4.00 | 0.0053 |
| Spermine | 2.68 | 0.0156 | 1.83 | 0.2000 | 2.22 | 0.1105 | 2.32 | 0.0274 |
| Spermidine | 2.34 | 0.0273 | 1.94 | 0.2066 | 2.24 | 0.1387 | 2.31 | 0.0383 |
| Xanthine | 1.80 | 0.0025 | 1.74 | 0.0198 | 1.56 | 0.0834 | 1.70 | 0.0071 |
| Taurocholate | 3.91 | 0.0361 | 5.11 | 0.0619 | 3.03 | 0.1194 | 3.03 | 0.0491 |
| O-Butanoyl Carnitine | 1.73 | 0.0361 | 1.78 | 0.0327 | 1.34 | 0.1194 | 1.61 | 0.0427 |
| S-Adenosyl-Homocysteine | 1.13 | 0.0688 | 1.11 | 0.1180 | 1.14 | 0.1547 | 1.15 | 0.0711 |
| Malate | 1.37 | 0.0456 | 1.46 | 0.1419 | 1.25 | 0.1337 | 1.31 | 0.0861 |
| Taurine | 1.32 | 0.0046 | 1.19 | 0.0520 | 1.34 | 0.0237 | 1.34 | 0.0071 |
| Aspartate | 1.14 | 0.1672 | 1.13 | 0.4797 | 1.13 | 0.4478 | 1.19 | 0.1465 |
| Glutamate | 1.00 | 0.7629 | 0.98 | 0.9197 | 0.91 | 0.7293 | 1.00 | 0.6201 |
| 2-Oxoglutarate | 1.18 | 0.0777 | 1.04 | 0.3343 | 1.13 | 0.1194 | 1.19 | 0.0508 |
| Prostaglandin G-2 | 2.15 | 0.0849 | 2.34 | 0.0434 | 2.18 | 0.0800 | 1.93 | 0.1506 |
| Metabolites of interest in the plasma (Range (Median)) tested for PMN Priming | ||||||||
| Metabolite | Healthy Plasma Concentration (μM) |
Trauma Plasma Concentration (μM) |
Concentration Range Tested (μM) |
Significant Priming? |
||||
| Succinate | 0-5 | 7.9-301.3 (29.8) | 1-1,000 | Yes (Fig 1) | ||||
| Creatine | 30-80* | Not measured | 100-1,000 | No | ||||
| Fumarate | 0.0-4.0* | 6.9-52.2 (23.67) | 100-1,000 | No | ||||
| Dehydroxy-ascorbate | 2.3-12.6* | Not measured | 10-100 | No | ||||
| Spermine | 0.01-0.13* | 0.04-97.0 (1.6) | 100-1,000 | Yes, ≥500 μM | ||||
| Spermidine | 0.07-10.3* | 0.1-47.5 (2.0) | 100-1000 | Yes, ≥500 μM | ||||
All metabolites in this table were tested for PMN priming as a proof of concept. Only succinate, spermine and spermidine primed the PMN oxidase.
Fold Change=Median ARDS/Median No ARDS; p-value=Mann Whitney 2-tailed t-test
Obtained through the Human Metabolome Database (http://www.hmdb.ca/metabolites/HMDB00254)
Succinate Receptor Expression:
PMNs were incubated with NS or succinate (500-1000 μM), fixed, smeared onto slides, incubated with a GPR91/SUCNR1 antibody (Novus Biological, Littleton, CO), and then with a species-specific fluorescent antibody followed by microscopic imaging at 100X magnification.24,38
SUCNR1 Inhibitor Synthesis and Inhibition of Succinate priming:
An inhibitor to SUCNR1, GPR91-2c, was synthesized by the Peptide and Protein Chemistry Core at the University of Colorado Anschutz Medical Campus.39 The GPR91-2c inhibitor was solubilized in DMSO with further dilutions in NS (DMSO-NS) or 1.25% fatty acid-free human serum albumin (HSA), with DMSO-NS or DMSO-HSA used as vehicle controls. PMNs were pre-incubated with vehicle or the GPR91-2c inhibitor [30 nM] for 5 minutes at 37°C, followed by DMSO/NS or succinate [500 μM] priming and activation with 1 μM fMLF.36
Two-event in vivo model of ARDS:
Male Sprague-Dawley rats underwent an established two-event in vivo model of ARDS under an approved protocol for the Animal Care and Use Committee (IACUC).24 Rats (330-430g) were anesthetized and given succinate [25-500 μM]FINAL, followed by IV Evans Blue Dye (EBD). After 6 hours the rats were infused with IV LPS (100 μg/kg, S.enteritides), a surrogate for infection and Toll-like receptor activation, or an equivalent volume of NS. Two hours later, blood was drawn and a bronchoalveolar lavage (BAL) performed.24,40 Lung leak was measured as EBD leak from the plasma to the BALF.24,40 For PMN depletion rats were injected IP with a rabbit anti-rat PMN-antibody (400 μL of Ab/100 g of rat weight).24 The GPR91-2c was solubilized in 1.25% HSA [30 nM] was transfused 30 min before the first event: NS or succinate.
Histology and PMN sequestration:
Histological changes were visualized using the two-event rat model on H&E stained lung sections (40X) read by a blinded observer.24,40 PMN sequestration was measured following the first event in flushed, fixed lungs as described.24,40
Measurement of BALF CINC-1, Lung MPO, Liver and Kidney Injury:
BALF chemokineinduced neutrophil chemoattractant (CINC-1) and MPO activity were quantified as described.24 Plasma alanine aminotransferase and urinary neutrophil gelatinase-associated lipocalin (NGAL) were measured as markers of liver and renal injury on the Orthoclinical Vitros 5600 or by commercial ELISA.41
Statistical Analysis:
Multivariate analyses were completed as described previously (see Methods, Supplemental Digital Content 1).7,29,42 Non-parametric data were analyzed between the ARDS and non-ARDS cohorts with the “screening” significance set at p<0.05 and ≥1.5-fold change between groups. Normally distributed data were analyzed using paired (in vitro) or nonparametric (in vivo) ANOVA with post-hoc Tukey’s or Dunn’s tests.
RESULTS
Circulating Metabolites in Injured Patients:
From the 144 patients enrolled in the COMBAT study, 67 patients initially satisfied the inclusion criteria. Twenty-one of these severely injured patients developed ARDS and survived 48 hours post-injury (Table 1).30 All patients diagnosed with post-injury ARDS satisfied the Berlin criteria, including: 1) PaO2/FiO2 <300, which was corrected for altitude such that the corrected ratio employed had a median of 263, with the highest PaO2/FiO2 of 215 in the post-injury ARDS group, 2) bilateral infiltrates on chest radiograph (CXR), and 3) respiratory insufficiency requiring mechanical ventilation (see Methods, Supplemental Digital Content 1).30 The two groups did not differ with regard to age, sex, BMI, mechanism of injury, admission heart rate, or admission blood pressure (Table 1). The ARDS group was more severely injured with a higher NISS, more severe chest injuries (Abbreviated Injury Severity Score, AIS), lower Glasgow Coma Scale (GCS) at admission, higher blood product requirements on day 1 with none being leukopenic (Table 1). Patients without ARDS had a statistically significant higher number of ventilator free days compared to patients with ARDS (p<0.0001), although there was no difference in mortality.
Prior to transfusion with cellular blood components, the plasma from the study patients was isolated from whole blood by centrifugation and snap-frozen, from “needle to nitrogen”, within a median of 150 minutes of arrival.15 A high throughput, untargeted metabolomics profile was completed on the first available plasma samples; the majority comprised of field samples (76%), and the rest collected in the ED within 4 hours of arrival (24%) with 90% of the whole blood drawn within 60 minutes of presentation. A manually selected, targeted strategy was completed for metabolites involved in energy and redox metabolism, which were validated against multiple quality control measures and quantified by spike-in stable isotope-labeled internal standards.31,32 From the many thousands of metabolites 138 were chosen and appear in Supplemental Digital Content 3, Table. The initial analysis revealed 18 metabolites that differed between groups (p<0.05 and ≥1.5-fold change) (Table 2). Further analyses excluded patients with baseline lung disease, severe traumatic brain injury (Head + Neck AIS >3), and severe chest wall trauma (Chest AIS>3), which in the absence of ARDS may affect the need for ventilatory support due to comorbid conditions at study entry, inability to extubate secondary to mental status, or severe chest wall injury with impaired pulmonary mechanics (Table 2).5 Four metabolites were significantly increased in the post-injury ARDS cohort versus injured patients who did not develop ARDS (Table 2). When head injury or chest injury were excluded, spermine and spermidine were not significantly increased in the ARDS cohort; however, because of their reported ability to prime the NADPH oxidase of PMNs further analysis was completed (Table 2). Fourteen additional metabolites, were also analyzed due to their structural similarity to succinate or because they also comprise molecules of the TCA cycle. Of the 18 metabolites tested for their ability to prime the PMN NADPH oxidase, only succinate primed at concentrations present in injured patients (Table 2 and Fig.1). Creatine, dehydro-ascorbate, and glycerol-3-phosphate were not quantified from the injured patients because of the lack of available standards; the concentrations used to screen metabolites for priming activity were determined by using the upper limit of the plasma concentration in healthy human subjects and multiplying it by the fold-change increase over these controls. Succinate plasma concentrations were independently associated with the development of ARDS in patients who survived 48 hours, versus the non-ARDS cohort (p<0.002), and did not increase over time up to 7 days postinjury (Supplemental Digital Content 2, Figure).
Figure 1. Priming of the PMN oxidase by selected Metabolites.
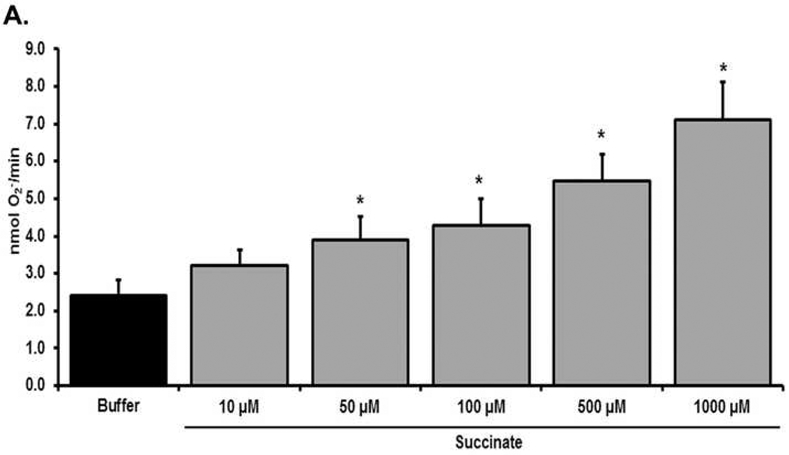
Treatment with succinate [10-1,000 μM] as compared to buffer (vehicle)-treated PMNs, primed the fMLF-activated respiratory burst in a concentration-dependent manner. Succinate concentrations <50 μM did not prime the respiratory burst vs. buffer controls (*=p<0.05 n=6).
Priming of the PMN oxidase:
Succinate alone significantly primed the fMLF-activated PMN oxidase compared to buffer-treated PMN controls at 50-1,000 μM (n=6, p<0.01), but not at lesser concentrations [1-20 μM] (Fig.1, Table 2, and data not shown). Importantly, succinate concentrations that primed PMNs approached the median (±SD) plasma succinate concentrations in the severely injured ARDS patients versus the severely injured non-ARDS patients: postinjury ARDS, 29.8±64.3 μM versus non-ARDS, 15.2±40.2 μM (p<0.002) and healthy controls, 0-5 μM. The counter-balancing cations spermine and spermidine significantly primed the PMN oxidase compared to buffer-treated controls at 500 and 1,000 μM: Spermine: 2.32±0.29 (buffer) vs 3.11±0.42 (500 μM), 3.67±0.46 (1000 μM); Spermidine: 2.32±0.29 vs 2.97±0.38 (500 μM), 3.32±0.48 (1000 μM) as reported.43 Thus, both positively and negatively charged metabolites primed PMNs in vitro. In a previous study of 500 trauma activation patients, the highest spermine and spermidine concentrations were 47.43 μM and 97.0 μM in two severely injured patients (NISS>40), with medians of 1.98 μM (IQR 1.08-3.58 μM) and 1.59 μM (IQR 0.9-3.05 μM), respectively (Table 2).35
Stimulation of the succinate receptor on the PMN membrane induces morphology changes:
Prior studies have yielded conflicting results on whether PMNs express the SUCNR1/GPR91 succinate receptor by mRNA expression.44 Incubation of PMNs with a fluorescently-labeled SUCNR1/GPR91 antibody demonstrated immunoreactivity near the membrane (Fig.2), and the mean fluorescent intensity (MFI) of the SUCNR1/GPR91 immunoreactivity did not change with succinate treatment (data not shown, n=3). This SUCNR1/GPR91 immunoreactivity was visualized at the PMN membrane by the co-localization of SUCNR1/GPR91 immunoreactivity (red) with the membrane (green) resulting in discrete co-localization (Fig.2, top row, yellow, arrows). Succinate also induced rapid changes in PMN morphology versus NS-treated controls, which are round with smooth membranes (Fig.2, Control) versus succinate-induced cellular elongation and membrane ruffling (Fig.2, 500 and 1,000 μM, 1 and 5 min incubations).
Figure 2. The succinate receptor (GPR91) is expressed on the surface of the PMN.

Micrographs (40X) illustrate that both in control and succinate-treated PMNs the GPR91 immunoreactivity is expressed on the PMN membrane as shown by co-localization with the membrane immunoreactivity (green) as discrete areas of yellow color (upper panel, arrows) which were unaffected by succinate treatment. The succinate treatment also primes PMNs at 500 μM and 1000 μM as illustrated by the changes in shape at 1 and 5 min compared to NS (upper panel). The lower panels show the GPR91 immunoreactivity (red) lower panels together with the chromatin stain (blue). Representative images are shown at 40X, n=3. Legend: GPR91: red Alexa Fluor 555, membrane: green, WGA-Alexa Fluor 488, chromatin: blue DAPI.
Succinate activates signal transduction in PMNs:
Previous reports have shown that succinate activation of its receptor, SUCNR1, elicits changes in cytosolic Ca2+ and the activation of protein kinase C (PKC).45 PMNs were pre-treated with BAPTA, AM [50 μM], a rapid cytosolic Ca2+ chelator, or Gö6983 [50 nM], a classical PKC inhibitor and then primed with 1000 μM succinate.36,37 Both inhibitors significantly decreased succinate priming of the PMN oxidase by 100±19% for BAPTA, AM and 88±8% for Gö6983, compared to DMSO pre-treated controls (p<0.05, n=5).
Succinate induces ARDS as the first event in a two-event rat model:
The ability of succinate to serve as one of two events required for ARDS was tested in a well-described, two-event rat model of acute lung injury/ARDS.24,40 In the initial experiments, the first event consisted of intravenous (IV) succinate [25-500 μM] or NS controls followed by IV lipopolysaccharide (LPS) or IV NS controls. As compared to NS/NS controls, succinate [25-500 μM]/NS or NS/LPS did not induce ARDS as quantified by EBD leak in the BALF (Fig.3). However, succinate [50 μM]/LPS and succinate [500 μM]/LPS resulted in statistically significant EBD leak (Fig.3A). In addition, the protein concentration in the BALF also increased with succinate [50 μM]/LPS (0.65±0.12 mg/ml) and succinate [500 μM]/LPS (0.78±0.23 mg/ml) compared to NS/NS (0.12±0.01 mg/ml) succinate [50 μM]/NS (0.13±0.01mg/ml) and NS/LPS (0.21±0.04 mg/ml) (p<0.01). The concentration of cytokine-induced neutrophil chemoattractant-1 (CINC-1) in the BALF, the analogue of IL-8 in rats, also significantly increased in the succinate [50 μM]/LPS and succinate [500 μM]/LPS versus the NS/NS, NS/LPS and succinate [50 and 500 μM]/NS treatment groups (Fig.3B).24,40 Histological evaluation of succinate/LPStreated lungs showed PMN infiltration, arcuate inflammation, thickening of the alveolar walls, and intra-alveolar debris as compared to the lungs from rats treated with NS/NS or succinate [50 μM]/NS (Fig.3C). Importantly, succinate [50 μM]/NS and NS/LPS both caused PMN sequestration compared to the NS/NS controls, identical to previous data which documented that LPS “alone” at these concentrations induced PMN sequestration but not ARDS (Fig.3C; data not shown).24,40 Previous data from this model has indicated that EBD leak and increased BALF protein correlate with increased interstitial fluid, another marker of ALI/ARDS.24
Figure 3. Succinate/LPS-induced lung injury.

Sprague-Dawley rats were transfused with 50-500 μM Succinate followed by an infusion of LPS (100 μg/kg) 6 hours later. Blood and BALF were taken to measure ALI. a) Succinate/LPS induces ALI, as measured by increased levels of EBD extravasation into BALF fluid compared to NS/NS, succinate/NS, NS/LPS (*=p<0.01 compared to all other groups). NS/LPS resulted in increased EBD compared to NS/NS and succinate/NS, but was not statistically significant (p>0.05). b) Succinate/LPS induces ALI, as measured in increased levels of BALF CINC-1 compared to NS/NS, Succinate/NS, NS/LPS (*p<0.01 compared to all other groups). No other groups show statistically significant differences. c) Representative images of lung sections stained with H&E from control (NS/NS), succinate/NS and succinate/LPS treatment groups are shown (40x). Control and succinate [50 μM]/NS animals showed little derangement in cellular architecture. The succinate [50 μM]/LPS group shows prominent neutrophil infiltration, pulmonary alveolar wall thickening, and arcuate inflammation.
LPS (2 mg/kg) IP or NS were then employed as the first event(s) followed by 20 or 50 μM succinate as the second event in this animal model of ARDS.24,40 Compared to rats with NS/NS, LPS/NS, and NS/succinate [50 μM], an LPS followed by [50 μM] succinate IV did not cause ARDS (EBD leak): NS/NS: 0.08±0.02%, NS/50 μM succinate: 0.15±0.04%, LPS/NS: 0.24±0.03%, LPS/20 μM succinate: 0.07±0.02%, and LPS/50 μM succinate: 0.06±0.01% (n=5 for each group).
Succinate causes PMN sequestration:
As shown in Fig.3A, succinate administered as a first event with NS as the second event was insufficient to produce ARDS; however, the combination of succinate infusion followed by IV LPS elicited lung injury, and succinate/NS did induce PMN sequestration as seen in the pulmonary histology (Fig.3C). Therefore, the ability of succinate to induce PMN sequestration in the lungs was investigated.24,26,40 Compared to the NS-treated rats, the succinate infusion resulted in increased PMN sequestration in the lung tissue, as measured by the ratio of PMNs to lung area (Fig.4, panels A and B). Importantly, there was no difference in total lung tissue area between groups (data not shown, p>0.05), and lung myeloperoxidase (MPO) levels increased in succinate-treated animals compared to NS-treated controls (p<0.05) (Fig.4C).
Figure 4. Succinate treatment generates PMN sequestration.

Lung samples were taken from rats that underwent a two-event model of ALI with 50 μM succinate followed by LPS 6 hours later being measured. A) Representative images of PMN sequestration in the lung tissue as a result of succinate [50 μM] as a first event (Left panel- NS treated rats, right panel- succinate [50 μM]/NS). There is an increase in PMNs (red, Alexa Fluor 555) at the membrane (green, WGA Alexa Fluor 488). The blue (DPAI) is a DNA marker. B) Succinate increases PMNs sequestration in lung tissue, as measured by the ratio of PMN/lung tissue area (*=p<0.001 compared to NS alone). There was no difference in the lung tissue area between groups (p>0.05). C) Succinate increases lung MPO activity compared to NS (*= p<0.05 compared to NS).
PMN depletion abrogates lung injury:
PMNs are required for most models of clinical ARDS.1,24-26,40 To investigate the role of PMNs in succinate/LPS-induced ARDS, rats were depleted of PMNs using a mouse antibody against the gr-1 antigen confirmed by manual differentials on blood smears resulting in 99±0.5% depletion of PMNs in the antibody-treated animals.24 PMN-depleted rats did not develop ARDS following succinate [50 μM]/LPS treatment versus non-antibody rats who received succinate [50 μM]/LPS: PMN Ab/Succinate/LPS, 0.07±0.01% vs. NS/Succinate/LPS, 1.90±0.65% EBD leak, respectively (p<0.01) (Fig.5A). Moreover, the PMN-depleted animals did not manifest increased BALF protein concentrations (PMN-depleted: 0.11±0.01 mg/ml vs. NS treated: 0.53±0.13 mg/ml) (p<0.001) or CINC-1 levels (PMN-depleted: 972.51±112.62 pg/ml vs. NS treated: 6,428,46± 1,626,28 pg/ml) (p<0.05). There were no statistical differences between rats treated with NS/NS and the PMN-depleted rats treated with succinate/LPS or NS in any ARDS measure (Fig 5A, p>0.05; data not shown).
Figure 5. PMN depletion and SUCNR1 inhibition eliminates succinate/LPS mediated lung injury and SUCNR1 inhibition abrogates PMN sequestration.

Panel A: As compared to 50 μM succinate/NS, 50 μM succinate/LPS caused increased EBD leak into the BALF. In rats pretreated with an anti-rat neutrophil antibody, the neutrophil depletion eliminates succinate [50 μM]/LPS induced ALI, as measured by levels of EBD extravasation into BALF fluid, and the EBD extravasation is not significant compared to NS/NS in the PMN depleted group (* p<0.05 compared to 50 μM succinate/LPS). Panel B: As compared to the HSA/NS/NS (Control, left panel), HSA/succinate [50 μM]/NS (middle panel) caused PMN infiltration/sequestration and an increase in the thickness of the pulmonary alveolar membranes. Pretreatment with the GPR91-2c inhibitor, GPR91-2c/succinate [50 μM]/NS decreased the numbers of the PMNs and decreased the thickness of the pulmonary alveolar membranes (right panel). The panel is representative of experiments performed in duplicate. The lung sections were stained with H&E and visualized at 40X. The bar graph is the histology score, membrane thickness of the histology. This figure represents the quantification of the pulmonary alveolar thickness of 10 images from two separate experiments, *=p<0.05 versus the lungs from NS/NS controls and †=p<.05 versus the lungs from succinate/NS treated rats. Panel C: Pretreatment for 30 min with the GPR91-2c [30 nM] inhibitor/NS did not elicit cause ARDS, as measured by EBD leak, in rats. Rats treated with the 1.25% HSA vehicle followed by 50 μM succinate/LPS or 500 μM succinate/LPS manifested ARDS. Pretreatment with the GPR91-2c abrogated ALI caused by 50 μM succinate/LPS or 500 μM succinate/LPS (*=p<0.05 vs. GPR91-2c/NS, GPR91-2c/LPS, HSA/50 μM succinate/NS and HSA/500 μM succinate/NS; †=p<0.05 versus HSA/50 μM succinate/LPS and HSA/500μM succinate/LPS, n=5 for each bar).
Inhibition of SUCNR1 prevents succinate-induced PMN priming, sequestration, and ARDS:
GPR91-2c, a specific SUCNR1 inhibitor, was synthesized as published.39 PMN pretreatment (5 min, 37°C) with GPR91-2c [30 nM] inhibited succinate [500 μM] priming of the fMLF-activated NADPH oxidase by 98±6%, respectively versus albumin-treated PMNs primed with succinate (p<0.05, n=7). Since succinate caused the first event in the two-event model of ARDS, rats were pretreated with GPR91-2c [30 nM], Succinate-elicited PMN sequestration was inhibited compared to rats pre-treated with the albumin vehicle (human serum albumin (HSA)) and infused with succinate [50 μM], as shown with histology (Fig.5B) and the increase in alveolar membrane thickness: (p<0.05, n=7) vs HSA/NS controls) vs. GPR91-2c [30 nM]/ succinate [50 μM] (p<0.05, n=7 vs. HSA/succinate [50 μM]) (Fig.5B, panel). Pretreatment with the succinate inhibitor GPR91-2c [30 nM] abrogated succinate [50 μM]/LPS- and succinate [500 μM]/LPS-induced lung injury vs. pre-treated HSA controls (vehicle control (p<0.05, n=4) (Fig.5C). Importantly, the GPR91-2c inhibitor alone did not induce lung injury in combination with LPS: HSA/NS/LPS: 0.10±0.01 versus GPR91-2c/NS/LPS: 0.08±0.02.
Succinate does not produce remote organ injury:
Post-injury multiple organ failure can occur following ARDS. Succinate [50 μM]/LPS treatment caused the development of ALI in rats, but was not sufficient to produce liver or kidney injury over controls. Liver injury was quantified by the measurement of plasma ALT concentrations: NS/NS: 151.4±16.8, succinate [50 μM]/NS: 181.9±33.0 U/L vs. succinate [50 μM]/LPS: 161.1±20.5 U/L. Kidney injury was interrogated by measuring urinary NGAL: NS/NS: 704.0±102.9 pg/ml, succinate [50 μM]/NS: 636.3±68.1 pg/ml vs. succinate [50 μM]/LPS: 816.6±153.6 pg/ml (p>0.05, n=5).
DISCUSSION
ARDS is the most common organ-specific dysfunction following severe injury affecting up 20-50% of trauma patients with substantial morbidity and >40% mortality.6,29,42 Using targeted metabolomics, we identified a profile of 4/138 metabolites in early blood samples, (drawn prior to transfusion of any cellular blood products: field, 76%), or <4 hours of ED arrival, 24%), that could directly precede the development of post-injury ARDS in severely injured patients. Of these, only elevated succinate appears to support a plausible mechanism through PMN priming.2,24-26,28,40 Succinate is present in rather small amounts [≤5μM] in the plasma from healthy volunteers, as well as RBC units in which it is unaffected by storage.46,47 Transfusion is unlikely to affect the plasma concentration as documented by the serial succinate measurements throughout patient resuscitation.46,47
Study criteria required that the patients survived 48 hours with appropriate monitoring to determine ARDS development. Succinate, which is negatively charged, primed PMNs in vitro and served as the first event in a well-described, in vivo rodent model of ARDS. Both PMN priming and ARDS were abrogated by pretreatment with SUCNR1 inhibitor, GPR91-2C.1,24-26,40 Shock, which was present in all severely injured study patients, was excluded from the animal model to investigate the primary role of succinate in ARDS.
In the critically ill, succinate accumulates due to a reversal of Respiratory Complex II activity directly due to ischemia in patients who undergo severe shock.9,11,12 Although all of the patients were severely injured (NISS>15), those who developed ARDS had higher NISS with increased head and chest injuries and received more blood products on day 1. Because succinate accumulation is driven by ischemia, the level and duration of shock directly results in increased plasma concentrations providing a mechanistic explanation of the first hit for ARDS in the most severely injured.9,11,12 Both post-injury ARDS and multiple organ failure (MOF) have been postulated to be the result of at least two events because many cases of ARDS in the injured do not involve acute infections.48-52 Additionally, these models of clinical ARDS/MOF stem from a two event construct in which injured patients are resuscitated and are in a vulnerable clinical state of hyperinflammation (priming of PMNs) during which a second innocuous event activates these primed PMNs leading to tissue destruction and end-organ, especially lung, injury.48-52 However, 5 patients with increased plasma succinate did not develop ARDS and thus, may not have received a second insult resulting in ARDS.1-3,24-26,28,48-52 Succinate may mediate ischemiareperfusion injury by generating reactive oxygen species through reverse electron flow, although its primary role in this process has been questioned.9,20,53 Succinate activation of the SUCNR1 receptor activates dendritic cells, has pro-inflammatory effects in the kidney and small intestine, and alters DNA methylation, post-translation modification of proteins, apoptosis, and turnorigenesis, with little data regarding its effects on neutrophils/innate immunity.9,11,20,53,54
This study has limitations, including a small cohort of patients from a single center at intermediate altitude, with the post-injury ARDS mortality lower than typically reported.4,5,7,42,55 Additionally, we focused the targeted metabolomics profile analytically on the polar fraction of the metabolome, and it is possible that non-polar lipids and other “untargeted” metabolites may play a role in ARDS, especially neutral lipids which are present in RBC units and have been linked to ARDS.40 We also studied the metabolites individually due to the complexity of testing combinations, and there may be synergistic effects from these combinations in severely injured patients.
Succinate is a predictor of early mortality in severely injured patients (NISS>15), as a marker of shock, global hypoxia, and a failure in energy production.15 In this study the succinate median level was 96±144 μM with a median lactate of 8.3±3.8 μM in those that died with 11 dying of exsanguination in the first 24 hours and one patient survived >48 hours before dying of exsanguination with concomitant ARDS.15 In the presented patient cohorts, the ARDS group had an initial median lactate of 5.1±1.3 mM versus 4.8±2.5 mM in the non-ARDS group, signifying their need for immediate resuscitation. Thus, severe ischemia/shock, as evidenced by base deficit, lactate levels, or blood transfusion requirements, causes increased plasma succinate concentrations, which, in turn, predisposes the severely injured to ARDS. The reported data provides for an alternate process, which explains post-injury ARDS with a systemic increase in succinate causing PMN sequestration through SUCNR1. Resuscitation employing a SUCNR1 antagonist may eliminate succinate-induced pulmonary PMN sequestration and inhibit the development of ARDS. SUCNR1 may represent a novel target for oxidative- and stress-related conditions, including severe shock, and provides a potential therapy for ARDS following severe injury.20
Supplementary Material
Supplemental Digital Content 3. Table of all the metabolites measured in the plasma of severely injured patients. doc
Supplemental Digital Content 2, Figure: Succinate levels over time in severely injured patients. Plasma samples from 67 severely injured (NISS>15) patients: 21 with ARDS and 46 without, were collected in the field or ED and during admission, up to 7 days. Succinate levels (left y-axis) were measured with metabolomics and the medians for patients that developed ARDS (blue dashed line, blue squares) and those that did not (solid gray line, gray circles) were plotted. Succinate levels were significant at the initial collection (field/ED, *p<0.05) and were decreased by hour 2. The numbers of patient tested in each group (right y-axis) are also quantified for each time point for critically injured patients with ARDS (n=21, blue horizontal hatch marks) and without ARDS (n=46, gray triangles). Only one critically ill patient died with ARDS just after hour 48, and no metabolomics samples were drawn once the patient left the Surgical Intensive Care Unit.
Supplemental Digital Content 1. Methods described in more detail. Doc
ACKNOWLEDGEMENTS
Additional Funding and Support were also provided by Vitalant Research Institute, Denver, CO.
Funding/Conflicts of Interest: This work was supported by grants #P50-GM049222, T32-GM008315, and 1RM1GM131968 from NIGMS, NIH, grant #UM1-HL120877 from NHLBI, NIH, grant #W81XWH-12-2-2008 from the Department of Defense. CCS and AD serve on the Scientific Advisory Board for Hemanext™. There are no other conflicts of interest.
Footnotes
Conflict of Interest: CCS and AD serve on the Scientific Advisory Board for Hemanext™. The other authors have declared that no conflicts of interest exist.
References
- 1.Fan E, Brodie D, Slutsky AS. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018; 319:698–710. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012; 122:2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellani G, Laffey JG, Pham T et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016; 315:788–800. [DOI] [PubMed] [Google Scholar]
- 4.Robinson BRH, Cohen MJ, Holcomb JB et al. Risk Factors for the Development of Acute Respiratory Distress Syndrome Following Hemorrhage. Shock 2018; 50:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salim A, Martin M, Constantinou C et al. Acute respiratory distress syndrome in the trauma intensive care unit: Morbid but not mortal. Arch Surg 2006; 141:655–658. [DOI] [PubMed] [Google Scholar]
- 6.Sperry JL, Guyette FX, Brown JB et al. Prehospital Plasma during Air Medical Transport in Trauma Patients at Risk for Hemorrhagic Shock. N Engl J Med 2018; 379:315–326. [DOI] [PubMed] [Google Scholar]
- 7.Sauaia A, Moore EE, Johnson JL et al. Temporal trends of postinjury multiple-organ failure: still resource intensive, morbid, and lethal. J Trauma Acute Care Surg 2014; 76:582–92, discussion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serkova NJ, Standiford TJ, Stringer KA. The emerging field of quantitative blood metabolomics for biomarker discovery in critical illnesses. Am J Respir Crit Care Med 2011; 184:647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chouchani ET, Pell VR, Gaude E et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014; 515:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Littlewood-Evans A, Sarret S, Apfel V et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 2016; 213:1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta 2016; 1857:1086–1101. [DOI] [PubMed] [Google Scholar]
- 12.D'Alessandro A, Moore HB, Moore EE et al. Early hemorrhage triggers metabolic responses that build up during prolonged shock. Am J Physiol Regul Integr Comp Physiol 2015; 308:R1034–R1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Human Protein Atlas. 2019. 12-15-2019. [Google Scholar]
- 14.D'Alessandro A, Slaughter AL, Peltz ED et al. Trauma/hemorrhagic shock instigates aberrant metabolic flux through glycolytic pathways, as revealed by preliminary (13)Cglucose labeling metabolomics. J Transl Med 2015; 13:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D'Alessandro A, Moore HB, Moore EE et al. Plasma succinate is a predictor of mortality in critically injured patients. J Trauma Acute Care Surg 2017; 83:491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhlen M, Oksvold P, Fagerberg L et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 2010; 28:1248–1250. [DOI] [PubMed] [Google Scholar]
- 17.Uhlen M, Fagerberg L, Hallstrom BM et al. Proteomics. Tissue-based map of the human proteome. Science 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 18.Hauser CJ, Otterbein LE. Danger signals from mitochondrial DAMPS in trauma and post-injury sepsis. Eur J Trauma Emerg Surg 2018; 44:317–324. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Raoof M, Chen Y et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464:104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills E, O'Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol 2014; 24:313–320. [DOI] [PubMed] [Google Scholar]
- 21.Biffl WL, Moore FA, Moore EE et al. Are corticosteroids salvage therapy for refractory acute respiratory distress syndrome? Am J Surg 1995; 170:591–595. [DOI] [PubMed] [Google Scholar]
- 22.Singh VK, Wise SY, Fatanmi OO et al. Alpha-tocopherol succinate- and AMD3100-mobilized progenitors mitigate radiation combined injury in mice. J Radiat Res 2014; 55:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Svennevig JL, Bugge-Asperheim B, Vaage J et al. Corticosteroids in the treatment of blunt injury of the chest. Injury 1984; 16:80–84. [DOI] [PubMed] [Google Scholar]
- 24.Kelher MR, Masuno T, Moore EE et al. Plasma from stored packed red blood cells and MHC class I antibodies causes acute lung injury in a 2-event in vivo rat model. Blood 2009; 113:2079–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Looney MR, Su X, Van Ziffle JA et al. Neutrophils and their Fc gamma receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest 2006; 116:1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salzer WL, McCall CE. Primed stimulation of isolated perfused rabbit lung by endotoxin and platelet activating factor induces enhanced production of thromboxane and lung injury. J Clin Invest 1990; 85:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyman TH, Bjornsen AJ, Elzi DJ et al. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol 2002; 283:C1592–C1603. [DOI] [PubMed] [Google Scholar]
- 28.Downey GP, Worthen GS, Henson PM et al. Neutrophil sequestration and migration in localized pulmonary inflammation. Capillary localization and migration across the interalveolar septum. Am Rev Respir Dis 1993; 147:168–176. [DOI] [PubMed] [Google Scholar]
- 29.Moore HB, Moore EE, Chapman MP et al. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: a randomised trial. Lancet 2018; 392:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ranieri VM, Rubenfeld GD, Thompson BT et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 2012; 307:2526–2533. [DOI] [PubMed] [Google Scholar]
- 31.D'Alessandro A, Reisz JA, Zhang Y et al. Effects of aged stored autologous red blood cells on human plasma metabolome. Blood Adv 2019; 3:884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemkov T, Reisz JA, Gehrke S et al. High-Throughput Metabolomics: Isocratic and Gradient Mass Spectrometry-Based Methods. Methods Mol Biol 2019; 1978:13–26. [DOI] [PubMed] [Google Scholar]
- 33.Stefanoni D, Shin HKH, Baek JH et al. Red blood cell metabolism in Rhesus macaques and humans: comparative biology of blood storage. Haematologica 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas T, Stefanoni D, Reisz JA et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samuels JM, Moore EE, Silliman CC et al. Severe traumatic brain injury is associated with a unique coagulopathy phenotype. J Trauma Acute Care Surg 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silliman CC, Elzi DJ, Ambruso DR et al. Lysophosphatidylcholines prime the NADPH oxidase and stimulate multiple neutrophil functions through changes in cytosolic calcium. J Leukoc Biol 2003; 73:511–524. [DOI] [PubMed] [Google Scholar]
- 37.Kelher MR, McLaughlin NJ, Banerjee A et al. LysoPCs induce Hck- and PKCdeltamediated activation of PKCgamma causing p47phox phosphorylation and membrane translocation in neutrophils. J Leukoc Biol 2017; 101:261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLaughlin NJ, Banerjee A, Khan SY et al. Platelet-activating factor-mediated endosome formation causes membrane translocation of p67phox and p40phox that requires recruitment and activation of p38 MAPK, Rab5a, and phosphatidylinositol 3-kinase in human neutrophils. J Immunol 2008; 180:8192–8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhuniya D, Umrani D, Dave B et al. Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg Med Chem Lett 2011; 21:3596–3602. [DOI] [PubMed] [Google Scholar]
- 40.Silliman CC, Kelher MR, Khan SY et al. Supernatants and lipids from stored red blood cells activate pulmonary microvascular endothelium through the BLT2 receptor and protein kinase C activation. Transfusion 2017; 57:2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akbas N, Gonzalez G, Edwards R et al. Assessment of liver function tests on Piccolo Xpress point of care chemistry analyzer in a pediatric hospital. Pract Lab Med 2015; 3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauaia A, Moore FA, Moore EE. Postinjury Inflammation and Organ Dysfunction. Crit Care Clin 2017; 33:167–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walters JD, Sorboro DM, Chapman KJ. Polyamines enhance calcium mobilization in fMet-Leu-Phe-stimulated phagocytes. FEBS Lett 1992; 304:37–40. [DOI] [PubMed] [Google Scholar]
- 44.Hakak Y, Lehmann-Bruinsma K, Phillips S et al. The role of the GPR91 ligand succinate in hematopoiesis. J Leukoc Biol 2009; 85:837–843. [DOI] [PubMed] [Google Scholar]
- 45.He W, Miao FJ, Lin DC et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004; 429:188–193. [DOI] [PubMed] [Google Scholar]
- 46.D'Alessandro A, Hansen KC, Silliman CC et al. Metabolomics of AS-5 RBC supernatants following routine storage. Vox Sang 2015; 108:131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.D’Alessandro A, Nemkov T, Kelher M et al. Routine storage of red blood cell (RBC) units in additive solution-3: a comprehensive investigation of the RBC metabolome. Transfusion 2015; 55:1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciesla DJ, Moore EE, Johnson JL et al. Decreased progression of postinjury lung dysfunction to the acute respiratory distress syndrome and multiple organ failure. Surgery 2006; 140:640–647. [DOI] [PubMed] [Google Scholar]
- 49.Gonzalez RJ, Moore EE, Ciesla DJ et al. Post-hemorrhagic shock mesenteric lymph activates human pulmonary microvascular endothelium for in vitro neutrophil-mediated injury: the role of intercellular adhesion molecule-1. J Trauma 2003; 54:219–223. [DOI] [PubMed] [Google Scholar]
- 50.Lomas-Neira JL, Heffernan DS, Ayala A et al. BLOCKADE OF ENDOTHELIAL GROWTH FACTOR, ANGIOPOIETIN-2, REDUCES INDICES OF ARDS AND MORTALITY IN MICE RESULTING FROM THE DUAL-INSULTS OF HEMORRHAGIC SHOCK AND SEPSIS. Shock 2016; 45:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Partrick DA, Moore FA, Moore EE et al. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz 1996; 4:194–210. [PubMed] [Google Scholar]
- 52.Rezende-Neto JB, Moore EE, Masuno T et al. The abdominal compartment syndrome as a second insult during systemic neutrophil priming provokes multiple organ injury. Shock 2003; 20:303–308. [DOI] [PubMed] [Google Scholar]
- 53.Andrienko TN, Pasdois P, Pereira GC et al. The role of succinate and ROS in reperfusion injury - A critical appraisal. J Mol Cell Cardiol 2017; 110:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nadjsombati MS, McGinty JW, Lyons-Cohen MR et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 2018; 49:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Howard BM, Kornblith LZ, Hendrickson CM et al. Differences in degree, differences in kind: characterizing lung injury in trauma. J Trauma Acute Care Surg 2015; 78:735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content 3. Table of all the metabolites measured in the plasma of severely injured patients. doc
Supplemental Digital Content 2, Figure: Succinate levels over time in severely injured patients. Plasma samples from 67 severely injured (NISS>15) patients: 21 with ARDS and 46 without, were collected in the field or ED and during admission, up to 7 days. Succinate levels (left y-axis) were measured with metabolomics and the medians for patients that developed ARDS (blue dashed line, blue squares) and those that did not (solid gray line, gray circles) were plotted. Succinate levels were significant at the initial collection (field/ED, *p<0.05) and were decreased by hour 2. The numbers of patient tested in each group (right y-axis) are also quantified for each time point for critically injured patients with ARDS (n=21, blue horizontal hatch marks) and without ARDS (n=46, gray triangles). Only one critically ill patient died with ARDS just after hour 48, and no metabolomics samples were drawn once the patient left the Surgical Intensive Care Unit.
Supplemental Digital Content 1. Methods described in more detail. Doc