

Abstract
EGb 761 has some protective effects on AD and can improve the cognitive functions of AD mice. However, the underlying molecular mechanisms are unknown. Here, we investigated the function of bilobalide, the effective component of EGb 761, in neuroinflammation and autophagy during AD. LPS-treated BV-2 cells were used as an in vitro model for neuroinflammation. The APP/PS1 AD mouse line was used to examine the function of bilobalide in AD. ELISA and qRT-PCR were used to measure the levels of proinflammatory cytokines, including TNF-α, IL-6 and IL-1β. Western blotting was employed to determine the protein levels of p-p65, iNOS, COX-2, LC3, beclin-1, p62 and p-STAT3. Immunostaining was applied to examine the number of autophagosomes. LPS treatment induced inflammatory responses and inhibited autophagy in BV-2 cells. Bilobalide suppressed LPS-induced neuroinflammation and promoted autophagy. Furthermore, bilobalide treatment increased the lincRNA-p21 levels, which suppressed STAT3 signalling. Knockdown of lincRNA-p21 reversed the effects of bilobalide. Overexpression of lincRNA-p21 promoted autophagy and inhibited neuroinflammation as well while STAT3 inhibitor blocked the effects of si-lincRNA-p21. In vivo experiments revealed that bilobalide improved the learning and memory capabilities of APP/PS1 AD mice. Bilobalide improves the cognitive functions of APP/PS1 AD mice. Mechanistically, bilobalide suppresses inflammatory responses and promotes autophagy possibly by upregulating lincRNA-p21 levels.
Keywords: Alzheimer’s disease, autophagy, bilobalide, lincRNA-p21, neuroinflammation
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease and the most common cause of dementia, which is characterized by a progressive and irreversible loss of cognitive functions, such as learning, memory and language [1,2]. AD patients usually require around-the-clock care and end up bed-bound, resulting in a huge burden on their family. At present, the disease is incurable and ultimately fatal despite numerous preclinical and clinical studies. The cellular and molecular mechanisms of AD are complex and remain elusive [3]. Therefore, further investigations of the underlying mechanisms are essential for future therapy development.
It has been widely acknowledged that AD is a multifactorial disorder, and risk factors including β-amyloid (Aβ) deposition, neurofibrillary tangle formation, and oxidative stress all contribute to AD development [1,4]. Over the past decades, it has been revealed that AD pathogenesis is not only restricted to neuron development but also involves interactions with immunological processes [5]. Increasing evidence suggests that neuroinflammation plays a crucial and even causal role in AD [6,7]. Many genes involved in the immune system are associated with AD [8,9]. Moreover, activation of microglial cells triggers innate immune responses and inflammatory cytokine release, which cause damage to neurons and contribute to AD progression and severity [10]. Thus, targeting neuroinflammation could be a potential way to treat AD.
The biggest hallmark of AD is the deposition of senile plaques that are composed mainly of Aβ and neurofibrillary tangles [1]. Plaque formation is crucially influenced by the autophagy process, which is a lysosome-dependent process that functions to degrade organelles and proteins [11]. Autophagy is a key regulator of Aβ generation and clearance, and many studies have indicated that impaired autophagy is an important mechanism involved in the pathology of Aβ metabolism in AD [12]. In addition, it has been suggested that there is cross-talk between autophagy and neuroinflammation in AD [13,14]. Autophagy decreases inflammation, although neuroinflammation can activate autophagy [15]. Upregulating the autophagy process might protect against AD through modulating the interaction with the inflammation process.
Our group and other groups have previously shown that EGb 761, an extract from the leaves of Ginkgo biloba, can improve the cognitive functions of AD mice [16,17], but the underlying molecular mechanisms are not clear. Bilobalide is the effective component of EGb761. Here, we sought to examine the mechanisms of how bilobalide protects against AD. We found that bilobalide could inhibit neuroinflammation and increase autophagy by upregulating lincRNA-p21, which leads to improved learning and memory capabilities in AD mice. This study helps shed light on the mechanisms of AD and, more importantly, provides avenues for future therapeutic approaches for AD.
Materials and methods
Animals and drug administration
All animal studies were approved by the Animal Care and Use Committee of The First Affiliated Hospital of Soochow University and performed under its guidance. APP/PS1 transgenic mice expressing human APP and PS1 mutant proteins were purchased from the Laboratory Animal Centre of Hunan Province (Changsha, Hunan, China). Transgenic mice and control wild-type C57 mice were housed in the standard university animal facility.
Bilobalide was dissolved in 10% DMSO in saline to make the stock solution, and bilobalide (4 mg/kg, 8 mg/kg) or vehicle was added to the food. APP/PS1 animals or control animals that were 2 months old were fed bilobalide containing food or vehicle for 6 months before sacrifice for further experiments.
BV-2 cell culture and LPS treatment
The murine microglia cell line (BV-2) was purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle Medium-high glucose (DMEM-HG) containing 10% foetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin-streptomycin. Cells were maintained in an incubator with 5% CO2 at 37°C.
To induce inflammation, lipopolysaccharide (LPS, 1 μg/ml) was added to the culture media for the indicated amount of time.
Plasmid and transfection
The lincRNA-p21 overexpression plasmid and empty vector (pIRES2-EGFP) were purchased from Shanghai GenePharma Co., Ltd. Transfection was performed using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer’s protocol.
Immunohistochemistry
Brain tissue was fixed in 4% formalin and embedded in paraffin. Paraffin sections (20 μm thick) were washed in PBS and briefly treated with 80% methanol containing 0.3% H2O2 followed by blocking with 10% normal goal serum for 2 h at room temperature. The anti-beta amyloid 1-42 primary antibody (1:200, Abcam, USA, ab10148) was incubated with the slices overnight at 4°C, and then the slices were incubated with HRP-labelled secondary antibody for 1 h at room temperature. The signals were analysed with 3,3’-diaminobenzidine substrate and counterstained with haematoxylin.
Morris water maze (MWM)
The MWM test was performed as previously described [18]. Briefly, animals were handled for 4 days before any training. Prior to the first training trial, mice were subjected to a single habituation trial without the platform to measure their basal swim speed. During the training (days 1-5), mice were randomly placed into the pool with the hidden platform in the same quadrant (target quadrant). The swim path and latency in finding the platform were tracked with a computerized video tracking system. Mice were trained for 4 trials per session for 2 sessions per day for 5 consecutive days. For the probe test (day 6), the platform was removed, and the preference for the target quadrant was measured.
Enzyme-linked immunosorbent assay (ELISA)
TNF-α, IL-6, IL-1β, and Aβ1-42 levels were determined with specific commercial ELISA kits (Invitrogen, CA, USA) according to the manufacturer’s protocols. The samples were extracted with the extraction buffer, and the concentrations of each cytokine were calculated based on the standard curve.
Western blotting
Total protein was extracted from cultured cells or brain tissues with RIPA buffer (Beyotime Institute of Biotechnology, Nantong, China) containing protease inhibitor cocktail. The protein concentration was determined by the Pierce BCA protein assay (Thermo Fisher Scientific, Massachusetts, USA). Equal amounts of protein from each sample were loaded and separated by SDS-PAGE followed by transfer to PVDF membranes (Millipore, Massachusetts, USA). The membranes were blocked for 0.5 h at room temperature and then incubated with primary antibodies at 4°C overnight. The next day, the membranes were incubated with secondary antibodies after 3 washes with PBS and subsequently analysed with an ECL kit (Thermo Fisher Scientific, Massachusetts, USA). The following primary antibodies were used: p-p65 (1:500; sc-3033s; Santa Cruz, USA); p65 (1:1000; sc-8242; Santa Cruz, USA); iNOS (1:1000; sc-12120; Santa Cruz, USA); COX-2 (1:500; sc-4842; Santa Cruz, USA); LC3-I/II (1:500; sc-4108; Santa Cruz, USA); Beclin-1 (1:500; sc-3738; Santa Cruz, USA); p62 (1:500; sc-5114S; Santa Cruz, USA); p-STAT3 (1:500; sc-9131; Santa Cruz, USA); STAT3 (1:500; sc-9139; Santa Cruz, USA); and β-actin (1:2000; sc-47778; Santa Cruz, USA).
RNA extraction and qRT-PCR
Total RNA was extracted from cultured cells or brain tissues using an RNeasy kit (Qiagen, USA) according to the manufacturer’s instructions. DNase I was used to avoid DNA contamination. One microgram of total RNA from each sample was used for reverse transcription with the SuperScript® IV First-Strand Synthesis System (Invitrogen, Massachusetts, USA) before PCR amplification using 1x Power SYBR® Green PCR Master Mix (Invitrogen, Massachusetts, USA). The relative expression levels of mRNAs or miRNAs were normalized to that of GAPDH mRNA or U6 small nuclear RNA (snRNA), respectively, as internal controls. The following primers were used: TNF-α forward primer: 5’-AGGCGCTCCCCAAGAAGACA-3’; TNF-α reverse primer: 5’-TCCTTGGCAAAACTGCACCT-3’; IL-6 forward primer: 5’-GGTACATCCTCGACGGCATCT-3’; IL-6 reverse primer: 5’-GT GCCTCTTTGCTGCTTTCAC-3’; IL-1β forward primer: 5’-AACCTGCTGGTGTGTGACGTTC-3’; IL-1β rever-se primer: 5’-CAGCACGAGGCTTTTTTGTTGT-3’; GAPDH forward primer: 5’-AGGTCGGTGTGAACGGATTTG-3’; GAPDH reverse primer: 5’-GGGGTCGTTGATGGCAACA-3’; lincRNA-p21 forward primer: 5’-CCTGTTCCACTCGCTTTCCA-3’; and lincRNA-p21 reverse primer: 5’-GGAACTGGAGACGGAATGTC-3’.
Immunostaining
Cells were fixed in 4% paraformaldehyde (PFA) at room temperature for 15 minutes and permeabilized with 0.1% Triton X-100 in PBS for half an hour at room temperature. Then, the cells were blocked with 1% BSA in PBS for 1 hour at room temperature followed by incubation with rabbit anti-LC3-II primary antibody (1: 500; sc-271625; Santa Cruz, USA) at 4°C overnight. Cells were then washed with PBS and incubated with TRITC-conjugated secondary antibody for 2 hours at room temperature. Images were acquired with a standard microscope. Cells containing >10 LC3 vacuoles were counted as LC3+ vacuole-positive cells. At least 100 cells were counted for each group.
Statistical analysis
All statistical analyses were performed in GraphPad Prism 7. Statistical significance was determined with an unpaired Student’s t test (two groups) or one-way ANOVA followed by Tukey’s post-test (more than two groups) as indicated in the figure legends. The data are presented as the mean ± SD.
Results
Bilobalide suppressed LPS-induced activation of BV-2 microglial cells
To investigate the molecular mechanisms underlying the effect of bilobalide on AD, we first examined the function of bilobalide in neuroinflammation, which has been shown to play a critical role in AD development and progression [7]. We employed the murine microglial cell line BV-2 and activated the cells via LPS treatment as previously described [19,20]. First, we measured the cytotoxicity of bilobalide using the MTT assay. BV-2 cells were treated with vehicle or bilobalide (10, 50, 100, 200, 500, or 1000 nM) for 12 hours, and the cell viabilities were measured. We observed no obvious cytotoxicity induced by bilobalide at concentrations lower than 500 nM, but 1000 nM bilobalide significantly decreased cell viability (Figure 1A).
Figure 1.

Bilobalide suppressed LPS-induced activation of BV-2 microglial cells. A. Cell viability following bilobalide treatment at various concentrations was measured using MTT assay. The viability was compromised only at high concentration (1000 nM). B-D. The secreted levels of TNF-α, IL-6 and IL-1β from cultured cells following LPS treatment with or without bilobalide incubation were examined by ELISA. LPS treatment alone increased those levels, but bilobalide suppressed those increases. E-G. The mRNA levels of TNF-α, IL-6 and IL-1β in cultured cells following LPS treatment with or without bilobalide incubation were measured by qRT-PCR. LPS treatment upregulated those levels while bilobalide treatment inhibited those upregulations. H. The protein levels of p-p65, iNOS and COX-2 in cultured cells following LPS treatment with or without bilobalide incubation were measured by western blotting. LPS treatment alone increased those protein levels while bilobalide suppressed those increases. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
To test the potential effects of bilobalide on proinflammatory responses, BV-2 microglial cells were first treated with LPS for 12 hours and then treated with vehicle or bilobalide (50, 100, or 200 nM) for another 12 hours. Proinflammatory cytokine levels were measured with ELISA. The results showed that LPS stimulation induced the release of proinflammatory cytokines, including TNF-α, IL-6 and IL-1β (Figure 1B-D). However, bilobalide (100, 200 nM) significantly reduced LPS-induced proinflammatory cytokine release, while 50 nM bilobalide treatment had no significant effects (Figure 1B-D). As an alternative approach, we used qRT-PCR to measure the expression levels of TNF-α, IL-6 and IL-1β. Consistent with the ELISA results, bilobalide (100, 200 nM) greatly inhibited the expression of proinflammatory cytokines induced by LPS treatment (Figure 1E-G). Western blotting was applied to determine the activation of the NF-κB signalling pathway. Again, we observed increased expression of p-p65, iNOS and COX-2 in BV-2 microglial cells following LPS stimulation, and bilobalide (100, 200 nM) significantly suppressed the upregulation of their expression induced by LPS, while 50 nM bilobalide did not have any effects (Figure 1H). Taken together, these data demonstrate that bilobalide inhibits LPS-induced BV-2 cell activation and proinflammatory processes.
Bilobalide enhanced autophagy and upregulated lincRNA-p21 in LPS-stimulated BV-2 cells
Next, we tested the effect of bilobalide on autophagy. As expected, LPS treatment decreased the number of LC3 puncta in BV-2 cells (Figure 2A). Bilobalide (100 or 200 nM) incubation significantly rescued the level of LC3 and inhibited the downregulation of autophagy induced by LPS (Figure 2A), while 50 nM bilobalide had no effects (Figure 2A). To further confirm this, we examined the levels of LC3, beclin-1, and p62 using Western blotting. Consistently, we observed a decreased LC3-II/LC3-I ratio and a reduced level of beclin-1 following LPS treatment (Figure 2B, 2C). In contrast, p62 and p-STAT3 were upregulated (Figure 2B, 2C). These results indicate that autophagy was greatly suppressed. Bilobalide (100, 200 nM) treatment rescued the levels of related proteins and upregulated autophagy (Figure 2B, 2C). We also examined the level of lincRNA-p21, a long noncoding RNA that has been shown to inhibit apoptosis [21]. We found that LPS treatment downregulated lincRNA-p21 levels, while bilobalide (100, 200 nM) reversed this downregulation (Figure 2D). Therefore, we conclude that bilobalide promotes autophagy, and this effect might be mediated by increases in lncRNA-p21 levels.
Figure 2.

Bilobalide increased autophagy by upregulating lincRNA-p21. A. Immunostaining images showed LC3 intensity and DAPI signal in cultured cells following LPS treatment with or without bilobalide incubation. The bar graph quantified the number of LC-3 positive autophagosomes in cells treated with or without bilobalide following LPS treatment. LPS alone decreased the number of LC3 puncta while bilobalide treatment recovered the number. B. The protein levels of LC3-I, LC3-II, beclin-1, and p62 in cultured cells following LPS treatment with or without bilobalide incubation were measured by western blotting. Bar graphs quantified the protein levels. LPS alone diminished the LC3II/LC3I ratio and Becline-1 level but increased p62 level while bilobalide treatment reversed those changes. C. The protein levels of p-STAT3 in cultured cells following LPS treatment with or without bilobalide incubation were examined by western blotting. The bar graph quantified the protein levels. LPS treatment alone increased pSTAT3 level while bilobalide suppressed the increase. D. lincRNA-p21 levels in cultured cells following LPS treatment with or without bilobalide incubation were analysed by qRT-PCR. LPS diminished lincRNA-p21 level while bilobalide recovered the level. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
Bilobalide regulated inflammation and autophagy via upregulating lincRNA-p21
To further investigate whether bilobalide modulates inflammation and autophagy through lincRNA-21, we silenced lncRNA-21 expression during the treatment of bilobalide. We found that the suppression in expression of proinflammatory cytokines including TNF-α, IL-6 and IL-1β mediated by bilobalide was lifted by si-lincRNA-p21 (Figure 3A-C). Similarly, the bilobalide-induced restrains in expression of p-p65, iNOS and COX-2 were also removed by si-lincRNA-p21 (Figure 3D). Furthermore, the effects of bilobalide on expression of autophagy-related proteins such as LC3-II/LC3-I ratio, beclin-1, and p62 were partially blocked by si-lincRNA-p21 (Figure 3E). These results indicate that bilobalide modulates inflammation and autophagy through upregulating lincRNA-p21.
Figure 3.

Bilobalide regulated inflammation and autophagy via upregulating lincRNA-p21. A-C. The mRNA levels of TNF-α, IL-6 and IL-1β in cells transfected with si-lincRNA-p21 or si-NC following LPS treatment with or without bilobalide incubation were analysed by qRT-PCR. LPS treatment upregulated those levels while bilobalide treatment inhibited those upregulations. Si-lincRNA-p21 blocked the effects of bilobalide. D. The protein levels of p-p65, iNOS and COX-2 cells transfected with si-lincRNA-p21 or si-NC following LPS treatment with or without bilobalide incubation were analysed were measured by western blotting. LPS treatment alone increased those protein levels while bilobalide suppressed those increases. Si-lincRNA-p21 blocked the effects of bilobalide. E. The protein levels of LC3-I, LC3-II, beclin-1, and p62 in cells transfected with si-lincRNA-p21 or si-NC following LPS treatment with or without bilobalide incubation were measured by western blotting. Bar graphs quantified the protein levels. LPS alone diminished the LC3II/LC3I ratio and Becline-1 level but increased p62 level while bilobalide treatment reversed those changes. Si-lincRNA-p21 blocked the effects of bilobalide. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
LincRNA-p21 inhibited LPS-induced activation of BV-2 cells
To further examine the underlying molecular mechanism, we studied the role of lincRNA-p21 in LPS-induced neuroinflammation. First, we observed a lower level of lincRNA-p21 in BV-2 cells after LPS treatment compared to that observed after vehicle treatment, and transfection of the lincRNA-p21 construct greatly increased the lincRNA-p21 level (Figure 4A). Moreover, overexpression of lincRNA-p21 significantly reduced LPS-induced release of proinflammatory cytokines, including TNF-α, IL-6 and IL-1β (Figure 4B-D). Using qRT-PCR, we also confirmed that the upregulation of proinflammatory factors whose expression was induced by LPS was suppressed by lincRNA-p21 (Figure 4E-G). Furthermore, lincRNA-p21 inhibited the LPS-induced activation of NF-κB (Figure 4H). Taken together, these results show that lincRNA-p21 restrains LPS-induced proinflammatory responses.
Figure 4.

LincRNA-p21 inhibited LPS-induced activation of BV-2 cells. A. LincRNA-p21 levels in cells transfected with the lincRNA-p21 or control vector following LPS treatment were measured by qRT-PCR. LPS treatment diminished lincRNA-p21 level. Overexpression of lincRNA-p21 in LPS treated cells recovered the level. B-D. The secreted levels of TNF-α, IL-6 and IL-1β by cells transfected with the lincRNA-p21 or control vector following LPS treatment were measured by ELISA. LPS treatment alone upregulated those levels while overexpression of lincRNA-p21 suppressed those upregulations. E-G. The mRNA levels of TNF-α, IL-6 and IL-1β in cells transfected with the lincRNA-p21 or control vector following LPS treatment were analysed by qRT-PCR. LPS treatment alone increased those levels while overexpression of lincRNA-p21 suppressed those increases. H. The protein levels of p-p65, iNOS and COX-2 in cells transfected with the lincRNA-p21 or control vector following LPS treatment were measured by western blotting. Bar graphs quantified the protein levels. LPS treatment alone increases those protein levels while lincRNA-p21 overexpression reversed those changes. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
LincRNA-p21 promoted autophagy and suppressed STAT3 signalling in LPS-stimulated BV-2 cells
To directly study whether lincRNA-p21 is involved in LPS-induced autophagy regulation, we overexpressed lincRNA-p21 and examined the ensuing effects. LPS treatment decreased the number of autophagosomes in BV-2 cells, while overexpression of lincRNA-p21 reversed this decrease (Figure 5A). Furthermore, the changes in the LC3-II/LC3-I ratio and the levels of other autophagy-related proteins, including beclin-1, p62 and p-STAT3, induced by LPS treatment were all reversed by lincRNA-p21 overexpression (Figure 5B, 5C). These data suggest that lincRNA-p21 promoted autophagyand that this effect might contribute to STAT3 signalling suppression.
Figure 5.

LincRNA-p21 promoted autophagy by suppressing STAT3 signalling. A. Immunostaining images showed LC3 intensity and DAPI signal in cells transfected with the lincRNA-p21 or control vector following LPS treatment. The bar graph quantified the number of LC-3 positive autophagosomes. LPS alone decreased the number of LC3 puncta while lincRNA-p21 overexpression recovered the number. B. The protein levels of LC3-I, LC3-II, beclin-1, and p62 in cells transfected with the lincRNA-p21 or control vector following LPS treatment were measured by western blotting. Bar graphs quantified the protein levels. LPS alone diminished the LC3II/LC3I ratio and Becline-1 level but increased p62 level while lincRNA-p21 overexpression reversed those changes. C. The protein levels of p-STAT3 in cells transfected with the lincRNA-p21 or control vector following LPS treatment were examined by western blotting. The bar graph quantified the protein levels. LPS treatment alone increased pSTAT3 level while lincRNA-p21 overexpression suppressed the increase. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
LincRNA-p21 modulated inflammation and autophagy via inhibiting STAT3 signalling
To further investigate whether lincRNA-p21 regulate inflammation and autophagy via STAT3, we silenced STAT3 expression during the knockdown of lincRNA-p21. Consistent with aforementioned results, knockdown of lincRNA-p21 further increased the levels of proinflammatory cytokines induced by LPS treatment, such as TNF-α, IL-6 and IL-1β (Figure 6A-C). However, STAT3 inhibitor blocked those increases (Figure 6A-C). In addition, si-lincRNA-p21 further upregulated the levels of p-p65, iNOS, and COX-2 while STAT3 inhibitor suppressed those upregulations (Figure 6D). Similarly, silencing lincRNA-p21 suppressed the LPS-induced autophagy while STAT3 inhibitor reversed the effects of si-lincRNA-p21 (Figure 6E). Therefore, we conclude that lincRNA-p21 regulates inflammation and autophagy via suppressing STAT3.
Figure 6.

LincRNA-p21 modulated inflammation and autophagy via inhibiting STAT3 signalling. A-C. The mRNA levels of TNF-α, IL-6 and IL-1β in cells transfected with si-lincRNA-p21 or si-NC following LPS treatment with or without STAT3 inhibitor (STAT3-IN-3) were analysed by qRT-PCR. LPS treatment upregulated those levels while si-lincRNA-p21 further increased those levels. STAT3-IN-3 blocked the effects of si-lincRNA-p21. D. The protein levels of p-p65, iNOS and COX-2 cells transfected with si-lincRNA-p21 or si-NC following LPS treatment with or without STAT3 inhibitor (STAT3-IN-3) were analysed were measured by western blotting. LPS treatment alone increased those protein levels while si-lincRNA-p21 further upregulated those levels. STAT3-IN-3 blocked the effects of si-lincRNA-p21. E. The protein levels of LC3-I, LC3-II, beclin-1, and p62 in cells transfected with the si-lincRNA-p21 or si-NC following LPS treatment with or without STAT3 inhibitor (STAT3-IN-3) were measured by western blotting. Bar graphs quantified the protein levels. LPS alone diminished the LC3II/LC3I ratio and Becline-1 level but increased p62 level while si-lincRNA-p21 exaggerated those changes. STAT3-IN-3 blocked the effects of si-lincRNA-p21. n = 3; *P < 0.05; **P < 0.01; ***P < 0.001.
Bilobalide improved the learning and memory of AD mice by decreasing Aβ accumulation
We showed that bilobalide suppressed the neuroinflammation process and promoted autophagy in LPS-stimulated BV-2 microglial cells. Next, we directly examined the function of bilobalide in AD using the APP/PS1 AD mouse line. We fed 2-month-old APP/PS1 mice food containing bilobalide for 6 months. First, we found that bilobalide feeding (4 or 8 mg/kg) greatly decreased Aβ accumulation in the brain compared to that in the vehicle group (Figure 7A). With ELISA, we showed that the accumulation of both soluble and insoluble Aβ1-42 protein were reduced following bilobalide feeding (4 and 8 mg/kg) (Figure 7B). Moreover, with the Morris water maze task, a learning and memory assay, we assessed the effect of bilobalide on cognitive functions. During the training and testing, the mice were automatically tracked, and their swim latencies and distances to the platform were measured. We found that wild-type (WT) mice learned the task quickly, and the latency to reach the platform and swimming distance gradually decreased with training, while APP/PS1 mice did not show evidence of this and failed to learn the task (Figure 7C, 7D). However, bilobalide-fed (4, 8 mg/kg) APP/PS1 mice learned a task similar to that of WT mice and had comparable latency and swimming distance during training (Figure 7C, 7D). There was no difference in swim velocity among groups of mice, suggesting that the differences were not caused by swim or motor capabilities (Figure 7E). Together, these results indicate that bilobalide can improve cognitive functions and decrease Aβ accumulation in AD mice.
Figure 7.
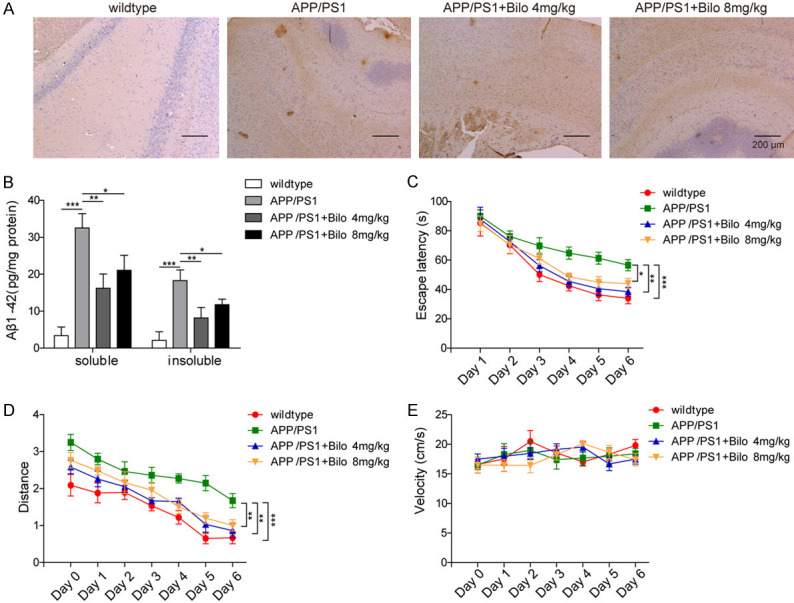
Bilobalide improved the learning and memory of AD mice by decreasing Aβ accumulation. A. IHC images showed the levels of Aβ accumulation in WT and APP/PS1 mice with or without bilobalide feeding. No Aβ accumulation was observed in WT mice while APP/PS1 mice exhibited high level of Aβ accumulation. Bilobalide treatment decreased the level of Aβ accumulation in APP/PS1 mice. B. The levels of soluble and insoluble Aβ1-42 in different groups of mice were analysed by ELISA. Compared to WT mice, APP/PS1 mice exhibited high levels of soluble and insoluble Aβ1-42. Bilobalide treatment decreased the levels. C. Learning curves based on the latency in finding the hidden platform during MWM training. APP/PS1 mice spent longer time in finding the hidden platform compared with other groups. D. Swimming distance before reaching the hidden platform during MWM training. APP/PS1 mice swam longer distances before reaching the hidden platform compared with other groups. E. Swim velocity of different groups of mice. No difference in swim velocity was observed among different groups. n = 6 for each group; *P < 0.05; **P < 0.01; ***P < 0.001.
Bilobalide inhibited neuroinflammation and promoted autophagy in vivo
Finally, we sought to confirm the molecular mechanisms of bilobalide function in vivo. We found that APP/PS1 AD mice had increased levels of TNF-α, IL-6 and IL-1β and that bilobalide feeding downregulated the mRNA levels of these proinflammatory factors (Figure 8A). Furthermore, autophagy was inhibited in APP/PS1 AD mice. The LC3-II/LC3-I ratio and beclin-1 were reduced, while p62 and p-STAT3 were upregulated in AD mice compared to WT mice (Figure 8B, 8C). However, bilobalide feeding rescued the levels of these autophagy-related proteins and promoted autophagy (Figure 8B, 8C). In addition, we measured the level of lincRNA-p21 in different groups of mice and found that lncRNA-p62 expression was reduced in APP/PS1 AD mice, but bilobalide feeding upregulated lincRNA-p21 levels (Figure 8D). Taken together, these in vivo results validated our findings in the in vitro model.
Figure 8.

Bilobalide inhibited neuroinflammation and promoted autophagy in vivo. A. The mRNA levels of TNF-α, IL-6 and IL-1β in different groups of mice were measured by qRT-PCR. APP/PS1 mice had higher levels compared to WT while bilobalide treatment downregulated those levels. B. The protein levels of LC3-I, LC3-II, beclin-1, and p62 in different groups of mice were measured by western blotting. Bar graphs quantified the protein levels. Compared with WT mice, APP/PS1 mice had lower levels of LC3II/LC3I ratio and Becline-1 but higher level of p62 while bilobalide treatment reversed those changes. C. The protein levels of p-STAT3 in different groups of mice were analysed by western blotting. Bar graph quantified the protein level. Compared with WT mice, APP/PS1 mice had higher level while bilobalide treatment reduced p-STAT3 level. D. LincRNA-p21 levels in different groups of mice were analysed by qRT-PCR. Compared with WT mice, APP/PS1 mice had lower lincRNA-p21 level while bilobalide treatment recovered the level. n = 6 for each group; *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
In the present study, we show that bilobalide inhibits the neuroinflammatory process but promotes autophagy, leading to improved cognitive function in AD mice. Mechanistically, we found that bilobalide upregulates lincRNA-p21 and suppresses the STAT3 signalling pathway. Our study reveals the molecular mechanisms underlying the protective effect of bilobalide on AD and further supports that neuroinflammation and autophagy play critical roles in AD development.
A great number of studies have indicated that EGb 761 has some protective effects on AD and can improve the cognitive functions of AD mice [16,17,22,23]. EGb 761 is a standard extract from Ginkgo biloba leaves and is made up of multiple components [24]. Some studies suggest that components of ginkgolide A, bilobalide, and flavonoids could promote autophagy to degrade p-Tao in lysosomes, while other components, such as ginkgolide B or C, do not have such effects [17]. However, the functions of each component and particularly the exact underlying mechanisms remain largely unknown, and further investigations are required. Here, we fully studied the function of bilobalide, the effective component of EGb 761. We show that feeding the mice bilobalide could significantly improve their learning and memory capabilities. Moreover, we examined its underlying molecular mechanisms. We show that bilobalide can increase lincRNA-p21 levels and promote autophagy while suppressing neuroinflammation. This will help shed light on the pathogenesis of AD and provide avenues for AD treatment.
The role of LPS in autophagy is controversial. Some studies reported that LPS induced autophagy [25]. However, other groups showed that LPS could suppress autophagy [26,27]. In our study, we did observe that LPS treatment significantly decreased LC3 puncta in BV-2 cells. Furthermore, the LC3-II/LC3-I ratio and beclin1 were diminished while p62 was upregulated following LPS treatment. These results provide strong evidence that LPS suppresses autophagy. This difference could result from the distinct treatment durations or concentrations and future investigation is necessary to examine that.
LncRNAs are long noncoding RNAs that can regulate specific gene expression [28,29]. An increasing number of studies have shown that many lncRNAs play a vital role in the progression of AD [30,31]. In this study, we showed that lincRNA-p21 expression is reduced in LPS-treated BV-2 cells as well as in APP/PS1 AD mice, suggesting that lincRNA-p21 is involved in AD. Moreover, overexpression of lincRNA-p21 could suppress neuroinflammation and promote autophagy, which is similar to the effects of bilobalide treatment, further supporting that it plays an important role in AD. The interaction between lincRNA-p21 and EGb 761 has been reported, which shows that EGb 761-induced upregulation of lincRNA-p21 could inhibit colorectal cancer metastasis [32]. Here, we observed a similar interaction in BV-2 cells and APP/PS1 AD mice and found that bilobalide treatment could increase lincRNA-p21 levels. Previous studies have shown that lincRNA-p21 directly binds to STAT3 and functions to block JAK2/STAT3 signalling [33]. Meanwhile, STAT3 phosphorylation could inhibit autophagy [34,35]. Therefore, taken together, our results suggest that bilobalide promotes autophagy and inhibits neuroinflammation by upregulating lincRNA-p21, which functions to suppress STAT3 signalling. In addition, lincRNA-p21 has been shown to regulate the HIF1-α/AKT/mTOR pathway, which is greatly involved in autophagy [36]. Future studies are necessary to examine the mechanism of how lincRNA-p21 affects autophagy in BV-2 cells and APP/PS1 AD mice.
In summary, our study provides detailed molecular mechanisms underlying the protective effect of bilobalide on AD and reveals for the first time that bilobalide improves the cognitive functions of AD mice by inhibiting inflammatory responses and promoting autophagy, in which lincRNA-p21 might be involved. Targeting these signalling pathways could be an avenue for AD treatment in the future.
Acknowledgements
This work was supported by Natural Science Foundation of Jiangsu Province [grant numbers: BK20170362]. All animal studies were approved by the Animal Care and Use committee of The First Affiliated Hospital of Soochow University and performed under the guidance of it.
The informed consent obtained from study participants.
Disclosure of conflict of interest
None.
References
- 1.Sharma P, Srivastava P, Seth A, Tripathi PN, Banerjee AG, Shrivastava SK. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog Neurobiol. 2019;174:53–89. doi: 10.1016/j.pneurobio.2018.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Magalingam KB, Radhakrishnan A, Ping NS, Haleagrahara N. Current concepts of neurodegenerative mechanisms in alzheimer’s disease. Biomed Res Int. 2018;2018:3740461. doi: 10.1155/2018/3740461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kocahan S, Dogan Z. Mechanisms of alzheimer’s disease pathogenesis and prevention: the brain, neural pathology, N-methyl-D-aspartate receptors, tau protein and other risk factors. Clin Psychopharmacol Neurosci. 2017;15:1–8. doi: 10.9758/cpn.2017.15.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crous-Bou M, Minguillon C, Gramunt N, Molinuevo JL. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9:71. doi: 10.1186/s13195-017-0297-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Calsolaro V, Edison P. Neuroinflammation in alzheimer’s disease: current evidence and future directions. Alzheimers Dement. 2016;12:719–732. doi: 10.1016/j.jalz.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 6.Sawikr Y, Yarla NS, Peluso I, Kamal MA, Aliev G, Bishayee A. Neuroinflammation in alzheimer’s disease: the preventive and therapeutic potential of polyphenolic nutraceuticals. Adv Protein Chem Struct Biol. 2017;108:33–57. doi: 10.1016/bs.apcsb.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in alzheimer’s disease. Alzheimers Dement (N Y) 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J Alzheimer Genetic Analysis Group. TREM2 variants in alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015;77:43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zare-Shahabadi A, Masliah E, Johnson GV, Rezaei N. Autophagy in Alzheimer’s disease. Rev Neurosci. 2015;26:385–395. doi: 10.1515/revneuro-2014-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, Stankiewicz AM. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:04. doi: 10.3389/fnagi.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bostanciklioglu M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene. 2019;705:157–166. doi: 10.1016/j.gene.2019.04.040. [DOI] [PubMed] [Google Scholar]
- 14.Bussi C, Peralta Ramos JM, Arroyo DS, Gaviglio EA, Gallea JI, Wang JM, Celej MS, Iribarren P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci Rep. 2017;7:43153. doi: 10.1038/srep43153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, Luo W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience. 2016;319:155–167. doi: 10.1016/j.neuroscience.2016.01.035. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Hao W, Qin Y, Decker Y, Wang X, Burkart M, Schotz K, Menger MD, Fassbender K, Liu Y. Long-term treatment with Ginkgo biloba extract EGb 761 improves symptoms and pathology in a transgenic mouse model of Alzheimer’s disease. Brain Behav Immun. 2015;46:121–131. doi: 10.1016/j.bbi.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Qin Y, Zhang Y, Tomic I, Hao W, Menger MD, Liu C, Fassbender K, Liu Y. Ginkgo biloba extract EGb 761 and its specific components elicit protective protein clearance through the autophagy-lysosomal pathway in tau-transgenic mice and cultured neurons. J Alzheimers Dis. 2018;65:243–263. doi: 10.3233/JAD-180426. [DOI] [PubMed] [Google Scholar]
- 18.Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You MM, Chen YF, Pan YM, Liu YC, Tu J, Wang K, Hu FL. Royal jelly attenuates LPS-induced inflammation in BV-2 microglial cells through modulating NF-kappaB and p38/JNK signaling pathways. Mediators Inflamm. 2018;2018:7834381. doi: 10.1155/2018/7834381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nam HY, Nam JH, Yoon G, Lee JY, Nam Y, Kang HJ, Cho HJ, Kim J, Hoe HS. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J Neuroinflammation. 2018;15:271. doi: 10.1186/s12974-018-1308-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Islam Khan MZ, Tam SY, Law HKW. Autophagy-modulating long non-coding RNAs (LncRNAs) and their molecular events in cancer. Front Genet. 2018;9:750. doi: 10.3389/fgene.2018.00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lautenschlager NT, Ihl R, Muller WE. Ginkgo biloba extract EGb 761(R) in the context of current developments in the diagnosis and treatment of age-related cognitive decline and Alzheimer’s disease: a research perspective. Int Psychogeriatr. 2012;24(Suppl 1):S46–50. doi: 10.1017/S1041610212001019. [DOI] [PubMed] [Google Scholar]
- 23.Muller WE, Eckert A, Eckert GP, Fink H, Friedland K, Gauthier S, Hoerr R, Ihl R, Kasper S, Moller HJ. Therapeutic efficacy of the Ginkgo special extract EGb761® within the framework of the mitochondrial cascade hypothesis of Alzheimer’s disease. World J Biol Psychiatry. 2019;20:173–189. doi: 10.1080/15622975.2017.1308552. [DOI] [PubMed] [Google Scholar]
- 24.EGb 761: ginkgo biloba extract, Ginkor. Drugs R D. 2003;4:188–193. doi: 10.2165/00126839-200304030-00009. [DOI] [PubMed] [Google Scholar]
- 25.Wang C, Wang Q, Lou Y, Xu J, Feng Z, Chen Y, Tang Q, Zheng G, Zhang Z, Wu Y, Tian N, Zhou Y, Xu H, Zhang X. Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J Cell Mol Med. 2018;22:1148–1166. doi: 10.1111/jcmm.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuang X, Yu Y, Jiang Y, Zhao S, Wang Y, Su L, Xie K, Yu Y, Lu Y, Lv G. Molecular hydrogen attenuates sepsis-induced neuroinflammation through regulation of microglia polarization through an mTOR-autophagy-dependent pathway. Int Immunopharmacol. 2020;81:106287. doi: 10.1016/j.intimp.2020.106287. [DOI] [PubMed] [Google Scholar]
- 27.Shao QH, Zhang XL, Chen Y, Zhu CG, Shi JG, Yuan YH, Chen NH. Corrigendum to “Anti-neuroinflammatory effects of 20C from Gastrodia elata via regulating autophagy in LPS-activated BV-2 cells through MAPKs and TLR4/Akt/mTOR signaling pathways” [Molecular Immunology 99 (2018) 115-123] . Mol Immunol. 2018;101:640. doi: 10.1016/j.molimm.2018.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Zampetaki A, Albrecht A, Steinhofel K. Long non-coding RNA structure and function: is there a link? Front Physiol. 2018;9:1201. doi: 10.3389/fphys.2018.01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandes JCR, Acuna SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long non-coding RNAs in the regulation of gene expression: physiology and disease. Noncoding RNA. 2019;5:17. doi: 10.3390/ncrna5010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riva P, Ratti A, Venturin M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. 2016;13:1219–1231. doi: 10.2174/1567205013666160622112234. [DOI] [PubMed] [Google Scholar]
- 31.Luo Q, Chen Y. Long noncoding RNAs and Alzheimer’s disease. Clin Interv Aging. 2016;11:867–872. doi: 10.2147/CIA.S107037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T, Zhang J, Chai Z, Wang G, Cui N, Zhou B. Ginkgo biloba extract EGb 761-induced upregulation of LincRNA-p21 inhibits colorectal cancer metastasis by associating with EZH2. Oncotarget. 2017;8:91614–91627. doi: 10.18632/oncotarget.21345. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Jin S, Yang X, Li J, Yang W, Ma H, Zhang Z. p53-targeted lincRNA-p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol Cancer. 2019;18:38. doi: 10.1186/s12943-019-0993-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.You L, Wang Z, Li H, Shou J, Jing Z, Xie J, Sui X, Pan H, Han W. The role of STAT3 in autophagy. Autophagy. 2015;11:729–739. doi: 10.1080/15548627.2015.1017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonchere B, Belanger A, Guette C, Barre B, Coqueret O. STAT3 as a new autophagy regulator. JAKSTAT. 2013;2:e24353. doi: 10.4161/jkst.24353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Y, Liu Y, Sun T, Yang W. LincRNA-p21 knockdown enhances radiosensitivity of hypoxic tumor cells by reducing autophagy through HIF-1/Akt/mTOR/P70S6K pathway. Exp Cell Res. 2017;358:188–198. doi: 10.1016/j.yexcr.2017.06.016. [DOI] [PubMed] [Google Scholar]