

Abstract
Renal clearance of many drugs is mediated by renal organic anion transporters OAT1/3 and inhibition of these transporters may lead to drug‐drug interactions (DDIs). Pyridoxic acid (PDA) and homovanillic acid (HVA) were indicated as potential biomarkers of OAT1/3. The objective of this study was to develop a population pharmacokinetic model for PDA and HVA to support biomarker qualification. Simultaneous fitting of biomarker plasma and urine data in the presence and absence of potent OAT1/3 inhibitor (probenecid, 500 mg every 6 h) was performed. The impact of study design (multiple vs. single dose of OAT1/3 inhibitor) and ability to detect interactions in the presence of weak/moderate OAT1/3 inhibitors was investigated, together with corresponding power calculations. The population models developed successfully described biomarker baseline and PDA/HVA OAT1/3‐mediated interaction data. No prominent effect of circadian rhythm on PDA and HVA individual baseline levels was evident. Renal elimination contributed greater than 80% to total clearance of both endogenous biomarkers investigated. Estimated probenecid unbound in vivo OAT inhibitory constant was up to 6.4‐fold lower than in vitro values obtained with PDA as a probe. The PDA model was successfully verified against independent literature reported datasets. No significant difference in power of DDI detection was found between multiple and single dose study design when using the same total daily dose of 2000 mg probenecid. Model‐based simulations and power calculations confirmed sensitivity and robustness of plasma PDA data to identify weak, moderate, and strong OAT1/3 inhibitors in an adequately powered clinical study to support optimal design of prospective clinical OAT1/3 interaction studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Pyridoxic acid (PDA) and homovanillic acid (HVA) have been identified as potential biomarkers for assessing OAT1/3‐mediated drug‐drug interactions (DDIs), based on sensitivity of their plasma concentrations following administration of potent OAT1/3 inhibitor probenecid.
WHAT QUESTION DID THIS STUDY ADDRESS?
A population pharmacokinetic model was developed to support qualification of PDA and HVA as endogenous biomarkers of OAT1/3. Comparison of different study designs was conducted to inform optimal design of prospective interaction studies.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This is the first study to estimate the synthesis and elimination of endogenous OAT1/3 biomarkers PDA and HVA using a modeling approach. Power calculations were performed for weak to moderate OAT1/3 inhibitors to guide optimal clinical DDI study design.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Modeling and simulation results obtained in this analysis support the utility of PDA as a selective endogenous biomarker for investigating weak to strong OAT1/3‐mediated DDIs.
INTRODUCTION
Uptake via organic anion transporters 1 and 3 (OAT1 and OAT3) expressed on the basolateral side 1 , 2 of the renal proximal tubule is considered to be a rate limiting step for renal disposition of many drugs (e.g., adefovir [OAT1], 3 , 4 benzylpenicillin [OAT3], 5 and furosemide [OAT1 and OAT3 6 ]). Inhibition of these transporters leads to drug‐drug interactions (DDIs) and reduced renal clearance. 4 , 7 Transporter‐mediated DDI risks are evaluated using in vitro‐in vivo extrapolation methods based on in vitro inhibition data, an approach supported by the regulatory authorities. 8 Although these methods are well‐established for prediction of metabolic DDIs, 9 there are challenges associated with prediction of transporter‐mediated DDIs. 10 , 11 , 12 , 13 Alternatively, over the past few years, interest has been to identify, characterize, and validate endogenous biomarkers as selective substrates of transporters in early phase clinical studies. 3 , 7 , 14 , 15 , 16 , 17 , 18 Such biomarkers could serve as early indicators of potential transporter‐mediated DDIs and inform the need for subsequent studies with clinical transporter probes. A number of factors, such as selectivity, sensitivity, and kinetic determinant of exposure, need to be verified before using these endogenous compounds as biomarkers of transporter function in vivo. 14
Modeling and simulation has increasingly been used as a tool to gain mechanistic insight into disposition of transporter biomarkers. 3 , 14 , 19 , 20 , 21 , 22 , 23 , 24 These techniques are helpful in understanding the formation, disposition, and elimination process of endogenous biomarkers to facilitate their qualification and inform optimal design of subsequent interaction studies. Most of the work so far has been reported for endogenous biomarkers of hepatic transporters (organic anion polypeptides OATP1B1 and OATP1B3), specifically coproporphyrin I (CPI). 3 , 19 , 20 , 22 , 24 In contrast, a very limited number of studies in the literature have investigated endogenous biomarkers of renal transporters, specifically OAT1/3 transporters. 25 , 26 , 27 , 28 Shen et al. 27 identified two organic anionic compounds, pyridoxic acid (PDA) and homovanillic acid (HVA), as promising biomarkers of OAT1/3 in cynomolgus monkeys and these were later evaluated in humans. 28 The authors reported that both PDA and HVA plasma concentrations in humans were sensitive to the effect of potent OAT1/3 inhibitor probenecid administered as a single dose (SD) of 1000 mg, and could be used for evaluation of OAT1/3 inhibition in early drug development. More recently, Willemin et al. (accepted for publication) investigated four endogenous compounds (PDA, HVA, taurine, and glycochenodeoxycholate sulfate [GCDCA‐S]) as potential biomarkers of OAT1/3 activity in humans. In contrast to Shen et al., 28 reported that probenecid was administered as multiple doses (MDs) of 500 mg 4 times in a day. Willemin et al. (accepted for publication) reported that PDA and HVA were the most sensitive biomarkers based on a significant increase in their area under the plasma concentration‐time curve from 0 to 24 h (AUC0‐24 h) observed following probenecid administration (fold change of 3.9 [PDA], 2.2 [HVA], 1.1 [taurine], and 1.9 [GCDCA‐S]). However, both PDA and HVA were indicated to be less selective than GCDCA‐S considering their overlapping substrate specificity for OAT1 and OAT3. Between subject variation in plasma concentrations both at baseline (without probenecid) and in the interaction phase (with probenecid) was lower in case of PDA and HVA compared with the other two endogenous compounds. Based on these findings, PDA and HVA have been selected for further evaluation as biomarkers of OAT1/3 activity in this study. Individual plasma and urine PDA and HVA data, together with probenecid plasma concentration data used in the modeling work, were from the study by Willemin et al. (in submission).
For verification of the utility of PDA and HVA as endogenous biomarkers of OAT1/3‐mediated DDIs, this study aimed to: (1) characterize the synthesis and elimination of these biomarkers in humans using population pharmacokinetic (PK) modeling of reported plasma and urine data both in the presence and absence of potent OAT1/3 inhibitor probenecid; (2) use clinical data to estimate in vivo OAT1/3 inhibitory constant (Ki) values of probenecid based on PDA and HVA as probes; (3) compare estimated probenecid in vivo Ki values with those obtained in vitro; (4) perform simulations to determine sensitivity of these endogenous biomarkers to identify DDI risk with moderate and weak OAT1/3 inhibitors; and (5) perform power calculations for a range of inhibitors administered either as SD or MDs to inform optimal design of prospective clinical studies with PDA and HVA as plasma OAT1/3 endogenous biomarkers.
METHODS
Clinical data
Individual plasma and urine data of PDA and HVA from six healthy subjects were obtained from the study reported in Willemin et al. (in submission). Details of the clinical study is provided in Supplementary Material. In short, this was a 2 phase, cross‐over study with a 21 day washout period between the 2 phases. A single dose of Janssen compound was administered to the healthy volunteers in phase I. In phase II, 2 doses of 500 mg probenecid with 5 h apart were given first on day 21 (relative to the phase I dosing) before MDs of 500 mg every 6 h for 7 days starting from day 22. In phase II, a single dose of Janssen compound was co‐administered on day 22. Plasma and urine sampling was performed at predefined timepoints over the 168 h period following administration of Janssen compound in both phases. The total number of plasma samples were 36, and 132 (66 on each occasion) for probenecid, and PDA/HVA, respectively, whereas the total number of urine samples was 108 (54 for each phase) for PDA/HVA. Plasma concentrations of PDA/HVA in the presence/absence of probenecid were used to calculate the AUC0‐tlast, where tlast is 168 h, using linear trapezoidal built‐in function in R Software. 29 Total fraction eliminated by renal transporters (fT) was calculated for PDA and HVA using Equation 1, as defined previously. 3
| (1) |
where AUC0‐tlast (control) and AUC0‐tlast (interaction) represent biomarker AUC0‐tlast in the control phase and in the presence of probenecid, respectively.
Structural and statistic models
Nonlinear mixed‐effects modeling software (NONMEM) with first order conditional estimation with interaction (FOCE‐I) method was used for modeling purposes. 30 Model performance evaluation, graphical, and statistical analyses were conducted in R. 29 An exponential model was used to describe the random effect parameters (interindividual variability [IIV], interoccasion variability [IOV]) with an assumption that these are normally distributed with mean value zero and the variance estimated during model fitting. Combined proportional and additive residual error model was used for the residuals. First, a population PK model was developed for probenecid plasma data (Supplementary Material). Individually estimated probenecid parameters were then fixed to the empirical Bayes estimates during PDA modeling in the second stage. Simultaneous fitting was performed for PDA plasma and urine data in the presence and absence of probenecid. A similar procedure was followed for HVA plasma and urine data.
PDA population PK model
Model structure describing the PDA plasma concentration was adopted from Barnett et al. 3 (Figure 1). It is a turnover model that includes synthesis rate (k syn) of PDA and its elimination by both renal and nonrenal routes. Inhibitory effect of probenecid on active secretory clearance via OAT1/3 was incorporated in the model as an effect on total renal clearance (CLr), assuming no effect of probenecid on glomerular filtration. In addition, probenecid was assumed to have no effect on k syn and nonrenal clearance (CLnr) of biomarkers. Probenecid showed negligible inhibition of MRP4 (11%) in vitro (Willemin et al. accepted for publication) and therefore the observed effect on PDA CLr was attributed to OAT1/3 inhibition. Equations 2 and 3 describe concentration in plasma (CPDA) and amount in urine (UPDA) compartments, respectively.
| (2) |
| (3) |
where C PDA is PDA plasma concentration, U PDA is PDA amount in urine, t is time, k syn is the zero‐order PDA synthesis rate, Ki is probenecid OAT1/3 inhibition constant using PDA data as a transporter probe, V PDA is the volume of distribution of PDA, and C probenecid is probenecid plasma concentration (Supplementary Material). Formal structural identifiability of the model was performed in DAISY 31 using PDA plasma and urine data in the presence and absence of probenecid. A similar procedure was done in the case of HVA.
FIGURE 1.
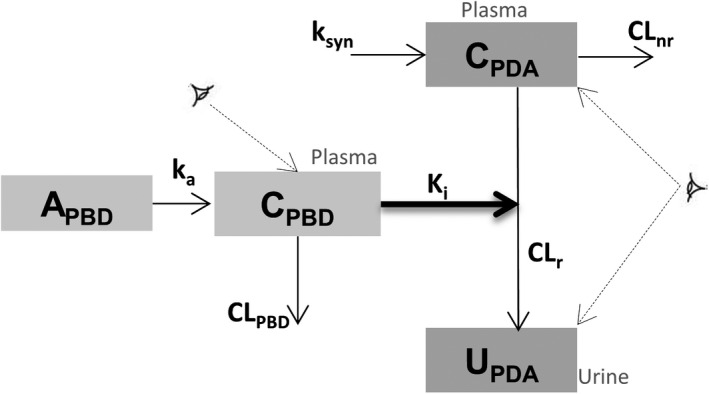
Schematic diagram of population pharmacokinetic (PK) models for probenecid (PBD) and pyridoxic acid (PDA) including interaction between PBD and PDA. A, amount; C, plasma concentration; CLnr, PDA nonrenal clearance; CLr, PDA renal clearance; k
a, absorption rate constant; K
i, probenecid OAT1/3 inhibitory constant; k
syn, PDA synthesis rate; U, urine;  , observed compartment)
, observed compartment)
Comparison of probenecid OAT K i with in vitro inhibition data
The estimated probenecid in vivo OAT Ki value with PDA as substrate was compared with probenecid in vitro half maximal inhibitory concentration (IC50) value for OAT1 and OAT3 obtained with PDA as substrate in the study (details of in vitro studies are in Supplementary Material). In addition, literature reported probenecid in vitro inhibition data were collated using a range of OAT1 and OAT3 probes (Table S1) and compared with estimated probenecid in vivo OAT1/3 K i value based on either PDA or HVA data.
PDA model verification
An independent clinical dataset with reported PDA plasma concentrations 28 was used for model verification. This verification dataset was an oral SD, randomized, 3‐phase, crossover study with 14 healthy volunteers receiving 1000 mg probenecid in the first phase, 40 mg furosemide (FSM; OAT1/3 substrate) in the second phase, and both probenecid and FSM together in the third phase. Mean ± SD PDA data were digitized using WebPlotDigitizer (https://automeris.io/WebPlotDigitizer/). Model simulations were performed for both cases (interaction and baseline) using the study design reported.
Power calculations
To assess the utility of PDA and HVA to identify weak and moderate OAT1/3 inhibitors, the probenecid and biomarker models were used to simulate biomarker plasma concentrations. Adjusted C probenecid/Ki ratios were applied in Equations 2 and 3; these ratios were modified to reflect different inhibitor potency or exposure relative to probenecid (ratio varied between 0.025 and 5, ratio of 1 representing equivalent to probenecid). Simulations were performed using estimated parameters from probenecid and biomarker model and AUC values were obtained using the trapezoidal function in R. 29 Simulations were based on the same study design as reported in Willemin et al. (accepted for publication), with a control phase (without probenecid) and interaction phase (with probenecid as 500 mg every 6 h). Power curves were generated for each modified I/Ki ratio by performing two‐samples paired t‐test on the log transformed AUC values (presence/absence of probenecid) following simulations at predefined sample sizes (5–30). At each sample size, 1000 simulations and t‐tests were performed and the power was calculated as the proportion of the simulations during which the null hypothesis was rejected at the significance level of 0.05 and 0.01.
Potential effect of transporter inhibitor on synthesis rate k syn
No information was available in the literature on the potential effect of probenecid on PDA k syn. Therefore, simulations were performed to assess the consequences of hypothetical scenarios and potential effect of transporter inhibitor on PDA k syn using Equation 4, as investigated previously on CPI. 3 PDA plasma concentrations were simulated using hypothetical relative ratios (C probenecid/K i)’ of 0, 0.1, and 10 of the probenecid effect on PDA CLr (C probenecid/K i), where the ratio of 0 corresponds to the scenario of no effect on PDA k syn and with inhibitory effect only on the OAT1/3 transporters. The values of 0.1 and 10 for (C probenecid/Ki)’ correspond to 10‐fold lower and higher, respectively inhibitory effect on k syn than the effect on OAT1/3‐mediated CLr of the biomarker.
| (4) |
RESULTS
Individual biomarker plasma data
Low IIV was observed in baseline levels of PDA (CV – 26.9%) and HVA (CV – 23.2%). In addition, low intra‐individual variability was evident, with no apparent prominent effect of circadian rhythm on concentrations of these biomarkers (Figure S1A and B). PDA was more sensitive to the effect of potent OAT1/3 inhibitor probenecid than HVA, as indicated by individual plasma AUC0‐tlast ratios ranging from 2.5 to 5.1 compared with a range of 2.1 to 3.7 observed for HVA. There was no correlation between the magnitude of PDA and HVA interaction with probenecid in the same individual (assessed by plasma AUC0‐tlast ratios; Figure S1C). Estimated individual fT values were higher for PDA (0.59–0.81) than HVA (0.53–0.73; Figure S1D). This estimate was interpreted to be OAT1/3‐driven, assuming that the uptake in the proximal renal tubule is the rate determining step for PDA and HVA renal disposition. 28
Structural PK models
The structural models used to describe plasma data of probenecid and both plasma and urine data of PDA/HVA are shown in Figure 1. The one‐compartment model best described the probenecid data, whereas a turnover model, analogous to model developed for CPI, 3 was used to describe the PDA and HVA data. Model parameters were successfully estimated for probenecid, PDA, and HVA and are reported in Table 1. Model performance and evaluation were examined by visual predictive check (VPC) plots and goodness of fit plots, as shown in Figure 2 and in Figures S2–S11.
TABLE 1.
Parameter estimates of the population PK model for probenecid, PDA, and HVA
| Drug/biomarker | Parameter | Estimates (SE%) | ||
|---|---|---|---|---|
| Population | IIV | IOV | ||
| Probenecid | CL, L/h | 0.82 (9) | 23 (23) | |
| V, L | 15 (22) | 40 (29) | ||
| ka/h | 0.74 (45) | |||
| Ki, µM (PDA data) | 54.5 (14) | |||
| Ki, µM (HVA data) | 137 (24) | |||
| σ prop, (%) | 18.8 (30) | |||
| σ add, µM | 0.001 (Fixed) | |||
| PDA | CLr, L/h | 15.2 (15) | 27 (15) | |
| CLnr, L/h | 3.23 (41) | |||
| V, L | 9.34 (56) | |||
| k syn, µg/h | 58.6 (26) | 32 (34) | 22 (57) | |
| σ prop (%) – plasma | 12.7 (7) | |||
| σ add, ng/ml – plasma | 0.213 (18) | |||
| σ prop (%) – urine | 32.1 (20) | |||
| σ add, µg – urine | 58 (78) | |||
| HVA | CLr, L/h | 20.4 (14) | 23 (26) | 5 (48) |
| CLnr, L/h | 1.29 (69) | |||
| V, L | 105 (35) | |||
| k syn, µg/h | 212 (16) | 27 (19) | ||
| σ prop (%) – plasma | 17.3 (8) | |||
| σ add, ng/ml – plasma | 0.001 Fixed | |||
| σ prop (%) – urine | 26.9 (14) | |||
| σ add, µg – urine | 373 (20) | |||
Values represents the total Ki estimated by the model. Correction for probenecid unbound fraction in plasma of 0.062 reported (28), resulted in probenecid in vivo OAT1/3 Ki values of 3.4 and 8.5 µM based on PDA and HVA data, respectively.
Abbreviations: σ add, additive residual error; σ prop, proportional residual error; CL, clearance; CLnr, nonrenal clearance; CLr, renal clearance; HVA, homovanillic acid; IIV, interindividual variability; IOV, interoccasion variability; k a, absorption rate constant; k i, inhibitory constant; k syn, synthesis rate; PDA, pyridoxic acid; PK, pharmacokinetic; V, volume.
FIGURE 2.
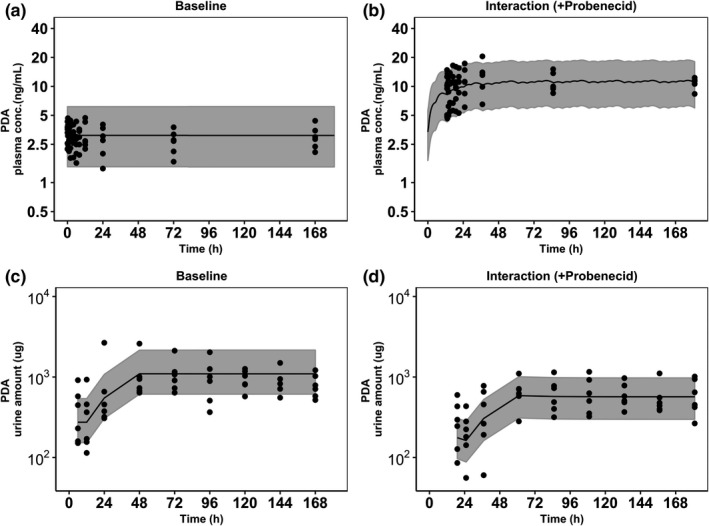
Visual predictive checks of the developed population pharmacokinetic pyridoxic acid (PDA) model superimposed with the observed data. Panels represent PDA plasma baseline (a), plasma data in the presence of probenecid (b), urine PDA baseline (c), and urine PDA data in the presence of probenecid (d). Shaded areas represent 90% prediction interval, black dots (●) represent observed data, and black lines (–) are the median lines
Probenecid population PK model
Population parameters of probenecid were successfully estimated with moderate standard errors (Table 1). IIV estimated for probenecid clearance (CL) and volume of distribution (V) was 23% and 40%, respectively. Model adequately described the observed data median trend and variability (Figures S2–S4).
Population PK model for OAT1/3 biomarkers
A model with inclusion of CLr and CLnr described successfully the observed data for PDA, with reasonable standard errors (15%–41%) for population parameter estimates. A similar performance was observed for HVA with slightly higher standard errors (14%–69%). Competitive OAT1/3 inhibition by probenecid was implemented on CLr, assuming no effect on k syn and CLnr. Parameters of this model were found to be globally identifiable following structural identifiability analysis. The results showed that renal elimination is the main route of elimination for both PDA (82%) and HVA (94%) and CLr values were comparable to the reported literature values. 26 IIV was estimated for PDA CLr (27%) and k syn (32%), with moderate standard errors of 15% and 34%, respectively. IOV between the two interaction phases was estimated only for k syn (22%). Similarly, parameter estimates were obtained for HVA and are shown in Table 1. Simultaneous fitting of all available PDA or HVA data ± probenecid allowed estimation of the in vivo probenecid OAT Ki value. The Ki corrected for probenecid fraction unbound in plasma of 0.062 28 was 3.4 µM based on PDA data as a transporter probe. This in vivo estimate was lower than experimentally obtained in vitro IC50 value for this inhibitor against OAT1 (21.7 µM) and OAT3 (4.2 µM) using PDA as substrate (Supplementary Material). This trend was consistent with literature collation of in vitro probenecid Ki using different OAT1/3 substrates (range of 1.3–29.8 µM; see Table S1). The probenecid in vivo Ki estimated from HVA data was higher (8.5 µM), but still within the range of reported in vitro probenecid inhibition data (Table S1). The model successfully captured the observed PDA plasma and urine data, including the variability both at baseline and during interaction phases, as shown in VPC plots in Figure 2. VPCs plots for HVA are reported in Figure S8.
PDA model verification
Considering higher sensitivity of PDA to OAT1/3 interaction and availability of additional clinical data, 28 the ability of the PDA model to predict the PDA plasma concentration‐time data from an independent study 28 following probenecid administration as a single dose was assessed. The model adequately predicted the magnitude of interaction and successfully captured the variation during both baseline and interaction phases (Figure 3). Simulated PDA plasma AUC0‐tlast ratios between control and interaction phases were in agreement with the observed individual AUC0‐tlast ratios (mean AUC0‐tlast ratio of 2.29 and 1.95 in simulated and observed data, respectively).
FIGURE 3.
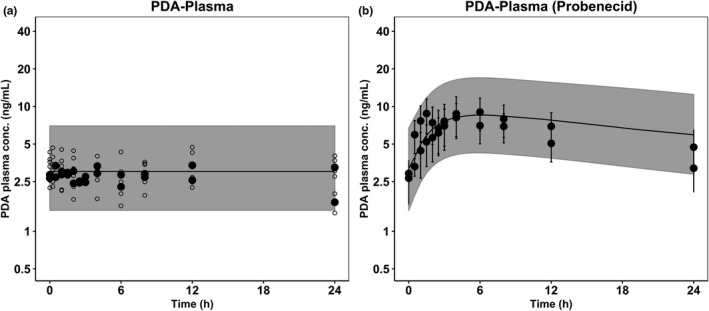
Verification of pyridoxic acid (PDA) model against independent clinical data. Simulated PDA plasma concentrations in control (a) and interaction phase (b). Shaded area represents 90% prediction interval, black circles (●) are the observed PDA literature data, 28 and open circles (o) are observed baseline data from the current study
Power calculations and study design
The PDA model performance and its verification using independent dataset confirmed validity of PDA plasma data as an endogenous biomarker for the identification of a potent OAT1/3 inhibitor, such as probenecid. Currently, there is a lack of clinical data on the effect of moderate or weak OAT1/3 inhibitors on PDA plasma concentrations. To address this gap, PDA plasma concentration data were simulated with varying inhibitor potency or exposure relative to probenecid (expressed as C probenecid/Ki ratios of 0.025–5). Furthermore, impact of different dosage regimens of inhibitors and duration of studies was evaluated (SD of 1000 mg probenecid, SD of 2000 mg, and MDs of 500 mg probenecid every 6 h). Power calculation analysis showed that reduced sample sizes are required for the MD study design (with sampling time over 168 h) compared to SD 1000 mg study design (with sampling time over 24 h) in order to detect significant interactions in case of weak‐to‐moderate inhibitors (I/Ki from 0.025 to 0.25 relative to probenecid; Figure 4a vs. b). Notably, the difference in the required sample size between SD and MD study designs was negligible when the same total daily dose of probenecid (2000 mg) was used (Figure 4a vs. c). For example, for detection of DDIs by weak OAT1/3 inhibitor (I/Ki =0.1 relative to probenecid), sample sizes of 9, 25, and 10 would be required for MDs (500 mg every 6 h), SD (1000 mg), and SD with the same total daily dose as in MDs (2000 mg), respectively. Power calculations at significance level of 0.01 are given in Figure S12. Similar calculations were performed for HVA and results are shown in Figures S13 and S14. No difference was found in power calculations between PDA and HVA to detect DDIs by strong OAT1/3 inhibitors, whereas in case of weak and moderate inhibitors, more subjects are needed to detect DDIs using HVA as an endogenous biomarker (Figure 4 vs. Figure S13). For example, in an SD study design of moderate OAT1/3 inhibitor (I/Ki = 0.25 relative to probenecid), 9 and 17 subjects were required to detect significant DDIs using PDA and HVA as endogenous biomarkers, respectively.
FIGURE 4.
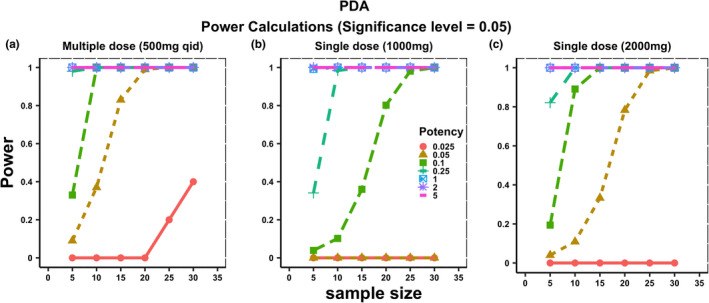
Pyridoxic acid (PDA) power curves at significance level (α = 0.05) representing relationship between sample size and power of detecting significant OAT1/3‐mediated drug‐drug interactions for inhibitors ranging from weak to strong (I/Ki of 0.025–5 relative to probenecid) and different study designs ((a) = 500 mg every 6 h q.i.d., (b) = 1000 mg single dose, and (c) = 2000 mg single dose)
DISCUSSION
Endogenous biomarkers have been proposed as a promising tool to evaluate transporter function in vivo and assess possible risk of transporter‐mediated interactions in early phases of drug development. However, mechanistic understanding of the disposition of biomarkers and factors contributing to IIV or intra‐individual variability in their baseline is still lacking for many. This is of particular relevance for endogenous biomarkers for renal transporters, in contrast to more advanced knowledge and evaluation of endogenous biomarkers for hepatic transporters. 3 , 15 , 20 , 24 In this study, a population PK model has been developed for PDA and HVA, two recently identified promising plasma‐based biomarkers of renal transporters OAT1/3.
Comparison of PDA and HVA
Between the two evaluated biomarkers, PDA showed more stable baseline plasma concentrations than HVA (Figure S1). PDA was also more sensitive to the effect of potent OAT1/3 inhibitor probenecid, as indicated by generally higher individual plasma AUC0‐tlast ratios than HVA (Figure S1c). No direct correlation could be established between the PDA and HVA magnitude of interaction with probenecid in the current dataset (Figure S1C), which may be attributed to differential OAT1/3 contribution to the renal disposition and relatively small sample size to establish any robust trends. Estimated unbound probenecid in vivo OAT1/3 Ki was lower based on PDA data (3.4 µM) than the value obtained using HVA data (8.5 µM). Power calculations indicated no difference between PDA and HVA with respect to required sample sizes to detect DDI with strong OAT1/3 inhibitors such as probenecid (Figure 4, Figure S13). However, in the case of weak to moderate inhibitors, a higher number of subjects was needed to detect DDIs when using HVA as a plasma OAT1/3 biomarker than PDA (Figure 4, Figure S13). Based on all the above, further model evaluation focused on PDA as a more relevant plasma endogenous biomarker of OAT1/3 activity.
PDA model development and verification
The PDA model was developed using both plasma and urine data (with and without probenecid); availability of data in both phases was crucial for the identifiability of model parameters. No direct relationship was found between individual’s baseline PDA plasma concentrations at time zero and the magnitude of probenecid interaction (data not shown). Therefore, based on the data available so far, it was not possible to establish whether individuals with lower initial baseline PDA concentrations would be more susceptible to OAT‐mediated interaction. The analysis has shown that renal excretion was the major elimination route for PDA, contributing 82% to its total CL. As probenecid is a potent inhibitor of both OAT1 and OAT3 32 and with no information on their relative contribution to PDA uptake, sensitivity of PDA was considered to be the net result of combined inhibition of OAT1 and OAT3. Therefore, use of plasma PDA data may result in underestimation of clinical DDI, if there is a specific inhibition of either of these transporters. Estimated unbound in vivo probenecid Ki value using PDA as OAT1/3 substrate was up to 6.4‐fold lower than in vitro OAT1/3 probenecid IC50 with PDA as a substrate, and also on the lower end of the literature reported in vitro probenecid inhibitory data for a range of OAT1 (3.9–28 µM) and OAT3 (1.3–29.8 µM) substrates (Table S1).
Low IIV and intra‐individual variability in the PDA baseline concentrations was important for obtaining reliable parameter estimates and to identify DDI risk with moderate and weak inhibitors. In the case of PDA, low intra‐individual variability over 168 h (Figure S1A), indicates negligible effect of circadian rhythm on its plasma concentrations. It is important to note that the clinical study used was designed in a way that there was no predose PDA concentration measurement; instead, a phase where Janssen compound was administered on its own was used as a baseline, assuming no effect on OAT1/3. This assumption was reasonable when PDA concentrations were compared with true predose mean PDA measurements reported previously. 28
PDA synthesis and diseased population
PDA is the end product of vitamin B6 33 and probenecid was considered to have no effect on its formation (defined in the model as k syn). However, the PDA k syn could be affected by external factors, such as disease or diet/vitamin intake, which may contribute to the variability in the biomarker baseline across different populations. Consequences of any potential effect of transporter inhibitor on the synthesis of PDA (in addition to its effect on OAT1/3) have been investigated using the PDA model developed (Figure 5). These simulations highlight potential bias in the interpretation of biomarker‐transporter interaction data in cases where biomarker synthesis is also affected by a transporter inhibitor. The extent of this effect is also linked to the study design, as the recovery of simulated PDA concentrations was rapid in case of SD of an OAT inhibitor in contrast to a MD study design. Certain diseases, for example, chronic kidney disease, are associated with decreased OAT1/3 activity 34 as a result of either the inhibitory effect of uremic solutes on transporters or reduced albumin‐facilitated transport. 35 This adds another level of complexity when interpreting PDA baseline and interaction data in such patient populations.
FIGURE 5.
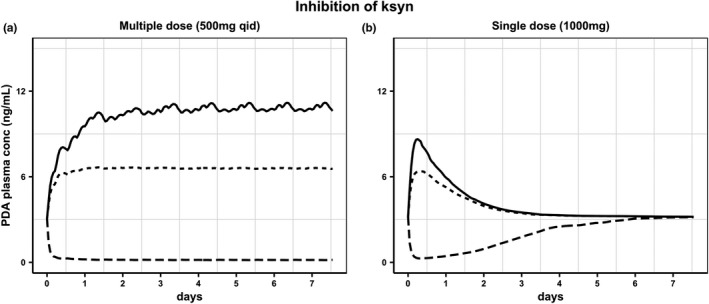
Simulated pyridoxic acid (PDA) plasma concentrations assuming the effect of OAT1/3 inhibitor also on biomarker synthesis (ksyn). Hypothetical ratio of 0 (continuous line) corresponds to no effect on PDAk syn. The values of 0.1 (dotted line), and 10 (dashed line) represent 10‐fold lower and higher, respectively, inhibitory effect on PDAk syn than the effect on OAT1/3‐mediated CLr of the biomarker. Scenarios simulated following (a) multiple doses of probenecid (500 mg every 6 h) or (b) single dose of probenecid (1000 mg)
Power calculations and interaction study design
Power calculations confirmed adequacy of previous clinical studies (MD study design with 6 subjects, as reported in Willemin et al. (accepted for publication) and SD with 14 subjects 28 ) and the suitability of PDA as OAT1/3 endogenous biomarker for the evaluation of strong OAT1/3 inhibitors, such as probenecid. To support biomarker qualification, it is essential to investigate the ability of PDA in detecting also weak and moderate inhibitors of OAT1/3. To address this gap, we have performed power calculations for different hypothetical inhibitors and under different study design/duration (MD 500 mg q.i.d., SD 1000 mg, and SD 2000 mg, same daily dose as in MD; Figure 4), assuming that the hypothetical inhibitors follow the same PK as probenecid (assuming no nonlinearity in protein binding at higher concentrations). Power calculations of weak and moderate OAT1/3 inhibitors showed the advantage of an MD study design (smaller sample size required) over SD (1000 mg) for OAT DDI detection (Figure 4a vs. b). The difference in required sample size between study designs was negligible for the same total daily dose (2000 mg) of an OAT1/3 inhibitor (Figure 4a vs. c). Therefore, an SD with maximum possible dose of an inhibitor may be preferable, as it requires a smaller number of subjects with short study duration (cost effective). In addition, this design is more in line with the first‐in‐human studies, which generally enroll a small number of subjects. In cases when an MD is necessary, a number of dosing frequencies (once, twice, or 4 times daily) were found to reach the same steady‐state concentrations (Figure S14), suggesting no difference in the required sample sizes for detection of OAT1/3‐mediated DDIs across different dosing frequencies.
In conclusion, this study confirms the sensitivity of plasma PDA data to identify OAT1/3 inhibitors with different potency (weak to strong). Critical evaluation based on multiple criteria showed that PDA is a more robust OAT1/3 biomarker than HVA. Power calculations and evaluation of different study designs performed in this work provide an invaluable tool to support the design of prospective OAT1/3 DDI studies in early phase clinical trials.
CONFLICT OF INTEREST
F.J. and J.S. are full‐time employees of Janssen Pharmaceutical. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
A.A., K.O., A.K., F.J., J.S., A.R.‐H., and A.G. wrote the manuscript. A.G. and J.S. designed the research. A.A. and K.O. performed the research. A.A. analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Thomas K. van der Made (University of Manchester) for collation of literature reported probenecid in vitro inhibition data with multiple OAT1 and OAT3 substrates. We acknowledge Marie‐Emilie Willemin (Janssen) for providing details of in vitro probenecid‐PDA inhibition experiments and individual clinical data for modeling purposes.
Funding information
This study was funded by the Centre for Applied Pharmacokinetic Research Centre at the University of Manchester.
REFERENCES
- 1. Zamek‐Gliszczynski MJ, Taub ME, Chothe PP, et al. Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin Pharmacol Ther. 2018;104:890‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giacomini KM, Shiew‐Mei H, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnett S, Ogungbenro K, Ménochet K, et al. Gaining mechanistic insight into Coproporphyrin I as endogenous biomarker for OATP1B‐mediated drug‐drug interactions using population pharmacokinetic modeling and simulation. Clin Pharmacol Ther. 2018;104:564‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maeda K, tian Y, Fujita T, et al. Inhibitory effects of p‐aminohippurate and probenecid on the renal clearance of adefovir and benzylpenicillin as probe drugs for organic anion transporter (OAT) 1 and OAT3 in humans. Eur J Pharm Sci. 2014;59:94‐103. [DOI] [PubMed] [Google Scholar]
- 5. Mathialagan S, Piotrowski MA, Tess DA, Feng B, Litchfield J, Varma MV. Quantitative prediction of human renal clearance and drug‐drug interactions of organic anion transporter substrates using in vitro transport data: a relative activity factor approach. Drug Metab Dispos. 2017;45:409‐417. [DOI] [PubMed] [Google Scholar]
- 6. Hasannejad H, Takeda M, Taki K, et al. Interactions of human organic anion transporters with diuretics. J Pharmacol Exp Ther. 2004;308:1021‐1029. [DOI] [PubMed] [Google Scholar]
- 7. Chu X, Chan GH, Evers R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter‐mediated drug‐drug interactions. J Pharm Sci. 2017;106:2357‐2367. [DOI] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration . Clinical Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Drug Interactions. Guidance for Industry. 2020. [Google Scholar]
- 9. Yoshida K, Zhao P, Zhang L, et al. In vitro–in vivo extrapolation of metabolism‐ and transporter‐mediated drug‐drug interactions—overview of basic prediction methods. J Pharm Sci. 2017;106:2209‐2213. [DOI] [PubMed] [Google Scholar]
- 10. Guo Y, Chu X, Parrott NJ, et al. Advancing predictions of tissue and intracellular drug concentrations using in vitro, imaging and physiologically based pharmacokinetic modeling approaches. Clin Pharmacol Ther. 2018;104:865‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zamek‐Gliszczynski MJ, Lee CA, Poirier A, et al. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport‐mediated PK and DDIs in humans. Clin Pharmacol Ther. 2013;94:64‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Snoeys J, Beumont M, Monshouwer M, Ouwerkerk‐Mahadevan S. Mechanistic understanding of the nonlinear pharmacokinetics and intersubject variability of simeprevir: a PBPK‐guided drug development approach. Clin Pharmacol Ther. 2016;99:224‐234. [DOI] [PubMed] [Google Scholar]
- 13. Gertz M, Cartwright CM, Hobbs MJ, et al. Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug‐drug interaction potential. Pharm Res. 2013;30:761‐780. [DOI] [PubMed] [Google Scholar]
- 14. Chu X, Liao M, Shen H, et al. Clinical probes and endogenous biomarkers as substrates for transporter drug‐drug interaction evaluation: perspectives from the International Transporter Consortium. Clin Pharmacol Ther. 2018;104:836‐864. [DOI] [PubMed] [Google Scholar]
- 15. Barnett S, Ogungbenro K, Ménochet K, Shen H, Humphreys WG, Galetin A. Comprehensive evaluation of the utility of 20 endogenous molecules as biomarkers of OATP1B inhibition compared with rosuvastatin and coproporphyrin I. J Pharmacol Exp Ther. 2019;368:125‐135. [DOI] [PubMed] [Google Scholar]
- 16. Jones NS, Yoshida K, Salphati L, Kenny JR, Durk MR, Chinn LW. Complex DDI by fenebrutinib and the use of transporter endogenous biomarkers to elucidate the mechanism of DDI. Clin Pharmacol Ther. 2020;107:269‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kunze A, Ediage EN, Dillen L, Monshouwer M, Snoeys J. Clinical investigation of coproporphyrins as sensitive biomarkers to predict mild to strong OATP1B‐mediated drug‐drug interactions. Clin Pharmacokinet. 2018;57:1559‐1570. [DOI] [PubMed] [Google Scholar]
- 18. Lai Y, Mandlekar S, Shen H, et al. Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J Pharmacol Exp Ther. 2016;358:397‐404. [DOI] [PubMed] [Google Scholar]
- 19. Yoshikado T, Toshimoto K, Maeda K, et al. PBPK modeling of coproporphyrin I as an endogenous biomarker for drug interactions involving inhibition of hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst Pharmacol. 2018;7:739‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshida K, Guo C, Sane R. Quantitative prediction of OATP‐mediated drug‐drug interactions with model‐based analysis of endogenous biomarker kinetics. CPT Pharmacometrics Syst Pharmacol. 2018;7:517‐524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scotcher D, Arya V, Yang X, et al. Mechanistic models as framework for understanding biomarker disposition: prediction of creatinine‐drug interactions. CPT Pharmacometrics Syst Pharmacol. 2020;9:282‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Asaumi R, Toshimoto K, Tobe Y, et al. Comprehensive PBPK model of rifampicin for quantitative prediction of complex drug‐drug interactions: CYP3A/2C9 induction and OATP inhibition effects. CPT Pharmacometrics Syst Pharmacol. 2018;7:186‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takita H, Scotcher D, Chinnadurai R, Kalra PA, Galetin A. Physiologically‐based pharmacokinetic modelling of creatinine‐drug interactions in the chronic kidney disease population. CPT Pharmacometrics Syst Pharmacol. 2020;9:695‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takita H, Barnett S, Zhang Y, et al. Application of PBPK model for coproporphyrin I to evaluate the impact of SLCO1B1 genotype, ethnicity, and sex on its inter‐individual variability. CPT Pharmacometrics Syst Pharmacol. 2021;10(2):137‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imamura Y, Tsuruya Y, Damme K, et al. 6β‐hydroxycortisol is an endogenous probe for evaluation of drug‐drug interactions involving a multispecific renal organic anion transporter, OAT3/SLC22A8, in healthy subjects. Drug Metab Dispos. 2014;42:685‐694. [DOI] [PubMed] [Google Scholar]
- 26. Tsuruya Y, Kato K, Sano Y, et al. Investigation of endogenous compounds applicable to drug‐drug interaction studies involving the renal organic anion transporters, OAT1 and OAT3, in humans. Drug Metab Dispos. 2016;44:1825‐1933. [DOI] [PubMed] [Google Scholar]
- 27. Shen H, Nelson DM, Oliveira RV, et al. Discovery and validation of pyridoxic acid and homovanillic acid as novel endogenous plasma biomarkers of organic anion transporter (OAT) 1 and OAT3 in cynomolgus monkeys. Drug Metab Dispos. 2018;46:178‐188. [DOI] [PubMed] [Google Scholar]
- 28. Shen H, Holenarsipur VK, Mariappan TT, et al. Evidence for the validity of pyridoxic acid (PDA) as a plasma‐based endogenous probe for OAT1 and OAT3 function in healthy subjects. J Pharmacol Exp Ther. 2019;368:136‐145. [DOI] [PubMed] [Google Scholar]
- 29. R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. https://www.R‐project.org/ [Google Scholar]
- 30. Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User’s Guides (1989–2009). Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- 31. Bellu G, Saccomani MP, Audoly S, D’Angiò L. DAISY: a new software tool to test global identifiability of biological and physiological systems. Comput Methods Programs Biomed. 2007;88:52‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steffansen B, Nielsen CU, Brodin B. In: Sugiyama Y, Steffansen, eds. Transporters in drug development: discovery, optimization, clinical study and regulation. New York, NY: Springer; 2015. [Google Scholar]
- 33. Ink SL, Henderson LM. Vitamin B6 metabolism. Annu Rev Nutr. 1984;4:455‐470. [DOI] [PubMed] [Google Scholar]
- 34. Hsueh CH, Yoshida K, Zhao P, et al. Identification and quantitative assessment of uremic solutes as inhibitors of renal organic anion transporters, OAT1 and OAT3. Mol Pharm. 2016;13:3130‐3140. [DOI] [PubMed] [Google Scholar]
- 35. van der Made TK, Fedecostante M, Scotcher D, et al. Quantitative translation of microfluidic transporter in vitro data to in vivo reveals impaired albumin‐facilitated indoxyl sulfate secretion in chronic kidney disease. Mol Pharm. 2019;16:4551‐4562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material